
Abstract
Plectropomus leopardus, as known as leopard coral grouper, is a valuable marine fish that has gradually been bred artificially. To promote future conservation, molecular breeding, and comparative studies, we generated an improved high-quality chromosomal-level genome assembly of leopard coral grouper using Nanopore long-reads, Illumina short reads, and the Hi-C sequencing data. The draft genome is 849.74 Mb with 45 contigs and N50 of 35.59 Mb. Finally, a total of 846.49 Mb corresponding to 99.6% of the contig sequences was anchored to 24 pseudo-chromosomes using Hi-C technology. A final set of 25,965 genes is annotated after manual curation of the predicted gene models, and BUSCO analysis yielded a completeness score of 99.5%. This study significantly improves the utility of the grouper genome and provided a reference for the study of molecular breeding, genomics and biology in this species.
Similar content being viewed by others


Background & Summary
Groupers (Family Epinephelidae, Subfamily Epinephelinae) are prominent marine fishes, mostly distributed in tropical and temperate marine areas, comprising 167 species that belong to 15 genera1. Due to their high protein, low fat, tender meat quality, and good taste, groupers are high-quality economic fish species in Asia2,3. Given the huge commercial interests at stake, groupers are highly susceptible to human-induced impacts, including overfishing, making them considered threatened by the International Union for Conservation of Nature (IUCN)4. Therefore, how to scientifically develop and protect their resources has become the top priority5.
The leopard coral grouper (Plectropomus leopardus) has a beautiful skin color and is a valuable marine fish that commands a higher price6,7,8. Wild populations are suffering sharp declines due to overfishing and the destruction of spawning aggregations9. In recent years, the increasing market demands have promoted the development of artificial breeding in leopard coral grouper10,11,12. A high-quality reference genome resource has become increasingly important to facilitate the genomic breeding program, biological phenomena investigation and germplasm conservation13,14. Although the leopard coral grouper genome has been released6,8,15, the completeness of genome assembly and annotations still need to be further improved. For examples, the reported chromosomal-scale assembly of the sequence contigs only anchored 87.7% of the whole genome sequence using Hi-C technology6. Additionally, a wide range of gene structure annotation errors existed in the previous versions15, or the annotation information is not released and accessible to the public8.
In the present study, we generated an improved high-quality chromosome-level genome assembly of leopard coral grouper using Nanopore long-reads, Illumina short reads, and the Hi-C sequencing data. Approximately 849.74 Mb genome was assembled, consisted of 45 contigs with the contig N50 length of 35.59 Mb. A total of 846.49 Mb (99.6%) of the assembled sequences were anchored to 24 pseudo-chromosomes with low missing bases, only about 2, 354 gaps. Based on this improved genome assembly, we have significantly improved upon previous gene annotations combining de novo prediction, homology-based searches and transcriptome-assisted methods. BUSCO alignment showed that our final assembly contained 4, 469 (97.5%) complete BUSCOs. Taken together, this high-quality reference genome provides a valuable basis for the conservation and utilization of germplasm resources, and the further genetic breeding program in leopard coral grouper.
Methods
De novo genome assembly
First, we estimated the genome size and heterozygosity of leopard coral grouper using GenomeScope v2.016 by k-mer analysis with clean Illumina short data. Program ontbc (https://github.com/FlyPythons/ontbc) was used to filter the Nanopore raw reads with parameters “-min_score 7 -min_length 1000”. Then, the filtered Nanopore reads self-corrected the base errors by the long-read assembler NextDenovo v2.3 (https://github.com/Nextomics/NextDenovo). Finally, clean long reads were assembled using NextDenovo v2.3 (https://github.com/Nextomics/NextDenovo) with the parameters: read_cutoff = 5k’ and ‘seed_cutoff = 40k’. We used purge_dups v1.2.517 to remove the haplotypic duplication after mapping the Nanopore reads with minimap2 v2.118. The assembly sequence was then polished using NextPolish v1.3.119 with default parameters based on Nanopore long reads. To ensure high accuracy of the genome assembly, Illumina paired-end clean reads were aligned to the assembly using BWA v0.7.1520, and the results were used to conduct another round of polishing by Pilon v1.2321 with the parameters:--fix SNPs, indels. The contig-level assembly covered 849.74 Mb of the genome consisted of 45 contigs with a contig N50 value of 35.59 Mb.
Hi-C analysis and chromosome assembly
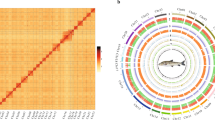
To obtain the chromosome-level genome, we further anchored all 45 contigs of the draft assembly onto 24 chromosomes using a 3D-DNA pipeline (version 201008)22 based on the published high-quality HiC reads15. The HiC reads were aligned to the polished genome using Juicer v1.5.7 software23 with default parameters. Mis-joins, order and orientation were corrected by the 3D-DNA pipeline22 with the following parameters: -r 2. After the first round of 3D-DNA, we manually adjusted the assembly with Juicebox23 and rerun the 3D-DNA. The Hi-C scaffolding resulted in 24 chromosome-length scaffolds (Fig. 1a).

Statistics on genome assembly and Comparison of four version annotations of the leopard coral grouper, Plectropomus leopardus. (a) Hi-C interaction heat map for Plectropomus leopardus. (b) BUSCO evaluation on the genome assembly completeness. (c) BUSCO evaluation on the predicted gene models.
Repeat annotation
De novo and structure-based searches were used to identify repetitive sequences with both RepeatModeler v224 (http://www.repeatmasker.org/RepeatModeler/) and RepeatMasker v4.0.925 (http://www.repeatmasker.org). Candidate LTR-RTs repetitive sequence library was identified using LTR_finder26 with parameters ‘-D 15000 -d 1000 -L 7000 -l 100 -p 20 -C -M 0.9’ and LTRharvest v1.5.827 with parameters ‘-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes’. The identified LTR-RT candidates were filtered with LTR_retriever v2.528 program with default parameters. RepeatScout v1.0.529 LTR_retriever v2.528 and RepeatModeler v224 were used to build de novo repeat libraries. The combined repeat library was used as the final library to identify repetitive sequences using RepeatMasker v4.0.925 with parameters ‘-q -no_is -norna -nolow -div 40’.
Gene prediction and annotation
To comprehensively annotate genes, protein-coding genes prediction was undertaken using the BRAKER v2.1.530 annotation pipeline which integrated different evidence, including de novo prediction, homology-based searches and transcriptome-assisted methods. First, for de novo gene prediction, we downloaded published RNA-seq (SRP20194331 and SRP32903132) and then mapped to the soft masked genome using Hi-SAT2 v. 2.1.033. Then, all mapping results were used to build transcript models using BRAKER v2.1.530 and StringTie v2.1.634. BRAKER v2.1.530 was run with Semi-HMM-based Nucleic Acid Parser (SNAP, v2013.11.29)35 and Augustus v3.3.336 which pre-trained using released gene models of P. leopardus6,15. Second, protein-coding sequences of from P. leopardus6,15, E. fuscoguttatus37, E. lanceolatus38, and E. moara39 were aligned to the genome assembly using TBLASTN and GeneWise v2.2.040. Third, Trinity v2.1.141 was used to generate the transcripts. The transcriptome data were further assembled using the PASA pipeline v2.5.242 with BLAT v3543 and GMAP (version 20150921)44 as the aligner. Finally, all evidences were merged to form a consensus gene set using EVidenceModeler v1.1.145. Finally, we identified a total of 25,965 protein-coding genes (Table 2). The noncoding RNA genes including rRNAs, tRNAs, snRNAs and miRNAs were screened using INFERNAL v 1.1.246 and tRNAscan-SE v1.447. Four types of noncoding RNAs, including 746 miRNAs, 1,224 tRNAs, 439 rRNAs and 596 sRNAs, were identified from the P. leopardus genome (Table 3).
In order to explore the function of predicted protein-coding genes in leopard coral grouper, InterPro30, Pfam32, PANTHER 14.1, Superfamily 1.75, Gene3D 4.2.0, SMART 7.1 and TrEMBL32 databases were respectively used to predict protein function based on the conserved protein domains by InterProScan v5.3648. We performed functional annotation by aligning the protein sequences to NCBI nr databases and SwissProt using BLASTP. The result showed more than 99.9% (25,927) of protein-coding genes were annotated (Table 4).
Data Records
The assembled genome has been deposited at GenBank under the accession GCA_026936395.149. Moreover, the whole genome sequence data reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center50,51, Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under accession number GWHBPCI00000000 that is publicly accessible at https://ngdc.cncb.ac.cn/gwh/Assembly/29542/show52. In addition, the genome annotation files had been submitted at the figshare53. The Nanopore long reads, Illumina genomic sequencing data and Hi-C data were downloaded from CNGBdb51,54 under the accession CNP000085955. Transcriptomic sequences can be retrieved under the following accession numbers: SRP20194331 and SRP32903132.
Technical Validation
To evaluate the quality of genome assembly, first, we assessed genome continuity with QUAST v5.0.256. Contig N50 (the length such that half of all sequence is in contigs of this size) has achieved a significant improvement to 35.59 Mb, which is much higher than other versions6,8,15 or closely related species (Epinephelus fuscoguttatus, Epinephelus lanceolatus, Epinephelus moara) assembled with long-read sequencing from 0.12 to 13.8 Mb. Meanwhile, in the latest version, there are very few gaps in the genome (2.77 per 100 kbp), which is remarkably less than the previous from 68.31 per 100 kbp to 1793.38 per 100 kbp6,8,15 (Table 1; Fig. 2). Second, Illumina paired-end clean reads and Nanopore long reads were mapped to the final reference genome assembly by using BWA v0.7.1520 and Minimap2 v2.118, respectively. The mapping rate of Illumina and Nanopore reads reached 99.18% and 99.95%. We only detected 6, 900 (0.0008%) conflicting sites in the final assembly, indicating that this is a high level of the complete genome (Fig. 2; Table 5). Finally, we evaluated the completeness of our genome assembly using Benchmarking Universal Single-Copy Orthologs (BUSCO, v3.0)57 with the actinopterygii_odb9 database. The actinopterygii_odb9 database contained 4,584 conserved core genes while our assembled genome contained 4,469 (97.5%) of the expected actinopterygii genes (including 4,393 (95.2%) single and 106 (2.3%) duplicated ones). Obviously, our data had complete gene coverage, and 48 (1.0%) were identified as fragmented, respectively, while 67 (1.5%) were missing in our assembled genome (Fig. 1b). Furthermore, we also used BUSCO to evaluate the completeness of gene annotations57, and only 22 (0.5%) genes were missing in the final annotation version (Fig. 1c) Table 5.

Global genome landscape of the leopard coral grouper, Plectropomus leopardus. From outer to inner circles: Density of genes with 500 kbp windows, ranging from 0 to 70; GC content with 500 kbp windows, ranging from 0.30 to 45; depth of coverage of Nanopore reads with 100 kbp windows, ranging from 20 to 150; depth of coverage of Illumina short reads with 100 kbp windows, ranging from 10 to 35; distribution of heterozygous SNPs with 500 kbp windows, ranging from 0 to 3,420; distribution of homozygous SNPs with 500 kbp windows, ranging from 0 to 3,420.
Code availability
The data analyses were performed according to the manuals by the developers of corresponding bioinformatics tools and all software, and codes used in this work are publicly available, with corresponding versions indicated in Methods.
References
Félix-Hackradt, F. C., Hackradt, C. W. & García-Charton, J. A. Biology and Ecology of Groupers. (CRC Press, 2022).
Fabinyi, M. Historical, cultural and social perspectives on luxury seafood consumption in China. Environ. Conserv. 39, 83–92 (2012).
Sale P. F. Coral reef fishes: dynamics and diversity in a complex ecosystem. (Academic Press, 2002).
Luiz, O. J., Woods, R. M., Madin, E. M. P. & Madin, J. S. Predicting IUCN Extinction Risk Categories for the World’s Data Deficient Groupers (Teleostei: Epinephelidae). Conserv. Lett. 9, 342–350 (2016).
Valderrama, S. P. et al. Marine protected areas in Cuba. B. Mar. Sci. 94, 423–442 (2018).
Zhou, Q. et al. De novo sequencing and chromosomal-scale genome assembly of leopard coral grouper, Plectropomus leopardus. Mol. Ecol. Resour. 20, 1403–1413 (2020).
Wang, L., Yu, C. P., Guo, L., Lin, H. R. & Meng, Z. N. In silico comparative transcriptome analysis of two color morphs of the common coral trout (Plectropomus leopardus). PLoS One 10, e0145868 (2015).
Yang, Y. et al. Whole-genome sequencing of leopard coral grouper (Plectropomus leopardus) and exploration of regulation mechanism of skin color and adaptive evolution. Zool. Res. 41, 328 (2020).
Agustina, S., Panggabean, A. S., Natsir, M., Retroningtyas, H. & Yulianto, I. Yield-per-recruit modeling as biological reference points to provide fisheries management of Leopard Coral Grouper (Plectropomus leopardus) in Saleh Bay, West Nusa Tenggara. IOP Conference Series: Earth and Environmental Science 278, 012005 (2019).
Ottolenghi, F., Silvestri, C., Giordano, P., Lovatelli, A. & New, M. B. Capture-based aquaculture: the fattening of eels, groupers, tunas and yellowtails. (FAO, 2004).
Nguyen, T. T. T., Davy, F. B., Rimmer, M. A. & De Silva, S. S. Use and exchange of genetic resources of emerging species for aquaculture and other purposes. Rev Aquacult. 1, 260–274 (2009).
Kongkeo, H., Wayne, C., Murdjani, M., Bunliptanon, P. & Chien, T. Current practices of marine finfish cage culture in China, Indonesia, Thailand and Vietnam. Aquac. Asia 15, 32–40 (2010).
Allendorf, F. W., Hohenlohe, P. A. & Luikart, G. Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709 (2010).
Mohanty, B. P. et al. Omics technology in fisheries and aquaculture. Adv. Fish Res. 7, 1–30 (2019).
Wang, Y. B. et al. Chromosome genome assembly of the leopard coral grouper (Plectropomus leopardus) with Nanopore and Hi-C sequencing data. Front. Genet. 11 (2020).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Hu, J., Fan, J. P., Sun, Z. Y. & Liu, S. L. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963 (2014).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Chen, N. S. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10.11–14.10.14 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2017).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Hoff, K. J., Lomsadze, A., Borodovsky, M. & Stanke, M. in Gene Prediction: Methods and Protocols (ed M., Kollmar) 65–95 (Springer New York, 2019).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP201943 (2021).
NCBI Sequence Read Archive https://identifiers.org/insdc.sra:SRP329031 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Yang, Y. et al. Whole-genome sequencing of brown-marbled grouper (Epinephelus fuscoguttatus) provides insights into adaptive evolution and growth differences. Mol. Ecol. Resour. 22, 711–723 (2022).
Zhou, Q. et al. A chromosome-level genome assembly of the giant grouper (Epinephelus lanceolatus) provides insights into its innate immunity and rapid growth. Mol. Ecol. Resour. 19, 1322–1332 (2019).
Zhou, Q., Gao, H. Y., Xu, H., Lin, H. R. & Chen, S. L. A Chromosomal-scale reference genome of the kelp grouper Epinephelus moara. Mar. Biotechnol. 23, 12–16 (2021).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Kent, W. J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57 (2016).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_026936395.1 (2022).
Chen, M. L. et al. Genome warehouse: a public repository housing genome-scale data. Genom. Proteom. Bioinforma. 19, 584–589 (2021).
CNCB-NGDC Members and Partners Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2021).
National Genomics Data Center https://ngdc.cncb.ac.cn/gwh/Assembly/29542/show (2022).
Han, W. Plectropomus leopardus genome. Figshare https://doi.org/10.6084/m9.figshare.21441396.v3 (2022).
FAIRsharing.org: CNGBdb; China National GeneBank DataBase; https://doi.org/10.25504/FAIRsharing.9btRvC.
Zhang X. & Institute of Biodiversity Conservation. leopard coral grouper genome. CNGBdb https://db.cngb.org/search/project/CNP0000859/ (2020).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Waterhouse, R. M. et al. BUSCO applications from quality Assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2017).
Acknowledgements
This research was funded by the National Key Research and Development Program of China (2022YFD2400501), the Project of Sanya Yazhouwan Science and Technology City Management Foundation (SKJC-2020-02-009), the Key R&D Project of Hainan Province (ZDYF2021XDNY133), and the China Postdoctoral Science Foundation (2021703030).
Author information
Authors and Affiliations
Contributions
J.H., B.W. and Z.B. conceived and designed the study. J.H. and B.W. coordinated and supervised the whole study. W.H. conducted the genome assembly and analysis. S.W., M.W., H.D. and M.W. participated in discussions and provided suggestions for manuscript improvement. W.H., B.W. and J.H. did most of the writing with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, W., Wu, S., Ding, H. et al. Improved chromosomal-level genome assembly and re-annotation of leopard coral grouper. Sci Data 10, 156 (2023). https://doi.org/10.1038/s41597-023-02051-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02051-z
This article is cited by
-
Chromosome-level genome assembly and annotation of the yellow grouper, Epinephelus awoara
Scientific Data (2024)
-
Genome sequencing and analysis of penicillin V producing Penicillium rubens strain BIONCL P45 isolated from India
International Microbiology (2024)
