
Abstract
Davidia involucrata Baill. is a rare plant endemic to China. Its exclusive evolutionary position and specific floral organs endow it with a high research value. However, a lack of genomic resources has constrained the study of D. involucrata functional genomics. Here, we report D. involucrata transcriptome reads from different floral tissues pooled from six individuals at two developmental stages using Illumina HiSeq technology and the construction of a high-quality reference gene set containing a total of 104,463 unigenes with an N50 of 1,693 bp and 48,529 high-quality coding sequences. The transcriptome data exhibited 89.24% full-length completeness with respect to the benchmarking universal single-copy (BUSCO) dataset and a PLAZA CoreGF weighted score of 98.85%. In total, 65,534 (62.73%) unigenes were functionally annotated, including 58 transcription factor families and 44,327 simple sequence repeats (SSRs). In addition, 96 known and 112 novel miRNAs were identified in the parallel small RNA sequencing of each sample. All these high-quality data could provide a valuable annotated gene set for subsequent studies of D. involucrata.
Measurement(s) | small RNA sequencing assay • transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | developmental stage |
Sample Characteristic - Organism | Davidia involucrata |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9772118
Similar content being viewed by others

Background & Summary
Davidia involucrata Baill., also called dove tree or handkerchief tree, is the sole species in the genus Davidia (Davidiaceae1 or Nyssaceae2) and is listed as a “first-grade” nationally protected plant in China3,4. It is a Tertiary paleotropical relic plant species that is rare in China and usually regarded as a “botanic living fossil”5. Its distribution is limited to the subtropical mountains of central to southwestern China; natural populations are often found in deciduous or evergreen broad-leaf forests at elevations of 1100–2600 m6. D. involucrata is not only an endangered and rare relic species but also famous as an ornamental plant by virtue of the pair of large white bracts that surround the small flowers and create the appearance of doves perching among its branches, giving the tree its common name7. The most unusual characteristics of D. involucrata are its floral organs, of which the inflorescences contain either a mixture of hermaphrodite and many male flowers or entirely male flowers; the flowers are without petals but have large, unequal, paired paper-like bracts instead. These intriguing bracts originally appear small and green, resembling leaves, but they increase in size and turn white as the flowers mature, and then, finally, turn to brown and yellow before being shed8,9. Bracts are special organs that appear in the reproductive development of plants, and D. involucrata is undoubtedly an ideal subject for the study of bracts and the developmental mechanisms of specific flower organs.
To date, studies of D. involucrata have mainly focused on the macroscopic and phenotypic levels, such as taxonomy, morphology, physiology, ecology, reproduction and so on9,10,11,12,13. However, research at the molecular level continues to progress slowly, which could result from the distribution of this species, which is intermittently scattered throughout southwest China, and its growth characteristics; D. involucrata is slow-growing, occurs in harsh growth conditions and has a low survival rate. Therefore, deeper research at the molecular level is critical for this endangered plant.
Studies of the molecular mechanisms underlying the growth and development of D. involucrata as well as its unique bracts and floral organs could further reveal the evolution of floral development, which remains unclear due to a lack of high-throughput data. To date, many molecular studies have focused on exploring and analysing traditional markers, such as simple sequence repeats (SSRs)14,15,16, random amplified polymorphic DNA (RAPD) markers17, microsatellites18 and chloroplast genes19, or utilized functional genes or factors, such as a MYB transcription factor from D. involucrata (DiMYB1)20, a cold-induced gene (DiRCI)21, a clathrin adaptor complex gene (DiCAC)22 and so on. At the same time, the transcriptome of the seed23 and the chloroplast genome24 have been reported. However, transcriptomic and sRNA data from some crucial tissues, for example, the floral organs and, specifically, the bracts, have not been examined in depth23,24,25.
Here, we established a complete gene set from multiple tissues of D. involucrata by means of next generation sequencing technology. This gene set would be useful for further studies. For example, it could be used as a reference for gene characterization, such as expression analysis, gene cloning and phylogenetic analysis, and it could also be used for gene model annotation once the genome is sequenced. In addition, the transcriptome annotations could be used to explore the crucial genes in flower development and bract development, which are of great significance to reveal the evolution of the floral organs in angiosperms. Furthermore, the small RNAs of each sample were sequenced and annotated in parallel to provide more information about sRNA-related regulatory mechanisms during floral organ development. The whole transcriptomes established herein lay a foundation for plant molecular marker-assisted breeding, evolutionary and developmental analysis, and even plant protection in the future.
Methods
Sample collection
The floral organ samples were collected from 6 blooming individuals at two developmental stages (3 individuals for each stage) in a wild population in the county town of Yingjing, Yaan, Sichuan Province, in April 2014 (Fig. 1a,b). Floral organ growth stages were defined based on the colour changes of the bracts. One stage was young inflorescences with small, green bracts (expanded to approximately <8 cm in length) resembling leaves, and the dark purple anthers were immature (Fig. 1a). The other stage was mature inflorescences with large white bracts (expanded to approximately >15 cm in length), and the anthers were mature (Fig. 1b). Each tissue sample collected from each plant was approximately thumbnail-sized and was immediately stored in RNA Fixer solution (Bioteke, China) for further use.

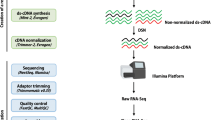
Schematic pipeline illustrating analysis of the whole transcriptome dataset. (a) The young inflorescences of D. involucrata. (b) The mature inflorescences of D. involucrata. (c) After library construction, mRNAs were sequenced with a PE90 strategy, and sRNAs were sequenced with an SE50 strategy. After low-quality read removal, the transcriptome of each sample was assembled using SOAPdenovo-trans, KGF and GapCloser. After clustering using TGICL, the final assemblies were obtained, and addition evaluation and annotation were performed.
The floral organ samples (Table 1) were named YB (young bracts: a pool of similar samples collected from the growing green bracts of three individuals), LB (mature bracts: a pool of similar samples collected from the mature white bracts of three individuals), LX (mature stamens: a pool of similar samples containing the complete, mature stamens from three individuals), LC (mature pistils: a pool of similar samples containing the complete, mature pistils from three individuals), YR (young mixed samples: a pool containing flowers at the early blooming stage including equal amounts of complete stamens and complete pistils from three individuals), ZR (mixed samples: a pool of all collected floral organ samples with added leaves, shoots, and roots at two periods from all six individuals).
Total RNA extraction
Total RNA extraction was performed using the TRIzol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions, followed by quality assessment on an Agilent 2100.
Transcriptome sequencing and filtering
The mRNA was extracted from the total RNA with oligo (dT)-attached magnetic beads, and a cDNA library with an insert size of 250 bp was constructed using the TruSeq RNA Sample Preparation Kit according to the manufacturer’s protocol (Illumina Inc., San Diego, CA). Library generation yielding 2 × 90 bp paired-end reads and sequencing (Illumina HiSeq 2000) were performed at BGI-Shenzhen. Low-quality reads matching one or more of the following criteria were filtered out using SOAPnuke (v1.5.6)26: reads containing adaptor contamination; reads including more than 5% of the unknown base “N”; reads including more than 20% bases with quality values lower than 10.
sRNA sequencing and filtering
RNA segments of different sizes from 18–30 nt were separated from the total RNA by 15% denaturing PAGE and selected for small RNA library construction using the Illumina TruSeq Small RNA Sample Preparation Kit according to the manufacturer’s protocols (Illumina Inc.). Library generation yielding 50 bp single-end reads and sequencing (Illumina HiSeq. 2000) were also performed at BGI-Shenzhen. The contaminant tags and low-quality tags were removed using SOAPnuke (v1.5.6)26: (1) tags with 5′ primer contaminants, oversized insertion, poly-A; (2) tags without 3′ primer or insert tags; (3) tags shorter than 18 nt. After the removal of low-quality reads, clean reads (Table 2) were retained and used in subsequent analyses (Fig. 1). The quality of all clean reads was assessed with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) and MultiQC (v1.8)27.
Transcriptome assembly
Transcriptome assembly was completed using a combined assembly strategy. Briefly, the RNA-Seq reads were assembled using SOAPdenovo-Trans (version 1.01)28 with the following settings: “-K 31-i 20 -e 2 -M 3-L 100”. The gaps were filled using KGF (v1.19) and the GapCloser tool (v1.12)29. All assemblies were merged into one large dataset using TGICL (v2.0.6)30 with the parameters “-l 40 -c 10 -v 25 -O ‘-repeat_stringency 0.95 -minmatch 35 -minscore 35’”.
To remove unreliably assembled transcripts, clean reads were aligned to the assembly using Bowtie231 with the parameters “–sensitive–score-min L,0,-0.1 -I 1 -X 1000–mp 1,1–np 1–no-mixed–no-discordant”, and the fragments per kilobase of exon model per million reads mapped (FPKM) values of the transcripts were calculated using the tool rsem-calculate-expression in RSEM (v1.2.21)32. The sequences with FPKM values of zero were removed from the assembly. Then, the lowest common ancestor (LCA) algorithm was applied to filter out contaminants. First, the assembly was used to search against the NCBI non-redundant protein database (Nr) using Diamond with e-value < 1e-5. The tool Blast2lca in MEGAN (v6.15.2)33 with the parameter “–minScore 75” was used to apply the LCA alignment and produce taxonomic classifications. Non-Viridiplantae sequences were removed according to their classifications. All unreliably unigenes of each individual were also filtered. Last, we obtained the final transcriptome assembly (Table 3).
Transcriptome Annotation
Functional annotation
Functional annotations were performed using a sequence-based search method (Table 4). First, the final assembly was annotated by searching against the NCBI non-redundant nucleotide (Nt) database34 using BLASTn (v2.4.0)35 with e-value < 1e-5, Viridiplantae-related non-redundant proteins in the Nr database, Swiss-Prot in UniProtKB36, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database37 and clusters of euKaryotic Orthologous Groups (KOG)38 using BLASTx (v2.4.0) with e-value < 1e-5, and the InterPro database39 using InterProScan (v5)40 with the default parameters36. Gene Ontology (GO)41 annotation was performed using Blast2GO (v2.5.0)42 based on the Nr annotation results.
Identification of coding gene set
First, the open reading frames (ORFs) of each sequence were predicted using TransDecoder (v3.0.1) as implemented in Trinity43. Then, the ORFs were searched against Swiss-Prot36 using Diamond Blastp (v0.8.31)44, and the output file was searched against Pfam45 using Hmmscan (v3)46. Finally, CDSs were predicted using TransDecoder with the results of the previous step (Table 5).
Identification of high-quality annotated coding gene set
To identify high-quality coding genes, we used the following pipeline. (1) All transcripts had matches with protein databases, and the best hit for each transcript (in the following order: Nr, Swiss-Prot, KEGG, KOG and InterPro) was extracted. (2) The annotated coding region and functional annotation for each transcript were selected from the best hit. (3) The transcript with the longest CDS was chosen if its annotated coding region could also be identified by TransDecoder. (4) After filtering out sequences with more than 5 Ns in the last 10 bases or more than 15 Ns in the last 50 bases, the eligible CDS of each transcript was extracted as the final coding gene. In total, 51,247 annotated coding genes with a minimal length of 297 nt and a maximal length of 12,852 nt were identified as high-quality coding genes (Table 5).
Transcription factor family classification
The open reading frame of each sequence was classified using getorf (EMBOSS:6.5.7.0)47, and the transcriptome factor (TF) family was identified based on plant TF domains in the plant TF database (PlnTFDB)48 using Hmmsearch46. In total, 3,064 genes were arranged in 58 TF families, and the top three annotated families were the Myb DNA-binding domain families MYB and MYB-related and the basic helix-loop-helix (bHLH) family (Fig. 2a).

Transcription factor family and simple sequence repeat statistics. (a) Transcription factor family classifications of the assembled sequences. (b) SSR statistics.
Simple sequence repeat detection
SSRs in the transcriptome sequences were detected using MISA (v1.043)49 with the definition parameters “1-12,2-6,3-5,4-5,5-4,6-4” and an interruption parameter of “100”. The total number of identified SSRs was 44,327, and the number of SSR-containing sequences was 20,739 (Fig. 2b).
sRNA Annotation
Small RNA annotation
Because of the lack of a D. involucrata special sRNA reference dataset, we used the microRNA datasets of Arabidopsis lyrata from miRBase (release 22)50 and RNA datasets from the Rfam database (v12.1)51 as the known small RNA reference databases. Clean reads were mapped to the miRBase database using Bowtie2 (v2.2.5)31 with “–sensitive -L 16” and the Rfam database using cmsearch in INFERNAL (v1.1.2)52 with “–noali” to obtain the annotations. Aligned tags were annotated after filtering reads with more than one mismatch out of each alignment. A total of 96 known miRNAs were annotated in this study.
Novel miRNA prediction
Because no more than 10% of the small RNA tags were annotated, miRA software53 was applied to predict novel miRNAs. First, the clean reads of each sample were mapped to the “genome” reference (the merged transcriptome) using Bowtie2 with the parameters “-L 16–rdg 1,10–rfg 1,10”. After removing the unreliable transcriptome sequences from the alignments, 45.28%, 44.08%, 43.62%, 44.21%, 38.90% and 54.74% tags could be mapped to the transcriptome for ZR, YR, YB, LB, LC and LX, respectively. After filtering out the annotated sRNA reads, the aligned read tags from all samples were combined into one FASTA file. Then, the combined FASTA file was mapped to the genome reference using Bowtie2 with “-f -l 16”, and the alignment file was filtered using SAMtools (v1.3.1)54 with “view -hS -F 4”. Finally, miRA (v1.2.0) with the default parameters was used to predict novel miRNAs. In total, 112 novel miRNAs were identified. The annotation and prediction results are summarized in Table 6 (Table 6, Fig. 3).

Length distributions of sRNA samples and sRNA annotation distribution of sample ZR. (a) Length distributions of sRNA. The peak of each sample was located at 24 nt. (b) sRNA annotation distribution of sample ZR.
miRNA target prediction
psRobot (v1.2)55 and TargetFinder (v1.0)56 with the default parameters were used to predict miRNA targets. The intersection of the target genes was extracted as the final prediction target result. The final intersection results included 172 miRNAs and 1,737 target genes.
Data Records
The final transcriptome and related data are published under the International Nucleotide Sequence Database Collaboration BioProject PRJNA513477 (https://identifiers.org/ncbi/bioproject:PRJNA513477) and CNGB Nucleotide Sequence Archive (CNSA) project CNP0000260 (https://db.cngb.org/search/project/CNP0000260). The read files were deposited in the NCBI Sequence Read Archive57. The final high-quality coding gene set and the transcriptome assemblies were deposited in NCBI Transcriptome Shotgun Assembly58 and CNSA. Annotation data set was uploaded to figshare59.
Technical Validation
Quality control and data statistics
To control the sequencing quality, the counts of clean reads, total bases, quality scores and GC content were calculated for each sample using FastQC and MultiQC (Tables 1, 2 and Fig. 4). The mean read counts per quality scores and the mean quality scores in each base position were higher than 30. The length distribution of clean small RNA tags showed that the peak of each sample was located at 24 nt (Fig. 3).

Quality assessment. Read count distributions by mean sequence quality of transcriptome reads (a) and sRNA reads (c). Mean quality score distributions of transcriptome reads (b) and sRNA reads (d).
Assembly quality control
Different tissues from different trees were collected in this study to comprehensively cover the D. involucrata transcriptome. Because reads were aligned to the merged assembly, we summarized the mapping percentages in Table 1 and then calculated the FPKM value for each sequence. The read mapping results showed that more than 90% of the reads were mapped to the transcriptome, and the sequences with FPKM values of 0 were removed. At the same time, the contaminant sequences were removed according to the LCA-based taxonomy classification method. The N50 and GC content for each assembly were also calculated using NGS QC Toolkit (v2.3.3)60 (Table 3).
We then employed BUSCO (v3)61 to evaluate the completeness of the final assembly using the 1,440 Embryophyta expected genes database (version 2). This analysis showed (Table 7) that 1,285 (89.24%) and 50 (3.47%) of the 1,440 expected Vertebrata genes were identified as complete and fragmented, respectively, while 105 (7.29%) genes were considered missing in the final assembly. BUSCO was also used to evaluate the completeness of the final gene set, and 87.99% of the 1,440 expected genes were identified (Table 7).
To assess the completeness of the core gene families (CoreGFs) within the green plant lineage, the CoreGF score was calculated using PLAZA (v2.5)62. First, the assembled transcriptome sequences were searched against the CoreGF gene set using BLASTx, and the protein sequences of high-quality CDSs were searched against the CoreGFs using BLASTp (V2.4.0) with e-value < 1e-5. Then, scores were calculated using the script coreGF_plaza2.5_geneset.py in PLAZA based on the BLAST hits. The CoreGF weighted score was 98.85%, and only 33 out of 2,928 CoreGFs were missing from the final assembly, while the score was 97.13%, and only 85 CoreGFs were missing in the high-quality coding gene set.
Contamination screening
The transcriptome sequences were subjected to contamination screening as mentioned in the methods, and the contaminant sequences were removed. The results of the contamination analysis also showed that (1) the main contaminants came from fungi or Arthropoda; (2) the most abundant fungi were Paraphaeosphaeria sporulosa in the Ascomycota; and (3) the transcriptome showed the highest similarity with Prunus avium, followed by Theobroma cacao and Nicotiana attenuata, in Viridiplantae (Fig. 5a).

Functional annotation. (a) Venn diagram of annotations based on the databases NR, KOG, KEGG, Swiss-Prot and InterPro. (b) Species distribution of annotated database NR sequences. (c) KEGG pathway annotations. Bars represent the numbers of unigenes clustered into KEGG Orthology (KO) hierarchies.
sRNAs were assembled using velvet (v1.2.10)63 with the parameters “velveth Assemout 15-short -fastq.gz” and “velvetg Assemout”. Assembled contigs with lengths greater than 100 bp were extracted to detect potential symbionts using the virus detection pipeline published previously64. In brief, a total of 115 queries with lengths greater than 100 bp were searched against the NCBI virus database using BLASTn with e-value < 1e-5. All matched sequences were searched against the Nt database using BLASTn with e-value < 1e-5 to identify false positives. After detection, none of the known viruses were detected in the sRNA data. In addition, sRNA reads mapped to the contaminant transcriptome sequences were removed from further analysis.
Annotation quality control
Statistical results of the functional annotation are summarized in Table 4 (Fig. 5b). A total of 65,534 (62.75%) unigenes were annotated. The distribution of KEGG pathway annotations is shown in Fig. 5c.
Statistical results for the predicted CDSs are summarized in Table 5. A total of 58,561 coding regions were detected by TransDecoder. The total, maximum and minimum lengths of the predicted CDSs were 60,633,393, 12,852 and 297. N50 and GC content were also calculated. Statistical results for the final high-quality coding genes are also summarized in Table 5.
Code Availability
All analyses were performed using open source software tools, and the detailed parameters for each tool are shown in the relevant methods.
References
Takhtajan, A. L. Outline of the classification of flowering plants (Magnoliophyta). The botanical review 46, 225–359 (1980).
Chase, M. W. et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181, 1–20 (2016).
Fu, L. & Jin, J. The Red Book of Chinese Plants–Rare and Endangered Plants. (Science Press, 1992).
Li, H. Davidia as the type of a new family Davidiaceae. Lloydia 17, 31 (1954).
Jiaxun, Z. Chinese Dovetree–Davidia involucrata. Journalof Plants 1, 008 (1988).
Fang, W.-p. & Chang, C. Y. Flora Reipublicae Popularis Sinicae: Angiospermae Dicotyledoneae. (Science Press, 1983).
Yeqin, Y. & Youyuan, X. A Preliminary Study On The Ecological Characteristics of Dovetree in Guizhou Province. Scientia Silvae Sinicae 22, 426–430 (1986).
Sun, J.-F., Gong, Y.-B., Renner, S. S. & Huang, S.-Q. Multifunctional bracts in the dove tree Davidia involucrata (Nyssaceae: Cornales): rain protection and pollinator attraction. The American Naturalist 171, 119–124 (2007).
Sun, J.-F. & Huang, S.-Q. White bracts of the dove tree (Davidia involucrata): Umbrella and pollinator lure. The Magazine of the Arnold Arboretum 68, 2–10 (2011).
Claßen-Bockhoff, R. & Arndt, M. Flower-like heads from flower-like meristems: pseudanthium development in Davidia involucrata (Nyssaceae). Journal of plant research 131, 443–458 (2018).
Jerominek, M., Bull-Hereñu, K., Arndt, M. & Claßen-Bockhoff, R. Live imaging of developmental processes in a living meristem of Davidia involucrata (Nyssaceae). Frontiers in plant science 5, 613 (2014).
Yi, Y., Luo, S., Li, X., Wang, L. & Xu, W. Studies on anatomical structure of dove tree stem and its formation of the callus. Journal of Hubei Institute for Nationalities (Natural Science) 18, 3–6 (2000).
Tang, C. Q. et al. Potential effects of climate change on geographic distribution of the Tertiary relict tree species Davidia involucrata in China. Scientific Reports 7, 43822 (2017).
Ma, Q. et al. Phylogeography of Davidia involucrata (Davidiaceae) inferred from cpDNA haplotypes and nSSR data. Systematic botany 40, 796–810 (2015).
Zhang, Y.-j., Li, M.-d., Shi, X. & Guan, P. Extraction of Genomic DNA and Optimization of ISSR-PCR Reaction System in Davidia involucrata [J]. Journal of Mountain Agriculture and Biology 3, 211–213 (2011).
Luo, S. et al. Genetic diversity and genetic structure of different populations of the endangered species Davidia involucrata in China detected by inter-simple sequence repeat analysis. Trees 25, 1063–1071 (2011).
Congwen, S. & Manzhu, B. Genetic diversity of RAPD mark for natural Davidia involucrata populations. Frontiers of Forestry in China 1, 95–99 (2006).
Du, Y. J. et al. Development of microsatellite markers for the dove tree, Davidia involucrata (Nyssaceae), a rare endemic from China. American journal of botany 99, e206–e209 (2012).
Chen, J.-M. et al. Chloroplast DNA phylogeographic analysis reveals significant spatial genetic structure of the relictual tree Davidia involucrata (Davidiaceae). Conservation Genetics 16, 583–593 (2015).
Dai, P., Ren, R., Dong, X., Li, M. & Cao, F. Bioinformatics Analysis of DiMYB1 Gene in Davidia involucrata Baill. Northern Horticulture 2, 027 (2017).
Ji, H. et al. Cloning and expression of a cold-induced gene (DiRCI) from Davidia involucrata (Davidiaceae). Acta Botanica Yunnanica 32, 151–157 (2010).
Ren, R. et al. Selection and validation of suitable reference genes for RT-qPCR analysis in dove tree (Davidia involucrata Baill.). Trees 33, 837–849 (2019).
Li, M. et al. De novo transcriptome sequencing and gene expression analysis reveal potential mechanisms of seed abortion in dove tree (Davidia involucrata Baill.). BMC plant biology 16, 82 (2016).
Yu, T., Lv, J., Li, J., Du, F. K. & Yin, K. The complete chloroplast genome of the dove tree Davidia involucrata (Nyssaceae), a relict species endemic to China. Conservation Genetics Resources 8, 263–266 (2016).
Li, Y.-X., Chen, L., Juan, L., Li, Y. & Chen, F. Suppression subtractive hybridization cloning of cDNAs of differentially expressed genes in dovetree (Davidia involucrata) bracts. Plant molecular biology reporter 20, 231–238 (2002).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, gix120 (2017).
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016).
Xie, Y. et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666 (2014).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1, 18 (2012).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323 (2011).
Huson, D. H. et al. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS computational biology 12, e1004957 (2016).
Coordinators, N. R. Database resources of the national center for biotechnology information. Nucleic acids research 44, D7 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Boutet, E., Lieberherr, D., Tognolli, M., Schneider, M. & Bairoch, A. In Plant bioinformatics 89–112 (Springer, 2007).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic acids research 36, D480–D484 (2007).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics 4, 41 (2003).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic acids research 43, D213–D221 (2014).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic acids research 33, W116–W120 (2005).
Consortium, G. O. The Gene Ontology (GO) database and informatics resource. Nucleic acids research 32, D258–D261 (2004).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494 (2013).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature methods 12, 59 (2014).
Finn, R. D. et al. Pfam: the protein families database. Nucleic acids research 42, D222–D230 (2013).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic acids research 41, e121–e121 (2013).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European molecular biology open software suite. Trends in genetics 16, 276–277 (2000).
Pérez-Rodríguez, P. et al. PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic acids research 38, D822–D827 (2009).
Thiel, T., Michalek, W., Varshney, R. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theoretical and applied genetics 106, 411–422 (2003).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research 42, D68–D73 (2013).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic acids research 43, D130–D137 (2014).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Evers, M., Huttner, M., Dueck, A., Meister, G. & Engelmann, J. C. miRA: adaptable novel miRNA identification in plants using small RNA sequencing data. BMC Bioinformatics 16, 370–370 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wu, H., Ma, Y., Chen, T., Wang, M. & Wang, X. PsRobot: a web-based plant small RNA meta-analysis toolbox. Nucleic Acids Research 40, 22–28 (2012).
Lavorgna, G., Guffanti, A., Borsani, G., Ballabio, A. & Boncinelli, E. TargetFinder: Searching annotated sequence databases for target genes of transcription factors. Bioinformatics 15, 172–173 (1999).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP178176 (2019).
Yang, H. TSA: Davidia involucrata, transcriptome shotgun assembly. GenBank, https://identifiers.org/ncbi/insdc:GHES00000000 (2019).
Yang, H. & Zhou, C. Reference gene set and small RNA set construction with multiple tissues from Davidia involucrata Baill. figshare, https://doi.org/10.6084/m9.figshare.8378594 (2019).
Patel, R. K. & Jain, M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one 7, e30619 (2012).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Van Bel, M. et al. Dissecting plant genomes with the PLAZA comparative genomics platform. Plant physiology 158, 590–600 (2012).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research 18, 821–829 (2008).
Zhou, C. et al. Characterization of viral RNA splicing using whole-transcriptome datasets from host species. Scientific reports 8, 3273 (2018).
Acknowledgements
This work was supported by the Science and Technology Department of Sichuan Province under Grants 2017SZ0181 and 2018NZDZX0003, the National Key R & D Programme of the People’s Republic of China under Grant 2018YFC1802605 and the Fundamental Research Funds for the Central Universities under Grant SCU2019D013.
Author information
Authors and Affiliations
Contributions
Y.Z. and R.W. conceived and designed the experiments. H.Y., C.Z., G.L. and J.W. collected the samples, H.Y. and C.Z. performed the bioinformatics analyses and wrote the article. R.W., M.W., Y.Z., P.G. and G.L. revised the article. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Yang, H., Zhou, C., Li, G. et al. Reference gene and small RNA data from multiple tissues of Davidia involucrata Baill. Sci Data 6, 181 (2019). https://doi.org/10.1038/s41597-019-0190-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0190-7
This article is cited by
-
Exploring the tymovirales landscape through metatranscriptomics data
Archives of Virology (2022)
