
Abstract
Nucleocytoplasmic transport (NCT) decline occurs with aging and neurodegeneration. Here, we investigated the NCT pathway in models of amyotrophic lateral sclerosis–fused in sarcoma (ALS–FUS). Expression of ALS–FUS led to a reduction in NCT and nucleoporin (Nup) density within the nuclear membrane of human neurons. FUS and Nups were found to interact independently of RNA in cells and to alter the phase-separation properties of each other in vitro. FUS–Nup interactions were not localized to nuclear pores, but were enriched in the nucleus of control neurons versus the cytoplasm of mutant neurons. Our data indicate that the effect of ALS-linked mutations on the cytoplasmic mislocalization of FUS, rather than on the physiochemical properties of the protein itself, underlie our reported NCT defects. An aberrant interaction between mutant FUS and Nups is underscored by studies in Drosophila, whereby reduced Nup expression rescued multiple toxic FUS-induced phenotypes, including abnormal nuclear membrane morphology in neurons.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


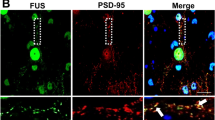
Data availability
No datasets that require mandatory deposition into a public database were generated during the current study. Any data generated and/or analyzed during the current study are available from the corresponding author on reasonable request. Source data are provided with this paper.
Code availability
The code used for the FRAP analysis of recombinant proteins is provided in the Supplementary Software (Supplemental_Script_FRAP.py).
References
Frey, S., Richter, R. P. & Görlich, D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science 314, 815–817 (2006).
Ryan, V. H. & Fawzi, N. L. Physiological, pathological, and targetable membraneless organelles in neurons. Trends Neurosci. 42, 693–708 (2019).
Schmidt, H. B. & Görlich, D. Transport selectivity of nuclear pores, phase separation, and membraneless organelles. Trends Biochem. Sci. 41, 46–61 (2016).
D’Angelo, M. A., Raices, M., Panowski, S. H. & Hetzer, M. W. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell 136, 284–295 (2009).
Mertens, J. et al. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Stem Cell 17, 705–718 (2015).
Daigle, N. et al. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 154, 71–84 (2001).
Toyama, B. H. et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 154, 971–982 (2013).
Sugiyama, K. et al. Calpain-dependent degradation of nucleoporins contributes to motor neuron death in a mouse model of chronic excitotoxicity. J. Neurosci. 37, 8830–8844 (2017).
Hutten, S. & Dormann, D. Nucleocytoplasmic transport defects in neurodegeneration—cause or consequence? Semin. Cell Dev. Biol. 99, 151–162 (2019).
Freibaum, B. D. et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133 (2015).
Zhang, K. et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525, 56–61 (2015).
Jovičić, A. et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229 (2015).
Boeynaems, S. et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6, 20877 (2016).
Coyne, A. N. et al. G4C2 repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron 107, 1124–1440 (2020).
Chou, C.-C. et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 21, 228–239 (2018).
Giampetruzzi, A. et al. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 10, 3827 (2019).
Sama, R. R. K., Ward, C. L. & Bosco, D. A. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 6, 1759091414544472 (2014).
Dormann, D. et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 29, 2841–2857 (2010).
Woerner, A. C. et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176 (2016).
Görlich, D. & Mattaj, I. W. Nucleocytoplasmic transport. Science 271, 1513–1518 (1996).
Kodiha, M., Chu, A., Matusiewicz, N. & Stochaj, U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 11, 862–874 (2004).
Bosco, D. A. et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175 (2010).
Baron, D. M. et al. Quantitative proteomics identifies proteins that resist translational repression and become dysregulated in ALS-FUS. Hum. Mol. Genet. 28, 2143–2160 (2019).
Cassany, A. & Gerace, L. Reconstitution of nuclear import in permeabilized cells. Methods Mol. Biol. 464, 181–205 (2009).
Tischbein, M. et al. The RNA-binding protein FUS/TLS undergoes calcium-mediated nuclear egress during excitotoxic stress and is required for GRIA2 mRNA processing. J. Biol. Chem. 294, 10194–10210 (2019).
Kaushansky, L. J. Investigating the Effects of Mutant FUS on Stress Response in Amyotrophic Lateral Sclerosis. MSc thesis, Univ. Massachusetts Medical School (2015).
Khosravi, B. et al. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 26, 790–800 (2016).
Wang, H. et al. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun. 9, 3683–18 (2018).
Kamelgarn, M. et al. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta 1862, 2004–2014 (2016).
Burke, K. A., Janke, A. M., Rhine, C. L. & Fawzi, N. L. Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell 60, 231–241 (2015).
Murakami, T. et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690 (2015).
Maharana, S. et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 (2018).
Protter, D. S. W. et al. Intrinsically disordered regions can contribute promiscuous interactions to RNP granule assembly. Cell Rep. 22, 1401–1412 (2018).
Davis, L. I. & Blobel, G. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc. Natl Acad. Sci. USA 84, 7552–7556 (1987).
Cordes, V. C., Reidenbach, S. & Franke, W. W. Cytoplasmic annulate lamellae in cultured cells: composition, distribution, and mitotic behavior. Cell Tissue Res. 284, 177–191 (1996).
Underwood, J. M., Becker, K. A., Stein, G. S. & Nickerson, J. A. The Ultrastructural signature of human embryonic stem cells. J. Cell. Biochem. 118, 764–774 (2017).
Sama, R. R. K. et al. ALS-linked FUS exerts a gain of toxic function involving aberrant p38 MAPK activation. Sci. Rep. 7, 115 (2017).
Lanson, N. A. et al. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 20, 2510–2523 (2011).
Casci, I. et al. Muscleblind acts as a modifier of FUS toxicity by modulating stress granule dynamics and SMN localization. Nat. Commun. 10, 5583 (2019).
Chen, Y. et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2, 477–486 (2011).
Rademakers, R. et al. Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42, 170–176 (2010).
Şahin, A. et al. Human SOD1 ALS mutations in a Drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205, 707–723 (2017).
Shi, K. Y. et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl Acad. Sci. USA 114, 1111–1117 (2017).
Vanneste, J. et al. C9orf72-generated poly-GR and poly-PR do not directly interfere with nucleocytoplasmic transport. Sci. Rep. 9, 15728 (2019).
Mann, J. R. et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8 (2019).
Kamelgarn, M. et al. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl Acad. Sci. USA 115, E11904–E11913 (2018).
Yamashita, T., Aizawa, H., Teramoto, S., Akamatsu, M. & Kwak, S. Calpain-dependent disruption of nucleo-cytoplasmic transport in ALS motor neurons. Sci. Rep. 7, 3643 (2017).
Ibarra, A. & Hetzer, M. W. Nuclear pore proteins and the control of genome functions. Genes Dev. 29, 337–349 (2015).
Archbold, H. C. et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 8, 920 (2018).
Steyaert, J. et al. FUS-induced neurotoxicity in Drosophila is prevented by downregulating nucleocytoplasmic transport proteins. Hum. Mol. Genet. 27, 4103–4116 (2018).
Ng, S.-Y. et al. Genome-wide RNA-seq of human motor neurons implicates selective ER stress activation in spinal muscular atrophy. Cell Stem Cell 17, 569–584 (2015).
Skarnes, W. C., Pellegrino, E. & McDonough, J. A. Improving homology-directed repair efficiency in human stem cells. Methods 164–165, 18–28 (2019).
Eftekharzadeh, B. et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 99, 925–940.e7 (2018).
Baron, D. M. et al. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol. Neurodegener. 8, 30 (2013).
Stoica, R. et al. ALS/FTD‐associated FUS activates GSK‐3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 17, 1326–1342 (2016).
An, H. et al. ALS-linked FUS mutations confer loss and gain of function in the nucleus by promoting excessive formation of dysfunctional paraspeckles. Acta Neuropathol. Commun. 7, 7 (2019).
Koulouras, G. et al. EasyFRAP-web: a web-based tool for the analysis of fluorescence recovery after photobleaching data. Nucleic Acids Res. 46, W467–W472 (2018).
Monahan, Z. et al. Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967 (2017).
Pandey, U. B. et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 (2007).
Ramesh, N. et al. RNA dependent suppression of C9orf72 ALS/FTD associated neurodegeneration by Matrin-3. Acta Neuropathol. Commun. 8, 1182 (2020).
Ortega, J. A. et al. Nucleocytoplasmic proteomic analysis uncovers eRF1 and nonsense-mediated decay as modifiers of ALS/FTD C9orf72 toxicity. Neuron 106, 90–107.e13 (2020).
Frost, B., Bardai, F. H. & Feany, M. B. Lamin dysfunction mediates neurodegeneration in tauopathies. Curr. Biol. 26, 129–136 (2016).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 (2008).
Lee, E. B. et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 134, 65–78 (2017).
Acknowledgements
We thank C. Fallini and S. Parekh for helpful discussion on the manuscript. We are thankful to the following investigators and facilities for providing iPSC lines: R. Baloh and the Induced Pluripotent Stem Cell Core at Cedars Sinai; K. Eggan (Harvard Medical School); F.-B. Gao (University of Massachusetts Medical School); N. Maragakis and the Ansari ALS Stem Cell Core at Johns Hopkins; L. Van Den Bosch (Vlaams Instituut voor Biotechnologie); and Target ALS and Rutgers University-based core facility (RUCDR). We gratefully acknowledge the contribution of the Cellular Engineering Service at The Jackson Laboratory for expert assistance in the gene editing of the iPSCs described in this manuscript. We thank J. McKeon for assistance with cell culture, A. Murthy for advice on the recombinant MBP–FUS protein, and M. Hammer for help with microscopy. We are grateful to the US National Institutes of Health (NIH), National Science Foundation (NSF) and other funding sources as follows to D.A.B: NIH R01 NS078145, R01 NS108769 (to D.A.B. and D.G.) and R21 NS091860; to U.B.P: NIH R01 NS081303, R21 NS101661, NS111768, AG064940 and NS100055, the Muscular Dystrophy Association, the ALS Association, the Robert Packard Center for ALS at Johns Hopkins; to N.L.F.: NIH R01 GM118530, the Human Frontier Science Program RGP0045/2018, NSF 1845734; to D.G.: NIH U01 DA047733, NIH GM123541, NSF 1917206; to E.B.L.: NIH P30 AG072979 and P01 AG066597; and to J.E.L.: NIH R01 NS073873 and R56 NS073873.
Author information
Authors and Affiliations
Contributions
Y.-C.L., M.S.K., N.R., U.B.P. and D.A.B. conceptualized and designed the project. Y.-C.L., M.S.K., N.R., E.N.A., A.T.N., B.K., S.C., J.A.M. and W.C.S. conducted the experiments. Y.-C.L., M.S.K., N.R., E.N.A., A.T.N., B.K., S.C., J.A.M., W.C.S., I.R.A.M., E.B.L., J.A.N., D.G., U.B.P. and D.A.B. analyzed the data. J.E.L. and N.L.F. provided critical intellectual input. R.L.-G. reprogrammed the control iPSC line designated as C1. D.A.B. wrote the paper with the co-first authors and contributions from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Neuroscience thanks Ludo Van Den Bosch, Liesbeth Veenhoff, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Methods, Supplementary Figures 1–19, Supplementary Table 1, Supplementary References.
Supplementary Software
A python script to normalize in vitro FRAP intensities
Supplementary Data 1
Unprocessed western blots for Supplementary Fig. 4
Supplementary Data 2
Unprocessed western blots for Supplementary Fig. 6
Supplementary Data 3
Unprocessed western blots and gels for Supplementary Fig. 7
Supplementary Video 1
Video of time lapse fluorescence images of the FRAP experiment on MBP-FUS droplets
Supplementary Video 2
Video of time lapse fluorescence images of the FRAP experiment on MBP-FUS/Nup62 assemblies
Source data
Source Data Fig. 1
Unprocessed western blots for Fig. 1
Source Data Fig. 3
Unprocessed western blots for Fig. 3
Rights and permissions
About this article

Cite this article
Lin, YC., Kumar, M.S., Ramesh, N. et al. Interactions between ALS-linked FUS and nucleoporins are associated with defects in the nucleocytoplasmic transport pathway. Nat Neurosci 24, 1077–1088 (2021). https://doi.org/10.1038/s41593-021-00859-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-021-00859-9
This article is cited by
-
Nuclear-import receptors as gatekeepers of pathological phase transitions in ALS/FTD
Molecular Neurodegeneration (2024)
-
Rapid and high-purity differentiation of human medium spiny neurons reveals LMNB1 hypofunction and subtype necessity in modeling Huntington’s disease
Inflammation and Regeneration (2024)
-
Molecular hallmarks of ageing in amyotrophic lateral sclerosis
Cellular and Molecular Life Sciences (2024)
-
Expression of ALS-PFN1 impairs vesicular degradation in iPSC-derived microglia
Nature Communications (2024)
-
Looking for answers far away from the soma—the (un)known axonal functions of TDP-43, and their contribution to early NMJ disruption in ALS
Molecular Neurodegeneration (2023)
