
Abstract
Proteogenomic methodologies have enabled the identification of protein sequences in wild species without annotated genomes, shedding light on molecular mechanisms affected by pollution. However, proteomic resources for sentinel species are limited, and organ-level investigations are necessary to expand our understanding of their molecular biology. This study presents proteomic resources obtained from proteogenomic analyses of key organs (hepatopancreas, gills, hemolymph) from three established aquatic sentinel invertebrate species of interest in ecotoxicological/ecological research and environmental monitoring: Gammarus fossarum, Dreissena polymorpha, and Palaemon serratus. Proteogenomic analyses identified thousands of proteins for each species, with over 90% of them being annotated to putative function. Functional analysis validated the relevance of the proteomic atlases by revealing similarities in functional annotation of catalogues of proteins across analogous organs in the three species, while deep contrasts between functional profiles are delimited across different organs in the same organism. These organ-level proteomic atlases are crucial for future research on these sentinel animals, aiding in the evaluation of aquatic environmental risks and providing a valuable resource for ecotoxicological studies.
Similar content being viewed by others
Background & Summary
The preservation of natural resources has become a crucial focus as our planet’s ecosystem faces significant challenges. In particular the increase of anthropogenic pollution, including toxic substances like heavy metals, nanoparticles, microplastics, pesticides, pharmaceutical drugs and other emerging contaminants poses a major threat to both human and ecosystem health1. The impact of the widespread exposure to these toxic substances on the health of living organisms, even at low doses, is largely unknown and difficult to manage from an environmental policy perspective. Their effects could even be exacerbated with global warming of our planet. Improving our understanding of the environment is more important than ever. The ‘One-Health’ concept aims to harmonize ecology, animal health, and human health knowledge and understanding of their inter-relationships for a better long-term quality of life2. In this context, the use of aquatic sentinel species viewed as scouts of water ecosystem health is gaining momentum to monitor the environmental quality with a great depth of integration, particularly to decipher and anticipate the impacts of chemical cocktail contamination on biodiversity.
Omics technologies such as genomics, transcriptomics, proteomics, and metabolomics offer a comprehensive molecular insight into the biological impact of environmental changes and pollution3. These technologies allow us to investigate changes in the genetic makeup, gene expression, protein levels, and metabolic pathways of sentinel species. The information gathered from these studies can enhance our understanding of the effects of environmental changes, including pollutions, on the health status of sentinel species. Development of molecular markers for precise biomonitoring is also an important outcome of such large scale molecular studies. It is also crucial to aid in developing conservation and management strategies to minimize their impact3. By means of combining data from RNA sequencing and shotgun mass spectrometry on proteins, proteogenomic methodologies have made possible to quickly identify protein sequences in a large variety of species without yet sequenced and annotated genomes4. In aquatic ecotoxicology, this approach can now be considered as standard for characterizing proteomes of sentinel species, as exemplified by pioneering studies such as the reproductive proteome of Gammarus fossarum and the immune proteome of Dreissena polymorpha5,6. Proteogenomics has also been used for deciphering the effects of contaminants on the proteomes of wild organisms. For example, these approaches have been employed to elucidate the basis of insecticide tolerance in the pollen beetle Brassicogethes aeneus7, as well as the effects of carbamazepine on the proteomes of the marine mussel Mytilus galloprovincialis8. However, proteomic resources for sentinel species remain limited, appealing to complement available information on biological functions impacted by pollutants or other environmental stressors that can be obtained through molecular investigations at the organ level.
The proteomic resources presented in this report correspond to proteogenomic analyses performed on relevant organs of three well-established aquatic sentinel species: Gammarus fossarum (Crustacean, Amphipod), Dreissena polymorpha (Mollusk, Bivalve), and Palaemon serratus (Crustacean, Decapod). These species are widely distributed in aquatic ecosystems (the two former are freshwater species and the latter is a marine species) and are found in most European countries9,10, and also in North America for the invasive bivalve. They have emerged as important ecotoxicological models for assessing the impact of pollution on freshwater wildlife and ecosystems. P. serratus, G. fossarum, and D. polymorpha are phylogenetically distant and come from the largest phyla of the animal kingdom (arthropods and mollusks)11. Using a combination of species-specific transcriptomes and shotgun proteomics, the proteomes of three organs (hepatopancreas/ hepatopancreatic caeca, hemolymph, and gills) from P. serratus and G. fossarum, as well as the gill proteome of D. polymorpha, were established and characterized. Overall, 3,891 and 2,419 proteins were experimentally validated from the three organs of P. serratus and G. fossarum, respectively, with a higher number of proteins identified in gills compared to digestive organs and hemolymph. In the case of D. polymorpha, proteogenomics identified 6,026 proteins in the gills. These comprehensive organ-level proteomic atlases were annotated in terms of putative function by sequence similarity, providing valuable insights into the biological pathways associated with each targeted organ. With over 90% of the proteins confidently annotated, the functional analysis revealed that analogous organs in the three species express partly functionally similar catalogues of proteins, while deep contrasts between functional profiles can be delimited across the different organs in the same organism. These observations highlight the value of such resources to capture biological processes at the molecular level in such sentinel non-model species. These proteomic atlases will serve as crucial references for future research on these sentinel animals and will aid in the assessment of aquatic environmental risks, contribute to a better understanding of molecular diversity in organisms, and provide a valuable resource for future ecotoxicological studies.
Methods
Study design
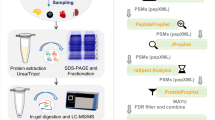
The proteomes of three organs (hepatopancreas/hepatopancreatic caeca, hemolymph, and gills) from two crustacean species (G. fossarum and P. serratus) and the gill proteome from D. polymorpha were investigated using proteogenomics with a RNA-transcriptome informed database (Fig. 1). The study involved extracting proteins from tissue samples, tryptic digestion, and analyzing the resulting peptides using liquid chromatography-tandem mass spectrometry (LC-MS/MS) in Data Dependent Acquisition (DDA) mode. The DDA strategy was preferred over Data Independent Acquisition mode in order to have the highest possible confidence in the identification of peptides. MS/MS spectra were interpreted using open-reading frame databases derived from transcriptome databases specific to each species. To gain first insight into the functional roles of each organ-oriented proteomes for each species, the experimentally validated protein sequences were annotated with gene ontology (GO) terms weighted with normalized spectral abundance factor (NSAF) and compared through clustered heatmap analysis.

Proteogenomic workflow for the characterization and functional profiling of sentinel species organ proteomes.
Construction of a paired-end RNA-Seq library and illumina sequencing
The reference transcriptomes used for G. fossarum and D. polymorpha were generated as described in Cogne et al.12 and Leprêtre et al.5, respectively. For P. serratus, RNA Seq library construction and sequencing were performed by Genotoul (INRAE, France), using the NovaSeq sequencing instrument (Illumina). The library construction was performed from RNAs coming from three distinct tissues: hepatopancreas, cephalon and muscle. The TrueSeq Stranded (Illumina) protocol was used to construct the libraries, and RNA were sequenced using the paired-end reads 2 × 150 bp method on a SP flowcell lane. The reads were then assembled using the Trinity pipeline (version 2.3.0).
Organ sampling from sentinel species for proteomic investigations
Adult specimens of P. serratus were collected by a fisherman (Prelev’Mar®), using specific traps, in the 2-nautical mile zone of Cherbourg (Normandy, France), in November 2020. The shrimps were transported to the laboratory within 3 h, in 30-liter plastic containers filled with natural seawater from the sampling site and kept oxygenated. Shrimps were kept some days into 80 L-aquariums, at density less than one specimen per liter, 33 of salinity, 18 ± 1 °C, with 16/8 h light/dark photoperiod, under constant oxygenation and filtration. During this period, shrimps were fed daily with a mix of krill and mysids (Europrix®). Male shrimps were kept in these containers until the organ sampling. A male specimen was anesthetized on ice. The hemolymph was collected from the pericardial sinus using a 1 mL syringe with a 1.5-inch, 23-gauge needle. Then, the hepatopancreas and gills were dissected. Samples were immediately snap-frozen in liquid nitrogen and stored at −80 °C.
For G. fossarum, adult specimens were collected in April 2021 from a bygone watercress farm in Saint-Maurice-de-Rémens (eastern central France) using 2- and 2.5-mm sieves. The organisms were stored in plastic bottles filled with ambient freshwater and immediately transferred to the laboratory. They were acclimated for two weeks in a 10-liter aquarium with a constant temperature of 12 ± 0.5 °C and a photoperiod of 8 h of darkness and 16 hours of light, with constant aeration. The organisms were fed with alder leaves (Alnus glutinosa) ad libitum. After two weeks, the hepatopancreatic caeca and gills from three male gammarids were dissected under an optical microscope. The hemolymph was sampled from the telson of gammarids using a modified microcapillary. The hemolymph from 25 organisms were pooled and transferred to 50 µl of Hanks’ Balanced Salt Solution (HBSS, Sigma). All samples were stored at −80 °C until proteomic investigation.
For D. polymorpha, zebra mussels between 20–25 mm long were collected from the lake of Der (Grand Est, France). The mussels were maintained in the laboratory for 24 h in 20-liter tanks filled with spring water Cristaline Aurèle (Jandun, France) with a constant temperature of 14 °C and controlled aeration. Gill tissues from individual mussel were dissected and stored at −80 °C for proteomic analysis.
For each organ of the different organisms the number of biological replicates used for MS/MS analyses are listed in the Table 1
Protein extraction and in gel digestion
For each organ except G. fossarum gills and caeca, 20 µL of LDS sample buffer (Thermo) was added per mg of organ. The samples were first mechanically homogenized as previously described in Cogne et al.13 by bead-beating with one 3.2 mm steel bead per tube with a Precellys instrument (Bertin). Then, the steel bead was removed and replaced with a mixture of 0.1 mm silica beads and 0.5/0.1 mm glass beads as reported earlier14 for another cycle of bead-beating. Proteins were subjected to SDS-PAGE onto a 4–12% gradient 10-well NuPAGE gel (Invitrogen) for a 10 min migration at 200 V. The proteome slightly resolved along the molecular weight was subdivided into five fractions of equal volume of polyacrylamide band (Table 1). The proteins in each polyacrylamide band were reduced with dithiothreitol (DTT), alkylated with iodoacetamide, washed, and proteolyzed with Sequencing Grade Trypsin (Roche) in the presence of 0.01% ProteaseMAX surfactant (Promega) for 1 h at 50 °C. The proteolysis was stopped with the addition of trifluoroacetic acid (TFA) 5% to reach a final concentration of 0.5%.and subjected to trypsin proteolysis. For the G. fossarum gills and caeca samples, the same protein extraction protocol was applied, but the SDS-PAGE was performed for 5 min. Each proteome was treated as a single polyacrylamide band.
NanoLC-MS/MS analysis
For all the samples except G. fossarum gills and hepatopancreatic caeca, the peptide samples extracted from one biological replicate and fractionated into five SDS-page bands were analysed in data-dependent acquisition mode with a Q-Exactive HF mass spectrometer (Thermo) coupled with an UltiMate 3000 LC system (Dionex-LC Packings). Peptides (200 ng) were desalted on-line and then resolved onto a nanoscale C18 PepMapTM 100 capillary column (LC Packings) with a 120-min gradient of CH3CN, 0.1% formic acid, at a flow rate of 0.2 µL/min. Peptides were analysed with scan cycles initiated by a full scan of peptide ions in the Orbitrap analyser, followed by high-energy collisional dissociation and MS/MS scans on the 20 most abundant precursor ions. Full scan mass spectra were acquired from m/z 350 to 1800 at a resolution of 60,000. Ion selection for MS/MS fragmentation and measurement was performed applying a dynamic exclusion window of 10 sec. Only 2+ and 3+ charged peptides were selected for fragmentation. Each peptide sample underwent duplicate analysis, with two technical replicates performed for each of the five SDS-page fractions (Table 1). So, ten nanoLC-MS/MS runs of 120 min were performed per organ to maximize the dataset size. For the G. fossarum gills and caeca samples, the peptides extracted from three biological replicate and fractionated into a unique SDS-page band were analysed with a single technical replicate using an Exploris 480 tandem mass spectrometer connected to a Vanquish Neo UHPLC (Thermo-Fisher) and operated in Data-Dependant Acquisition mode with parameters similar to those previously described15. The applied gradient was developed for 120 min (5%–25% of 0.1% HCOOH/99.9% CH3CN against 0.1% HCOOH/99.9% H2O), followed by a 5 min wash (25%–40%) and re-equilibration. Ions with 2+ or 3+ charges were selected for fragmentation applying a dynamic exclusion of 10 s.
Proteogenomics database design and MS/MS interpretation
For all the samples except G. fossarum gills and caeca, proteomics data were interpreted against specific transcriptome databases following a two-step database strategy. A first round of search was performed to identify the most probable proteins and construct a more specific database by selecting proteins with at least one unique peptide. The second round of search was performed on this resulting database to confidently identify the proteins and obtain improved quantitation. The P. serratus initial database comprised 52,782 entries totalling 25,079,966 residues. The P. serratus final database comprised 7,203 entries totalling 4,585,752 residues. The D. polymorpha initial database was obtained after predicting with Transeq the most probably open reading frames from a transcriptomics database with 941,470 entries. A total of 246,689 protein sequences totalling 42,459,289 residues were selected. The D. polymorpha final database comprised 11,978 entries totalling 4,916,414 residues. The G. fossarum database was with 80,589 entries totalling 15,753,148 residues. For these searches, the mascot search engine was used with a mass tolerance of 5 ppm for parent ions and 0.02 Da for MS/MS fragment analysis, allowing for up to two missed cleavages and full-trypsin specificity. Modifications considered were carboxyamidomethylated cysteine (+57.0215) as static, and oxidized methionine (+15.9949) and deamidation of asparagine/glutamine (+0.9848) as dynamic modifications. Peptides with a p-value lower than 0.05 were validated. A protein was validated in the final search if two unique peptide sequences were detected in a specific organ. The false positive rate for protein identification was estimated to be less than 1% using a reverse decoy database and the same parameters.
Estimation of protein abundances
For each proteome, protein abundances were estimated using the normalized spectral abundance factor (NSAF) method. This involved summing the spectral counts of protein-derived peptides obtained from peptide-to-spectrum assignments. NSAF values were calculated by dividing the spectral count of each protein by its molecular mass expressed in kDa, as previously described16. To express the NSAF values as a ratio, each protein’s NSAF was divided by the sum of all NSAF values in the organ proteome, resulting in a percentage of NSAF (%NSAF) for each protein which is a proxy of its molar abundance.
Functional analysis of proteomes
Proteomic sequences validated by proteogenomics were annotated as follows. BLAST sequence similarity searches were carried out using the BLASTp module of the OmicsBox software (version 2.0) against the SWISSPROT database with no restrictions on taxonomy. From these results, gene ontology (GO) and GO-slims annotations were retrieved using the GO-mapping tool of OmicsBox. The validity of all functional annotations was ensured with an E-value threshold of 1 × 10−3. Finally, KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways were predicted using the EggNOG-mapper tool, using default parameters as described by Huerta-Cepas et al. (2017).
To gain insight into functional differences between targeted organs, a comprehensive analysis of GO annotations was performed. For this purpose, functional GO-slims annotations were weighted with the %NSAF values calculated for each identified protein. To visually compare frequencies of GO-slim annotations between organs and species, a clustered heatmap was generated using the R package pheatmap.
Data Records
Transcriptomic databases
Transcriptomic data and ORF data for G. fossarum can be accessed from Cogne et al.12. Specifically, the transcriptome has been deposited in GenBank with the identifiers GHDA0100000017, while the ORF database referred as “T-GFBM” can be downloaded as FASTA files from figshare within the folder “YC02_Transcriptome translated ORFs”18. Read sequences were deposited in the European Nucleotide Archive under the accession number SRR80897219 for P. serratus and the NCBI Sequence Reads Archive under the accession number SRP44865620 for D. polymorpha. The open-access data repository Recherche Data Gouv21 provides reference transcriptomes for P. serratus and D. polymorpha, available as FASTA files named “Palaemon_serratus_transcriptome” and “Dreissena_polymorpha_transcriptome.” Additionally, the repository offers FASTA files of the translated coding sequence regions of contigs, which were generated by Transdecoder and used for MS/MS spectrum assignments in P. serratus and D. polymorpha. These files are named “Palaemon_serratus_ORF” and “Dreissena_polymorpha_ORF” respectively21.
Proteomic data
All mass spectrometry data are readily accessible through the ProteomeXchange Consortium, available via the PRIDE partner repository22,23,24,25. The dataset identifiers corresponding to these data can be found in Table 2. Information regarding the number of biological replicates and technical replicates associated with the Pride repository can be found in Table 1. A synthetic Excel table (named “PRIDE_descriptions”) available from the data repository Recherche Data Gouv21 provides a comprehensive description of all the file names and their associated replica types in the PRIDE databases.
For detailed insights into the identified proteins, their functional annotations, and estimated abundances based on NSAF values, Tables named “PS_proteins”, “GF_proteins”, and “DP_proteins” have been provided for organs from P. serratus, G. fossarum, and D. polymorpha, respectively. The excel files can be downloaded from data repository Recherche Data Gouv21 and include accession names of proteins linked to ORFs accessions, protein scores, percent peptide coverage of proteins, spectral counts of analytical replicates, NSAF values, Blast annotations, GO-terms and KEGG pathway assignments for each identified protein.
Technical Validation
Mass spectrometry quality control
Mass Spectrometry Quality Controls based on Pierce HeLa protein digest standard (Thermo) were incorporated at multiple points in the worklist and the performances of reproducibility were assessed in terms of peptide and protein identification. The correct calibration of the instrument was checked before starting the worklist and was verified by the average error done on the measures for each sample. The calibration error was in average below 0.5 ppm (0.44 ppm for P. serratus proteomes, 0.43 ppm for D. polymorpha proteomes, and 0.26 ppm for G. fossarum proteomes). The efficiency of proteolysis of samples, defined as less than 7% of 2 miss-cleavage, was tested for each sample.
The reproducibility of analytical replicates was also evaluated by comparing the set of proteins identified in each replicate (Fig. 2). For each sample tested in duplicate, over 90% of the proteins validated from a specific organ (see section below) with a unique peptide sequence were identified in both replicates. Using linear regression correlations, the repeatability of the protein abundance estimations between replicate was also evaluated by comparing the %NSAF values of shared proteins calculated independently in each analytical replicate. Results showed that the lowest correlation coefficient (R2) between %NSAF values was observed for the replicates of P. serratus hepatopancreas samples (Fig. 2). However, even in this case, the R2 was greater than 98%, indicating a high level of reproducibility. Overall, these results showed that the protein identification and abundance estimation were highly reproducible between analytical replicates, which enhances the reliability and robustness of MS investigations.

Percentage of validated proteins from species organs identified in both (shared) or single (specific) analytical replicates with unique peptides. Linear coefficient correlation (R2) between the %NSAF values of shared proteins, calculated in each analytical replicate, are also noted.
Peptide and protein validation
Decoy databases were generated to assess the false discovery rate (FDR) of identified peptides and proteins. A decoy database is a database of protein sequences generated by reversing or shuffling the target database of protein sequences. The decoy database contained the same number of proteins and similar properties as the target database but was composed of entirely random protein sequences. When searching for a shotgun experiment dataset against a decoy database, identified peptides that match to proteins in the decoy database represented false positives. In this study, an FDR of 1% was considered acceptable for peptide and protein identification in analyzed samples. In addition to controlling the FDR, the identification of at least two unique peptides (unambiguous in the whole database) was required for validating the identification of a protein in organs. Two unique peptides increase confidence in the identification of the protein since it is unlikely that two or more unrelated peptides will match to a single protein by chance. Moreover, identifying two or more unique peptides also allows for the estimation of the abundance of the protein, which can aid in understanding the biological function of the protein in the context of the sample being analyzed. In summary, the use of decoy databases and the identification of at least two unique peptides for validating protein identification in shotgun analyses ensured a high level of confidence in the identification of proteins in each organ of sentinel species.
Using this confident strategy, a greater number of peptides and proteins were identified in the gills of D. polymorpha compared to the other samples investigated (Table 3). When comparing the proteomes of the three organs investigated in P. serratus and G. fossarum, a higher number of peptides and proteins were identified in the gill tissues compared to the digestive organs (hepatopancreas and hepatopancreatic caeca), followed by the hemolymphatic compartment with less than 500 proteins identified (Table 3). When mapping the proteins identified in different organs within a species, a small number of proteins were shown to be shared between organs, while the majority were unique to each organ. For example, in P. serratus, out of the 3,891 of proteins identified, only 132 were found to be common across all organs and more than 30% of the proteins were specific to each organ (Fig. 3a). The same trend was observed for the organ proteomes of G. fossarum, as shown in the Fig. 3b. These results underline that proteogenomics allowed to capture the proteome specificity of the various organs of species, proteomes that are logically tailored for performing the specific functions of each organ.

Venn diagram mapping shared or organ-specific proteins in P. serratus (a) and G. fossarum (b).
Functional validity of organ proteomes
Functional analyses were performed to assess the functional significance of the proteomes established in the different target organs. For each proteome, functional profiling was conducted using ‘biological process’ related GO-slims annotated to about 90% of identified proteins with E-value cutoffs of 1.10−3 (Table 3). GO-slims were weighted with %NSAF values and compared across organ proteomes and species using a clustered heatmap analysis (Fig. 4). The heatmap showed that analogous organs in different species had similar functional profiles, grouping into three clusters of organs. The gills of all three species formed the first cluster, while the second cluster included the hepatopancreatic caeca and hepatopancreas from G. fossarum and P. serratus, and both hemolymph proteomes from G. fossarum and P. serratus were grouped together. The heatmap also revealed unique functional profiles specific to each organ type, distinguished by three functional annotation clusters. Overall, the functional profiles of proteomes are consistent with the expected biological functions of these organs, reinforcing the functional validity of the analyzed proteomic data.

Clustered heatmap analysis performed on GO-slim annotations of the biological process class, weighted by the %NSAF calculated from each proteome. The rows were compared with the heatmap, and clustering was performed using the Euclidian method.
The first functional annotation cluster (cl) showed that most of the GO-slims were found in higher abundance in the gill tissues of sentinel species compared to other organs (cl 1, Fig. 4). This cluster includes the GO-term ‘respiratory system process’, which is unsurprising given the crucial role that gills play in the respiration of aquatic organisms. Additionally, several GO-terms related to cellular transport mechanisms, such as transmembrane, intracellular, vesicle-mediated transport, and autophagy processes, were also identified. Cell transport mechanisms are vital for maintaining both osmoregulation and homeostasis in living organisms, as they enable cells to regulate the movement of molecules and ions in and out of cells, which is essential for maintaining stable internal conditions and proper physiological function26,27. Because gills are in direct contact with the external environment, they are known to play a critical role in osmoregulation and homeostasis in aquatic animals, regulating the exchange of gases, including oxygen and carbon dioxide, as well as the balance of ions and water26,27. The functional analysis also revealed that gill tissues exhibited a higher representation of GO-terms related to cell cycle mechanisms such as those involved in mitosis or DNA replication processes, suggesting that the gills have a high cell turnover rate compared to other organs28,29. Additionally, the over-representation of the GO-slim term “DNA repair” in gill tissues indicates that this organ may be subject to higher levels of genotoxic stress than other organs. As pinpointed by previous studies, this may be related to the direct exposure of gill tissues to environmental toxins and pollutants present in water30,31. A second cluster was characterized by a higher abundance of proteins annotated to GO-slims related to immune system processes in hemolymph compared to other organs (cl 2, Fig. 4). The hemolymphatic compartment, consisting mainly of immune cells (hemocytes) and plasma, plays multiple roles in the transport of immune effectors and the regulation of the immune response in invertebrate species5. Finally, the third group showed a higher abundance of GO-slims in the hepatopancreas and hepatopancreatic caeca of P. serratus and G. fossarum, respectively (cl 3, Fig. 4). As expected in these organs, these GO-slims were mainly related to metabolic processes, including the metabolism of amino acids, lipids, carbohydrates, and vitamins32. The GO-slim ‘detoxification’ was also overrepresented, indicating the role of these organs in removing harmful substances from the body also confirmed by the literature33.
Usage Notes
Despite the significant findings obtained from the proteogenomic analyses conducted in this study, it is important to acknowledge certain limitations. One such limitation is the utilization of few biological and technical replicates for the MS/MS analysis of P. serratus, G. fossarum and D. polymorpha samples. This limitation arises due to practical constraints associated with the complex and resource-intensive nature of the experimental design. Future studies with larger sample sizes and multiple biological replicates would be valuable to validate and build upon the findings reported here. Such studies have the potential to provide a more comprehensive understanding of the proteomic landscape and further elucidate the variations that may occur within and between individuals of the same species.
Despite these limitations, it is important to emphasize that the present analyses represent an initial exploratory investigation that lays the groundwork for future studies. The findings of this study provide crucial insights into the proteomic profiles of the selected sentinel species and establish a strong foundation for future ecotoxicological investigations and environmental monitoring efforts.
Code availability
No custom code has been used to process the data.
The search engine used to match MS/MS spectra to peptide sequences was Mascot Daemon 2.6.1 version (Matrix Science).
References
Gogoi, A. et al. Occurrence and fate of emerging contaminants in water environment: A review. Groundw. Sustain. Dev. 6, 169–180 (2018).
Destoumieux-Garzón, D. et al. The One Health Concept: 10 Years Old and a Long Road Ahead. Front. Vet. Sci. 5, (2018).
Ebner, J. N. Trends in the Application of “Omics” to Ecotoxicology and Stress Ecology. Genes 12, 1481 (2021).
Armengaud, J. et al. Non-model organisms, a species endangered by proteogenomics. J. Proteomics 105, 5–18 (2014).
Leprêtre, M. et al. The immune system of the freshwater zebra mussel, Dreissena polymorpha, decrypted by proteogenomics of hemocytes and plasma compartments. J. Proteomics 202, 103366 (2019).
Trapp, J. et al. Proteogenomic insights into the core-proteome of female reproductive tissues from crustacean amphipods. J. Proteomics 135, 51–61 (2016).
Kocourek, F. et al. Proteogenomic insight into the basis of the insecticide tolerance/resistance of the pollen beetle Brassicogethes (Meligethes) aeneus. J. Proteomics 233, 104086 (2021).
Dumas, T. et al. An integrated metabolomics and proteogenomics approach reveals molecular alterations following carbamazepine exposure in the male mussel Mytilus galloprovincialis. Chemosphere 286, 131793 (2022).
Stepien, C. et al. Limiting Environmental Factors and Competitive Interactions between Zebra and Quagga Mussels in North America. in Quagga and Zebra Mussels (CRC Press, 2013).
Wattier, R. et al. Continental-scale patterns of hyper-cryptic diversity within the freshwater model taxon Gammarus fossarum (Crustacea, Amphipoda). Sci. Rep. 10, 16536 (2020).
Zhang, Z.-Q. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148, 99–103 (2011).
Cogne, Y. et al. De novo transcriptomes of 14 gammarid individuals for proteogenomic analysis of seven taxonomic groups. Sci. Data 6, 184 (2019).
Cogne, Y. et al. Comparative proteomics in the wild: Accounting for intrapopulation variability improves describing proteome response in a Gammarus pulex field population exposed to cadmium. Aquat. Toxicol. 214, 105244 (2019).
Hayoun, K. et al. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 10, 1985 (2019).
Lozano, C. et al. Mass spectrometry detection of monkeypox virus: Comprehensive coverage for ranking the most responsive peptide markers. Proteomics 23, e2200253 (2023).
McIlwain, S. et al. Estimating relative abundances of proteins from shotgun proteomics data. BMC Bioinformatics 13, 308 (2012).
TSA. Gammarus fossarum breed In natura, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GHDA01000000 (2019).
Cogne, Y. et al. YC02. figshare. https://doi.org/10.6084/m9.figshare.c.4568087.v1 (2019).
European Nucleotide Archive http://identifiers.org/insdc.sra:SRR18114569 (2022).
NCBI Sequence Read Archive http://identifiers.org/ncbi/insdc.sra:SRP448656 (2023).
Degli Esposti, D. et al. Transcriptome assembly, open reading frames (ORF) predictions and protein catalogues of two aquatic sentinel species: Palaemon serratus and Dreissena polymorpha. Recherche Data Gouv https://doi.org/10.57745/5TDKA4 (2023).
Armengaud, J. Proteogenomic analysis of Palaemon serratus. PRIDE Archive https://doi.org/10.6019/PXD031698 (2022).
Armengaud, J. Proteogenomic analysis of gills and caeca from Gammarus fossarum. PRIDE Archive https://doi.org/10.6019/PXD040344 (2023).
Armengaud, J. Proteogenomic analysis of hemolymph from Gammarus fossarum. PRIDE Archive https://doi.org/10.6019/PXD031777 (2022).
Armengaud, J. Proteogenomic analysis of gills from the freshwater zebra mussel Dreissena polymorpha. PRIDE Archive https://doi.org/10.6019/PXD031681 (2022).
Pritchard, J. B. The gill and homeostasis: transport under stress. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 285, R1269–R1271 (2003).
McNamara, J. C. & Faria, S. C. Evolution of osmoregulatory patterns and gill ion transport mechanisms in the decapod Crustacea: a review. J. Comp. Physiol. B 182, 997–1014 (2012).
Lyndon, A. R. & Houlihan, D. F. Gill Protein Turnover: Costs of Adaptation. Comp. Biochem. Physiol. A. Mol. Integr. Physiol. 119, 27–34 (1998).
Piquet, B. et al. Regionalized cell proliferation in the symbiont-bearing gill of the hydrothermal vent mussel Bathymodiolus azoricus. Symbiosis 82, 225–233 (2020).
Flórez-Barrós, F., Prado-Alvarez, M., Méndez, J. & Fernández-Tajes, J. Evaluation of genotoxicity in gills and hemolymph of clam Ruditapes decussatus fed with the toxic dinoflagellate Prorocentrum lima. J. Toxicol. Environ. Health A 74, 971–979 (2011).
Leveroni, F. A., Caffetti, J. D. & Pastori, M. C. Genotoxic response of blood, gill and liver cells of Piaractus mesopotamicus after an acute exposure to a glyphosate-based herbicide. Caryologia 70, 21–28 (2017).
Rőszer, T. The invertebrate midintestinal gland (“hepatopancreas”) is an evolutionary forerunner in the integration of immunity and metabolism. Cell Tissue Res. 358, 685–695 (2014).
Ahearn, G. A., Mandal, P. K. & Mandal, A. Mechanisms of heavy-metal sequestration and detoxification in crustaceans: a review. J. Comp. Physiol. B 174, 439–452 (2004).
Acknowledgements
This study also received financial support from the program ‘OFB action DCE-biomarqueurs’ and the APPROve project funded by the French National Research Agency (ANR-18-CE34-0013-01). This work benefitted from the French GDR ‘Aquatic Ecotoxicology’ framework which aims at fostering stimulating scientific discussions and collaborations for more integrative approaches. The authors also thank Rémi Recoura-Massaquant for his help with the data repositories.
Author information
Authors and Affiliations
Contributions
M.L., D.D.E., K.S., R.C., C.B.L., B.X., M.P.L., A.G., O.G., J.A. and A.C. conceived and designed the analysis. D.D.E., N.D., A.D., B.X., M.P.L. and I.B. collected the biological samples. M.L., D.D.E., K.S., A.E. and J.C.G. contributed to data analysis. M.L., A.C. and J.A. draft the manuscript. The study was supervised by A.C. and J.A. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leprêtre, M., Degli Esposti, D., Sugier, K. et al. Organ-oriented proteogenomics functional atlas of three aquatic invertebrate sentinel species. Sci Data 10, 643 (2023). https://doi.org/10.1038/s41597-023-02545-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02545-w
