
Abstract
Dysregulation of peptide-activated pathways causes a range of diseases, fostering the discovery and clinical development of peptide drugs. Many endogenous peptides activate G protein-coupled receptors (GPCRs) — nearly 50 GPCR peptide drugs have been approved to date, most of them for metabolic disease or oncology, and more than 10 potentially first-in-class peptide therapeutics are in the pipeline. The majority of existing peptide therapeutics are agonists, which reflects the currently dominant strategy of modifying the endogenous peptide sequence of ligands for peptide-binding GPCRs. Increasingly, novel strategies are being employed to develop both agonists and antagonists, to both introduce chemical novelty and improve drug-like properties. Pharmacodynamic improvements are evolving to allow biasing ligands to activate specific downstream signalling pathways, in order to optimize efficacy and reduce side effects. In pharmacokinetics, modifications that increase plasma half-life have been revolutionary. Here, we discuss the current status of the peptide drugs targeting GPCRs, with a focus on evolving strategies to improve pharmacokinetic and pharmacodynamic properties.
Similar content being viewed by others
Introduction
G protein-coupled receptors (GPCRs) mediate a wide range of signalling processes and are targeted by one third of the drugs in clinical use1. Although most GPCR-targeting therapeutics are small molecules2, the endogenous ligands for many GPCRs are peptides (comprising 50 or fewer amino acids), which suggests that this class of molecule could be therapeutically useful.
GPCRs are divided into families based on structural similarities. The largest group is the class A (rhodopsin-like) family, followed by the class B (secretin) family. Although other families exist, including class C and the frizzled and adhesion classes, therapeutics have predominantly targeted class A and B GPCRs, so this Review is focused on these two groups. The International Union of Basic and Clinical Pharmacology (IUPHAR) Guide to Pharmacology3 currently lists 197 class A receptors with known ligands (excluding olfactory, vision, taste and vomeronasal sensory receptors), where 64 (32%) of these bind to endogenous peptides3. In GPCR class B, there are 20 receptors activated by 15 endogenous peptides. These GPCRs are grouped in the following families, based on the ligand to which they bind: calcitonin, corticotropin-releasing factor, glucagon, parathyroid hormone (which is generally considered to be a peptide, despite its 84-amino-acid length), vasoactive intestinal peptide (VIP) or pituitary adenylate cyclase-activating peptide (PACAP).
A further 87 ‘orphan’ receptors from different families — for which the endogenous ligand is not yet known — have been identified in the human genome. In the cases of 54 of these orphans, at least one publication has proposed an endogenous ligand, and some of these ligands are peptides4. The ‘de-orphanization’ of these receptors is ongoing. For example, G protein-coupled receptor 171 (GPR171) and GPR83 were recently found to interact with the neuropeptides PEN and LEN, respectively, which are abundant in mouse brain (and highly conserved in humans). Initial studies suggest that these GPCRs may be functionally coupled in the regulation of feeding, and if substantiated, the receptors could be new potential drug targets5,6.
Endogenous peptides that bind to GPCRs on cell surfaces spatiotemporally span paracrine and autocrine signalling, from long-acting hormones to locally released mediators of cellular functions and neurotransmitters. Peptides are one of the largest and most ancient classes of intercellular chemical messengers7. The pioneering development, using these naturally occurring peptides as therapeutics, was the use of insulin in the 1920s. Insulin principally targets a tyrosine kinase receptor8, and the development of this therapeutic exploited the remarkable pharmacodynamic properties of peptides: their high affinity, selectivity and potency. In line with the observed effects of insulin, most other peptide therapies are well tolerated, with few off-target effects.
Naturally occurring peptides, however, do not typically make good therapeutics. The development of peptides as drugs has been limited by poor pharmacokinetics (short half-life, rapid degradation and high levels of clearance) and by a lack of oral bioavailability due to a combination of low gastrointestinal stability and poor permeability. Therefore, strategies need to be developed that can address these aspects before most peptides can become effective medicines.
Peptide drugs occupy a structural space between biologics (antibodies and proteins) and small molecules. Whereas endogenous signalling peptides usually have 50 or fewer residues, the FDA defines peptide drugs9 as having a maximum of 40 residues (a few exceptions have been noted), not limited to the 20 genetically encoded amino acids. These therapeutics are undergoing a renaissance. Emerging novel strategies include half-life extension platforms, stapling and resistance to proteolysis, all of which significantly improve pharmacokinetics and oral bioavailability. These strategies are perhaps best exemplified by the development of glucagon-like peptide 1 (GLP-1) receptor agonists that have increased resistance to proteolytic degradation and reduced renal clearance. Several successfully marketed products and a multitude of preclinical novel approaches (for example, stapling, cyclization and glycosylation) have come from these efforts. This success has also fostered the development of multifunctional peptides that combine agonism for two or more GPCRs in the same peptide, based on relatively high sequence homology and similar binding sites. Complementary pharmacodynamic strategies are extending the repertoire of drugs that act at a given GPCR.
Peptide ligands are also emerging that can selectively activate downstream signalling pathways — for example, G proteins or β-arrestins (the two main pathways downstream of GPCRs) — towards which these ligands are described as being biased. These pathways may be linked to distinct physiological or pathophysiological responses, either beneficial or detrimental, so biased ligands can, in theory, be designed to have the optimum therapeutic activity, but with reduced side effects or receptor internalization.
The application of structure-based design has significantly altered all aspects of small-molecule drug discovery, including drugs targeting GPCRs10,11. Peptides are also amenable to structure-based design strategies. X-ray crystallography or cryo-electron microscopy (cryo-EM) structures of the binding domains of over 27 peptide-binding or protein-binding GPCRs have been reported; amongst these 65 unique receptor–ligand complexes, 22 contain a peptide ligand. The rapidly expanding repertoire of receptor structures will substantially advance understanding of the basic peptide–receptor structure–activity relationship (SAR) and of the more subtle aspects of conformational biased signalling, thereby enabling rational agonist or antagonist design to activate or block, respectively, a pathophysiological process.
In this Review, we discuss the clinically approved and preclinical GPCR-targeting peptide therapeutics (Fig. 1) and outline the challenges in the field. We also highlight key strategies to improve pharmacokinetics (mainly via increases in plasma half-life) and pharmacodynamics (via increased potency), as well as ligand bias.

The numbers of peptide drugs that are approved, in phase III or in phase II clinical trials are shown for G protein-coupled receptors (GPCRs) from class A (blue), class B (red) and class C (green).
Approved peptide therapeutics
The majority of GPCR-targeting peptide drugs that are either approved for clinical use or in development function as agonists and are used to replace or enhance low levels of endogenous peptides. In class A, the major receptor targets include μ and κ opioid receptors (abbreviated MOR and KOR, respectively) to relieve pain; oxytocin and vasopressin receptors for the induction of labour; and apelin and angiotensin receptors in cardiovascular disease (Tables 1,2). More specialized targets include the somatostatin receptor, for acromegaly and Cushing’s disease. Peptide therapies that target class B receptors are dominated by GLP-1 receptor agonists for the treatment of type 2 diabetes mellitus (T2D), along with synthetic or modified versions of peptide hormones.
Few antagonists have made it to the clinic. Most of those that have made this leap target class A GPCRs. Antagonists that block the action of gonadotrophin-releasing hormone, such as degarelix and abarelix, or agonists that desensitize the receptor in order to have the same effect, such as buserelin, are used for the treatment of cancer.
Synthetic endogenous peptide analogues
Oxytocin12,13 and vasopressin were the first chemically synthesized variants of endogenous peptides for class A receptors to enter clinical use, and they did so in the 1950s. These two peptides are amongst the ten peptides that are usually synthesized chemically or by recombinant technology but that have sequences identical to those of their endogenous peptide equivalents, and that have been approved for clinical use in one or more countries (Table 1). Evaluation of those ten peptides provides an opportunity to examine the properties of native peptides that may limit or enable their widespread therapeutic application, as well as to investigate the impact of those properties on drug development strategies.
The peptides that target class A GPCRs are mainly agonists that bind with both high affinity (median negative log of the dissociation constant (pKi) = 8.4) and potency (median negative log of the half maximal effective concentration (pEC50) = 8.5), compared with the average clinically used drug14. However, they typically demonstrate very short plasma half-lives following intravenous administration, with a median value of ~5.3 min, reflecting their rapid degradation by peptidases and/or their high rates of excretion, particularly by the kidney. Theoretically, peptide levels will have therefore declined to <1% of the original dose within 6 half-lives, which is about 32 min. Consistent with their polar, hydrophilic chemical properties, they have low median volumes of distribution (~9.8 l, which is close to the expected total volume of interstitial fluid), indicating that the protein is restricted to the fluid compartment, with negligible distribution into tissues. Finally, endogenous peptides typically have low protein plasma binding, which ensures that the majority of the circulating peptide is in the unbound state and is available to directly bind to and activate its cognate receptor. However, unbound peptide is also highly susceptible to renal elimination and cleavage by serum or tissue proteases; both of these processes decrease plasma half-life.
The endogenous ligands for all class B GPCRs are peptides. For peptides that interact with class B GPCRs, the median pharmacodynamic and pharmacokinetic values are similar to those for peptides that target class A receptors: high potency (pEC50 = 9.9), combined with short plasma half-life (15 min) and low volume of distribution (~9.9 l). Calcitonin (isolated from salmon in the 1970s) was one of the first native peptides to be used clinically and was used to treat Paget disease in patients who were unresponsive to first-line treatments. Human calcitonin is used in patients who develop antibodies to the salmon calcitonin. The goal of treatment is to inhibit bone resorption by osteoclasts, and therefore to increase bone mass15.
Paradoxically, at least some of the peptides in both classes are likely successful because their short plasma half-lives, which would usually be an undesirable trait, are exploited for clinical benefit. At one extreme, the peptide therapeutic with the shortest half-life is angiotensin II, first synthesized in 1947 but only approved in 2017 (as Giapreza). This synthetic analogue of angiotensin II is used for the treatment of critically ill patients with septic shock, in whom an abnormal distribution of blood to the smallest blood vessels results in inadequate systemic blood supply, which can be fatal. The drug binds to vascular smooth muscle AT1 receptors and takes a median time of 5 min to adequately increase blood pressure following intravenous dosing16. The short half-life — less than 1 min — ensures that hypertension resulting from an overdose is very unlikely. Similarly, oxytocin, which was sequenced and synthesized in the 1950s13,14, is used to induce labour and strengthen contractions immediately after intravenous administration, but the effects subside within an hour, reflecting a short half-life of a few minutes.
Modified peptides
Twenty-six synthetic peptides (20 agonists and 6 competitive antagonists) targeting 8 class A receptor families have been approved for clinical use (Table 2). The few antagonists that have been developed (such as icatibant and cetrorelix) usually have sub-nanomolar affinity, which is higher than the affinity of the endogenous peptide. This may be an important requirement for a clinically successful peptide antagonist, given the high potency and affinity of the endogenous peptide agonists, suggesting that effective antagonists require high levels of receptor occupancy in order to maintain efficacy. In this regard, peptides targeting the gonadotropin-releasing hormone (GnRH) receptor in the pituitary are particularly intriguing for non-steroidal manipulation of the reproductive endocrine axis. For example, the agonist buserelin acts by an unusual pharmacological mechanism, as it desensitizes the GnRH receptor, reducing the amount of gonadotropin released from the pituitary gland, thereby inhibiting testosterone secretion in males and oestrogen secretion in females. This agonist peptide effectively switches off the GnRH receptor by removing receptors from the cell surface and reducing further stimulation. Whereas transient administration of buserelin — for example, in the setting of in vitro fertilization — suppresses the premature surge of luteinizing hormone, unwanted side effects are caused by the initial agonist activity of this peptide, such as hyperstimulation of the ovaries, which has led to the development of GnRH receptor antagonists such a ganirelix17 (Fig. 2). To date, no peptide inverse agonists or allosteric modulators have been reported in clinical use; most of the current drugs are based on endogenous peptides, and therefore bind to the orthosteric site.

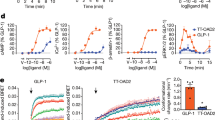
Peptides are shown as beads, based on the 3D position of Cα atoms of the structural templates of these peptides bound to experimentally determined seven-transmembrane domain (7TM) structures of the associated G protein-coupled receptors (GPCRs) (Supplementary Table 1). The orientations of the 3D peptide bead strings are consistent with their binding mode in the GPCR binding site (Fig. 4), assuming a view in which transmembrane domain 1 (TM1) is on the left, TM5 is on the right, and the extracellular side is facing up. See Supplementary Fig. 2 for further structures of peptide drugs that target class A and B GPCRs.
In class B, all 13 existing therapeutics are agonists that target one of three receptor families (principally the GLP-1 receptor in the glucagon family) and, like the class A-targeting synthetic peptides, have a high median affinity and potency (pKi = 8.9; pEC50 = 9.7) and a comparatively low volume of distribution (~16 l) following subcutaneous injection. The median plasma half-life is around 1 h (but this value excludes peptides modified specifically to have very long half-lives, measured in days), which has been achieved by a combination of selective amino acid substitutions at known sites of enzymatic cleavage and conjugation to ligands that bind serum proteins, such as albumin, which protects the peptides from enzymatic cleavage and reduces renal clearance18,19,20,21,22.
Plasma protein binding of the remaining synthetic peptides in Table 2 is, in many cases, also high, despite a lack of specific modifications that link the peptides to plasma proteins. According to the free-drug hypothesis, only unbound drug is available to bind to and act at physiological sites of action. Therefore, plasma protein binding can influence both the pharmacodynamic effects and the pharmacokinetic properties of peptides. Liu et al.23 have argued that, because many small-molecule drugs (~30%) have high plasma binding, this is not necessarily an undesirable trait. Smith et al.24 have maintained that increasing plasma binding, which increases metabolic stability and reduces clearance by organs such as the kidney, will lead to better drugs. The effects of plasma binding on the elimination (and therefore the half-life) of peptides can be complex. For peptides excreted in their intact form by renal glomerular filtration (such as cetrorelix, 86% of which is plasma-bound), increasing plasma binding would be expected to decrease the rate of elimination, because only the free peptide is filtered. Conversely, buserelin (15% of which is plasma-bound) is metabolized by proteolytic enzymes; therefore, for this peptide, reducing proteolytic cleavage is expected to have a greater effect on plasma exposure than would increasing plasma protein binding (Table 2).
Lessons from recent clinical approvals
During the last 3 years, 16 of the 195 FDA-approved new drugs have been peptides25,26,27. Abaloparatide, which was approved for postmenopausal osteoporosis in 2017, was the first analogue of human parathyroid hormone-related protein to be developed. Although this molecule is distinct from teriparatide (a parathyroid hormone analogue), both target the PTH1 receptor for the same clinical condition and have similar side effects; however, abaloparatide induces a greater increase in bone density28. Abaloparatide binds to the receptor with an affinity that is two orders of magnitude higher than that of teriparatide. In addition, abaloparatide binds with higher selectivity to a G protein-dependent receptor conformation (called RG), which results in transient responses and favours bone formation, than to a second conformation (called R0), which results in comparatively prolonged binding and favours unwanted bone resorption29; R0 is bound by parathyroid hormone and analogues such as teriparatide.
The year 2017 also saw the approval of semaglutide30, the fifth GLP-1 receptor agonist to be approved for T2D. Of note, semaglutide is one of several drugs that have a significantly increased half-life, in this case around 168 h. This increase was achieved by using a free fatty acid linker that allows the molecule to have a non-covalent reversible interaction with albumin, which reduces renal excretion. A different protein-linking strategy was used in the design of albiglutide and dulaglutide. These two GLP-1 receptor agonists become covalently linked (irreversibly) to large proteins — albumin and an Fc fragment of human IgG4, respectively. Importantly, an α-aminobutyric acid was engineered into semaglutide at position 8 in order to reduce metabolism by dipeptyl peptidase 4 (DPP4), a cell-surface protein that cleaves numerous circulating peptides. The GLP-1 sequence in position 8 of albiglutide has also been modified, by substituting glycine for alanine adjacent to the DPP4 hydrolysis site, to reduce metabolism. Modifications to GLP-1 receptor agonists have led to substantial increases in plasma half-lives, reducing the frequency of dosing from twice daily to weekly, which should improve patient compliance.
Although it does not target a GPCR, plecanatide, an engineered peptide agonist of guanylate cyclase C, is notable in that it is administered orally and acts directly in the gastrointestinal tract, demonstrating that local targeting approaches are feasible for intestinally restricted targets. GPCRs are abundant in the gut, so this could provide a novel approach for future therapeutic strategies.
Other peptide therapeutics have been approved for novel indications or uses. In 2018, the second melanocortin agonist, bremelanotide, was approved, but the approval was for a new clinical indication, sexual arousal disorder31. Lutathera (lutetium Lu 177 dotatate) was also approved in 2018 for the treatment of neuroendocrine tumours and is the most recent example of a peptide receptor radionuclide therapy. In this compound, the peptide dotatate, a somatostatin receptor agonist, is labelled with lutetium 177. After binding to somatostatin receptors, which are found at a high density on the surface of tumours, the radiation emitted by the peptide causes tumour cell death but, owing to the limited particle range, it causes little toxicity to adjacent healthy tissue32.
In 2019, difelikefalin was approved as a ‘first-in-class’ KOR agonist that has a high degree of selectivity for KOR over other opioid receptors and is used in the treatment of pain after abdominal surgery. Importantly, this treatment has few of the central nervous system (CNS) side effects, such as sedation, dysphoria, and hallucinations, that are associated with small-molecule analgesics33. This is consistent with the fact that little or no CNS permeability has been observed for difelikefalin, making this an example of a therapeutic for which lack of brain penetrance, as often occurs with peptides, is an advantage over a small molecule. The development of compounds selective for peripheral KORs is crucial for managing pain in the context of the emerging opiate crisis.
Peptides in the pipeline
Phase III
Three of the peptides currently in phase III development are potentially first in their class (Table 3). BL-8040 is the first peptide antagonist for CXC-chemokine receptor 4 (CXCR4), and if it progresses to clinic, BL-8040 would compete with an established small-molecule antagonist (plerixafor) for the same clinical indication — the mobilization of haematopoietic stem cells to peripheral blood prior to collection for autologous cell transplantation34. The interaction between the chemokine CXCL12 (also known as SDF1) and its receptor, CXCR4, plays a key role in haematopoietic stem cell mobilization.
Livoletide is a first-in-class analogue of unacylated ghrelin (UAG) and is being evaluated for the treatment of Prader–Willi syndrome, a rare genetic disease, to reduce hyperphagia (excessive eating) and obesity35,36. Ghrelin, the ‘hunger hormone’, stimulates appetite, increases food intake and promotes fat storage, all of which are severely upregulated in patients with Prader–Willi syndrome. Interestingly, livoletide is a fully cyclized (cyclo(Ser-Pro-Glu-His-Gln-Arg-Val-Gln)) variant, which protects this peptide from metabolism (at least in vitro in human blood) and extends its plasma half-life35,36. However, the exact mechanism of action is still unclear. A number of studies suggest that when UAG is co-administered with ghrelin it acts as a functional antagonist or acts via another receptor37. Other studies suggest that UAG is a full agonist38, so the data from late-stage clinical trials could be informative.
Setmelanotide39 is proposed as a possible first-in-class MC4 receptor agonist, as it has a modest ~20-fold selectivity over the MC3 receptor, which could potentially allow it to avoid cardiovascular side effects. The MC4 receptor is a key regulator in the hypothalamic control of food intake. The synthetic peptide is in development for the treatment of individuals with genetic variants in the pro-opiomelanocortin and leptin receptors, which cause severe early-onset obesity and hyperphagia, in order to restore signalling in the MC4 receptor pathway.
Phase II
In ongoing phase II trials, novel therapeutic strategies are exemplified by EP-100, which contains synthetic GnRH, consisting of 10 amino acids, attached to an 18-amino-acid cationic α-helical lytic peptide (CLIP-71) without a linker40. This lytic domain interacts with the negatively charged membrane to induce cell death. Cancerous cells overexpress GnRH1 receptors compared with normal tissue, and therefore are thought to bind the GnRH peptide, so binding of EP-100 to these receptors would target the lytic CLIP-71 domain preferentially to cancer cells. One potential advantage of this strategy is that the lytic peptide does not need to be released from GnRH by cleavage of a linker, which avoids possible toxicity.
Among class B receptor ligands, the naturally occurring peptide stresscopin (urocortin 3) targets the corticotropin-releasing factor CRF2 receptor and has short-term, dose-dependent efficacy in improving cardiac index and systemic vascular resistance41. Stresscopin (as RT-400) is in clinical trials for acute decompensated heart failure, a major cause of hospitalization.
Table 3 also summarizes peptides that have been discontinued in phase II or phase III clinical trials. The reasons why trials have not progressed are often not disclosed in peer-reviewed articles and are mainly found on the relevant company websites. With this important caveat, the predominant reason cited for termination of a trial has been futility (the inability to achieve clinical objectives) rather than adverse side effects. In some cases the phase III trials were testing new peptides and clinical indications. For example, foot ulcers affect one in four patients with diabetes, and there is currently an unmet need for new treatments. Angiotensin II has been shown to promote wound healing42; however, aclerastide, an angiotensin II receptor agonist, failed to meet the primary efficacy end-point of confirmed complete wound closure of the target ulcer within 12 weeks of the start of treatment. For others, peptides lacked efficacy against established targets. Terlipressin has been used in the treatment of hypotension and septic shock (Table 2) since 2006; surprisingly, in 2018, a large (868-patient) phase IIB/III clinical trial of selepressin, which targets the same receptors, in sepsis43 was terminated early for futility44.
Strategies to improve peptide design
Potency, half-life and administration
What pharmacokinetic and pharmacodynamic properties contribute to an efficacious peptide drug? Currently there is no consensus, but a trend is emerging amongst recently approved drugs for high sustained target potency combined with increased plasma half-life and reduced enzymatic metabolism and renal elimination. Synthetically modified peptides targeting class A GPCRs have maintained a high affinity (median pKi = 8.8), similar to that observed for native peptides, but have a substantially increased median plasma half-life (3 h, excluding compounds with flip-flop kinetics, compared with ~5 min for native peptide equivalents). Flip-flop kinetics have also been used to increase plasma half-life. For example, degarelix (Table 2) is a synthetic derivative of GnRH that blocks binding of the endogenous peptide to receptors in the pituitary gland and is used to treat prostate cancer. Following subcutaneous administration, degarelix forms a depot at the site of injection, from which the drug is slowly released into circulation to produce a plasma half-life from 42–70 days. Clearance is unaffected, occurring mainly via hydrolysis in the hepatobiliary system and excretion of the unchanged drug by the kidney45,46. Similarly, lanreotide, a somatostatin agonist that acts mainly by binding to the SST2 and SST5 receptors and is used to inhibit growth hormone release to treat acromegaly, also forms a drug depot at the site of injection, giving a plasma half-life of 22 days47, and also contains unnatural amino acids (Box 1).
The majority of the peptides that target GPCRs are administered by injection, although other routes (for example, intranasal administration is used for desmopressin) are increasingly being exploited. Charged and hydrophilic molecules such as peptides are typically not orally bioavailable. After several decades of synthesizing modified peptides, the inherent disadvantages of low membrane permeability, which limits oral bioavailability and tissue distribution, including to the CNS, are still applicable to most peptide drugs. However, there are a number of encouraging examples of the application of permeation enhancer strategies48, and detailed SARs are being explored, especially for cyclic and conformationally constrained peptides49. The most advanced, and exciting, of these approaches has been pioneered by Emisphere using a pharmaceutically inactive small-molecule enhancer N-[8-(2-hydroxybenzoyl)amino] caprylate (SNAC) co-formulated with semaglutide (a GLP-1 receptor agonist already approved as an injectable for the treatment of T2D). The resulting drug, oral semaglutide50, had increased transcellular permeability and bioavailability of ~4%51. The highest dose tested, 40 mg administered orally once daily, resulted in efficacy comparable to that of 1 mg of semaglutide injected once weekly52. Oral semaglutide was approved by the FDA in 2019. For drugs with high hydrophilicity and poor membrane permeability, such as peptides, absorption enhancers can promote membrane permeability and improve oral bioavailability.
Experimental approaches to enhance brain permeability include linking neurotensin to a brain-penetrant peptide, angiopep-2, thereby increasing transport across the blood–brain barrier via receptor-mediated transcytosis by about tenfold. This was sufficient to reverse pain behaviours in animal models of neuropathic and bone cancer pain53.
Desmopressin is one of the few examples of a peptide that can be administered orally. Cyclization contributes to its resistance to metabolism, and its hydrophobic nature enhances cellular absorption across the gut. Bioavailability is very low by this route (0.08–0.16%)54, but this level is sufficient to achieve a plasma concentration that is clinically effective. The success of desmopressin has proven the potential of engineering peptides to have oral activity. As a result of the range of strategies outlined above, a main area of growth for peptide drugs is in targeting peripheral peptide receptors, particularly those linked to metabolic diseases (Table 2).

Strategies for GLP-1 receptor agonists
The history of the development of GLP-1 receptor agonists exemplifies the broad range of approaches used to address the pharmacokinetic challenges in peptide development (Fig. 2; Table 4). To date, seven peptide GLP-1 receptor agonists have been approved for the treatment of T2D, with projected global sales in 2020 of at least US$10 billion (see EvaluatePharma in Related links).
GLP-1 has multiple effects that are beneficial in the treatment of T2D. Despite this, the natural peptide has a very short plasma half-life (~2 min) because of rapid enzymatic cleavage and enzymatic inactivation by DPP4, which precludes its use as an effective therapeutic treatment. Additional studies have confirmed high plasma clearance following the subcutaneous route of administration55,56.
Exenatide (Table 4) was the first GLP-1 receptor agonist approved for clinical use. In 1992, Eng et al.57 identified exendin-4, a new peptide hormone from the saliva of the Gila monster (Heloderma suspectum). Exendin-4 had many of the same pharmacological properties as GLP-1: it increased insulin secretion and reduced plasma glucose levels. However, unlike GLP-1, exendin-4 is resistant to cleavage and inactivation by DPP4. Exendin-4, as exenatide, received approval for the treatment of T2D in 2005 as an adjunctive therapy, and in 2009 as a monotherapy58. Although exenatide has been widely used for the treatment of T2D, its short plasma half-life requires frequent (twice daily) subcutaneous injection, which limits its efficacy, results in poor patient compliance and increases the risks of additional side effects, such as infection at the sites of injection.
Following the success of exenatide, multiple strategies were employed to increase plasma stability and half-life, reduce renal elimination and improve oral bioavailability for GLP-1 receptor agonists. These strategies can be broadly divided into two approaches. One is based on extending the plasma half-life of exenatide, leading to Exenatide once weekly (QW) and lixisenatide. Exenatide QW59 is a reformulation of exenatide into microspheres consisting of a biodegradable polymer, poly-(D,L-lactide-co-glycolide), to extend the dosing interval to weekly administration. In lixisenatide60, modification of the exenatide sequence, including addition of a C-terminal lysine tail, conferred resistance to DPP4 cleavage and increased plasma half-life.
The other strategy focused on modifying the native GLP-1 peptide, leading to liraglutide, albiglutide, dulaglutide and, most recently, semaglutide (Fig. 2; Table 4). Because of the short half-life of native GLP-1, as a direct result of both cleavage by DPP4 and rapid renal elimination owing to its relatively small size, a combination of strategic solutions has been employed. Liraglutide61 was approved in 2009 and is a human GLP-1 receptor agonist based on the native GLP-1 peptide, with one amino acid substitution (Lys34Arg) and a C16 palmitic acid side chain attached via a glutamyl spacer at position 26. These modifications increase its binding to serum albumin, which significantly reduces renal elimination and DPP4 cleavage. The active modified GLP-1 is released from albumin at a slow, constant rate, resulting in slower degradation and reduced renal elimination compared, for example, to that of the mature endogenous form of GLP-1, GLP-17–37. Semaglutide62, which was approved in 2017, incorporates the unnatural amino acid α-aminoisobutyric acid (Box 1) at position 8, to reduce DPP4 cleavage, and contains an alternative C18 fatty diacid at Lys26, which provides strong albumin binding. Albiglutide22 and dulaglutide63 are variants of peptide fusion proteins. Both are protected from DPP4 cleavage because of substitutions at position 8, and both contain two copies of GLP-1, either fused to human serum albumin (albiglutide) or covalently linked to a human IgG4–Fc heavy chain by a small peptide linker (dulaglutide), to increase the half-life of the molecule, owing to recycling by the neonatal Fc receptor (FcRn) and/or increased molecular weight.
All of these GLP-1 receptor agonists require subcutaneous administration. However, several promising oral delivery approaches are in various stages of development, including the recently approved oral semaglutide, which is co-formulated with SNAC as described in Potency, half-life and administration.
Ligand bias
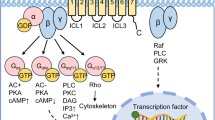
Traditional drug–receptor theory posits that drugs have two properties: affinity and intrinsic efficacy. Affinity is the quantifiable measure of how tightly a drug binds to its target and is constant for each drug–receptor pair, supporting medicinal-chemistry-led SAR investigations and the application of concepts such as drug selectivity for target over non-target receptors. However, affinity says nothing about the functional consequences of a drug–receptor interaction. This is defined by the term ‘intrinsic efficacy’, which describes the effect a drug has on receptor activity. Using this original definition, drugs are either agonists (with a combination of affinity and intrinsic efficacy) or antagonists (with affinity but no intrinsic efficacy), an approach that has underpinned drug development for the past few decades. However, there is increasing evidence that the simple concepts of agonism and antagonism only scratch the surface of the drug–receptor signalling landscape. We now know that receptors adopt a range of conformational states, thus giving rise to important new pharmacological concepts such as constitutive activity and biased signalling (or functional selectivity). Because of their relatively large size, peptides often interact at multiple key positions within both the extracellular and transmembrane domains of GPCRs. These contemporary pharmacological concepts may have major implications for the design and optimization of new peptide drugs, especially when used in combination with the structural biology techniques described later in this article.
A receptor may be able to engage with a spectrum of downstream signalling pathways, but a ligand with affinity for that receptor may affect only a subset of these pathways, and likewise may be an agonist of some and an antagonist of others. This observation underpins the concept of ligand bias and is the definition of a biased peptide ligand used in this Review. This principle has been used as a criterion to identify biased peptides from the literature (Box 2). Bias is usually examined in the context of the two most thoroughly characterized GPCR signalling pathways, those initiated by the binding of either β-arrestin or G proteins to the GPCR complex. Importantly, however, biased signalling can refer to any signalling pathway measured — for example, those involving different subtypes of G proteins — and can be considered to be specific to both the context and the pathway. As such, multiple drugs acting at a single receptor might all be characterized as agonists, but each might have a different functional selectivity profile for the cellular pathways regulated by that receptor.
How important is bias in a particular pathway? Physiologically, this is exemplified with the GnRH1 receptor, which is unusual amongst the peptide-binding GPCRs because it lacks the C-terminal intracellular domain. Prolonged agonist stimulation of GPCRs usually results in phosphorylation of residues in the intracellular C terminus, which then interact with β-arrestin64. This interaction induces receptor endocytosis, which terminates receptor signalling and results in desensitization of the receptor to the peptide. The GnRH1 receptor does not undergo agonist-induced phosphorylation or couple to β-arrestin, and is therefore slowly internalized64. By contrast, the GnRH2 receptor, which is found in other vertebrates, retains the C terminus, and stimulation induces phosphorylation of C-terminal residues, β-arrestin binding, receptor internalization and rapid receptor desensitization.
How important is biased agonism in drug discovery? This dichotomy outlined for the GnRH receptors suggests that biasing compounds away from β-arrestin recruitment and receptor internalization will reduce desensitization that would limit drug efficacy. Bias may have additional clinically important consequences. For example, β-arrestin-mediated respiratory depression is an adverse consequence of treatment with MOR agonists, which could potentially be reduced with biased agonists65. Stretch-induced myopathy in the heart occurs as a consequence of the apelin receptor acting as a mechano-sensor in the absence of endogenous apelin66, which is downregulated in disease. Replacing the lost apelin with a biased agonist might avoid activating the deleterious pathway (see The apelin receptor).
Biased signalling could herald a new, more specific pharmacological strategy for GPCR agonists, some examples of which are described in the following sections. However, the vast majority of existing examples of biased signalling have been defined using relatively simple in vitro cellular outputs. Predicting clinical benefit will require an understanding of the relevant cellular mechanisms that contribute to disease and the identification of biased ligands from appropriate in vitro cell-signalling assays. Many receptors — including those activated by GnRH67, opioids68,69, chemokines70, neuropeptide S71, proteinases72 or parathyroid hormone73 — also exhibit bias but are not discussed in the following sections.

Angiotensin II and AT1 receptor
Biased peptide agonists that target the angiotensin (AT1) receptor are the most extensively studied of the peripheral GPCR targets. This peptide–receptor pair is important in regulating blood pressure; the AT1 receptor is targeted by the ‘sartan’ class of small-molecule antagonists, which are used as antihypertensive agents.
Pioneering studies revealed a synthetic angiotensin II analogue, SII, that bound with high affinity and was able to internalize the AT1 receptor (presumably by β-arrestin recruitment, but this was not measured) and activate the β-arrestin effector mitogen-activated protein kinase (MAPK), but did not induce the G protein-mediated production of inositol triphosphate (IP3)74. More potent compounds that stimulated β-arrestin-mediated signalling but that competitively antagonized G protein coupling were subsequently developed, including TRV027 (Sar-Arg-Val-Tyr-Ile-His-Pro-d-Ala-OH; Table 3). In rats, TRV027 antagonized AT1 receptor-mediated G protein signalling and reduced mean arterial pressure, similar to the antihypertensive agent losartan. Crucially, its effect on cardiac contractility was opposite to that of losartan: TRV027 induced the β-arrestin-mediated activation of kinase pathways and increased endothelial nitric-oxide synthase phosphorylation75, with a resulting increase in cardiac contractility. This pharmacological profile, of an antihypertensive action combined with an increase in cardiac output, was demonstrated to be beneficial in a dog model of heart failure, in which TRV027, when co-administered with the commonly used loop diuretic furosemide, was shown to preserve furosemide-mediated natriuresis and diuresis76.
This molecule was then evaluated in patients with acute heart failure, with the objective of reducing afterload while increasing cardiac performance and maintaining stroke volume77. In individuals with elevated plasma renin levels (indicative of acute heart failure), a short, reversible and modest (5 mmHg) reduction in blood pressure was reported, but no change was observed in volunteers with normal renin levels78. However, no benefits were observed over those of standard-of-care drugs through a 30-day follow-up in a phase IIb randomized, double-blind clinical trial79. The reasons for this lack of efficacy are unclear. Insufficient target engagement seems unlikely, as the peak plasma concentration (Cmax) at the highest dose was ~580 nM which, combined with low plasma binding and high affinity (16 nM), would be expected to result in significant receptor occupancy78. Signalling pathways may be subtly altered in conditions such as heart failure, which could affect drug efficacy. Angiotensin II, SII and TRV027 have distinct downstream phosphorylation events and gene expression profiles80, which emphasizes the need for comprehensive analyses of the signalling pathways for biased peptide ligands, some of which are being evaluated for clinical use. Peptides have also been developed as tool compounds whose bias is reversed compared to that of TRV027, and which are presumed, therefore, to be deleterious, which should allow further insights into this signalling pathway.
The apelin receptor
The apelin system physiologically antagonizes angiotensin II signalling. Although it is not currently targeted by any approved drug, this class A GPCR and its ligands, apelin and elabela (also called apelin receptor early endogenous ligand or toddler), might play a role in the physiological regulation of the cardiovascular system. Dysregulation of the apelin system and loss of endogenous peptides have been proposed to contribute to a number of conditions, such as pulmonary arterial hypertension81,82,83 and heart failure84,85, indicating potential for more precise targeting of apelin signalling pathways using biased ligands. Specifically, a G protein biased agonist, if used to replace the missing ligand, would show a reduced propensity to desensitize the apelin receptor with repeated use. Interestingly, mice lacking the apelin receptor were protected from cardiac hypertrophy and heart failure associated with chronic pressure overload, whereas mice lacking apelin itself were not66. In the heart, apelin normally stimulates Gαi-mediated protective responses. However, the cardiac apelin receptor, in the absence of apelin, acts via β-arrestin pathways as a mechano-sensor of stretching; cardiomyocytes from apelin receptor knockout mice have a reduced hypertrophic stretch response66. Therefore, apelin receptor ligands that are G protein-biased or that preferentially block β-arrestin signalling may be beneficial in patients with heart failure.
Preproapelin is a 77-amino-acid peptide that is predicted to be cleaved into biologically active peptides, including apelin-36 (corresponding to amino acids 42–77), apelin-17, apelin-13 (corresponding to amino acids 65–77) and a pyroglutamate-modified form, [Pyr1]apelin-13. In human cardiovascular tissues in vitro, apelin-13, [Pyr1]apelin-13 (which was identified as the predominant isoform) and apelin-36 were found to be equipotent as vasodilators and inotropes86; however, apelin-13 and apelin-36 elicited different patterns of receptor internalization in cell-based assays87,88. These data suggest that putative endogenous apelin isoforms may demonstrate unique signalling profiles in vivo, but. whether this might have physiological or pathophysiological consequences is not yet known. However, specific pathway bias has been described in vitro: for example, in cAMP and β-arrestin assays, compared to [Pyr1]apelin-13, the N-terminally extended apelin-17 demonstrated an ~70-fold bias towards β-arrestin89. Modified peptides based on the apelin sequence have also demonstrated pathway bias. Compared to apelin-17, a truncated apelin-17 that lacks the C-terminal phenylalanine (Lys16Pro), or versions of apelin-17 and [Pyr1]apelin-13 in which the C-terminal phenylalanine is replaced by an alanine (Lys17Ala and pGlu13Ala, respectively), retained similar binding affinity and potency in inhibiting cAMP but did not induce receptor internalization90. Indeed, interactions between the C-terminal phenylalanine and residues in an aromatic pocket (Phe255 and Trp259 in the rat apelin receptor) are required for apelin-mediated internalization91. In terms of downstream signalling events, apelin-17 stimulates extracellular-signal-regulated kinase 1/2 (ERK1/2) phosphorylation in both a Gαi- and a β-arrestin-dependent manner, whereas the action of Lys16Pro is G protein-dependent but β-arrestin-independent92. The highly conserved Ser348 in the C terminus of the apelin receptor is critical for interaction with G protein-coupled receptor kinase (GRK) and for β-arrestin-mediated signalling, but replacing Ser348 with Ala did not alter the cell surface expression of the receptor, binding of apelin ligands or activation of the Gαi or Gαq pathway93.
The SAR of biased signalling at the apelin receptor has been assessed with a novel series of cyclic peptides based on the apelin-13 structure94. The main conclusion was that, consistent with the data for linear peptides, the C-terminal amino acid is important for receptor binding, β-arrestin recruitment and receptor internalization. In addition, the [Pyr1]apelin-13 sequence incorporates an N-terminal RPRL motif that is absolutely necessary for receptor binding. Earlier SAR studies demonstrated that His7 and Met11 substitutions did not affect the binding or function of the ligand95. From these SAR studies, it is apparent that modified peptides can be designed that may show G protein or β-arrestin signalling bias, as is exemplified by the macrocyclic peptide MM07 (ref.96). MM07 has induced vasodilatation and increased cardiac output in both rats and human volunteers, which may be desirable in the clinical setting, and importantly, the receptor was not desensitized on repeated application. This peptide has subsequently been shown to have efficacy in a rat model of pulmonary arterial hypertension97. Further modification of MM07 or of similarly G protein-biased peptides, as we described above for GLP-1, may result in a peptide with improved plasma half-life to take forward to proof-of-principle clinical studies.
The ghrelin receptor
As we mentioned above, ghrelin is a gut hormone with a role in hunger signalling, which has made the ghrelin receptor a potential target for anti-obesity drugs. However, the physiological actions of ghrelin are diverse, including effects on gastric motility, growth hormone release, reward behaviour and mood. Therefore, drugs that mimic or block ghrelin may have a number of therapeutic uses, but the impetus for developing such agents is hampered by the potential for undesirable on-target side effects. The discovery of compounds that can distinguish between the ghrelin responses now suggests that it may be possible to develop biased ligands that, for example, selectively reduce body weight. Modification of the ghrelin inverse agonist [d-Arg1,d-Phe5,d-Trp7,9,Leu11]substance P generated compounds containing an N-terminal d-Trp-Phe-d-Trp (wFw) motif with differing C-terminal peptide-mimetic spacers98. One of these, wFw–isonipecotic acid, was the first ghrelin receptor biased agonist, as unlike known ghrelin mimetics, it did not signal through the serum-response element (SRE) pathways (presumably downstream of Gα12/13) but did activate the Gαq and ERK1/2 pathways. A likely explanation was revealed by mutagenesis and modelling studies: wFw–isonipecotic acid does not interact with receptor residues that are important for the binding and function of ghrelin or other ghrelin mimetics98. wFw–isonipecotic acid did not stimulate feeding in rats, which may reflect the relative importance of SRE pathways for this function. If confirmed, these data imply that biased ghrelin ligands could be designed that distinguish its effects on growth hormone release from its effects on food intake99. For another ghrelin-targeted peptide, ulimorelin (Table 3), the prokinetic effect on gut motility was sustained, unlike the tachyphylaxis seen with other ghrelin agonists. Perhaps ulimorelin does not activate β-arrestin signalling, which would limit receptor desensitization with continued use. Additionally, ulimorelin mimics the orexigenic and gut motility effects of ghrelin, but it is not a growth hormone secretagogue. Whether this unique pharmacology is explained by pathway bias is unclear. Unfortunately, despite ulimorelin’s apparent advantages over other ghrelin mimetics, a phase III trial in postoperative ileus was discontinued owing to a lack of efficacy over placebo.
The ghrelin receptor exhibits high constitutive activity, independent of the cellular environment100, that is abolished in a naturally occurring human mutant receptor (Ala204Glu)101 that is associated with short stature. This mutation increases the probability that the C-terminal section of extracellular loop 2 (ECL2) will form an extended α-helix, thereby constraining this part of the protein and resulting in the loss of constitutive activity101. Additionally, whereas ghrelin-stimulated Gαq and Gα12/13 signalling were essentially unaffected by the mutation, β-arrestin responses were substantially reduced, indicating that ECL2 is also important for determining ligand bias. A role for constitutive receptor activity in fasting-induced hyperphagia in mice102 has been proposed. Biased signalling at the ghrelin receptor can also be achieved with inverse agonists103, which could inform future drug discovery efforts.
GLP-1 receptors
Class B receptors are also tractable to ligand bias. Insulin secretion and regulation of blood glucose in response to GLP-1 may result from receptor engagement with a number of different G proteins and several signalling pathways. Pathway preference may be determined by the agonist and/or the cell type. Endogenous peptides derived from the proglucagon peptide include both full-length (GLP-11–36, GLP-11–37) and truncated (GLP-17–36, GLP-17–37; the mature isoforms) forms of GLP-1, each of which can each also exist in an amidated form, as well as oxyntomodulin. The effects of the endogenous peptide agonists, as well as the clinically used peptide mimetic exenatide, have been compared in physiologically relevant assays that measure cAMP, Ca2+ mobilization and ERK1/2 phosphorylation. Using GLP-11–37 amide as the reference ligand, the shorter GLP-1 isoforms and exenatide had a similar pathway activation profile, whereas the longer GLP-1 isoforms and oxyntomodulin exhibited some pathway bias104. Subsequently, from a peptide library screen, an N-terminally modified version of exenatide, exendin-P5, was identified. P5 was shown to be G protein-biased (lacking β-arrestin activity) and was more effective than exenatide in a chronic mouse model of T2D105, suggesting that G protein-biased ligands may be advantageous in this condition.
A novel strategy to generate biased versions of GLP-1 involves the replacement of particular α-amino acids in the peptide backbone with β residues or with non-proteinogenic α-amino acids106,107. The resulting α–β peptides are resistant to degradation by endogenous peptidases. For example, α-aminoisobutyric acid, a strong helix inducer that occurs rarely in nature, protects the N terminus from degradation by DPP4 and neprilysin. Thioamidation limits degradation, and thioamidated GLP-1 analogues have an in vitro half-life of many hours, compared to 2 min for the native peptide108. Thioamides do not have appreciable β-arrestin agonist activity and are therefore also G protein-biased. An alternative, but challenging, strategy may be to develop allosteric modulators that affect both endogenous peptide-binding kinetics and signalling bias109.
Calcitonin receptors
Experiments using the calcitonin (CT) receptor were some of the first to crystallize the understanding that differences in relative agonist potencies among different tissues did not necessarily mean that these tissues expressed different receptor subtypes110. Both human and salmon calcitonin have FDA approval for treatment of Paget disease (Tables 1,2). Interestingly, they exhibit distinct binding kinetics, affinity and functional efficacy in different G protein pathways, implying that they stabilize different active conformations of the CT receptor111,112. The response to activation of the human receptor is complicated by the existence of two major splice variants that exhibit tissue-specific expression patterns and that couple to different signalling pathways113. The predominant human receptor isoform, CT(a) receptor, lacks a 16-amino-acid insert in the first ECL that is present in the less abundant CT(b) receptor isoform114. Both variants bind calcitonin peptides with comparable affinity; however, the CT(b) receptor is not internalized well and preferentially activates Gαs over Gαq, relative to the CT(a) receptor115. Mutational data116 mapped with molecular dynamic simulations have highlighted that ECL2 was important in conformational propagation linked to the Gαs–cAMP pathway, which was distinct from the ligand-specific and pathway-specific effects propagated by ECL3. These observations highlighted differences in the mechanisms of ligand interaction and receptor activation of the CT receptor compared to another class B receptor, GLP-1 (ref.116).
Oxytocin receptors
Atosiban, described as an oxytocin receptor antagonist, is used clinically to prevent preterm labour, by blocking a Gαq-linked increase in intracellular Ca2+ that normally promotes uterine contractility. However, atosiban is more correctly identified as a biased ligand, as it promotes coupling of the oxytocin receptor to Gαi (which is linked, via MAPK, to the inhibition of cell proliferation), in addition to antagonizing Gαq signalling117. This is of interest because oxytocin receptors are overexpressed in several cancers, and therefore atosiban could be repurposed as a chemotherapy. Interestingly, whilst atosiban can activate or inhibit the pathways downstream of different Gα proteins, it has little effect on β-arrestin signalling: receptor internalization was markedly attenuated following exposure to atosiban, whereas these receptors are rapidly and profoundly lost in response to oxytocin117. Furthermore, oxytocin-triggered IP3 accumulation was competitively blocked by pre-exposure to atosiban117. It has subsequently been confirmed that atosiban does not recruit β-arrestin to the oxytocin receptor and shows selectivity for Gαi3 over other Gαi isoforms118.
Insights from structural studies
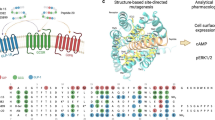
Experimental X-ray crystallography or cryo-EM structures have been reported for the 7-transmembrane domains of 62 GPCRs, covering 212 distinct GPCR–ligand complexes and 200 unique ligands119,120. Of these GPCRs with solved structures, 27 bind peptides or proteins with 65 solved unique receptor–ligand complexes115,116,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157. So far, 22 of the experimentally determined GPCR structures (10%) (see the GPCR database in Related links) contain a peptide ligand, covering class A and B GPCRs (Figs 3,4; Supplementary Table 1). The peptide-bound structures of these receptors provide structural templates and detailed insights into the structural determinants of ligand binding and functional activity (Box 3; Supplementary Text and Supplementary Table 1). About half of the clinically relevant peptides reported in Tables 1,2,4 have been structurally modelled on the basis of homologous peptides and/or receptors. These peptides serve as potential templates for the structure-based optimization and design of novel GPCR peptide therapeutics.

Peptide G protein-coupled receptors (GPCRs) for which seven-transmembrane domain (7TM) or full-length structures are available in complex with peptide ligands (red) or only in complex with non-peptide ligands (turquoise) are distinguished from peptide GPCRs for which only extracellular domain (ECD) structures are available (green) and non-peptide GPCRs for which 7TM or full-length structures are available (blue). For details, see Supplementary Table 1. Related GPCR families — adhesion, glutamate, frizzled and taste (TAS2) — are shown for reference. Receptors are classified as orphans when endogenous ligands are not yet established. The Class A receptor family is shown classified into four groups: the α-group includes amine, peptide and prostaglandin receptors; the β-group includes only receptors that bind peptides; the γ-group contains chemokine receptors, some receptors that bind peptides such as somatostatins, galanin, and opioids, and receptors that bind other types of ligands; and the δ-group includes olfactory, purine and glycoprotein receptors.

The panels summarize the structural interactions for aligned binding-site residues of class A and class B peptide G protein-coupled receptors (GPCRs), forming polar H-bond or ionic interactions (red), or only lipophilic interactions (grey), with the peptide ligands shown in the individual binding-mode figure panels. Amongst the class A GPCRs, the binding sites of AMG3054-bound apelin receptor (Protein Data Bank identifier (PDB ID): 5VBL)121, octapeptide partial agonist (Ang II [Sar1, Ile8]) bound AT1 receptor (PDB: 6DO1)122, PMX53-bound complement C5a1 receptor (PDB: 6C1Q)124, endothelin-bound ETB receptor (PDB: 5GLH)125, neurotensin-bound NTS1 receptor (PDB: 3ZEV)127, UR-MK299-bound neuropeptide NPY1 receptor (PDB: 5ZBQ)141, DAMGO-bound μ opioid receptor (MOR) (PDB: 6DDE)128, and CCL5-bound CCR5 (PDB: 5UIW)129 are shown. Amongst the class B GPCRs, the calcitonin gene-related peptide (CGRP) bound to the CGRP receptor (CRLR) and complexed with Gαs (PDB: 6E3Y)134, NN1702-bound glucagon receptor (PDB: 5YQZ)135, peptide 5-bound (PDB: 5NX2)136 and exendin-P5-bound (PDB: 6B3J)135 GLP-1 receptor, and ePTH-bound PTH1 receptor139 are shown. All views are focused on the seven-transmembrane domain and are consistent with the orientations of the peptide diagrams shown in Fig. 2. The binding pocket surfaces (grey mesh) are contoured at 1 kcal mol–1 using the carbon sp3 (C3) GRID probe199, whereas lipophilic areas are defined using the C1 = (lipophilic) probe contoured at –2.8 to –3.0 kcal mol–1, customized to the GPCR binding sites200. Generic GPCR residue numbers201 are provided that are based on the Ballesteros–Weinstein class A GPCR (apelin receptor, AT1, C5a1, CCR5, ETB, MOR, NPY1)202 and the Wootten203 class B GPCR (CGRP, glucagon, GLP-1, PTH1) numbering schemes. According to these schemes, the first number (1–7) denotes the transmembrane helix, and the following number indicates the residue position relative to the most conserved amino acid in the helix (which is assigned the number 50), considering numbering offset due to helical bulges or constrictions201.
Peptide ligands bind to GPCRs with numerous binding modes, reflecting the diverse chemical structures and properties of both the ligands and the receptor binding sites. Class A GPCR peptide ligands generally bind to ECL2 and a polar or ionic interaction site at the top of ECL3 (the portion of the protein between transmembrane region 6 (TM6) and TM7) and bind differentially located and shaped lipophilic regions deeper in the receptor pocket. Most class B GPCR ligands are helical; the helical ligands have a lipophilic interaction with a site in the region between TM1, TM2 and TM7 that is less accessible in class A GPCRs, and some of the class B ligands’ specificity is determined by polar interaction networks that can form with different relative orientations of the extracellular N-terminal domains (ECDs) and the transmembrane domains149.
Class B GPCRs contain an ECD of 120–160 residues and a transmembrane domain of 310–420 residues. In addition to the transmembrane-domain-only and full-length structures of class B GPCRs that have been described, several structures of isolated class B GPCR ECDs have been solved149 (Supplementary Table 1). These ECD–peptide complexes have conserved hydrophobic interactions between conserved lipophilic residues in the C-terminal part of the ligand and hydrophobic interaction sites in the ECD of the corresponding receptor.
Considerable progress has been made in elucidating the 3D structures of key regions for peptide recognition and selectivity by GPCRs. Methodological and technical improvements to cryo-electron microscopy are expanding the role of this technique, alongside X-ray crystallography, in solving GPCR structures. Emerging information on the different activation states and structural features responsible for activation or inhibition are being exploited to guide drug discovery. This is particularly important for class B, in which all endogenous ligands are peptides and there is the potential to discover new compounds based on exploiting the allosteric binding sites revealed by structural studies. Future studies will help unravel the importance of receptor dimerization and contribute to the rational, rather than empirical, design of biased peptide ligands, as our knowledge expands of the key residues involved in the kinetics and dynamics of signalling by means such as β-arrestin.

Perspectives and conclusions
Nearly all of the peptide drugs approved for clinical use to date function as full agonists. This probably reflects the predominant strategy for the discovery of clinical candidates, which is based on structural modifications to naturally occurring peptide sequences, rather than high-throughput screens against target receptors, which are often used to identify small-molecule therapeutics. This situation may, however, be set to change. The number of deduced structures of the GPCRs that bind peptides is rapidly increasing using crystallography (Supplementary Table 1), which will enable the rational, structure-guided design of peptides. This approach will expand further because of cryo-EM, from which, crucially, structures can be determined from GPCRs bound to an agonist in an active state, as has been successfully done for class B receptors133,137,140. Indeed, peptide allosteric modulators have been proposed for the urotensin II receptor that block urotensin II-mediated contraction of aortic rings, but that have no effect on the activity of urotensin II-related peptide, the second endogenous agonist that binds the receptor158. Structure-guided design may therefore enable peptides drugs to selectively distinguish between and modulate the action of two endogenous peptides that act at the same receptor, one of which causes a detrimental pathophysiological action, perhaps owing to differences in spatial or temporal signalling. Strategies such as screening phage display peptides have also been effective in discovering novel peptide ligands; for example, antagonists of the class B VIP2 receptor that have nanomolar affinity were identified using this approach159.
How do we use this information to synthesize a better peptide drug? This Review has highlighted one major trend over the last two decades: the successful exploitation of unnatural amino acids and chemical modifications to manipulate physicochemical properties, principally to improve pharmacokinetics but also, to a lesser extent, to improve pharmacodynamics. Another, earlier-stage trend stems from the discovery of peptides that can be biased towards G protein-dependent or G protein-independent β-arrestin-mediated pathways.
Biased ligands have enormous potential to selectively activate pathways that produce beneficial clinical effects, while reducing signalling via pathways that may cause unwanted on-target side effects. Biased peptide agonists have been identified for a number of receptor families, and clinical proof-of-concept studies are emerging94. As proof of principle, but subsequent to clinical approval, the OT receptor antagonist atosiban was identified as being biased, in that it does not activate β-arrestin, thereby reducing internalization, which is an important aspect of its mechanism of action118.
Particularly compelling evidence for the need for biased agonists has emerged from studies of the MC4 receptor. Remarkably, gain-of-function mutations identified in humans that were associated with reduced body mass index and protection from T2D and coronary artery disease were biased towards the β-arrestin pathway. This suggests that β-arrestin-biased MC4 agonists that act at the native receptor may be a new strategy for the treatment of obesity160. Individuals from the 1000 Genomes Project had, on average, 68 missense variations that occurred within the coding regions of one-third of the GPCR drug targets; only 8 of these variants had previously known clinical associations with altered drug response161. In data from ~68,000 individuals in the Exome Aggregation Consortium (EXAC), variants were found in the drug binding sites of 108 GPCRs162. Interestingly, variants of MOR and CCKA, which binds cholecystokinin, were shown experimentally to have an altered drug response. These results suggest that mining databases such as that from the 100,000 Genomes Project163, in which disease phenotypes are linked with whole-genome sequencing and patients can be recalled, will yield additional variants that experimentally lead to a loss or gain of function that can be used to identify new GPCR targets for novel treatments.
Dual agonist peptides that activate two different GPCRs are also emerging. The focus to date has been on combinations that target the GLP-1 receptor along with either the glucagon164 or glucose-dependent insulinotropic polypeptide (GIP)165,166 receptor. Interestingly, a compound that binds to all three receptors is in phase I testing166. In a phase II trial, a molecule that contained both GLP-1 and GIP sequences significantly improved glycaemic control and reduced body weight in patients with T2D167. The rationale was that the two peptides account for most of the effects of incretins. However, both are degraded by DPP4. Therefore, designing a peptide that contains both peptide sequences but that lacks DPP4 cleavage sites, in order to enhance plasma half-life, could mimic the beneficial effects of incretins. Clearly, a single molecule is an attractive strategy for synergy and patient compliance, but empirical determination of the optimum relative balance between the potency of the two agonistic effects of this single molecule is challenging. Stapled peptides that constrain α-helices to lock a peptide in a particular (often active) conformation are being explored — for example, as modified orexin168 and oxyntomodulin169 ligands. Similarly, pepducins, which are derived from short sequences of intracellular loops of GPCRs and are lipidated to penetrate cells so they can access allosteric sites and stabilize GPCR conformations, are being developed170, and some are being tested in experimental medicine studies in the clinic171.
In pharmacokinetics, modifications to dramatically increase plasma half-life from a few minutes to days have been the most revolutionary advances, and a range of strategies can effectively reduce metabolism and/or renal excretion. Sustained-release formulations are another key development. Theoretically, these innovations can be applied to virtually any peptide.
Promising experimental or early-stage trials include linking genetically engineered exendin-4 to a single-domain albumin-binding antibody (AlbudAb), which prolonged plasma half-life to 6–10 days while maintaining agonist activity (measured as reduced postprandial glucose and insulin levels, and delayed gastric emptying)172. An alternative strategy has been employed to chemically link an apelin peptide analogue to a single-domain antibody173. The advantage of using a chemical link is that non-genetically encoded amino acids can be introduced, so this agonist has high affinity for the apelin receptor but is also resistant to peptidase-mediated degradation. Genetically engineered peptide–AlbudAb conjugates, for example, cannot be made resistant to peptidases in the same way.
Nanotechnology strategies have been applied to induce reversible peptide self-assembly to prolong the bioactivity of a peptide in vivo. As proof of concept, Ouberai et al.174 demonstrated that oxyntomodulin self-assembled into a stable nanofibril formulation, which subsequently dissociated in vivo so as to release active peptide, thereby prolonging detectable activity in the plasma from 4 h to 5 days. Oral (albeit with low bioavailability) and nasal delivery are being used in the clinic to avoid daily injections, and these strategies will be increasingly explored. The challenge of engineering peptides that cross the blood–brain barrier remains, and many GPCRs with peptide ligands reside in the brain. Linking GPCR-targeting peptides to a brain-penetrant peptide in order to transport compounds across the blood–brain barrier is being investigated experimentally. Finally, the number of potential new GPCR targets is expanding, as orphan GPCRs continue to be paired with peptide ligands175.
It is not yet clear where the balance lies between the costs of developing clinical candidates based on a peptide versus a small molecule. However, the development of new GPCR peptide drugs continues on an upward trajectory, with seven approved by the FDA in 2017–2019, and more than ten in the pipeline in phases II and III, which is mirrored by a rise in the estimated global value of the industry to US$25.4 billion.
References
Santos, R. et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 16, 19–34 (2017). This detailed annotation of current FDA-approved drugs includes peptides targeting ~670 human genome-derived proteins mapped to therapeutic indications.
Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017). This report presents a comprehensive analysis of all FDA-approved drugs targeting GPCRs, lists novel agents in clinical trials and identifies potential new GPCRs without an approved drug.
Alexander, S. P. et al. The Concise Guide to Pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 176, S21–S141 (2019). This comprehensive, curated database incorporates the IUPHAR recommendations for the nomenclature of drug targets, including GPCRs, as well as quantitative information on interactions with approved medicines and experimental agents.
Davenport, A. P. et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol. Rev. 65, 967–986 (2013).
Wardman, J. H. et al. Identification of a small-molecule ligand that activates the neuropeptide receptor GPR171 and increases food intake. Sci. Signal. 9, ra55 (2016).
Bobeck, E. N. et al. The BigLEN-GPR171 peptide receptor system within the basolateral amygdala regulates anxiety-like behavior and contextual fear conditioning. Neuropsychopharmacology 42, 2527–2536 (2017).
Rode, B. M. Peptides and the origin of life. Peptides 20, 773–786 (1999).
Lau, J. L. & Dunn, M. K. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 26, 2700–2707 (2018).
US Food and Drug Administration. Impact story: developing the tools to evaluate complex drug products: peptides. FDA https://www.fda.gov/Drugs/ScienceResearch/ucm578111.htm (2019).
Congreve, M., Dias, J. M. & Marshall, F. H. Structure-based drug design for G protein-coupled receptors. Prog. Med. Chem. 53, 1–63 (2014).
Cooke, R. M., Brown, A. J., Marshall, F. H. & Mason, J. S. Structures of G protein-coupled receptors reveal new opportunities for drug discovery. Drug Discov. Today 20, 1355–1364 (2015).
Du Vigneaud, V., Ressler, C. & Trippett, S. The sequence of amino acids in oxytocin, with a proposal for the structure of oxytocin. J. Biol. Chem. 205, 949–957 (1953).
Du Vigneaud, V., Ressler, C., Swan, J. M., Roberts, C. W. & Katsoyannis, P. G. The synthesis of oxytocin. J. Am. Chem. Soc. 76, 3115–3121 (1954).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006).
Alonso, N., Calero-Paniagua, I. & Del Pino-Montes, J. Clinical and genetic advances in Paget’s disease of bone: a review. Clin. Rev. Bone Min. Metab. 15, 37–48 (2017).
Khalique, S. C. & Ferguson, N. Angiotensin II (Giapreza): a distinct mechanism for the treatment of vasodilatory shock. Cardiol. Rev. 27, 167–169 (2019).
Tarlatzis, B. C. & Kolibianakis, E. M. GnRH agonists vs antagonists. Best Pract. Res. Clin. Obstet. Gynaecol. 21, 57–65 (2007).
Malm-Erjefalt, M. et al. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab. Dispos. 38, 1944–1953 (2010).
Bern, M., Sand, K. M., Nilsen, J., Sandlie, I. & Andersen, J. T. The role of albumin receptors in regulation of albumin homeostasis: implications for drug delivery. J. Control. Rel. 211, 144–162 (2015).
Jensen, L. et al. Absorption, metabolism and excretion of the GLP-1 analogue semaglutide in humans and nonclinical species. Eur. J. Pharm. Sci. 104, 31–41 (2017).
Lau, J. et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J. Med. Chem. 58, 7370–7380 (2015).
Rendell, M. S. Albiglutide: a unique GLP-1 receptor agonist. Expert Opin. Biol. Ther. 16, 1557–1569 (2016).
Liu, X., Wright, M. & Hop, C. E. Rational use of plasma protein and tissue binding data in drug design. J. Med. Chem. 57, 8238–8248 (2014).
Smith, D. A., Di, L. & Kerns, E. H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 9, 929–939 (2010).
Torre, B. G. & Albericio, F. The pharmaceutical industry in 2016. An analysis of FDA drug approvals from a perspective of the molecule type. Molecules 22, 368 (2017).
Al Musaimi, O., Al Shaer, D., de la Torre, B. G. & Albericio, F. 2017 FDA peptide harvest. Pharmaceuticals 11, E52 (2018).
Al Shaer, D., Al Musaimi, O., Albericio, F. & de la Torre, B. G. 2018 FDA peptide harvest. Pharmaceuticals 12, E42 (2019).
Leder, B. Z. et al. Effects of abaloparatide, a human parathyroid hormone-related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J. Clin. Endocrinol. Metab. 100, 697–706 (2015).
Hattersley, G., Dean, T., Corbin, B. A., Bahar, H. & Gardella, T. J. Binding selectivity of abaloparatide for PTH-Type-1-receptor conformations and effects on downstream signaling. Endocrinology 157, 141–149 (2016).
O’Neil, P. M. et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet 392, 637–649 (2018).
Clayton, A. H., Lucas, J., DeRogatis, L. R. & Jordan, R. Phase I randomized placebo-controlled, double-blind study of the safety and tolerability of bremelanotide coadministered with ethanol in healthy male and female participants. Clin. Ther. 39, 514–526.e14 (2017).
Naraev, B. G., Ramirez, R. A., Kendi, A. T. & Halfdanarson, T. R. Peptide receptor radionuclide therapy for patients with advanced lung carcinoids. Clin. Lung Cancer 20, e376–e392 (2019).
Cowan, A., Kehner, G. B. & Inan, S. Targeting Itch with ligands selective for Kappa opioid receptors. Handb. Exp. Pharmacol. 226, 291–314 (2015).
Abraham, M. et al. Single dose of the CXCR4 antagonist BL-8040 induces rapid mobilization for the collection of human CD34+ cells in healthy volunteers. Clin. Cancer Res. 23, 6790–6801 (2017).
Allas, S. et al. AZP-531, an unacylated ghrelin analog, improves food-related behavior in patients with Prader-Willi syndrome: a randomized placebo-controlled trial. PLOS ONE 13, e0190849 (2018).
Allas, S. et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZP-531, a first-in-class analogue of unacylated ghrelin, in healthy and overweight/obese subjects and subjects with type 2 diabetes. Diabetes Obes. Metab. 18, 868–874 (2016).
Delhanty, P. J., Neggers, S. J. & van der Lely, A. J. Des-acyl ghrelin: a metabolically active peptide. Endocr. Dev. 25, 112–121 (2013).
Gauna, C. et al. Unacylated ghrelin is not a functional antagonist but a full agonist of the type 1a growth hormone secretagogue receptor (GHS-R). Mol. Cell Endocrinol. 274, 30–34 (2007).
Chen, K. Y. et al. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J. Clin. Endocrinol. Metab. 100, 1639–1645 (2015).
Curtis, K. K. et al. Novel LHRH-receptor-targeted cytolytic peptide, EP-100: first-in-human phase I study in patients with advanced LHRH-receptor-expressing solid tumors. Cancer Chemother. Pharmacol. 73, 931–941 (2014).
Gheorghiade, M. et al. Haemodynamic effects, safety, and pharmacokinetics of human stresscopin in heart failure with reduced ejection fraction. Eur. J. Heart Fail. 15, 679–689 (2013).
Yahata, Y. et al. A novel function of angiotensin II in skin wound healing. Induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J. Biol. Chem. 281, 13209–13216 (2006).
Lewis, R. J. et al. Rationale and design of an adaptive phase 2B/3 clinical trial of selepressin for adults in septic shock: selepressin evaluation programme for sepsis-induced shock-adaptive clinical trial. Ann. Am. Thorac. Soc. 15, 250–257 (2018).
Rodriguez, R. et al. Novel vasopressors in the treatment of vasodilatory shock: a systematic review of angiotensin II, selepressin, and terlipressin. J. Intensive Care Med. 35, 327–337 (2020).
Steinberg, M. Degarelix: a gonadotropin-releasing hormone antagonist for the management of prostate cancer. Clin. Ther. 31, 2312–2331 (2009).
Anderson, J. Degarelix: a novel gonadotropin-releasing hormone blocker for the treatment of prostate cancer. Future Oncol. 5, 433–443 (2009).
Kyriakakis, N., Chau, V., Lynch, J., Orme, S. M. & Murray, R. D. Lanreotide autogel in acromegaly — a decade on. Expert Opin. Pharmacother. 15, 2681–2692 (2014).
Maher, S. et al. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Delivery Rev. 106, 277–319 (2016).
Nielsen, D. S. et al. Orally absorbed cyclic peptides. Chem. Rev. 117, 8094–8128 (2017).
Knudsen, L. B. & Lau, J. The discovery and development of liraglutide and semaglutide. Front. Endocrinol. 10, 155 (2019).
Buckley, S. T. et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl Med. 10, eaar7047 (2018). Semaglutide co-formulated in a tablet with the absorption enhancer SNAC is absorbed in the stomach to achieve therapeutically relevant plasma concentrations. This clinically transformative study demonstrates how therapeutic peptides can be transformed from injectable to tablet-based oral therapies.
Davies, M. et al. Effect of oral semaglutide compared with placebo and subcutaneous semaglutide on glycemic control in patients with type 2 diabetes: a randomized clinical trial. JAMA 318, 1460–1470 (2017).
Demeule, M. et al. Conjugation of a brain-penetrant peptide with neurotensin provides antinociceptive properties. J. Clin. Invest. 124, 1199–1213 (2014).
Medicines and Healthcare products Regulatory Agency. Desmopressin acetate 100 microgram tablet (MHRA, 2007)
Ritzel, R., Orskov, C., Holst, J. J. & Nauck, M. A. Pharmacokinetic, insulinotropic, and glucagonostatic properties of GLP-1 [7-36 amide] after subcutaneous injection in healthy volunteers. Dose-response-relationships. Diabetologia 38, 720–725 (1995).
Nauck, M. A. et al. Effects of subcutaneous glucagon-like peptide 1 (GLP-1 [7-36 amide]) in patients with NIDDM. Diabetologia 39, 1546–1553 (1996).
Eng, J., Kleinman, W. A., Singh, L., Singh, G. & Raufman, J. P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 267, 7402–7405 (1992).
Furman, B. L. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 59, 464–471 (2012).
Charokopou, M., Vioix, H., Verheggen, B. G., Bratt, T. & Franks, D. The impact of long-term clinical evidence on cost-effectiveness of exenatide once weekly (Bydureon(R)) versus insulin glargine for patients with type 2 diabetes mellitus (T2dm) from a UK NHS perspective. Value Health 17, A343 (2014).
Leon, N., LaCoursiere, R., Yarosh, D. & Patel, R. S. Lixisenatide (Adlyxin): a once-daily incretin mimetic injection for type-2 diabetes. P T 42, 676–711 (2017).
Jackson, S. H., Martin, T. S., Jones, J. D., Seal, D. & Emanuel, F. Liraglutide (Victoza): the first once-daily incretin mimetic injection for type-2 diabetes. P T 35, 498–529 (2010).
Dhillon, S. Semaglutide: first global approval. Drugs 78, 275–284 (2018).
Pozzilli, P. et al. Placebo-controlled, randomized trial of the addition of once-weekly glucagon-like peptide-1 receptor agonist dulaglutide to titrated daily insulin glargine in patients with type 2 diabetes (AWARD-9). Diabetes Obes. Metab. 19, 1024–1031 (2017).
McArdle, C. A., Franklin, J., Green, L. & Hislop, J. N. The gonadotrophin-releasing hormone receptor: signalling, cycling and desensitisation. Arch. Physiol. Biochem. 110, 113–122 (2002).
DeWire, S. M. et al. G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 344, 708–717 (2013).
Scimia, M. C. et al. APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398 (2012).
Riccetti, L. et al. Human luteinizing hormone and chorionic gonadotropin display biased agonism at the LH and LH/CG receptors. Sci. Rep. 7, 940 (2017).
Gach-Janczak, K. et al. In vitro and in vivo activity of cyclopeptide Dmt-C[D-Lys-Phe-Asp]NH2, a µ opioid receptor agonist biased toward β-arrestin. Peptides 105, 51–57 (2018).
Thompson, G. L. et al. Biased agonism of endogenous opioid peptides at the µ-opioid receptor. Mol. Pharmacol. 88, 335–346 (2015).
Ziarek, J. J. et al. Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Sci. Signal. 10, eaah5756 (2017).
Liao, Y. et al. Human neuropeptide S receptor is activated via a Galphaq protein-biased signaling cascade by a human neuropeptide S analog lacking the C-terminal 10 residues. J. Biol. Chem. 291, 7505–7516 (2016).
Hollenberg, M. D. et al. Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br. J. Pharmacol. 171, 1180–1194 (2014).
Bohinc, B. N. & Gesty-Palmer, D. Biased agonism at the parathyroid hormone receptor: a demonstration of functional selectivity in bone metabolism. Mini Rev. Med. Chem. 12, 856–865 (2012).
Daniels, D., Yee, D. K., Faulconbridge, L. F. & Fluharty, S. J. Divergent behavioral roles of angiotensin receptor intracellular signaling cascades. Endocrinology 146, 5552–5560 (2005).
Violin, J. D. et al. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 335, 572–579 (2010).
Boerrigter, G. et al. TRV120027, a novel β-arrestin biased ligand at the angiotensin II type I receptor, unloads the heart and maintains renal function when added to furosemide in experimental heart failure. Circ. Heart Fail. 5, 627–634 (2012).
Felker, G. M. et al. Heart failure therapeutics on the basis of a biased ligand of the angiotensin-2 type 1 receptor: rationale and design of the BLAST-AHF study (Biased Ligand of the Angiotensin Receptor Study in Acute Heart Failure). JACC Heart Fail. 3, 193–201 (2015).
Soergel, D. G., Subach, R. A., Cowan, C. L., Violin, J. D. & Lark, M. W. First clinical experience with TRV027: pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 53, 892–899 (2013).
Pang, P. S. et al. Biased ligand of the angiotensin II type 1 receptor in patients with acute heart failure: a randomized, double-blind, placebo-controlled, phase IIB, dose ranging trial (BLAST-AHF). Eur. Heart J. 38, 2364–2373 (2017).
Santos, G. A. et al. Comparative analyses of downstream signal transduction targets modulated after activation of the AT1 receptor by two β-arrestin-biased agonists. Front. Pharmacol. 6, 131 (2015).
Alastalo, T. P. et al. Disruption of ppargamma/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J. Clin. Invest. 121, 3735–3746 (2011).
Falcao-Pires, I. et al. Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 296, H2007–H2014 (2009).
Yang, P. et al. Elabela/toddler is an endogenous agonist of the Apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135, 1160–1173 (2017).
Foldes, G. et al. Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure. Biochem. Biophys. Res. Commun. 308, 480–485 (2003).
Pitkin, S. L., Maguire, J. J., Kuc, R. E. & Davenport, A. P. Modulation of the apelin/APJ system in heart failure and atherosclerosis in man. Br. J. Pharmacol. 160, 1785–1795 (2010).
Maguire, J. J., Kleinz, M. J., Pitkin, S. L. & Davenport, A. P. [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease. Hypertension 54, 598–604 (2009).
Zhou, N. et al. Cell–cell fusion and internalization of the CNS-based, HIV-1 co-receptor, APJ. Virology 307, 22–36 (2003).
Lee, D. K., Ferguson, S. S., George, S. R. & O’Dowd, B. F. The fate of the internalized apelin receptor is determined by different isoforms of apelin mediating differential interaction with β-arrestin. Biochem. Biophys. Res. Commun. 395, 185–189 (2010).
Yang, P. et al. [Pyr(1)]Apelin-13(1-12) is a biologically active ACE2 metabolite of the endogenous cardiovascular peptide [Pyr(1)]apelin-13. Front. Neurosci. 11, 92 (2017).
El Messari, S. et al. Functional dissociation of apelin receptor signaling and endocytosis: implications for the effects of apelin on arterial blood pressure. J. Neurochem. 90, 1290–1301 (2004).
Iturrioz, X. et al. By interacting with the C-terminal Phe of apelin, Phe255 and Trp259 in helix VI of the apelin receptor are critical for internalization. J. Biol. Chem. 285, 32627–32637 (2010).
Ceraudo, E. et al. Biased signaling favoring GI over β-arrestin promoted by an apelin fragment lacking the C-terminal phenylalanine. J. Biol. Chem. 289, 24599–24610 (2014).
Chen, X., Bai, B., Tian, Y., Du, H. & Chen, J. Identification of serine 348 on the apelin receptor as a novel regulatory phosphorylation site in apelin-13-induced G protein-independent biased signaling. J. Biol. Chem. 289, 31173–31187 (2014).
Murza, A. et al. Structure–activity relationship of novel macrocyclic biased apelin receptor agonists. Org. Biomol. Chem. 15, 449–458 (2017).
Medhurst, A. D. et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem. 84, 1162–1172 (2003).
Brame, A. L. et al. Design, characterization, and first-in-human study of the vascular actions of a novel biased apelin receptor agonist. Hypertension 65, 834–840 (2015).
Yang, P. et al. A novel cyclic biased agonist of the apelin receptor, MM07, is disease modifying in the rat monocrotaline model of pulmonary arterial hypertension. Br. J. Pharmacol. 176, 1206–1221 (2019)
Sivertsen, B. et al. Unique interaction pattern for a functionally biased ghrelin receptor agonist. J. Biol. Chem. 286, 20845–20860 (2011).
Fraser, G. L., Hoveyda, H. R. & Tannenbaum, G. S. Pharmacological demarcation of the growth hormone, gut motility and feeding effects of ghrelin using a novel ghrelin receptor agonist. Endocrinology 149, 6280–6288 (2008).
Damian, M. et al. High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J. Biol. Chem. 287, 3630–3641 (2012).
Mokrosinski, J., Frimurer, T. M., Sivertsen, B., Schwartz, T. W. & Holst, B. Modulation of constitutive activity and signaling bias of the ghrelin receptor by conformational constraint in the second extracellular loop. J. Biol. Chem. 287, 33488–33502 (2012).
Fernandez, G. et al. Evidence supporting a role for constitutive ghrelin receptor signaling in fasting-induced hyperphagia in male mice. Endocrinology 159, 1021–1034 (2018).
M’Kadmi, C. et al. Agonism, antagonism, and inverse agonism bias at the ghrelin receptor signaling. J. Biol. Chem. 290, 27021–27039 (2015).
Koole, C. et al. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: implications for drug screening. Mol. Pharmacol. 78, 456–465 (2010).
Zhang, H. et al. Autocrine selection of a GLP-1R G-protein biased agonist with potent antidiabetic effects. Nat. Commun. 6, 8918 (2015).
Hager, M. V., Johnson, L. M., Wootten, D., Sexton, P. M. & Gellman, S. H. β-arrestin-biased agonists of the GLP-1 receptor from β-amino acid residue incorporation into GLP-1 analogues. J. Am. Chem. Soc. 138, 14970–14979 (2016).
Hager, M. V., Clydesdale, L., Gellman, S. H., Sexton, P. M. & Wootten, D. Characterization of signal bias at the GLP-1 receptor induced by backbone modification of GLP-1. Biochem. Pharmacol. 136, 99–108 (2017).
Chen, X. et al. Thioamide substitution selectively modulates proteolysis and receptor activity of therapeutic peptide hormones. J. Am. Chem. Soc. 139, 16688–16695 (2017).
Jones, B. J. et al. Potent prearranged positive allosteric modulators of the glucagon-like peptide-1 receptor. ChemistryOpen 6, 501–505 (2017).
Watson, C. et al. The use of stimulus-biased assay systems to detect agonist-specific receptor active states: implications for the trafficking of receptor stimulus by agonists. Mol. Pharmacol. 58, 1230–1238 (2000).
Hilton, J. M., Dowton, M., Houssami, S. & Sexton, P. M. Identification of key components in the irreversibility of salmon calcitonin binding to calcitonin receptors. J. Endocrinol. 166, 213–226 (2000).
Furness, S. G. B. et al. Ligand-dependent modulation of G protein conformation alters drug efficacy. Cell 167, 739–749 (2016).
Beaudreuil, J. et al. Molecular characterization of two novel isoforms of the human calcitonin receptor. Gene 343, 143–151 (2004).
Poyner, D. R. et al. International union of pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol. Rev. 54, 233–246 (2002).
Dal Maso, E. et al. Characterization of signalling and regulation of common calcitonin receptor splice variants and polymorphisms. Biochem. Pharmacol. 148, 111–129 (2018).
Dal Maso, E. et al. Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy. Biochem. Pharmacol. 150, 214–244 (2018).
Reversi, A. et al. The oxytocin receptor antagonist atosiban inhibits cell growth via a ‘biased agonist’ mechanism. J. Biol. Chem. 280, 16311–16318 (2005).
Busnelli, M. et al. Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J. Biol. Chem. 287, 3617–3629 (2012).
Vass, M. et al. Chemical diversity in the G protein-coupled receptor superfamily. Trends Pharmacol. Sci. 39, 494–512 (2018).
Pandy-Szekeres, G. et al. GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res. 46, D440–D446 (2018).
Ma, Y. et al. Structural basis for apelin control of the human apelin receptor. Structure 25, 858–866 e854 (2017).
Wingler, L. M., McMahon, C., Staus, D. P., Lefkowitz, R. J. & Kruse, A. C. Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176, 479–490.e12 (2019).
Zhang, H. et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature 544, 327–332 (2017).
Liu, H. et al. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 25, 472–481 (2018).
Shihoya, W. et al. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 537, 363–368 (2016).
White, J. F. et al. Structure of the agonist-bound neurotensin receptor. Nature 490, 508–513 (2012).
Fenalti, G. et al. Structural basis for bifunctional peptide recognition at human delta-opioid receptor. Nat. Struct. Mol. Biol. 22, 265–268 (2015).
Koehl, A. et al. Structure of the µ-opioid receptor-Gi protein complex. Nature 558, 547–552 (2018).
Zheng, Y. et al. Structure of CC chemokine receptor 5 with a potent chemokine antagonist reveals mechanisms of chemokine recognition and molecular mimicry by HIV. Immunity 46, 1005–1017.e1005 (2017).
Wu, B. et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 (2010).
Qin, L. et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 347, 1117–1122 (2015).
Burg, J. S. et al. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 347, 1113–1117 (2015).
Liang, Y. L. et al. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546, 118–123 (2017).
Liang, Y. L. et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561, 492–497 (2018).
Zhang, H. et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 553, 106–110 (2018).
Jazayeri, A. et al. Crystal structure of the GLP-1 receptor bound to a peptide agonist. Nature 546, 254–258 (2017). A thermo-stable GLP1 receptor construct encompassing the extracellular and trans-membrane domains, complexed with peptide ligands that exhibit multiple non-native substitutions that lack GLP1 residues 18–37, and so employs alternative receptor contacts but retains agonist activity.
Zhang, Y. et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546, 248–253 (2017). This landmark paper uses cryo-EM to study the full-length GLP1(7–37) peptide, bound in an active conformation to GLP1 receptor and its G protein-signalling domain. The key contact for the ligand–receptor interactions underlying signal transduction is determined.
Liang, Y. L. et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature 555, 121–125 (2018).
Ehrenmann, J. et al. High-resolution crystal structure of parathyroid hormone 1 receptor in complex with a peptide agonist. Nat. Struct. Mol. Biol. 25, 1086–1092 (2018).
Zhao, L. H. et al. Structure and dynamics of the active human parathyroid hormone receptor-1. Science 364, 148–153 (2019).
Yang, Z. et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 556, 520–524 (2018).
Che, T. et al. Structure of the nanobody-stabilized active state of the Kappa opioid receptor. Cell 172, 55–67 e15 (2018).
Thompson, A. A. et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 (2012).
Johansson, E. et al. Type II turn of receptor-bound salmon calcitonin revealed by X-ray crystallography. J. Biol. Chem. 291, 13689–13698 (2016).
Hollenstein, K. et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013). This study presents the first crystal structure of a class B GPCR, CRF1, in complex with small-molecule inhibitor that binds to a very deep pocket in the intracellular half of the receptor core. It was published contemporaneously with the crystal structure of a glucagon receptor (see reference 154).
Pal, K., Swaminathan, K., Xu, H. E. & Pioszak, A. A. Structural basis for hormone recognition by the Human CRFR2 G protein-coupled receptor. J. Biol. Chem. 285, 40351–40361 (2010).
Grace, C. R. et al. NMR structure and peptide hormone binding site of the first extracellular domain of a type B1 G protein-coupled receptor. Proc. Natl Acad. Sci. USA 101, 12836–12841 (2004).
Kusano, S. et al. Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding. Protein Sci. 21, 199–210 (2012).
de Graaf, C. et al. Extending the structural view of class B GPCRs. Trends Biochem. Sci. 42, 946–960 (2017).
Arimont, M. et al. Structural analysis of chemokine receptor–ligand interactions. J. Med. Chem. 60, 4735–4779 (2017).
Miles, T. F. et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. eLife 7, e35850 (2018).
Parthier, C. et al. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proc. Natl Acad. Sci. USA 104, 13942–13947 (2007).
Runge, S., Thogersen, H., Madsen, K., Lau, J. & Rudolph, R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J. Biol. Chem. 283, 11340–11347 (2008).
Underwood, C. R. et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J. Biol. Chem. 285, 723–730 (2010).
de Graaf, C. et al. Glucagon-like peptide-1 and its class B G protein-coupled receptors: a long march to therapeutic successes. Pharmacol. Rev. 68, 954–1013 (2016).
Siu, F. Y. et al. Structure of the human glucagon class B G-protein-coupled receptor. Nature 499, 444–449 (2013). This study presents the first crystal structure of a class B GPCR (glucagon receptor), published contemporaneously with the crystal structure of a CRF1 receptor (see reference 143).
Gardella, T. J. & Vilardaga, J. P. International Union of Basic and Clinical Pharmacology. XCIII. The parathyroid hormone receptors — family B G protein-coupled receptors. Pharmacol. Rev. 67, 310–337 (2015).
Billard, E., Hebert, T. E. & Chatenet, D. Discovery of new allosteric modulators of the rotensinergic system through substitution of the urotensin II-related peptide (URP) phenylalanine residue. J. Med. Chem. 61, 8707–8716 (2018).
Sakamoto, K., Koyama, R., Kamada, Y., Miwa, M. & Tani, A. Discovery of artificial VIPR2-antagonist peptides possessing receptor- and ligand-selectivity. Biochem. Biophys. Res. Commun. 503, 1973–1979 (2018).
Lotta, L. A. et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 177, 597–607.e9 (2019). Variants in the melanocortin 4 receptor gene, identified in 0.5 million individuals as causing a gain of function associated with protection from obesity, are found to be biased towards β-arrestin signalling. Biased agonists may represent a new therapeutic strategy at the melanocortin 4 receptor.
1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Hauser, A. S. et al. Pharmacogenomics of GPCR drug targets. Cell 172, 41–54.e19 (2018).
Genomics England. The 100,000 Genomes Project. Genomics England https://www.genomicsengland.co.uk/about-genomics-england/the-100000-genomes-project/ (2019).
Tillner, J. et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes. Metab. 21, 120–128 (2019).
Frias, J. P. et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 392, 2180–2193 (2018). This double-blind, randomized phase II study, in patients with T2D, demonstrates the better efficacy, compared with dulaglutide, of dual GIP/GLP1 receptor agonists combined in the same molecule for controlling glucose and for weight loss.
Sanchez-Garrido, M. A. et al. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia 60, 1851–1861 (2017).
Frias, J. P. et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. 26, 343–352 e342 (2017).
Karhu, L. et al. Stapled truncated orexin peptides as orexin receptor agonists. Peptides 102, 54–60 (2018).
Tian, Y. et al. Design of stapled oxyntomodulin analogs containing functionalized biphenyl cross-linkers. Tetrahedron 75, 286–295 (2019).
Chaturvedi, M. et al. Emerging paradigm of intracellular targeting of G protein-coupled receptors. Trends Biochem. Sci. 43, 533–546 (2018).
Gurbel, P. A. et al. Cell-penetrating pepducin therapy targeting PAR1 in subjects with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 36, 189–197 (2016).
O’Connor-Semmes, R. L. et al. GSK2374697, a novel albumin-binding domain antibody (AlbudAb), extends systemic exposure of exendin-4: first study in humans—PK/PD and safety. Clin. Pharmacol. Ther. 96, 704–712 (2014).
Read, C. et al. Apelin peptides linked to anti-serum albumin domain antibodies retain affinity in vitro and are efficacious receptor agonists in vivo. Basic Clin. Pharmacol. Toxicol. https://doi.org/10.1111/bcpt.13227 (2019).
Ouberai, M. M. et al. Controlling the bioactivity of a peptide hormone in vivo by reversible self-assembly. Nat. Commun. 8, 1026 (2017).
Foster, S. R. et al. Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell 179, 895–908.e21 (2019). The protein sequences and structures of ~300 human class A GPCRs are used to identify the universal characteristics of peptidergic signalling systems. Novel pairings of 17 endogenous ligands proposed for 5 orphan GPCRs are associated with genetic, neoplastic, nervous and reproductive system disorders.
Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 17, 134–143 (2015).
Millar, R. P. et al. Gonadotropin-releasing hormone receptors. Endocr. Rev. 25, 235–275 (2004).
Anthony, L. & Freda, P. U. From somatostatin to octreotide LAR: evolution of a somatostatin analogue. Curr. Med. Res. Opin. 25, 2989–2999 (2009).
Murray, R. D. & Melmed, S. A critical analysis of clinically available somatostatin analog formulations for therapy of acromegaly. J. Clin. Endocrinol. Metab. 93, 2957–2968 (2008).
Weckbecker, G., Briner, U., Lewis, I. & Bruns, C. SOM230: a new somatostatin peptidomimetic with potent inhibitory effects on the growth hormone/insulin-like growth factor-I axis in rats, primates, and dogs. Endocrinology 143, 4123–4130 (2002).
Madsbad, S. Review of head-to-head comparisons of glucagon-like peptide-1 receptor agonists. Diabetes Obes. Metab. 18, 317–332 (2016).
Urban, J. D. et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13 (2007).
Galandrin, S., Oligny-Longpre, G. & Bouvier, M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol. Sci. 28, 423–430 (2007).
van der Westhuizen, E. T., Breton, B., Christopoulos, A. & Bouvier, M. Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol. Pharmacol. 85, 492–509 (2014).
Wootten, D., Christopoulos, A., Marti-Solano, M., Babu, M. M. & Sexton, P. M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 19, 638–653 (2018).
Smith, J. S., Lefkowitz, R. J. & Rajagopal, S. Biased signalling: from simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 17, 243–260 (2018).
Wootten, D. et al. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism. Cell 165, 1632–1643 (2016).
Wootten, D. et al. A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: lessons from class B crystal structures. Mol. Pharmacol. 89, 335–347 (2016).
Tehan, B. G., Bortolato, A., Blaney, F. E., Weir, M. P. & Mason, J. S. Unifying family A GPCR theories of activation. Pharmacol. Ther. 143, 51–60 (2014).
Venkatakrishnan, A. J. et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536, 484–487 (2016).
Bonde, M. M. et al. Biased signaling of the angiotensin II type 1 receptor can be mediated through distinct mechanisms. PLOS ONE 5, e14135 (2010).
Katritch, V. et al. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 39, 233–244 (2014).
Wingler, L. M. et al. Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176, 468–478 e411 (2019).
Zhang, H. et al. Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem. 290, 29127–29139 (2015).
Zhang, H. et al. Structure of the angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–844 (2015).
Ban, T. et al. GPCR structure and function relationship: identification of a biased apelin receptor mutant. Biochem. J. 475, 3813–3826 (2018).
Koole, C. et al. Second extracellular loop of human glucagon-like peptide-1 receptor (GLP-1R) has a critical role in GLP-1 peptide binding and receptor activation. J. Biol. Chem. 287, 3642–3658 (2012).
Song, G. et al. Human GLP-1 receptor transmembrane domain structure in complex with allosteric modulators. Nature 546, 312–315 (2017).
Goodford, P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 28, 849–857 (1985).
Mason, J. S. High end GPCR design: crafted ligand design and druggability analysis using protein structure, lipophilic hotspots and explicit water networks. Silico Pharmacol. 1, 23 (2013).
Isberg, V. et al. Generic GPCR residue numbers — aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 36, 22–31 (2015).
Konvicka, K., Guarnieri, F., Ballesteros, J. A. & Weinstein, H. A proposed structure for transmembrane segment 7 of G protein-coupled receptors incorporating an Asn-Pro/Asp-Pro motif. Biophys. J. 75, 601–611 (1998).
Wootten, D., Christopoulos, A. & Sexton, P. M. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat. Rev. Drug Discov. 12, 630–644 (2013).
Acknowledgements
The authors are funded by Wellcome Trust [WT107715/Z/15/Z], Cambridge Biomedical Research Centre Biomedical Resources Grant, University of Cambridge.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
C.C.G.S., C.d.G. and A.J.H.B. are employees of Sosei Heptares. A.P.D. and J.J.M. are in receipt of an ORBIT award from Sosei Heptares.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov
DrugBank: https://www.drugbank.ca
Electronic Medicines Compendium: https://www.medicines.org.uk/emc
EvaluatePharma: https://www.evaluate.com/
GlobalData (2019): https://www.globaldata.com/store/search/pharmaceuticals
GPCR database: https://gpcrdb.org
RCSB Protein Data Bank: https://www.rcsb.org
RxList: https://www.rxlist.com
Supplementary information
Glossary
- Cryo-electron microscopy
-
(cryo-EM). Electron microscopy technique that allows near-atomic resolution of biomolecules such as G protein-coupled receptors (GPCRs) in samples cooled to low temperatures and embedded in an environment of vitreous water, avoiding the crystallization that is a prerequisite for X-ray crystallography.
- Flip-flop kinetics
-
A property of compounds that are administered by subcutaneous injection and slowly absorbed, resulting in fairly continuous release of the compound into the blood.
- Afterload
-
The pressure against which the heart must work to eject blood.
- Tachyphylaxis
-
The rapidly diminishing response to repeated doses of a therapeutic agent.
- Non-proteinogenic
-
Not naturally encoded in any known organism.
- Incretins
-
Metabolic hormones, induced upon eating, that decrease blood glucose by stimulating insulin release. The two known incretins are glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP).
Rights and permissions
About this article

Cite this article
Davenport, A.P., Scully, C.C.G., de Graaf, C. et al. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat Rev Drug Discov 19, 389–413 (2020). https://doi.org/10.1038/s41573-020-0062-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-020-0062-z
This article is cited by
-
Kisspeptin-10 binding to Gpr54 in osteoclasts prevents bone loss by activating Dusp18-mediated dephosphorylation of Src
Nature Communications (2024)
-
Ligand recognition mechanism of the human relaxin family peptide receptor 4 (RXFP4)
Nature Communications (2023)
-
Effects of site-directed mutagenesis of GLP-1 and glucagon receptors on signal transduction activated by dual and triple agonists
Acta Pharmacologica Sinica (2023)
-
Therapeutic strategies for COVID-19: progress and lessons learned
Nature Reviews Drug Discovery (2023)
-
Structure–function relationship and physiological role of apelin and its G protein coupled receptor
Biophysical Reviews (2023)
