
Abstract
The function of many biological systems, such as embryos, liver lobules, intestinal villi, and tumors, depends on the spatial organization of their cells. In the past decade, high-throughput technologies have been developed to quantify gene expression in space, and computational methods have been developed that leverage spatial gene expression data to identify genes with spatial patterns and to delineate neighborhoods within tissues. To comprehensively document spatial gene expression technologies and data-analysis methods, we present a curated review of literature on spatial transcriptomics dating back to 1987, along with a thorough analysis of trends in the field, such as usage of experimental techniques, species, tissues studied, and computational approaches used. Our Review places current methods in a historical context, and we derive insights about the field that can guide current research strategies. A companion supplement offers a more detailed look at the technologies and methods analyzed: https://pachterlab.github.io/LP_2021/.
Similar content being viewed by others

Main
It has long been recognized that in biological systems ranging from the Drosophila embryo to the hepatic lobule, many genes need to be properly regulated in space for the system to function. To study the spatial patterns of gene expression, many different spatial transcriptomics methods, which produce spatially localized quantification of messenger RNA (mRNA) transcripts as proxies for gene expression, have been developed. Thanks to growing interest in the field, several reviews have been written in the past 5 years, providing overviews of experimental techniques for data collection1,2, and describing how such techniques can be applied to specific biological systems, for example tumors3, brain4, and liver5. These reviews typically begin with either laser capture microdissection (LCM) or single-molecule fluorescent in situ hybridization (smFISH) in the late 1990s, although the quest to profile the transcriptome in space is much older.
Unlike the previous reviews, this paper presents a database of literature dating back to 1987 comprehensively documenting the historical evolution and current development in data collection and analysis in spatial transcriptomics. In addition, we have analyzed the literature metadata from the database to show trends in the field. Key highlights from the database and analyses are presented in this paper, and more details are presented in our book-length supplement: https://pachterlab.github.io/LP_2021/. Section and figure numbers of the supplement in this paper refer to those in the DOI PDF version, while those in the online HTML version are subject to change, as it is continuously updated to reflect changes in the field. This database was curated by searching keywords such as “spatial transcriptomics” and “Visium” on PubMed and bioRxiv, and manually screening literature citing influential papers in the field. Literature metadata collected include the date published or posted and the institution of the first author. In addition, metadata for publications concerning new datasets include the species and tissue from which the data were collected, the experimental techniques used to collect the data, and the programming languages used to analyze the data. Metadata for publications concerning new data-analysis methods include the programming languages used in the implementation, the code repository of the implementation, and whether the code is packaged and documented. The database is continuously updated by manually screening RSS feeds from PubMed and bioRxiv for relevant keywords, or by submission via a Google Form.
Prequel era
By “spatial transcriptomics”, we mean attempts to quantify mRNA expression of large numbers of genes within the spatial context of tissues and cells. Some important technologies enabling spatial transcriptomics date back to the 1970s (Chapter 2 in Supplementary Information). Various forms of in situ hybridization (ISH) have been used for a long time to visualize gene expression in space. Radioactive ISH was first introduced in 1969, visualizing ribosomal RNA6 and DNA7 in Xenopus laevis oocytes, and was first used to visualize transcripts of specific genes (globin) in 1973 (ref. 8) (Fig. 1a). Non-radioactive fluorescent or colorimetric ISH was developed in the 1970s and the early 1980s, improving spatial resolution, enabling three-dimensional (3D) staining, and shortening required exposure times9,10 (Fig. 1a). Early ISH was performed in tissue sections, making it challenging to apply to blastulas and to reconstruct 3D tissue structures; whole-mount ISH (WM ISH) was first introduced in Drosophila in 1989 (ref. 11) and was soon adapted to other species, such as mice, in the early 1990s (ref. 12).

a, Development of prequel era technologies. References: 1969 radioactive ISH of ribosomal RNA (rRNA)6,7, 1973 radioactive ISH of goblin mRNAs8, 1977 FISH of rRNA10, 1982 immunological FISH with biotin-labeled probe9, 1982 FISH of actin mRNA135, 1987 Drosophila enhancer trap14, 1989 WM ISH in Drosophila11, 1989 ES cell enhancer and gene trap in mice15, 1991 in situ reporter in Caenorhabditis elegans136. b, Major WM ISH atlases and gene expression pattern databases. References: 1994 scaling up WM ISH in C. elegans137, 1995 first mouse WM ISH138, 1998 AXelDB139, 1999 mouse: GXD140, 2000 Maboya Gene Expression patterns and Expression Sequence Tags (MAGEST)141, 2001 the Nematode Expression Pattern Database (NEXTDB)142, 2001 Ghost143, 2002 GenePaint144, 2002 D. melanogaster: Berkeley Drosophila Genome Project (BDGP)24, 2003 Medaka Expression Pattern Database (MEPD)145, 2003 Zebrafish Information Network (ZFIN)31, 2004 Gallus Expression In Situ Hybridization Analysis (GEISHA)25, 2005 miRNA atlas29, 2006 Allen26, 2006 BDTNP146, 2007 Fly-FISH147, 2007 Xenbase148, 2011 mouse Genitourinary Development Molecular Anatomy Project (GUDMAP)27, 2017 LungMAP28, 2020 ZEBrA149. c, Development of current-era technologies and their notable precursors, colored by type of technology. References: 1976 LCM17, 1988 ligase-mediated single -nucleotide variant (SNV) detection150, 1989 single-cell cDNA amplification151,152, 1989 FISH with combinatorial barcoding21, 1995 cDNA microarray153, 1996 commercial LCM18,19, 1998 smFISH23, 1999 LCM + microarray154, 2002 combinatorial FISH22, 2008 RNA-seq155, 2012 Tomo-array156 (the cDNA microarray predecessor of Tomo-seq), 2013 high-throughput RCA + ISS59, 2014 seqFISH20, 2015 MERFISH50, 2016 ST87, and 2019 GeoMX DSP44.
Another strand of development in early spatial transcriptomics was the enhancer and gene trap screen, which was developed in the 1980s when DNA sequencing throughput was increasing13 and metazoan genomes were newly opened frontiers. The first screens in Drosophila14 and mice15 were performed in the late 1980s in order to visualize expression of untargeted, and often previously unknown, genes. With increasing throughput, enhancer and gene traps became the technology of choice for spatial transcriptomics in the 1990s, until the rise of WM ISH in the late 1990s, which leveraged automation. WM ISH also avoided the need for transgenic lines, and was facilitated by the availability of reference genomes in the early 2000s for computational probe design. Although now eclipsed by newer methods, enhancer trap, gene trap, and in situ reporter methods have been used to build reference databases of gene expression and enhancer usage patterns in transgenic lines throughout the 2000s and 2010s16.
The foundation for many current-era technologies was built in the decades between the 1970s and the 2000s (Fig. 1c). For example, ultraviolet (UV) laser was first used to cut tissue in 1976 (ref. 17). Popular infrared (IR) and UV LCM systems were first reported in 1996 (refs. 18,19) and were soon commercialized. Some highly multiplexed smFISH technologies, such as sequential FISH (seqFISH)20, rely on combinatorial barcoding, that is encoding each gene with a combination of colors so transcripts of more genes with easily discernible colors (up to 5) can be quantified simultaneously. Combinatorial barcoding was first reported in immunological DNA FISH in 1989 (ref. 21) and was first used for transcripts in 2002 (ref. 22). The first unequivocal demonstration of smFISH showing each mRNA molecule as a spot was reported in 1998 (ref. 23). Highly multiplexed smFISH would not have been possible without the development of these technologies.
WM ISH was the technology of choice in the late 1990s and the 2000s, before the rise of highly multiplexed, high-resolution, and more quantitative technologies, and has been used to create gene expression atlases in embryos of several species such as Drosophila melanogaster24, Mus musculus, and Gallus gallus25; in various mouse organs such as the brain26, genitourinary tract27, and lung28; and for specific types of genes, such as microRNAs (miRNAs)29 (Fig. 1b). For miRNAs and many species other than mice and humans, the only spatial transcriptomics resources currently available are, for the most part, WM ISH atlases. Model-organism databases collecting proliferating gene expression patterns from various sources were also established in this period, such as Gene Expression Database (GXD)30 and Zebrafish Information Network31 (Fig. 1b). The golden age of WM ISH seems to have ended in the 2010s (Fig. 1b), perhaps due to some of the technology’s disadvantages, such as requiring stereotypical tissue structure, the need for thousands of animals to generate an atlas, and the largely qualitative nature of results.
Early motivating applications for spatial transcriptomics included identification of genes with restricted patterns that indicated function in development, identification of novel cell-type markers, and identification of novel cell types not evident from tissue morphology14,15. In the 1980s and 1990s, analyses were typically done manually, although more recently automated methods have been developed (Chapter 3 in Supplementary Information). Convergence of strands of technologies, including more powerful computing infrastructure, decreasing cost of sequencing, and the generation of more quantitative data, have mainstreamed and revolutionized spatial transcriptomics and opened up new possibilities. However, the legacy of the prequel era still lives on in the usage of prequel resources, such as referencing the Allen Brain Atlas (ABA)32 and the Allen Mouse Common Coordinate Framework33, and in institutions such as the Allen Brain Institute and the Jackson Laboratory, which are contributing to the current era of research34,35.
Data collection
Current-era technologies broadly fall into five categories in terms of how spatial information is acquired: region of interest (ROI) selection (Section 5.1 in Supplementary Information), smFISH (Section 5.2 in Supplementary Information), in situ sequencing (ISS) (Section 5.3 in Supplementary Information), next-generation sequencing (NGS) with spatial barcoding (Section 5.4 in Supplementary Information), and methods not requiring a priori spatial locations (Section 5.6 in Supplementary Information). Developers of such technologies often seek to enable a trifecta of transcriptome-wide profiling, single-cell resolution, and high gene-detection efficiency. Although this achievement appears to be increasingly within reach, current-era technologies are characterized by trade-offs between these goals.
ROI selection
Spatial locations can be obtained by selection and isolation of ROIs of known locations and shapes, which can be performed by physical (Section 5.1.3 in Supplementary Information) and optical marking of ROIs for isolation (Section 5.1.4 in Supplementary Information). The isolated ROIs can then be analyzed with complementary DNA (cDNA) microarray or RNA sequencing (RNA-seq), or dissociated into single cells for single-cell RNA-seq (scRNA-seq).
Physical microdissection includes LCM, 2000s voxelation36, and Tomo-seq37, which sections a tissue with a cryotome along an axis of interest, followed by RNA-seq on each section. Since 1999, by far the most widely used microdissection technology is LCM, which has been used in various biological fields, such as oncology, neuroscience, immunology, developmental biology, and botany (see Chapter 6 in the Supplementary Information for topic modeling of PubMed and bioRxiv LCM literature). In LCM, ROIs in the tissue section are dissected by either UV laser cutting (lasers manufactured by Zeiss and Leica) or fusion of tissue with a membrane by IR laser (manufactured by Arcturus, Fig. 2a); the two are combined in recent versions of Arcturus, in which IR fusion removes the ROI cut using UV. Combining LCM and Tomo-seq, the spatial transcriptome in 3D can be profiled as in geographical position sequencing (Geo-seq)38, albeit with limited spatial resolution. An innovative physical microdissection method is STRP-seq39, which slices adjacent tissue sections into stripes at different angles and reconstructs gene expression patterns in 3D with an algorithm inspired by ray-based computerized tomography. Manual dissection is commonly used to profile gene expression along one spatial axis of interest in plants40.

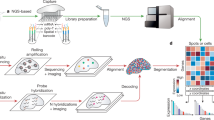
a, IR LCM. b, GeoMX DSP. The purple circle in step 2 is the UV-illuminated ROI. c, seqFISH barcoding and error correction scheme: if signal from one round of hybridization is missing, the remaining rounds can still uniquely identify the gene barcoded. d, MERFISH Hamming distance 4 barcoding and error correction scheme: from the design of the barcodes, if signal from one round of hybridization is missing, the correct barcode can be recovered. If two rounds are missing, the remaining signals are equidistant to two different barcodes so the original barcode cannot be recovered. e, Cartana ISS with cPAL sequencing: many copies of the gene barcode are made with RCA for signal amplification, which are then sequenced in situ with cPAL. The orange line stands for the RCA amplicon. Short blue lines stand for the gene barcode. Brown stands for the probe; bases not labeled are degenerate. Gray stands for primer matching constant region. f, NGS barcoding techniques. In Visium, the spots are arranged in a hexagonal grid, 100 µm apart center to center and 55 µm in diameter. In DBiT-seq, positional barcodes are deposited in microfluidic channels and spatial resolution is determined by the width (down to 10 µm) and spacing of the channels. In Slide-seq, barcoded beads 10 µm in diameter are spread in a single layer on a slide. In XYZeq, spatial barcodes are conferred on multiple cells in wells 500 µm in diameter, which are then dissociated for scRNA-seq. In Seq-Scope, the tissue is mounted on a repurposed Illumina flow cell with barcoded polony spots 0.6 µm apart on average. For Visium and Slide-seq, the lines represent oligonucleotides attached to the slide or bead. For DBiT-seq, red and green lines represent the flow in microfluidic channels carrying barcoding oligonucleotides. For Seq-Scope, the tissue (pink block) is mounted on repurposed Illumina flow cell with bridge-amplified polonies each with its own spatial barcode represented by different colors. For XYZeq, different colors of the cells represent different spatial barcodes in the microwells, and the cells are dissociated for scRNA-seq. t-SNE, t-distributed stochastic neighbor embedding. g, Data-analysis workflow: upstream analysis is technology specific, and includes image processing for smFISH and ISS-based technologies, and FASTQ file processing, quality control of the gene count matrix, and data normalization for NGS-based technologies. Non-spatial scRNA-seq data can be integrated by mapping cells to locations with landmark genes in the smFISH or ISS data or deconvolving cell types in Visium spots. Downstream analyses tend to be technology agnostic, and include finding spatially variable genes, transcriptionally defined spatial regions, and cell–cell interactions. Created with BioRender.com.
Optical marking of ROIs includes NICHE-seq41, which uses two-photon irradiation to mark ROIs in tissue from transgenic mice expressing photoactivatable green fluorescent protein (PA-GFP), and then uses fluorescence-activated cell sorting (FACS) to isolate cells with activated PA-GFP for scRNA-seq. Similar to NICHE-seq but without transgenic mice is spatially photoactivatable color encoded cellular address tags (SPACECAT)42, which stains cultured live cells or organoids with photocaged fluorophores and photoactivates ROIs for FACS and scRNA-seq. Also using photocaging, ZipSeq43 attaches anchor oligonucleotides with photocaged overhangs to tissue with antibodies or lipid insertion, and adds spatial ‘zipcodes’ to photoactivated ROIs hybridizing to the overhangs. A more popular commercial optical ROI-selection technique is the GeoMX Digital Spatial Profiler (DSP)44 and whole-transcriptome atlas (WTA)45 of Nanostring (Fig. 2b), which shines UV light on ROIs to release photo-cleavable gene barcodes for quantification with either nCounter or NGS. As GeoMX uses predefined gene panels rather than poly-A capture, Nanostring provides the Cancer Transcriptome Atlas (CTA) gene panel with over 1,800 genes, as well as human and mouse whole-transcriptome panels with over 18,000 genes.
Single-molecule FISH
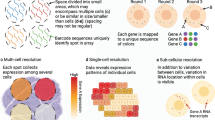
Chronologically, the next technology developed in the current era is highly multiplexed single-molecule FISH (smFISH), which began with a 2012 prototype (seqFISH) that relied on super-resolution microscopy (SRM) to simultaneously profile 32 genes in yeast by hybridizing probes with different colors to transcripts, and then deducing the relative locations of the colors present46. SRM is no longer needed; in 2014, seqFISH20 was published, in which one color per gene is visualized per round of hybridization, and the probes are stripped before the next round for the next color in the barcode. All transcripts of the same gene have the same barcode. Four colors and 8 rounds of hybridization (48 = 65,536) are more than enough to encode all genes in the human or mouse genome. In practice, an error-correcting round of hybridization is performed, so that genes can still be distinguished if signal from one round of hybridization is missing47 (Fig. 2c). More recently, in a version of seqFISH based on RNA sequential probing of targets (RNA SPOTs)48, the ‘colors’ themselves are one-hot encoded by a sequence of hybridizations, expanding the palette to 20 ‘colors’ per channel and enabling the profiling of 10,000 genes49.
Another smFISH technique is multiplexed error-robust FISH (MERFISH)50, which uses a different barcoding strategy, in which each gene is encoded by a binary code. The color codes in each experiment must be separated by a Hamming distance (HD) of four to allow for correction of missing signal in one round, and by two to identify error without the facility for correcting it (Fig. 2d). The length of barcodes can be increased to encode 10,000 genes51. As only the fluorophores are removed but the probes are not stripped, numerous rounds of hybridization in MERFISH are less time consuming than those in seqFISH. Most other smFISH-based techniques, such as hybridization-based ISS (HybISS)52 and split-FISH53, use either seqFISH-like or MERFISH-like barcoding.
smFISH faces a number of challenges, which have been addressed by various methods. Signal-to-noise ratio can be improved with rolling circle amplification (RCA)52, branched DNA (bDNA)54, hybridization chain reaction (HCR)47, primer exchange reaction55, and tissue clearing56. With an increasing number of genes profiled, the transcript spots are increasingly likely to overlap, causing optical crowding. This can be mitigated by expansion microscopy (ExM)57, only imaging a subset of probes at a time and using computational super-resolution49, imaging highly expressed genes without combinatorial barcoding50, and computationally resolving overlapping spots58.
In situ sequencing
ISS methods yield spatial transcriptome information by sequencing, typically by ligation (SBL), gene barcodes (targeted), or short fragments of cDNAs (untargeted) in situ. Such methods rely on ligase joining only two pieces of DNA—a primer with known sequence and a probe—if they match the template, and non-matching probes are washed away. The probes used are degenerate except for one or two query bases encoded by a color. RCA is commonly used for signal amplification. The 2013 ISS59, later commercialized by Cartana, and barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses (BOLORAMIS)60 use one query base per probe, as in combinatorial probe anchor ligation (cPAL)61, to sequence gene barcodes (Fig. 2e). In cPAL, each probe queries one base in the gene barcode. Fluorescence ISS (FISSEQ)62 and a later adaptation with ExM, called ExSeq63, use SOLiD, which uses two query bases per probe to sequence circularized and RCA-amplified cDNAs. In spatially resolved transcript amplicon readout mapping (STARmap)56, gene barcodes are sequenced by sequencing with error-reduction by dynamic annealing and ligation (SEDAL), in which SOLiD-like two query bases are used to reject error, but one-base encoding can also be used. Barcode analysis by sequencing (BAR-seq) also RCA amplifies probes with gene barcodes, but uses sequencing by synthesis (SBS) instead of SBL to sequence the barcodes64.
NGS with spatial barcoding
Spatial locations of transcripts can also be preserved by capturing the transcripts from tissue sections on in situ arrays. Such arrays can be manufactured by printing spot barcodes, unique molecular identifiers (UMIs), and poly-T oligonucleotides on commercial microarray slides to capture polyadenylated transcripts, as in the spatial transcriptomics (ST) and Visium technologies (Fig. 2f). They can also be Drop-seq-like beads65 with split pool barcodes, UMIs, and poly-T oligonucleotides spread on slides in a single layer (for example, Slide-seq66) or confined in wells etched on the slides (for example, high-definition spatial transcriptomics (HDST)67), with bead barcodes subsequently located using in situ SBL. Alternatively, in deterministic barcoding in tissue for spatial omics sequencing (DBiT-seq)68, an array is generated by microfluidic channels, which are used to deposit one type of barcode in one direction and then another in a perpendicular direction, with the orthogonal barcodes ligated so each spot can be identified with a unique pairwise combination. While NGS barcoding techniques are typically designed for 3′-end Illumina sequencing, Visium has been adapted to Nanopore long-read sequencing69.
NGS barcoding techniques have been applied to large areas of tissue33, and their use is increasing (Fig. 4b). Nevertheless, they do not have single-cell spatial resolution. The commonly used Visium has spots in a hexagonal array 100 µm center to center and 55 μm in diameter (Fig. 2f). Bead diameter is 10 μm in Slide-seq and 2 μm in HDST (Fig. 2f). Slide-seq and HDST use bead sizes smaller than single cells, but they may not always provide single-cell resolution because one bead can span two or more cells. Resolution of DBiT-seq is determined by channel width (either 50, 25, or 10 μm, Fig. 2f). More recently, the spot size can be reduced to below 1 μm, with RCA-amplified DNA nanoballs as small as 0.22 μm across, with spot barcodes deposited in wells that are 0.5 or 0.715 μm apart in Stereo-seq70, and in Seq-Scope polymerase colonies (polonies) with spatial barcodes ~0.6 μm center to center on an Illumina flow cell that has been repurposed to capture transcripts from tissue sections71 (Fig. 2f). Another polony-based method, PIXEL-seq, achieves a spot diameter of about 1.22 μm, but unlike in the flow cell, polony (or DNA cluster)-indexed library-sequencing (PIXEL-seq) does not have much spacing around each polony72. Techniques such as XYZeq73 and sci-Space74 have been developed to dissociate the single cell or nuclei in spatially barcoded spots for scRNA-seq, so the data have single-cell transcriptomic, but not spatial, resolution (Fig. 2f).
De novo reconstruction of spatial information
Some technologies have been developed to preserve information necessary to computationally reconstruct spatial gene expression patterns without knowing or collecting spatial locations. One such technology is DNA microscopy75,76, which records proximity between cDNAs. This information can be used to reconstruct relative locations of transcripts. At the cellular level, gene expression in rare cell types can be reconstructed by deliberately assaying multiplets and then mapping them to locations in a spatial reference on the basis of gene expression of cells from common cell types attached to cells from the rare cell types77. Variants of the term “spatial transcriptomics” have also been used to describe techniques localizing transcripts to organelles (for example, APEX-seq78), although no spatial coordinates are recorded.
Multi-omics
The transcriptome is only one aspect of cell function. Other aspects, such as the proteome, neuronal connectome, and 3D chromatin conformation are also important to cell function, and some methods have been developed to profile them along with the transcriptome in the same cells (Section 5.8 of Supplementary Information). For the proteome, oligonucleotide-tagged antibodies are used to detect proteins of interest, and the oligonucleotide signifying the protein species can be detected with smFISH-based methods. Such antibody panels have been combined with transcriptomics, such as in DBiT-seq68, SM-Omics79, GeoMX DSP44, and MERFISH80. With the oligonucleotide barcode, over 100 antibodies can be used, such as when using all available antibody panels for GeoMX DSP. For 3D chromatin conformation, MERFISH and seqFISH+ have been adapted to visualize chromatin structure, by targeting DNA genomic loci81 or introns of nascent transcripts81,82. For the neuronal connectome, multiplexed transcript quantification can also be combined with neuron projection tracing. For instance, cholera toxin subunit b (CTb) retrograde tracing has been used in conjunction with MERFISH to visualize axons83. Also, BAR-seq was originally designed to use ISS for axon tracing by sequencing neuron-specific barcodes introduced by a virus injected into the brain, but was later adapted to sequence gene barcodes64 as well. In addition, while not an -ome per se, electrophysiology has been recorded prior to transcriptome profiling in the same cells, such as with a patch–clamp in explanted human neurons, followed by HCR–smFISH84, and with extracellular electrodes in cultured cardiomyocytes, followed by STARmap in electro-seq85.
Comparison across categories
In this section, we discuss trade-offs, among high detection efficiency, transcriptome-wide profiling, high spatial resolution, and sometimes larger tissue area, made by different types of technologies, as well as practical factors relevant to selection of technology, such as FFPE compatibility and cost/usability.
Detection efficiency
Detection efficiency is commonly estimated by performing non-barcoded smFISH with near 100% sensitivity for select marker genes on the same cell type and comparing the average number of transcripts detected for each gene per cell for techniques in which cells can be segmented, or per unit tissue area for techniques without single-cell resolution. For NGS-based techniques with UMI, sometimes the number of UMIs and genes detected per cell or unit area is compared with that of other techniques with UMI. Note that comparisons of efficiencies are confounded by different tissues and methods used to estimate efficiencies in different studies and by different sequencing depths in NGS.
Highly multiplexed smFISH techniques tend to excel in this area, with ~95% for Hamming distance 4 MERFISH86 compared with non-barcoded smFISH; multiple rounds of hybridization tend to decrease the efficiency, in part because barcodes with incorrigible errors are discarded. NGS barcoding techniques tend to have lower efficiency. For select genes in the same tissue type, ST detected around 6.9% as many UMIs as transcript spots detected by non-multiplexed smFISH per unit area analyzed87, comparable to the detection efficiency of scRNA-seq per cell analyzed. Visium’s efficiency seems to be moderately higher than that of ST, and DBiT-seq’s is even higher, at ~15.5% per area compared with smFISH68. Efficiencies of the submicrometer techniques, in the number of UMIs per unit area in the same tissue, might be comparable to that of Visium72. ISS tends to be less efficient, in part because of inefficiency of reverse transcription (RT) and SBL. Whereas the detection efficiency of scRNA-seq techniques is between 3% and 25% (refs. 65,88,89,90,91), the detection efficiencies of Cartana ISS and FISSEQ92 are ~5% and ~0.005% respectively, with STARmap being only marginally better than scRNA-seq. However, compared with smFISH, ExSeq claims up to 62% efficiency per cell for genes tested63. Newer technologies tend to skip RT and make ligation of the padlock probe on an RNA template more efficient, such as in BOLORAMIS and hybridization-based RNA ISS (HybRISS)93, or to substitute SBL with seqFISH-like barcoding, as in HybISS, to improve detection efficiency.
Transcriptome-wide profiling
Techniques not targeting specific genes with a panel of known probes are transcriptome wide, such as ROI selection followed by NGS, and NGS barcoding, where NGS is performed on poly-A captured transcripts, as well as untargeted ISS, such as FISSEQ and untargeted ExSeq. However, these transcriptome-wide techniques tend to have lower detection efficiency. It is possible to use certain techniques that require gene probe panels to quantify transcripts of over 10,000 genes, such as seqFISH+, MERFISH, and GeoMX WTA, although unlike in NGS, novel transcripts not targeted by the probes cannot be detected. While GeoMX WTA has been used in some studies outside Nanostring, where GeoMX originated94, the number of overall genes profiled with smFISH-based techniques per dataset has not increased over time (Fig. 3g). Instead, in studies using smFISH- and ISS-based techniques, a smaller number of genes is profiled, and the smFISH or ISS dataset is complementary to a transcriptome-wide scRNA-seq dataset95.

a, Number of institutions that have published papers or preprints with each technique, excluding LCM literature too vast to be manually curated. Only techniques used by at least three institutions are shown. b, Number of publications for each healthy organ in humans (male shown here, as there is no study on healthy female-specific organs in humans at present). c, Number of publications for pathological organs in humans (female shown here, but there are two studies on prostate cancer). d, Number of publications per species. e, Number of publications per healthy organ in mice. f, Number of publications for pathological organs in mice. g, Number of genes per dataset over time. Gray ribbon in g and h stands for 95% confidence interval. The slope is not significantly different from 0 in g (t test). In g and h, the y axis is log-transformed. h, Total number of cells per study profiled by smFISH-based techniques over time among studies that reported the number of cells. IceFISH, intron chromosomal expression FISH; C-FISH, consecutive FISH; MOSAICA, multi-omic single-scan assay with integrated combinatorial analysis; SGA, spatial genomic analysis; corrFISH, correlation FISH; EASI-FISH, expansion-assisted iterative FISH; par-seqFISH, parallel seqFISH; CISI, composite in situ imaging; SCRINSHOT, single-cell-resolution in situ hybridization on tissue; coppaFISH, combinatorial padlock-probe-amplified FISH. i, Number of publications for data collection using each of the fice most popular programming languages for downstream data analysis. j, Number of publications for data analysis using each of the five most popular programming languages for package development. In both i and j, each icon stands for 20 publications. Note that multiple programming languages can be used in one publication.
The number of genes that can be detected by highly multiplexed smFISH is limited by optical crowding, and expansion microscopy was used to address this issue in MERFISH and ExSeq. However, expansion reduces the amount of tissue covered per field of view, thus limiting imaging throughput.
Spatial resolution
smFISH- and ISS-based techniques have single-cell and single-molecule resolution, although cell segmentation can be challenging. In addition, smFISH- and ISS-based techniques can be applied to cleared thick tissue sections80, although the number of genes profiled in this case is much smaller than in most two-dimension (2D) highly multiplexed smFISH studies. All other types of techniques require tissue sections and are thus limited to 2D, or 3D with z resolution limited to section thickness, which is usually at least 10 μm for frozen sections. Although there are submicrometer-resolution NGS barcoding techniques, and the ROIs of LCM and GeoMX can in principle be single-cell or smaller, these types of techniques, as they are most commonly used, tend to have lower spatial resolution, such as 55 μm for Visium and several hundred micrometers across for GeoMX (for example 700 × 800 μm in ref. 94), owing to insufficient sensitivity of transcript detection at single-cell or subcellular resolution96.
Tissue area
Overall, techniques with lower detection efficiencies tend to be better at profiling larger tissue areas, and for smFISH, there seems to be a trade-off between the number of cells and the number of genes. In current-era spatial transcriptomics, a tissue section several millimeters across, such as a substantial portion of a mouse brain coronal section, which can fit into a Visium or ST tissue capture area, is considered large, and increasing tissue area and sequencing depth for sensitivity would increase sequencing cost. Cartana ISS and HybISS have also been used to profile large areas of tissue several millimeters across, but only around 100 genes97. An advantage of HybISS here is strong RCA signal and less optical crowding, thanks to lower detection efficiency facilitating lower magnification (×20; MERFISH uses ×60) and thus faster imaging. While most highly multiplexed smFISH datasets remain at hundreds of genes (Fig. 3g), among studies that reported the number of cells, the total number of cells per study has increased (Fig. 3h, P < 0.001, two-sided t-test). ROI-selection techniques are generally used for small numbers of ROIs, as it’s labor intensive to select very large numbers of ROIs and process them separately without spatial barcoding. However, when high spatial resolution is not as crucial or practical, ROIs with very low resolution can be selected to cover more tissue, as in the LCM dataset in the Allen Human Brain Atlas98.
Usability
While most techniques were originally developed for frozen sections, some are compatible with FFPE, which, as this is a common tissue archive, may at times be the only type of tissue available. Among smFISH-based techniques, ACD’s RNAscope99 is FFPE compatible but can profile only 12 genes at a time in FFPE, compared with 48 in frozen sections. Among NGS barcoding techniques, Visium100 and DBiT-seq101 are FFPE compatible, but owing to crosslinking and RNA fragmentation in archival storage, detection efficiency as number of UMIs and genes detected per spot in FFPE tissues is about 5 to 10 times lower than in their frozen counterparts. LCM has long been applied to FFPE tissues, even at single-cell resolution with the sensitive SMART-3Seq102. GeoMX is not only FFPE compatible, but also predominantly used on pathological human FFPE tissues (Figure 5.8 in Supplementary Information).
While many new techniques have been developed, most never spread beyond their institutions of origin (Figure 4.9 in Supplementary Information). Among those that have spread far, the most popular tend to have commercial platforms, such as LCM, 10X Visium and its precursor ST, Cartana ISS (acquired by 10X), and Nanostring GeoMX (Fig. 3a). In addition, many major institutions have core facilities for NGS, if not LCM, Visium, and GeoMX (for example, the TPCL at the University of California, Los Angeles, and the Advanced Genomics Core at University of Michigan, Ann Arbor), reducing the cost of purchasing new equipment and training personnel in individual laboratories. Tomo-seq has also spread, perhaps because of its ease of implementation with standard equipment. In contrast, smFISH-based techniques have not spread as widely thus far, perhaps due to the complicated home-built fluidic system, long imaging time, terabytes of images, and expensive probes. However, some smFISH techniques are being commercialized with automated imaging and fluidic platforms, such as MERFISH, commercialized as MERSCOPE by Vizgen, and another smFISH-based technique, Resolve Biosciences’s Molecular Cartography platform. In addition, Rebus Esper can be programmed to automate different smFISH technologies and can process images online as in Illumina sequencing, and has been used to automate cyclic-ouroboros smFISH (osmFISH)103. With the new automated commercial platforms, the popularity of smFISH-based techniques might rise, especially if such platforms are adopted by core facilities.
Data analysis
The processing and analysis of high-throughput spatial transcriptomics data requires new methods and tools, especially for problems such as image preprocessing, spatial reconstruction of scRNA-seq data, cell-type deconvolution of NGS barcoding data, identification of spatially variable genes, and inference of cell–cell interactions (Fig. 2g).
Upstream
Upstream data analysis converts raw data into forms more amenable to biological interpretation and is dependent on the data-collection technology.
For smFISH- and ISS-based methods, the raw data consist of images of fluorescent spots, which must be processed to identify transcript spots, match spots to genes, and assign spots to cells (Section 7.1 of Section Information). smFISH and ISS studies often use classical image-processing tools, such as top-hat filtering, to remove background, translation to align images from different rounds of hybridization, and watershed for cell segmentation47,56,86. Machine learning in Ilastik, deep learning packages like DeepCell104, and alternative tools incorporating scRNA-seq data105 can also be used for cell segmentation. However, without visualizing the plasma membrane, accuracy of cell segmentation is limited. Some analyses, such as identification of tissue regions, can be performed without cell segmentation105. Until 2019, image processing was typically performed with poorly documented and technique-specific code written in the proprietary language MATLAB, but more recently, such code is increasingly written in the open-source language Python. The package starfish106 was developed as an attempt to provide a unified and well-documented user interface to process images from different techniques, such as seqFISH, MERFISH, and ISS, but it has not been widely adopted.
Improvements in scRNA-seq technology have inspired new methods for leveraging the complementary nature of high-resolution transcriptome quantification with spatial transcriptomics data. For smFISH and ISS data that are not transcriptome wide, expression patterns of genes not profiled in the spatial data can be imputed with scRNA-seq data, either by mapping dissociated scRNA-seq cells to the spatial reference or by directly imputing gene expression in space using expression profiles from scRNA-seq (Section 7.3 in Supplementary Information). Cells can be mapped to spatial locations on an existing spatial dataset with genes shared by the two datasets, with an ad hoc score favoring similarity between cell and location107 or via optimal transport modeling108. While ad hoc scoring is simple to implement, the results tend to be qualitative. Gene expression in space can also be imputed from scRNA-seq without explicitly mapping scRNA-seq cells to locations. A common approach is to project the spatial and scRNA-seq data into a shared low-dimensional and batch-free latent space, and to subsequently estimate gene expression by projecting the spatial cells into the latent space. Examples of this approach include Seurat3 (ref. 32) and gimVI109. These methods may also be used to add spatial context to single-cell multi-omics data when spatial techniques for some of the multi-omics data are not available.
In spatial data that are not single-cell resolution, such as those derived from ST and Visium, scRNA-seq data can inform cell-type composition of the spots or voxels (Section 7.4 of Supplementary Information). Negative binomial models and non-negative least squares (NNLS) are common principles underlying cell-type deconvolution methods. Negative binomial models are typically parameterized with rate and dispersion, and the rate is modeled as a weighted sum of cell-type signatures from scRNA-seq, with scaling factors for library size and technology sensitivity; the non-negative weights may be normalized to sum up to 1 as cell-type proportions per spot. Negative-binomial-based methods include stereoscope110 and cell2location111. Simpler than negative binomial, gene expression is modeled as Poisson instead in RCTD112. Cell-type deconvolution can also be performed by modeling gene expression at each spot as a weighted sum of cell-type signatures outside the rate parameter of negative binomial distributions, and the weights are inferred with NNLS. For example, AdRoit113 uses the means of negative binomial distributions fitted to spot gene expression and to scRNA-seq cell-type signatures. The cell-type signatures can be non-negative matrix factorization (NMF) cell factors from scRNA-seq assigned to cell types, as in NMFreg66 and SPOTlight114. The cell-type weights can be regularized or thresholded to limit the number of cell types assigned to each spot. Parallels can also be drawn between cell-type deconvolution and topic modeling in text mining; cell types are analogous to topics, and genes are analogous to words. Latent Dirichlet allocation (LDA) from topic modeling has been adapted to cell-type deconvolution, such as in spatial transcriptomics deconvolution by topic modeling (STRIDE)115 and STdeconvolve116; the latter is unsupervised and does not require a scRNA-seq reference.
Downstream
Downstream analyses most often apply to the gene count matrix and cell or spot locations, and are thus largely independent of data-collection technologies.
Given the relevance of scRNA-seq to spatial data, and how spatial data are often analyzed like scRNA-seq data in exploratory data analysis (EDA), popular scRNA-seq EDA ecosystems, such as Seurat32, SCANPY (which spatical single-cell analysis in Python (Squidpy) is built on)117, and SingleCellExperiment (extended by SpatialExperiment)118, have added functionalities for spatial data, such as updates to data containers and functions to facilitate visualization of gene expression and cell or spot metadata at spatial locations (Section 7.2 of Supplementary Information). EDA packages dedicated to spatial data with beautiful graphics and good documentation have also been written, such as Giotto119 and STUtility120. Seurat and Giotto also implement basic methods to identify spatially variable genes. In addition, Giotto implements methods to identify cell-type enrichment in ST and Visium spots, to identify gene coexpression and association between gene expression and cell-type colocalization, and to identify spatial regions121.
Spatially variable genes are genes whose expression is associated with spatial location (Section 7.5 of Supplementary Information). Three approaches are commonly used for these genes: Gaussian process regression (GPR)122 and its generalization to Poisson123 and NB124, Laplacian score125, and Moran’s I. GPR-based methods model normalize gene expression or the rate parameter of Poisson or NB gene expression as a GPR and find whether the model better describes the data with the spatial term than without. Laplacian-score-based methods identify genes whose expression better reflects the structure of a spatial neighborhood graph. The locations of cells can also be modeled as a spatial point process with gene expression as marks; spatially variable genes can be identified as marks associated with locations126. Fitting GPR models to numerous genes can be time consuming, especially when a Bayesian approach with Markov chain Monte Carlo is used. Permutation testing used in Laplacian-score-based methods can also be time consuming. As both GPR- and Laplacian-score-based methods seek to identify spatial autocorrelation, sometimes the classic spatial autocorrelation metric Moran’s I is directly used to identify spatially variable genes, as in Seurat v3 and above. MERINGUE127 uses a local version of Moran’s I. Moran’s I and its significance testing are implemented in established geospatial packages and are easy and fast to run, but may have less statistical power than model-based methods123.
Spatial information also enables identification of potential cell–cell interactions (Supplementary Section 7.8). This is commonly done with knowledge of ligand–receptor (L–R) pairs, and can test which L–R pairs are more likely to be expressed in neighboring cells or spots128 or whether two cell types each expressing the ligand and the receptor are more likely to colocalize127. The cross-type L function from a spatial point process can be used to find cell types that colocalize129. Expression of genes of interest can also be modeled, including a term for cell–cell colocalization; a gene is considered associated with cell–cell colocalization if the model better describes the data with this term than without130.
There are many other types of downstream analysis that are useful for spatial transcriptomics analysis, including identification of archetypal gene patterns (Section 7.6 of Supplementary Information), spatial regions defined by the transcriptome (Section 7.7 of Supplementary Information), inferring gene–gene interactions (Section 7.9 of Supplementary Information), subcellular transcript localization (Section 7.10 of Supplementary Information), and gene expression imputation from H&E images (Section 7.11 of Supplementary Information).
Trends in the spatial transcriptomics field
The quality versus quantity trade-off inherent in existing technologies means that there is no single “best” solution currently available, and the difficulty in implementing methods has resulted in many technologies never spreading beyond their institutions of origin. LCM, Visium, ST, GeoMX DSP, and Tomo-seq have been the most widely adopted (Fig. 3a), and in almost all cases in the United States and western Europe (Figures 4.12, 5.27, and 5.33 in Supplementary Information). In terms of tissues analyzed, multiplexed current-era techniques have been used widely to characterize human tissues131, tumors87 (especially breast tumors), and pathological tissues that don’t necessarily have a stereotypical structure132 (Fig. 3b,c). In the SARS-CoV-2 pandemic, GeoMX DSP has been used for spatial transcriptomic profiling in lung autopsies of people who died due to COVID-19 (ref. 94).
Some of the processed data, and associated spatially variable genes, can be downloaded and visualized from SpatialDB133. Excluding LCM literature too vast to manually curate, the vast majority of current-era studies were performed in either humans or mice (Fig. 3d), and the brain is the most studied healthy organ while the lungs (particularly due to COVID-19) and breast tumors are also often studied in humans (Fig. 3b,e,f). In particular, the international project Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative—Cell Census Network (BICCN) is constructing a multi-modal atlas for human, mouse, and non-human primate brains, including spatial data such as those from MERFISH and seqFISH34.
All packages mentioned in the “Data analysis” section are open source and written in languages such as R, Python, and Julia. Downstream analyses in studies primarily concerning new data anddata-analysis packages predominantly use open-source programming languages, such as R, Python, and C++ (Fig. 3i,j). While MATLAB is still popular, its use has not risen, as was the case for R and Python (Figure 7.12 in Supplementary Information). While R is more popular for downstream analyses and EDA, Python, and C++ are more popular for package development (Fig. 3i,j). Most of the packages are not hosted on standard repositories, such as the Comprehensive R Archive Network (CRAN), Bioconductor, pip, or conda (Figure 7.13 in Supplementary Information). While most packages using R, Python, and C++ are well-documented, many MATLAB packages are not (Figure 7.12 in Supplementary Information). The standard repositories and documentation make packages more usable; this is discussed in more detail in Section 7.12 of the Supplementary Information.
Future perspective
While technologies of the past are rapidly depreciating, the ideas and methods that underlie them are fundamental to current-era spatial transcriptomics. The field has dramatically expanded over the past 5 years (Fig. 4a), with a plethora of new techniques and the popularization of Visium driving growth (Fig. 4b and Figures 4.9, 5.38, and 8.1 of Supplementary Information).

a, Number of publications over time for current-era data collection and data analysis. Bin width is 120 days; the curves drop because the plot was made at the beginning of a new bin. Non-curated LCM literature is excluded. b, The data collection curve in a, broken down by category of techniques. The colors are stacked and sorted in descending order of total number of publications using techniques in that category.
What lies ahead of the rising curves (Fig. 4)? First, more can be done to improve data-collection techniques. For example, most current-era techniques require tissue sections. Highly multiplexed whole-mount smFISH and tissue clearing protocols, and more efficient computational tools that will align multiple sections that may come from multiple individuals or even developmental stages, should be developed to extend current-era techniques to 3D and to spatiotemporal analysis. Future techniques may also extend the current era from the scale of millimeters to centimeters and across other modalities, such as epigenomics and metabolomics, to give a fuller picture of cellular function. Furthermore, smFISH and ISS techniques, with signal amplification to reduce the number of probes per transcript, can be adapted to target isoform-specific exons or untranslated regions, rather than all transcripts of a gene.
Second, current-era data have not yet been integrated into comprehensive databases. Prequel databases, such as GXD and e-Mouse Atlas and Gene Expression (EMAGE)134, include data from multiple sources and can be queried by gene symbol and developmental and spatial ontologies. In addition, ABA26 and EMAGE aligned ISH images to common coordinates and can be queried with expression patterns. While some current-era authors provide online interactive visualization of datasets from their studies33, comprehensive databases integrating, querying, and visualizing data from multiple sources, as in the prequel era, have not yet been developed. Furthermore, while prequel ontologies are still used in current-era studies, such ontologies may be improved with the transcriptome-wide quantitative data from the current era.
Third, outside of LCM, the current era is highly focused on humans and mice, while potential spatial transcriptomics investigations of other species, such as plants and invertebrates, lag behind. Technological modernization of prequel consortia for organisms other than humans and mice holds much promise for the development of useful spatial transcriptomics atlases.
Fourth, an open-source, well-documented, interoperable, and scalable workflow with an integrated, easy-to-use interface would greatly simplify spatial transcriptomics data collection and analysis. At present, for tasks beyond EDA, users still often need to learn new syntax, convert object types, and even learn new languages to use some data-analysis tools. Finally, our survey of methods shows that spatial transcriptomics methods need to be more open and accessible so that they become adopted around the world and are not restricted to elite Western institutions.
Data availability
The database of spatial transcriptomics literature can be accessed at https://docs.google.com/spreadsheets/d/1sJDb9B7AtYmfKv4-m8XR7uc3XXw_k4kGSout8cqZ8bY/edit#gid=1363594152. The version used as of writing is in the metadata.xlsx file in the frozen DOI version of the GitHub repository to reproduce the figures in this paper and render the supplementary website: https://doi.org/10.5281/zenodo.5774128
Code availability
All code used to generate figures in this paper and render the supplementary website is in the GitHub repository: https://github.com/pachterlab/LP_2021. The frozen DOI version of the repository as of final submission of this paper is on Zenodo: https://doi.org/10.5281/zenodo.5774129.
Change history
19 April 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41592-022-01494-3
References
Liao, J., Lu, X., Shao, X., Zhu, L. & Fan, X. Uncovering an organ’s molecular architecture at single-cell resolution by spatially resolved transcriptomics. Trends Biotechnol. 39, 43–58 (2021).
Asp, M., Bergenstråhle, J. & Lundeberg, J. Spatially resolved transcriptomes—next generation tools for tissue exploration. Bioessays 42, e1900221 (2020).
Smith, E. A. & Hodges, H. C. The spatial and genomic hierarchy of tumor ecosystems revealed by single-cell technologies. Trends Cancer Res. 5, 411–425 (2019).
Lein, E., Borm, L. E. & Linnarsson, S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017).
Saviano, A., Henderson, N. C. & Baumert, T. F. Single-cell genomics and spatial transcriptomics: discovery of novel cell states and cellular interactions in liver physiology and disease biology. J. Hepatol. 73, 1219–1230 (2020).
Gall, J. G. & Pardue, M. L. Formation and detection of RNA–DNA hybrid molecules in cytological preparations. Proc. Natl Acad. Sci. USA 63, 378–383 (1969).
John, H. A., Birnstiel, M. L. & Jones, K. W. RNA–DNA hybrids at the cytological level. Nature 223, 582–587 (1969).
Harrison, P. R., Conkie, D., Paul, J. & Jones, K. Localisation of cellular globin messenger RNA by in situ hybridisation to complementary DNA. FEBS Lett. 32, 109–112 (1973).
Langer-Safer, P. R., Levine, M. & Ward, D. C. Immunological method for mapping genes on Drosophila polytene chromosomes. Proc. Natl Acad. Sci. USA 79, 4381–4385 (1982).
Rudkin, G. T. & Stollar, B. D. High resolution detection of DNA–RNA hybrids in situ by indirect immunofluorescence. Nature 265, 472–473 (1977).
Tautz, D. & Pfeifle, C. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98, 81–85 (1989).
Rosen, B. & Beddington, R. S. Whole-mount in situ hybridization in the mouse embryo: gene expression in three dimensions. Trends Genet. 9, 162–167 (1993).
Giani, A. M., Gallo, G. R., Gianfranceschi, L. & Formenti, G. Long walk to genomics: history and current approaches to genome sequencing and assembly. Comput. Struct. Biotechnol. J. 18, 9–19 (2020).
O’Kane, C. J. & Gehring, W. J. Detection in situ of genomic regulatory elements in Drosophila. Proc. Natl Acad. Sci. USA 84, 9123–9127 (1987). This is the oldest entry in our database. It also gives a glimpse into the early motivations behind profiling gene expression in space.
Gossler, A., Joyner, A. L., Rossant, J. & Skarnes, W. C. Mouse embryonic stem cells and reporter constructs to detect developmentally regulated genes. Science 244, 463–465 (1989).
Jenett, A. et al. A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2, 991–1001 (2012).
Meier-Ruge, W. et al. The laser in the Lowry technique for microdissection of freeze-dried tissue slices. Histochem. J. 8, 387–401 (1976).
Emmert-Buck, M. R. et al. Laser capture microdissection. Science 274, 998–1001 (1996).
Becker, I. et al. Single-cell mutation analysis of tumors from stained histologic slides. Lab. Invest. 75, 801–807 (1996).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). This is the original publication for non-SRM seqFISH. Some later smFISH-based methods used seqFISH-like barcoding to profile transcripts of more genes than easily distinguishable colors.
Nederlof, P. M. et al. Multiple fluorescence in situ hybridization. Cytometry 11, 126–131 (1990).
Levsky, J. M., Shenoy, S. M., Pezo, R. C. & Singer, R. H. Single-cell gene expression profiling. Science 297, 836–840 (2002).
Femino, A. M., Fay, F. S., Fogarty, K. & Singer, R. H. Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998).
Tomancak, P. et al. Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 3, RESEARCH0088 (2002).
Bell, G. W., Yatskievych, T. A. & Antin, P. B. GEISHA, a whole-mount in situ hybridization gene expression screen in chicken embryos. Dev. Dyn. 229, 677–687 (2004).
Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). This is the publication for the ABA and the original CCF, which greatly influenced data analysis in the prequel era, and remains influential in the current era.
Harding, S. D. et al. The GUDMAP database—an online resource for genitourinary research. Development 138, 2845–2853 (2011).
Ardini-Poleske, M. E. et al. LungMAP: The Molecular Atlas of Lung Development Program. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L733–L740 (2017).
Wienholds, E. MicroRNA expression in zebrafish embryonic development. Science 309, 310–311 (2005).
Ringwald, M. et al. A database for mouse development. Science 265, 2033–2034 (1994).
Sprague, J. et al. The Zebrafish Information Network (ZFIN): the zebrafish model organism database. Nucleic Acids Res. 31, 241–243 (2003).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–190 (2019).
Ortiz, C. et al. Molecular atlas of the adult mouse brain. Sci. Adv. 6, eabb3446 (2020).
BRAIN Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature 598, 86–102 (2021).
Baker, D. et al. A cellular reference resource for the mouse urinary bladder. Preprint at bioRxiv https://doi.org/10.1101/2021.09.20.461121 (2021).
Brown, V. M. et al. Multiplex three-dimensional brain gene expression mapping in a mouse model of Parkinson’s sisease. Genome Res. 12, 868–884 (2002).
Junker, J. P. et al. Genome-wide RNA tomography in the zebrafish embryo. Cell 159, 662–675 (2014). While this is not the first attempt to profile transcriptomes from samples microdissected with a microtome, later Tomo-seq works adapted the protocol from this paper. Tomo-seq is the most popular current era technique after LCM, Visium/ST, and GeoMX DSP.
Peng, G. et al. Spatial transcriptome for the molecular annotation of lineage fates and cell identity in mid-gastrula mouse embryo. Dev. Cell 55, 802–804 (2020).
Schede, H. H. et al. Spatial tissue profiling by imaging-free molecular tomography. Nat. Biotechnol. 39, 968–977 (2021).
Hufnagel, B. et al. High-quality genome sequence of white lupin provides insight into soil exploration and seed quality. Nat. Commun. 11, 492 (2020).
Medaglia, C. et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 358, 1622–1626 (2017).
Genshaft, A. S. et al. Live cell tagging tracking and isolation for spatial transcriptomics using photoactivatable cell dyes. Nat. Commun. 12, 4995 (2021).
Hu, K. H. et al. ZipSeq: barcoding for real-time mapping of single cell transcriptomes. Nat. Methods 17, 833–843 (2020).
Merritt, C. R. et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 38, 586–599 (2020). This is the original publication for GeoMX DSP, which is the most popular current era technique after LCM and Visium, and has been used in several COVID studies.
Roberts, K. et al. Transcriptome-wide spatial RNA profiling maps the cellular architecture of the developing human neocortex. Preprint at bioRxiv https://doi.org/10.1101/2021.03.20.436265 (2021).
Lubeck, E. & Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 9, 743–748 (2012).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Eng, C.-H. L., Shah, S., Thomassie, J. & Cai, L. Profiling the transcriptome with RNA SPOTs. Nat. Methods 14, 1153–1155 (2017).
Eng, C.-H. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). This is the original publication for MERFISH, which has been used to collect data for the BICCN. Some later smFISH-based techniques use MERFISH-like barcoding to profile transcripts of more genes than easily distinguishable colors.
Xia, C., Fan, J., Emanuel, G., Hao, J. & Zhuang, X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl Acad. Sci. USA 116, 19490–19499 (2019).
Gyllborg, D. et al. Hybridization-based In Situ Sequencing (HybISS): spatial transcriptomic detection in human and mouse brain tissue. Nucleic Acids Res. 48, e112 (2020).
Goh, J. J. L. et al. Highly specific multiplexed RNA imaging in tissues with split-FISH. Nat. Methods 17, 689–693 (2020).
Battich, N., Stoeger, T. & Pelkmans, L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat. Methods 10, 1127–1133 (2013).
Kishi, J. Y. et al. SABER amplifies FISH: enhanced multiplexed imaging of RNA and DNA in cells and tissues. Nat. Methods 16, 533–544 (2019).
Wang, X. et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018).
Chen, F., Tillberg, P. W. & Boyden, E. S. Optical imaging. Expansion Microsc. Sci. 347, 543–548 (2015).
Coskun, A. F. & Cai, L. Dense transcript profiling in single cells by image correlation decoding. Nat. Methods 13, 657–660 (2016).
Ke, R. et al. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 (2013). This ISS technique, which has been commercialized by Cartana, is the most popular current era technique after LCM, Visium/ST, GeoMX DSP, and Tomo-seq. The RCA in this technique is also used in several later techniques such as STARmap and BOLORAMIS.
Liu, S. et al. Barcoded oligonucleotides ligated on RNA amplified for multiplexed and parallel in situ analyses. Nucleic Acids Res. 49, e58 (2021).
Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
Lee, J. H. et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014).
Alon, S. et al. Expansion sequencing: spatially precise in situ transcriptomics in intact biological systems. Science 371, eaax2656 (2021).
Sun, Y.-C. et al. Integrating barcoded neuroanatomy with spatial transcriptional profiling reveals cadherin correlates of projections shared across the cortex. Nat. Neurosci. 24, 873–885 (2021).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual. cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Vickovic, S. et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019).
Liu, Y. et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681 (2020).
Lebrigand, K. et al. The spatial landscape of gene expression isoforms in tissue sections. Preprint at bioRxiv https://doi.org/10.1101/2020.08.24.252296 (2022).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball patterned arrays. Preprint at bioRxiv https://doi.org/10.1101/2021.01.17.427004 (2021).
Cho, C.-S. et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572.e22 (2021).
Fu, X. et al. Continuous polony gels for tissue mapping with high resolution and RNA capture efficiency. Preprint at bioRxiv https://doi.org/10.1101/2021.03.17.435795 (2021).
Lee, Y. et al. XYZeq: Spatially resolved single-cell RNA sequencing reveals expression heterogeneity in the tumor microenvironment. Sci. Adv. 7, eabg4755 (2021).
Srivatsan, S. R. et al. Embryo-scale, single-cell spatial transcriptomics. Science 373, 111–117 (2021).
Weinstein, J. A., Regev, A. & Zhang, F. DNA microscopy: optics-free spatio-genetic imaging by a stand-alone chemical reaction. Cell 178, 229–241.e16 (2019).
Hoffecker, I. T., Yang, Y., Bernardinelli, G., Orponen, P. & Högberg, B. A computational framework for DNA sequencing microscopy. Proc. Natl Acad. Sci. USA 116, 19282–19287 (2019).
Halpern, K. B. et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 36, 962–970 (2018).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178, 473–490 (2019).
Vickovic, S. et al. SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 795 (2022).
Wang, G., Moffitt, J. R. & Zhuang, X. Multiplexed imaging of high-density libraries of RNAs with MERFISH and expansion microscopy. Sci. Rep. 8, 4847 (2018).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659.e26 (2020).
Shah, S. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376 (2018).
Zhang, M. et al. Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137–143 (2021).
Kim, M.-H. et al. Molecular and genetic approaches for assaying human cell type synaptic connectivity. Preprint at bioRxiv https://doi.org/10.1101/2020.10.16.343343 (2020).
Li, Q. et al. In situ electro-sequencing in three-dimensional tissues. Preprint at bioRxiv https://doi.org/10.1101/2021.04.22.440941 (2021).
Moffitt, J. R. & Zhuang, X. RNA imaging with multiplexed error-robust fluorescence in situ hybridization (MERFISH). Methods Enzymol. 572, 1–49 (2016).
Ståhl, P. L. et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). This is the precursor of Visium, which is the most popular current era method perhaps after LCM.
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Hashimshony, T. et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-seq. Genome Biol. 17, 77 (2016).
Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Lee, J. H. et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 10, 442–458 (2015).
Lee, H., Salas, S. M., Gyllborg, D. & Nilsson, M. Direct RNA targeted transcriptomic profiling in tissue using hybridization-based RNA in situ sequencing (HybRISS). Preprint at bioRxiv https://doi.org/10.1101/2020.12.02.408781 (2020).
Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021).
La Manno, G. et al. Molecular architecture of the developing mouse brain. Nature 596, 92–96 (2021).
Zimmerman, S. M. et al. Spatially resolved whole transcriptome profiling in human and mouse tissue using digital spatial profiling. Preprint at bioRxiv https://doi.org/10.1101/2021.09.29.462442 (2021).
Qian, X. et al. Probabilistic cell typing enables fine mapping of closely related cell types in situ. Nat. Methods 17, 101–106 (2020).
Hawrylycz, M. J. et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012).
Wang, F. et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn. 14, 22–29 (2012).
Villacampa, E. G. et al. Genome-wide spatial expression profiling in formalin-fixed tissues. Cell Genomics 1, 100065 (2021).
Liu, Y., Enninful, A., Deng, Y. & Fan, R. Spatial transcriptome sequencing of FFPE tissues at cellular level. Preprint at bioRxiv https://doi.org/10.1101/2020.10.13.338475 (2020).
Foley, J. W. et al. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 29, 1816–1825 (2019).
Bhaduri, A. et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204 (2021).
Van Valen, D. A. et al. Deep learning automates the quantitative analysis of individual cells in live-cell imaging experiments. PLoS Comput. Biol. 12, e1005177 (2016).
Petukhov, V., Soldatov, R. A., Khodosevich, K. & Kharchenko, P. V. Bayesian segmentation of spatially resolved transcriptomics data. Preprint at bioRxiv https://doi.org/10.1101/2020.10.05.326777 (2020).
Perkel, J. M. Starfish enterprise: finding RNA patterns in single cells. Nature 572, 549–551 (2019).
Karaiskos, N. et al. The embryo at single-cell transcriptome resolution. Science 358, 194–199 (2017).
Nitzan, M., Karaiskos, N., Friedman, N. & Rajewsky, N. Gene expression cartography. Nature 576, 132–137 (2019).
Lopez, R. et al. A joint model of unpaired data from scRNA-seq and spatial transcriptomics for imputing missing gene expression measurements. Preprint at https://arxiv.org/abs/1905.02269 (2019).
Andersson, A. et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol. 3, 565 (2020).
Kleshchevnikov, V. et al. Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics. Preprint at bioRxiv https://doi.org/10.1101/2020.11.15.378125 (2020).
Cable, D. M. et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00830-w (2021).
Yang, T. et al. AdRoit is an accurate and robust method to infer complex transcriptome composition. Commun. Biol. 4, 1218 (2021).
Elosua, M., Nieto, P., Mereu, E., Gut, I. & Heyn, H. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50 (2021).
Sun, D. et al. STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing. Preprint at bioRxiv https://doi.org/10.1101/2021.09.08.459458 (2021).
Miller, B. F., Huang, F., Atta, L., Sahoo, A. & Fan, J. Reference-free cell-type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data. Preprint at bioRxiv https://doi.org/10.1101/2021.06.15.448381 (2021).
Palla, G. et al. Squidpy: a scalable framework for spatial omics analysis. Nat. Methods 19, 171–178 (2022).
Righelli, D. et al. SpatialExperiment: infrastructure for spatially resolved transcriptomics data in R using Bioconductor. Preprint at bioRxiv https://doi.org/10.1101/2021.01.27.428431 (2021).
Dries, R. et al. Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78 (2021).
Bergenstråhle, J., Larsson, L. & Lundeberg, J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genomics 21, 482 (2020).
Zhu, Q., Shah, S., Dries, R., Cai, L. & Yuan, G.-C. Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nat. Biotechnol. 36, 1183–1190 (2018).
Svensson, V., Teichmann, S. A. & Stegle, O. SpatialDE: identification of spatially variable genes. Nat. Methods 15, 343–346 (2018).
Sun, S., Zhu, J. & Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 17, 193–200 (2020).
BinTayyash, N. et al. Non-parametric modelling of temporal and spatial counts data from RNA-seq experiments. Bioinformatics 37, 3788–3795 (2021).
Govek, K. W., Yamajala, V. S. & Camara, P. G. Clustering-independent analysis of genomic data using spectral simplicial theory. PLoS Comput. Biol. 15, e1007509 (2019).
Edsgärd, D., Johnsson, P. & Sandberg, R. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 15, 339–342 (2018).
Miller, B. F., Bambah-Mukku, D., Dulac, C., Zhuang, X. & Fan, J. Characterizing spatial gene expression heterogeneity in spatially resolved single-cell transcriptomic data with nonuniform cellular densities. Genome Res. 31, 1843–1855 (2021).
Pham, D. et al. stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. Preprint at bioRxiv https://doi.org/10.1101/2020.05.31.125658 (2020).
Canete, N. P. et al. spicyR: Spatial analysis of in situ cytometry data in R. Preprint at bioRxiv https://doi.org/10.1101/2021.06.07.447307 (2021).
Arnol, D., Schapiro, D., Bodenmiller, B., Saez-Rodriguez, J. & Stegle, O. Modeling cell–cell interactions from spatial molecular data with spatial variance component analysis. Cell Rep. 29, 202–211.e6 (2019).
Maynard, K. R. et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci. 24, 425–436 (2021).
Lundmark, A. et al. Gene expression profiling of periodontitis-affected gingival tissue by spatial transcriptomics. Sci. Rep. 8, 9370 (2018).
Fan, Z., Chen, R. & Chen, X. SpatialDB: a database for spatially resolved transcriptomes. Nucleic Acids Res. 48, D233–D237 (2020).
Armit, C. et al. eMouseAtlas: an atlas-based resource for understanding mammalian embryogenesis. Dev. Biol. 423, 1–11 (2017).
Singer, R. H. & Ward, D. C. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc. Natl Acad. Sci. USA 79, 7331–7335 (1982).
Hope, I. A. ‘Promoter trapping’ in Caenorhabditis elegans. Development 113, 399–408 (1991).
Seydoux, G. & Fire, A. Soma-germline asymmetry in the distributions of embryonic RNAs in Caenorhabditis elegans. Development 120, 2823–2834 (1994).
Bettenhausen, B. & Gossler, A. Efficient isolation of novel mouse genes differentially expressed in early postimplantation embryos. Genomics 28, 436–441 (1995).
Gawantka, V. et al. Gene expression screening in Xenopus identifies molecular pathways, predicts gene function and provides a global view of embryonic patterning. Mech. Dev. 77, 95–141 (1998).
Ringwald, M., Mangan, M. E., Eppig, J. T., Kadin, J. A. & Richardson, J. E. GXD: a gene expression database for the laboratory mouse. The Gene Expression Database Group. Nucleic Acids Res. 27, 106–112 (1999).
Kawashima, T., Kawashima, S., Kanehisa, M., Nishida, H. & Makabe, K. W. MAGEST: MAboya gene expression patterns and sequence tags. Nucleic Acids Res. 28, 133–135 (2000).
Maeda, I., Kohara, Y., Yamamoto, M. & Sugimoto, A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol. 11, 171–176 (2001).
Satou, Y. et al. Gene expression profiles in Ciona intestinalis tailbud embryos. Development 128, 2893–2904 (2001).
Carson, J. P., Thaller, C. & Eichele, G. A transcriptome atlas of the mouse brain at cellular resolution. Curr. Opin. Neurobiol. 12, 562–565 (2002).
Henrich, T. et al. MEPD: a Medaka gene expression pattern database. Nucleic Acids Res. 31, 72–74 (2003).
Luengo Hendriks, C. L. et al. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution I: data acquisition pipeline. Genome Biol. 7, R123 (2006).
Lécuyer, E. et al. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131, 174–187 (2007).
Bowes, J. B. et al. Xenbase: a Xenopus biology and genomics resource. Nucleic Acids Res. 36, D761–7 (2008).
Lovell, P. V. et al. ZEBrA: Zebra finch Expression Brain Atlas—a resource for comparative molecular neuroanatomy and brain evolution studies. J. Comp. Neurol. 528, 2099–2131 (2020).
Landegren, U., Kaiser, R., Sanders, J. & Hood, L. A ligase-mediated gene detection technique. Science 241, 1077–1080 (1988).
Belyavsky, A., Vinogradova, T. & Rajewsky, K. PCR-based cDNA library construction: general cDNA libraries at the level of a few cells. Nucleic Acids Res. 17, 5883–5883 (1989).
Van Gelder, R. N. et al. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl Acad. Sci. USA 87, 1663–1667 (1990).
Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470 (1995).
Luo, L. et al. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat. Med. 5, 117–122 (1999).
Lister, R. et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536 (2008).
Okamura-Oho, Y. et al. Transcriptome tomography for brain analysis in the web-accessible anatomical space. PLoS ONE 7, e45373 (2012).
Acknowledgements
This work was supported by a grant from the National Institute of Mental Health (NIMH), National Institute of Health (NIH), of the U.S. Department of Health & Human Services (number U19MH114830, L.P.). We thank the following people for providing feedback for earlier versions of this paper and the supplement: D. Furth from the Cold Spring Harbor Laboratories, L. Cai from the California Institute of Technology, and G. Victora from the Rockefeller University.
Author information
Authors and Affiliations
Contributions
L.P. suggested the project. L.M. curated the database, performed the analyses of the metadata, and wrote the manuscript and the supplement, which have been proofread and edited by L.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks Sten Linnarsson, Quan Nguyen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Lei Tang was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
PDF version of the book-length supplement
Rights and permissions
About this article

Cite this article
Moses, L., Pachter, L. Museum of spatial transcriptomics. Nat Methods 19, 534–546 (2022). https://doi.org/10.1038/s41592-022-01409-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-022-01409-2
This article is cited by
-
Library size confounds biology in spatial transcriptomics data
Genome Biology (2024)
-
scGIST: gene panel design for spatial transcriptomics with prioritized gene sets
Genome Biology (2024)
-
SRT-Server: powering the analysis of spatial transcriptomic data
Genome Medicine (2024)
-
Spatial multi-omics: novel tools to study the complexity of cardiovascular diseases
Genome Medicine (2024)
-
Decoder-seq enhances mRNA capture efficiency in spatial RNA sequencing
Nature Biotechnology (2024)
