
Abstract
Osteosarcoma (OS) is a primary bone tumor with high malignancy and the mechanism of hematogenous metastasis in OS is still not clear. The plasma exosomes derived from osteosarcoma play a key role in the process of tumor metastasis. Here, we established RNA-seq dataset for lncRNAs, circRNAs and mRNAs in plasma exosomes from 10 OS patients and 5 healthy donors. A total of 329.52 Gb of clean data was obtained. Besides, 1754 lincRNAs, 7096 known and 1935 new circRNA was identified. Finally, gene expression profiles and differentially expressed genes (DEGs) were analyzed among these 15 samples. There were 331 DEGs of mRNA, 132 of lincRNA and 489 of circRNA was obtained, respectively. This data set provides a significant resource for relevant researchers to excavate potential dysregulated lncRNAs, circRNAs and mRNAs of plasma exosomes in OS versus normal conditions.
Similar content being viewed by others

Background & Summary
Osteosarcoma (OS), which mainly occur in children and adolescents, is a primary bone tumor with high malignancy1. Despite great efforts have made in the past 20 years, it is still difficult to exceed the survival rate of OS to 60%. The main reason for this poor prognosis is that OS is prone to lung metastasis via hematogenous channel2. However, the mechanism of hematogenous metastasis in OS is still not clear.
Exosomes, a tiny extracellular vesicle with a diameter of 40–100 nm, can be released by various cells3. Exosomes contains many substances, such as long non-coding RNAs (lncRNAs), microRNAs (miRNA), circular RNAs (circRNAs) and protein. Thus, exosomes are regarded as the medium of intercellular communication, which are closely related to the many kinds of diseases4. It should be noted that tumor-derived exosomes can enter into blood and lead to tumor metastasis. Researches show that the plasma exosomes derived from osteosarcoma play a key role in the process of tumor metastasis5. However, the specific biological function of plasma exosomes to cancer metastasis remains unclear.
LncRNAs present linear chain structure with transcripts ≥200 nucleotides in length, while circRNAs is an enclosed circle structure without 3′-end and 5′-end. Although both lncRNAs and circRNAs belong to non-coding RNAs, they make a critical difference to regulation of mRNA6. LncRNAs, circRNAs and mRNAs are the main small molecular substance in exosomes, playing a crucial role in biological function of exosomes. A growing number of evidence show that exosomes secreted by primary tumor cells can carry these three types of molecules to the metastatic lesions and regulate the biological behavior of metastatic cells7. Thus, to make a profound study on these three types of RNA in plasma exosomes is essential to control cancer metastasis. RNA-seq is an effective and comprehensive method to explore RNA expression in exosomes. Data set of RNA seq of blood exosomes in various disease has been established8. However, transcriptome profiling of plasma exosomes in OS has not been systematically explored.
Here, we established RNA-seq dataset for lncRNAs, circRNAs and mRNAs in plasma exosomes from 10 OS patients and 5 healthy donors. Although miRNA is not captured in the present study for the small sample and technical defects, this data set still provides a significant resource for relevant researchers to excavate potential dysregulated lncRNAs, circRNAs and mRNAs of plasma exosomes in OS versus normal conditions.
Methods and Result
Ethical approval
This study was supported by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University (2019(KY-E-162)). All steps are carried out according to the standards of the Ethics Committee. Patients and healthy volunteers in this research were informed of the study, including statement that their sequencing data would be shared. Both patients and healthy volunteers signed the informed consent.
Subjects and sample collection
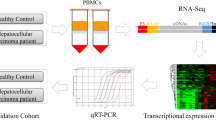
The experimental design and all workflow were presented in Fig. 1. To make the results more comparable, we developed elaborate inclusion and exclusion criteria for both patients and controls. Inclusion criteria: 1) patients were diagnosed with OS by postoperative pathological examination of resected specimens. 2) patients with tumor located in the extremities. 3) patients with initial treatment. Exclusion criteria: 1) patients and controls with infectious diseases, such as fever, hepatitis B infection, HIV, autoimmune disease and etc. 2) patients and controls with hemopathy, endocrine disease, and so on. 10 OS patients (6 female and 4 male) and 5 age- and sex-matched healthy controls were ultimately included in this study. According to surgical grading of bone tumor, 2 patients were classified into III, 5 patients into IIB, 2 patients into IB, and 1 patient into IIA, respectively (Table 1). 10 ml peripheral blood was collected from patients and controls in the morning with an empty stomach. Peripheral blood samples will be centrifuged at 300 min for 15 min to obtain blood plasma.

The experimental design and all workflow of this study.
Isolation of plasma exosomes
Firstly, plasma sample from individuals would be underwent a procedure of ultracentrifuged, which is similar to previous reports9. 7-fold phosphate-buffered saline (PBS) was added to the plasma sample to dilute the sample, centrifuged under condition of 13,000 × g for 30 minutes, and removed large particles via a 0.22 μm filter. The above supernatant would be centrifuged again for 2 hours at at 100,000 × g, 4 °C and then the exosomes pellet was obtained. The pellet was re-suspended with PBS and centrifuged again at 100,000 × g 4 °C for 2 h. Finally, 100 µl PBS were put into pellet to re-suspende the exosomes.
Identification of exosomes
The isolated exosomes should be identified at three levels: nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and western blot analysis (WB).
Nanoparticle tracking analysis (NTA)
We used ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) equipped with 405 nm laser to determine the size and quantity of isolated particles. A video with a duration of 60 seconds was shot at a frame rate of 30 frames per second, and particle motion was analyzed using NTA software (ZetaView 8.02.28). Our result showed the mean diameter of exosomes was 105.3 nm and the main peak of diameter was located in 85.2 nm with a proportion 98.4%. Besides, the concentration was 1.5E + 12 particles/ mL (Fig. 2a and Table 2).

Identification of exosomes. (a) The result of Nanoparticle tracking analysis (NTA) showed that the mean diameter of exosomes was 105.3 nm and the Wave Crest of diameter was located in 85.2 nm. (b) Transmission electron microscopy (TEM) showed that cup sharp of exosomes were clearly visible. (c) Western blot analysis (WB) demonstrated that compared to positive control group (CL), positive protein (CD63, HSP70, TSG101) was high expressed in our identified exosomes, while negative protein (calnexin) didn’t express.
Transmission electron microscopy (TEM)
10 µl exosomes sample of was placed on the copper mesh of electron microscope at room temperature for 1 min. After absorbing the floating liquid with filter paper, 15 µl of 2% uranyl acetate solution was took to stain for 1 min. The sample was then moved to an incandescent lamp and grilled for 2 minutes. The copper mesh was observed and photographed under a TME (H-7650, Hitachi Ltd., Tokyo, Japan). The morphology of exosomes in this study was observed in Fig. 2b.
Western blot analysis (WB)
WB was used to detect the exosomes marker proteins CD63, HSP90, TSG101 and calnexin. The sample stock solution was diluted according to the detected protein concentration. The sample was transferred to PVDF membrane after SDS-PAGE electrophoresis. Rabbit polyclonal antibody of above exosomes marker were added to the samples, and then incubated at 4 °C for 1 hour. Finally, IgG secondary antibody (all diluted according to 1:5000) and enhanced chemotherapy (ECL) was added to develop images. The result of WB analysis demonstrated that compared to positive control group (CL), positive protein (CD63, HSP70, TSG101) was high expressed in our identified exosomes, while negative protein (calnexin) didn’t express (Fig. 2c).
ExoRNA isolation and RNA analyses
MiRNeasy Plasma Advanced Kit (Qiagen, cat. No. 217204), which can purify total RNA from a small amount of plasma, was used in this study. Briefly, dissolve the serum sample in QIAzol lysis reagent, add chloroform, and centrifuge. Take the upper aqueous phase, add ethanol and RNeasy Minelute centrifuge column, and the total RNA will be bound to the membrane. Then we used Agilent Bioanalyzer 2100 System (Agilent Technologies, CA, USA) to perform the quality control of RNA sample.
Library preparation and sequencing
Ribosomal RNA accounts for a high proportion (~90%) of total RNA, which need to be removed before library preparation. SMARTer Stranded Total RNA Seq Kit V2 (Takara Bio USA, Inc.), which is designed to efficiently prepare Illumina sequencing library from picogram level total RNA (250 pg-10 ng), was used in this study according to the manufacturer’s instructions. The evaluation of library quality was performed on the Agilent Bioanalyzer 2100 and Quantitative real-time PCR. The clustering of the index-coded samples was performed on acBot Cluster Generation System using TruSeq PE Cluster Kitv3-cBot-HS (Illumina, San Diego, CA, USA). The resulting library will then be sequenced via an Illumina Hiseq platform and paired-end reads were generated.
Quality validation of the raw data and library
Clean data (clean reads) were obtained by removing reads containing adapter, reads containing ploy-N and low-quality reads from raw data using Cutadapt software of version 2.10 (https://cutadapt.readthedocs.io/en/stable/). At the same time, Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. The percentage of Q30 of each sample was not less than 88.74% in the present study, which released that the raw data in the present study was accredited (Table 3). After quality control of raw data, 329.52 Gb clean data was obtained.
In order to evaluate the quality of the library, we firstly estimate dispersion degree of estimated insert size. As showed in Fig. 3a, the main peak of the curve falls near 200 bp and does not deviate from the target area, indicating a small degree of dispersion in the length of the inserted fragment, and the selection of the inserted fragment size is normal. Subsequently, randomness distribution map was plotted to understand the degradation of mRNA. As we can see in Fig. 3b, a 3′ bias was found. This is due to the discontinuous nature of RNA of exosomes, but not related to mRNA degradation. Finally, to evaluate whether the data is sufficient and meet subsequent analysis, saturation testing is performed on the gene numbers. The result demonstrated that the number of genes detected in each sample is saturated, indicating that the sequencing depth of this study is sufficient (Fig. 4).

Quality validation of library. (a) Dispersion degree of estimated insert size. The result showed that the main peak of the curve falls near 200 bp. (b) Randomness distribution map. The result showed that a 3′ bias was present, which is due to the discontinuous nature of RNA of exosomes, but not related to mRNA degradation.

The saturation testing of the library. The result demonstrated that the number of genes detected in each sample is saturated, indicating that the sequencing depth of this study is sufficient.
Read alignment and transcript assembly
We used the designated genome GRCh38 (ftp://ftp.ensembl.org/pub/release-101/fasta/homo_sapiens/) as a reference for sequence alignment and subsequent analysis. For lncRNAs and mRNAs, we used the HISAT2 software (version 2.2.1.0)10 to align the reads and StringTie software (version 2.1.3)11 to assemble the aligned reads. Considering the feature of circRNA sequencing data, we used Burrows-Wheeler-Alignment Tool (https://sourceforge.net/ projects/bio-bwa/files/) to complete sequence alignment. The read mapping results of lncRNA, mRNA and circRNA were summarized in Table 3.
lncRNA gene prediction and identification
Cuffcompare program from the Cufflinks package v2.2.1 was firstly applied to compare the assembled transcripts with the existing gene annotation. We compared the results of assembled transcripts with the known lncRNA, and removed the known transcripts (class_code = c). The unknown transcripts were used to screen for putative lncRNAs. Four computational software for coding capability prediction, including CPC12, CNCI13, Pfam14 and CPAT15, were combined to sort non-protein coding RNA candidates from putative protein-coding RNAs in the unknown transcripts. The candidate lncRNA should meet the following conditions: (1)Transcription length ≥ 200 nt, (2) having more than two exons, (3) FPKM ≥ 0.1, 4) and class_code were “i”,“x”,“u”,“o”,“e”. Menwhile, further screened using above four computational approaches that have the power to distinguish the protein-coding genes from the non-coding genes. As well as the different types of lncRNAs include lincRNA, intronic lncRNA, anti-sense lncRNA were selected using cuff compare. The intersection of prediction results of several tools was taken as the prediction result of new lncRNAs. We ultimately obtained 1754 lincRNAs (Fig. 5). Supplementary Table 1 showed the prediction of the names of the lincRNA.

Venn diagrams of lncRNA gene prediction and identification by CPC, CNCI, Pfam and CPAT.
CircRNA prediction and identification
Two algorithms of CIRI2 and find_circ were used to predict circRNA. In CIRI216, by utilizing BWA software (Version: 7.12-r1039), the clean data was aligned to the reference gene set GRCh38 and the SAM files could be obtained. These SAM files were analyzed by CIRI2 software v2.0.5 (https://sourceforge.net/projects/ciri/files/CIRI2/) and ultimately gained the result of RNA prediction. Find_circ software (https://github.com/marvin- jens/find_circ/archive /v1.2.tar.gz) used bowtie2 (version 2.2.3) to complete the procedure of aligning and obtained predictive circRNA. The intersection gene obtained by the two algorithms will be used as the final circRNA. The predictive circRNA would be aligned to circBase database (http://www.circbase.org/) to identify known and new circRNA. We ultimately dentified 7096 known and 1935 new circRNA. Supplementary Tables 2, 3 showed the prediction of the known and new names of the circRNA, respectively.
Expression abundance quantification of mRNA and lincRNA
Stringtie (https://ccb.jhu.edu/software/stringtie/index.shtml#pub) was applied to evaluate FPKMs of all mRNA and lncRNA genes in each sample. Gene FPKMs was computed by summing the FPKMs of transcripts in each gene group. FPKM means fragments per kilo-base of exon per million fragments mapped, calculated based on the length of the fragments and reads count mapped to this fragment. The formula of FPKMs: FPKM = cDNA fragments/Mapped fragments (Millions)*Transcript Length(kb).
Expression abundance quantification of circRNA
Junction reads were used as circRNA expression level. We used TPM to normalize the expression abundance quantification. The formula of TPMs: TPMi = (Ni/Li)*1000000/sum(Ni/Li+………+ Nm/Lm). Ni is the junction reads of gene i and Li present exon length of gene i.
Differential expression analysis of mRNA, lincRNA and circRNA
The determination of differentially expressed genes (DEGs) between OS patients and normal samples was assessed by Deseq 217. The screening criteria were |log2FC| ≥ 2 and pvalue < 0.05. Finally, there were 331 DEGs of mRNA (Fig. 6a and Supplementary Table 4), 132 of lincRNA (Fig. 6b and Supplementary Table 5) and 489 of circRNA (Fig. 6c and Supplementary Table 6), respectively. Besides, the DEGs and the name of top 20 DEGs of mRNA, lincRNA and circRNA were also showed in the volcano plot (Fig. 6d–f).

Heatmap of differentially expressed genes (DEGs) between OS patients and normal samples. (a) mRNA, (b) lincRNA, (c) circRNA. The DEGs and the name of top 20 DEGs of mRNA (d), lincRNA (e) and circRNA (f) were showed by the volcano plot.
Data Records
The FASTQ files for the raw data and BAM files have been deposited in NCBI Sequence Read Archive (SRA) under BioProject accession of PRJNA904243 and SRP40918518. The transcript abundance was deposited in the NCBI Gene Expression Omnibus (GEO) and the accession number was GSE21852619. The GEO accession number of each sample was listed in Table 1. The files of RNA prediction, expression abundance quantification and DEGs analysis was also deposited in figshare20.
Technical Validation
RNA integrity assessment
NanoDrop spectrophotometer was utilize to quickly determine the OD value of RNA samples at 260 and 280 nm. The ratio of A260/A280 range 1.8–2.0 was considered acceptable, otherwise it would be resampled. Subsequently, Agilent 2100 Bioanalyzer was used to evaluate RNA integrity.
Quality validation of RNA data
In order to obtain clean data, Cutadapt software of version 2.10 (https://cutadapt.readthedocs.io/en/stable/) was first used to remove reads containing adapter. At the same time, Q20, Q30, GC-content and sequence duplication level of the clean data were calculated. Low quality read that met the following conditions would be excluded: 1) The reads in which the number of undetermined base pairs was greater than 10%; 2) Q30 < 88.74% .
Code availability
Most steps were completed base on the public-domain software, except for the calculation of differential genes. All analytical code of DEGs is available on the GitHub repository (https://github.com/tangaode/Plasma-exosomes). The provided R code was run and tested using R 4.1.0. The name and the links of all database depositories was showed in Table 4.
References
Suehara, Y. et al. Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin Cancer Res. 25(21), 6346–6356 (2019).
Subbiah, V. et al. Alpha Particle Radium 223 Dichloride in High-risk Osteosarcoma: A Phase I Dose Escalation Trial. Clin Cancer Res. 25(13), 3802–3810 (2019).
Sato, K., Meng, F., Glaser, S. & Alpini, G. Exosomes in liver pathology. J Hepatol. 65(1), 213–221 (2016).
Dong, Z. et al. Profiling of Serum Exosome MiRNA Reveals the Potential of a MiRNA Panel as Diagnostic Biomarker for Alzheimer’s Disease. Mol Neurobiol. 58(7), 3084–3094 (2021).
Wortzel, I., Dror, S., Kenific, C. M. & Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell. 49(3), 347–360 (2019).
Yao, P. et al. Research Progress of Exosome-Loaded miRNA in Osteosarcoma. Cancer Control. 29, 1–10 (2022).
Yuan, X. et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics. 11(3), 1429–1445 (2021).
Zhang, X. et al. Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer Commun (Lond). 42(4), 287–313 (2022).
Thery, C., Amigorena, S., Raposo, G. & Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 3, 22 (2006). Chapter 3, Unit.
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12(4), 357–60 (2015).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc. 11(9), 1650–67 (2016).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic acids research. 35, W345–9 (2007).
Sun, L. et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41(17), e166 (2013).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42(Database issue), D222–30 (2014).
Wang, L. et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41(6), e74 (2013).
Gao, Y., Wang, J. & Zhao, F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 16, 4 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq 2. Genome Biol. 15(12), 550 (2014).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP409185 (2023).
Liu, Y., Lu, J. & Tang, H. Long non-coding RNA, circular RNA and coding RNA profling of plasma exosomes of osteosarcoma by RNA-seq. Gene Expression Omnibus https://identifiers.org/geo/GSE218526 (2022).
Tang, H. Long non-coding RNA, circular RNA and coding RNA profiling of plasma exosomes of osteosarcoma by RNA-seq. figshare https://doi.org/10.6084/m9.figshare.22659328.v1 (2023).
Acknowledgements
This study was supported by Natural Science Foundation of Guangxi Province (2020GXNSFAA259088), the National Natural Science Foundation of China (81960768), the National Natural Science Foundation of China (82260814), Youth Science and Technology Project of the First Affiliated Hospital of Guangxi Medical University (201903038) and “Medical Excellence Award” Funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University.
Author information
Authors and Affiliations
Contributions
L.J.L., Y.L., Z.X.L. and W.Q.J. designed and managed study. H.N.G., L.C. and M.J.F. collected the tissue samples and clinical data, T.H.J. and T.W. were responsible for isolation and identification of exosomes. L.J.L., L.S.J., T.W. and T.H.J. performed sequencing projects and data analysis. L.K., L.F.C. and L.S.Y. reviewed related article and summarized background knowledge. T.H.J., X.T.Y. and F.W.Y. analyzed the data and wrote the paper. All authors had made contributions to this article and approved to submit manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Tang, H., Li, C. et al. Long non-coding RNA and circular RNA and coding RNA profiling of plasma exosomes of osteosarcoma by RNA seq. Sci Data 10, 395 (2023). https://doi.org/10.1038/s41597-023-02295-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02295-9
