
Abstract
The maintenance of iron homeostasis is essential for proper cardiac function. A growing body of evidence suggests that iron imbalance is the common denominator in many subtypes of cardiovascular disease. In the past 10 years, ferroptosis, an iron-dependent form of regulated cell death, has become increasingly recognized as an important process that mediates the pathogenesis and progression of numerous cardiovascular diseases, including atherosclerosis, drug-induced heart failure, myocardial ischaemia–reperfusion injury, sepsis-induced cardiomyopathy, arrhythmia and diabetic cardiomyopathy. Therefore, a thorough understanding of the mechanisms involved in the regulation of iron metabolism and ferroptosis in cardiomyocytes might lead to improvements in disease management. In this Review, we summarize the relationship between the metabolic and molecular pathways of iron signalling and ferroptosis in the context of cardiovascular disease. We also discuss the potential targets of ferroptosis in the treatment of cardiovascular disease and describe the current limitations and future directions of these novel treatment targets.
Key points
-
The death of terminally differentiated cardiomyocytes is an important pathogenic contributor to the development of several forms of cardiovascular disease.
-
Ferroptosis is a newly characterized form of regulated cell death driven by iron-dependent lipid peroxidation and linked to cardiovascular disease.
-
Ferroptosis involves various metabolic processes, including iron, lipid and glutathione metabolism.
-
Both in vitro and in vivo evidence supports the pathophysiological role of ferroptosis in myocardial ischaemia–reperfusion injury, anthracycline-mediated cardiotoxicity, sepsis‑induced heart injury, hypertrophic cardiomyopathy and diabetic cardiomyopathy.
-
Targeting ferroptosis with specific inhibitors might provide new therapeutic opportunities for previously untreatable cardiovascular conditions.
Similar content being viewed by others
Introduction
Various forms of regulated cell death, such as apoptosis, necroptosis, pyroptosis and autophagy, have been implicated in the pathogenesis of cardiovascular diseases1. In the past 10 years, a growing number of studies support the notion that ferroptosis — an iron-dependent form of non-apoptotic cell death that involves the accumulation of lipid hydroperoxides — has a pathophysiological role in the development of cardiovascular diseases, including doxorubicin-induced cardiomyopathy, myocardial ischaemia–reperfusion injury, myocardial infarction and heart failure1,2.
As an essential trace element that is present in nearly all forms of life, iron is involved in many biological processes, including energy metabolism and nucleotide synthesis and repair3. In humans, iron deficiency is the most prevalent malnutrition-related condition and affects up to 75% of patients with heart failure4. Conversely, both primary and secondary forms of iron overload can cause heart disease via oxidative damage, but the exact mechanisms underlying this process are not clear5. Excess iron in cardiomyocytes has been shown to directly induce ferroptosis via the accumulation of phospholipid hydroperoxides in the cell membrane6.
In addition to altered iron homeostasis, excessive production of reactive oxygen species (ROS) or reactive nitrogen species can also directly induce ferroptosis in cardiomyocytes by catalysing the oxidation of phospholipids in the cell membrane. Indeed, oxidative stress has been linked to the development of cardiovascular disease, and targeting the molecular and metabolic pathways that regulate cellular defence against oxidative stress — particularly the glutathione-dependent antioxidant system — has been shown to prevent cardiomyopathy in animal models7,8.
The evidence to date suggests that the development of many forms of cardiovascular disease is driven by ferroptosis. For example, high levels of ferroptosis mediated by distinct signalling and metabolic pathways can contribute to ischaemic heart disease, cardiac injury, heart failure and cardiomyopathy. In this Review, we summarize the mechanisms involved in the regulation of iron homeostasis, glutathione synthesis and lipid metabolism in cardiomyocytes; we discuss newly identified putative targets of ferroptosis in heart disease; and we provide critical perspectives on the potential of new clinical therapies that target ferroptosis in the heart.
Discovery of ferroptosis
Although ferroptosis was first reported in 2012 as a novel form of cell death that could be inhibited by the iron-chelating agent deferoxamine2, various forms of cell death involving iron and oxidative stress had already been known for decades9. The concept of ferroptosis might have been derived from our knowledge of cysteine depletion-induced cancer cell death and glutamate-induced cytotoxicity10,11. Interestingly, oxytosis was first reported in neurons as far back as 2001, and is characterized by oxidative stress-induced non-apoptotic and non-excitotoxic pathways that promote glutamate-induced cell death, although the underlying mechanism remains largely unknown12. Since its initial identification, ferroptosis has been defined as an iron-dependent form of regulated cell death that involves the iron-catalysed accumulation of lethal lipid peroxides13.
Molecular and metabolic drivers of ferroptosis
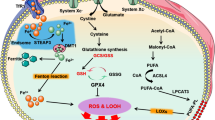
Cells that undergo ferroptosis have genetic, biochemical, morphological and metabolic features that are distinct from those of other known forms of cell death, including apoptosis, necroptosis and pyroptosis14. Interestingly, unlike other forms of cell death identified to date, ferroptosis can propagate rapidly through cell populations in a wave-like manner15,16. In terms of morphological changes, cells undergoing ferroptosis have mitochondrial abnormalities that can be visualized using electron microscopy, including swelling, changes in density and rupture of the outer membrane2,17,18. Finally, the pathways involved in iron, glutathione and lipid metabolism converge to control the initiation and execution of ferroptosis, particularly in cardiomyocytes (Fig. 1). The roles of these metabolic pathways in mediating ferroptosis and cardiovascular disease are discussed below.

At the cellular level, ferroptosis is driven primarily by iron-dependent lipid peroxidation. Many aspects of iron metabolism such as the absorption, storage and utilization of iron have important roles in regulating ferroptosis. In addition, the activation of long-chain fatty-acid CoA ligase 4 (LACS4), lysophospholipid acyltransferase 5 (LPLAT5), lipoxygenase (LOX) or NADPH oxidase (NOX) in the lipid metabolic pathway promotes lipid peroxidation and ferroptosis. The canonical ferroptosis-suppressing pathway involves the uptake of cystine (Cys) via the cystine–glutamate antiporter (system xc−), which results in glutathione (GSH) biosynthesis. Using GSH as a cofactor, the glutathione peroxidase 4 (GPX4) reduces phospholipid hydroperoxides to their corresponding alcohols. The peroxidation of phospholipids is also kept in check by the ferroptosis suppressor protein 1 (FSP1)–coenzyme Q10 (CoQ10) system. Ferroptosis is also regulated by the iron metabolism pathway that involves iron absorption, transport, storage and utilization. At the cellular level, non-haem iron is transported into cells by either transferrin receptor protein 1 (TFR1)-mediated, transferrin (TF)-bound iron uptake or metal transporter solute carrier family 39 member 14 (SLC39A14; also known as metal cation symporter ZIP14)-mediated, non-TF-bound iron uptake. In addition, haem degradation and nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy can increase the labile iron pool (LIP), thereby sensitizing cells to ferroptosis via the Fenton reaction. FPN, ferroportin; Glu, glutamate; GSSG, glutathione disulfide; HO1, haem oxygenase 1; KEAP1, Kelch-like ECH-associated protein 1; ML1, mucolipin 1; NRAMP2, natural resistance-associated macrophage protein 2; NRF2, nuclear factor-erythroid 2-related factor 2; PUFA, polyunsaturated fatty acid; PUFA–CoA, coenzyme A-activated polyunsaturated fatty acid; PUFA–PL, polyunsaturated fatty acid-containing phospholipid; RNF217, E3 ubiquitin protein ligase RNF217; STEAP3, metalloreductase STEAP3.
Iron metabolism and ferroptosis in the heart
Regulation of iron homeostasis in the cardiovascular system
The cellular uptake of iron is mediated by the binding of iron-bound transferrin (which contains two ferric iron molecules) to its receptor (transferrin receptor protein 1 (TFR1)), which triggers clathrin-dependent endocytosis of the entire holo-complex19. The endosome is then acidified by vacuolar ATPase, leading to the reduction of ferric iron to ferrous iron by the STEAP (six-transmembrane epithelial antigen of prostate) family of metalloreductases19. Ferrous iron is then released from the endosome into the cytoplasm via natural resistance-associated macrophage protein 2 (NRAMP2; also known as DMT1), and apo-transferrin and TFR1 are shuttled back to the cell surface to be reused by the cell20. Mice lacking Tfr1 in the heart have severe cardiomyopathy with cardiac iron deficiency and die in the second week of life21. Non-transferrin-bound iron was thought to be transported into cardiomyocytes by voltage-dependent calcium channels20,21, but a study published in 2021 raised doubts about the role of calcium channels in mediating the cardiac uptake of non-transferrin-bound iron22.
In the cytoplasm, ferrous iron is oxidized to its ferric state by cytoplasmic ferritin, and the resulting ferritin-bound iron can be either degraded for use in enzymatic reactions or stored for later use23. Iron-saturated ferritin is degraded by nuclear receptor coactivator 4 (NCOA4)-mediated autophagy — a process known as ferritinophagy24 — which leads to the degradation of lysosomal ferritin, with subsequent release of its iron content25 and its export to the cytosol via lysosomal NRAMP2 (ref.26). Cardiac-specific deletion of Fth1 (encoding ferritin heavy chain) leads to iron dysregulation and increased oxidative stress in the heart, resulting in increased susceptibility to iron overload-induced tissue injury27. Conversely, deletion of Ncoa4 in mouse hearts improved cardiac function and attenuated ferritinophagy-mediated ferritin degradation that was induced by pressure overload28.
Ferroportin is the only known iron exporter in vertebrate cells, and cardiac iron overload, impaired heart function and a shortened lifespan have been observed in mice with early cardiomyocyte-specific deletion of the gene encoding ferroportin (Fpn)29,30. Hepcidin, a peptide hormone primarily synthesized in the liver, inhibits ferroportin via E3 ubiquitin protein ligase RNF217-mediated ubiquitination in the gut and spleen, which regulates iron absorption and iron recycling, respectively31,32. Genetic hepcidin deficiency causes the most severe form of systemic iron overload in both mice and humans33,34. However, loss of hepcidin specifically in cardiomyocytes results in a fatal cardiomyopathy as a consequence of cardiac iron deficiency, highlighting a cell-autonomous role of the hepcidin–ferroportin axis in cardiac iron homeostasis35 (Fig. 2a).

a | Iron uptake in cardiomyocytes is dependent on the endocytosis of diferric transferrin (TF) bound to its receptor transferrin receptor protein 1 (TFR1). To maintain the levels of iron in the cytoplasm, iron can be released from TF in endolysosomes and exported to the cytoplasm by natural resistance-associated macrophage protein 2 (NRAMP2) after a metalloreductase STEAP3-mediated reduction. Excess iron is either bound to ferritin heavy chain (FTH) or exported by ferroportin (FPN), the only iron exporter. In addition, iron can be released from FTH via nuclear receptor coactivator 4 (NCOA4)-mediated autophagic degradation of ferritin, a process known as ferritinophagy. b | Mitoferrin 1 (also known as SLC25A37) and mitoferrin 2 (also known as SLC25A28) mediate the transport of iron across the mitochondrial membrane. Iron is primarily used to synthesize iron–sulfur (Fe–S) clusters and haem in the mitochondria. Excess iron can be stored in the mitochondria-specific form of ferritin (FTMT). FLVCR1B (feline leukaemia virus subgroup C receptor-related protein 1B) promotes haem efflux into the cytoplasm, whereas the export of Fe–S clusters into the cytoplasm might require iron–sulfur clusters transporter ABCB7, mitochondrial (ABCB7) and ABCB8 (also known as mitochondrial potassium channel ATP-binding subunit). Ala, 5-aminolevulinate; ALAS, aminolevulinic acid synthase; Apo-TF, apo-transferrin; CO, carbon monoxide; Cys, cysteine; ETC, electron transport chain; HO1, haem oxygenase 1; Holo-TF, holo-transferrin; LIP, labile iron pool; ML1, mucolipin 1; NFS1, cysteine desulfurase, mitochondrial; OXPHOS, oxidative phosphorylation; RNF217, E3 ubiquitin protein ligase RNF217; ROS, reactive oxygen species; TCA, tricarboxylic acid.
Cellular iron levels are regulated at the post-transcriptional level by iron regulatory protein 1 (IRP1) and IRP2 (refs.36,37). These cytoplasmic proteins bind to the 3′-untranslated region (UTR) of target transcripts such as TFR1 mRNA, stabilizing the transcripts and increasing translation. By contrast, binding of IRP1 or IRP2 to the 5′-UTR of target mRNAs such as FPN and FTH1 blocks ribosomal entry and prevents their translation36,37. Holo-IRP1 attached to iron–sulfur (Fe–S) clusters functions as a cytosolic aconitase and cannot bind to mRNA molecules. However, when cellular iron levels decrease, the Fe–S clusters dissociate from IRP1, which can then bind to the ribosome entry sites. IRP2 is constitutively active; however, when cellular iron levels are sufficient, IRP2 is ubiquitinated and degraded via a process that requires F-box/LRR-repeat protein 5 (refs.36,37). Therefore, when iron levels are sufficient, IRP1 contains an Fe–S cluster and IRP2 is degraded, thereby inhibiting the IRP system.
When the IRP system fails to maintain adequate cellular iron levels during severe iron deficiency, another iron regulatory pathway can be activated. This pathway is known as the ‘iron conservation pathway’ and is distinct from the ‘iron acquisition pathway’ mediated by the IRP system38. During a state of iron deficiency, levels of the mRNA-binding protein tristetraprolin increase, paradoxically reducing TFR1 levels to conserve cellular iron. Tristetraprolin degrades its target mRNAs by binding to the CNOT1 (CCR4–NOT transcription complex subunit 1) deadenylase complex39. The induction of tristetraprolin during iron deficiency is crucial for cell survival, because tristetraprolin-deficient mice develop cardiac dysfunction in response to iron deficiency40, and depletion of tristetraprolin in vitro causes cell death under low iron conditions38. In the heart, tristetraprolin binds to and causes the degradation of mRNA transcripts that encode proteins involved in several processes, including Fe–S cluster-containing proteins in the mitochondrial electron transport chain. Consequently, under iron-deficient conditions, excess numbers of apo-proteins that would otherwise bind to iron are not synthesized, allowing the limited amount of iron to be used by essential proteins40.
Iron metabolism in ferroptosis
Iron availability is a crucial factor in driving ferroptosis. Mice fed a high-iron diet, as well as haemojuvelin-deficient mice and SMAD family member 4-deficient mice, have severe iron overload and increased hepatic ferroptosis41. By contrast, mice lacking Hfe (encoding hereditary haemochromatosis protein homologue) have only moderate iron overload without hepatic ferroptosis41. Of note, the accumulated cellular iron — particularly labile ferrous iron — can react directly with cellular oxidants to produce cytotoxic hydroxyl radicals via the Fenton reaction, which in turn promotes ferroptosis. By contrast, the binding of free ionic iron to proteins such as transferrin or ferritin can have a protective effect against the Fenton reaction and ferroptosis27,42. In addition, peroxidation of polyunsaturated fatty acids (PUFAs) by lipoxygenases via the phosphorylase kinase G2-dependent iron pool is required for initiating ferroptosis43. Therefore, numerous proteins involved in regulating cellular iron homeostasis can affect the sensitivity of the cell to ferroptosis44 (Table 1).
Iron uptake via TFR1, solute carrier family 39 member 14 (SLC39A14; also known as metal cation symporter ZIP14) and/or NRAMP2 can dictate cellular sensitivity to ferroptosis42,45,46,47. Cytosolic ferritin confers resistance to ferroptosis by controlling iron availability27,48,49; therefore, selective autophagy of ferritin via NCOA4 can increase the susceptibility of the cell to ferroptosis50,51. The iron chaperone poly(rC)-binding protein 1 (PCBP1) binds and delivers Fe2+ to ferritin52. Moreover, Pcbp1-knockout mouse hepatocytes have increased levels of labile iron and lipid peroxidation, suggesting that PCBP1 might have a key role in preventing ferroptosis-related disease52. By contrast, knocking down the expression of iron or ferritin exporters such as ferroportin and prominin 2 has been shown to promote ferroptosis53,54. Caeruloplasmin helps ferroportin to leave cells via its ferroxidase activity, and loss of ceruloplasmin promotes both erastin-induced and transcription factor RSL3-induced ferroptosis, whereas overexpression of caeruloplasmin suppresses ferroptosis in tumour cells55,56.
The maintenance of mitochondrial iron homeostasis also has an important role in preventing ferroptosis. Mitoferrin 1 (also known as SLC25A37) and mitoferrin 2 (also known as SLC25A28) are key mitochondrial iron importers involved in haem and Fe–S biogenesis57. Deletion of mitoferrin 2 reduces erastin-induced cell death, whereas overexpression of mitoferrin 2 increases ferroptosis58. Activation of haem oxygenase 1 (HO1), a mitochondrial enzyme that catalyses the degradation of haem to produce ferrous iron, causes mitochondrial iron overload and increases ferroptosis both in vitro and in vivo6,59,60. However, mild upregulation of HO1 might actually be cytoprotective61. Like cytosolic ferritin, mitochondrial ferritin has a protective role against ferroptosis. For example, Drosophila and cells overexpressing mitochondrial ferritin are resistant to erastin-induced ferroptosis62.
Several Fe–S proteins have a role in lipid peroxidation during ferroptosis. For example, suppressing NFS1 (cysteine desulfurase, mitochondrial), which uses sulfur from cysteine to synthesize Fe–S clusters, sensitizes cancer cells to ferroptosis63. In addition, the Fe–S-binding proteins mitoNEET (also known as CISD1) and NAF1 (also known as CISD2) have been shown to participate in mitochondrial iron transportation, increasing the tolerance of cancer cells to ROS-induced cell death61,62. Increased expression of mitoNEET prevented erastin-induced ferroptosis in human hepatocellular carcinoma cells64, and NAF1 overexpression similarly conferred resistance to sulfasalazine-induced ferroptosis in a mouse tumour xenograft model65.
Iron overload-associated cardiac disorders
Iron overload occurs when the body stores excess iron. The heart is particularly susceptible to damage induced by accumulation of iron66. Two general types of iron overload have been described. Primary iron overload is caused by genetic disorders that cause dysregulated absorption of dietary iron, whereas secondary iron overload develops as a result of repeated blood transfusions, drug-induced toxicity or excess consumption of iron67. However, given the lack of robust biomarkers for measuring ferroptosis in patients, the link between ferroptosis and these diseases remains poorly understood.
Hereditary haemochromatosis is one of the most common inherited diseases among white populations68. A wide range of mutations affecting either the production or function of the iron-regulating peptide hepcidin can cause various degrees of iron overload in a number of organs, including the heart. Therefore, heart failure is a common complication among patients with hereditary haemochromatosis and is more prevalent in those with juvenile forms of the disease69. Carriers of the C282Y mutation in HFE (encoding hereditary haemochromatosis protein) have a higher risk of acute myocardial infarction and cardiovascular death than non-carriers70,71. Similarly, impaired endothelial function and increased intima–media thickness have been associated with altered iron status in patients with hereditary haemochromatosis; iron depletion therapy can therefore reduce the risk of cardiovascular events in these patients72.
Friedreich ataxia is an autosomal recessive neurodegenerative disease that affects both the nervous system and non-neural tissues, including the heart and pancreas73. Friedreich ataxia is caused by a homozygous expansion of the GAA triplet repeat in the first intron of the FXN gene, which encodes the protein frataxin (also known as mitochondrial frataxin)74. Frataxin is essential for mitochondrial function owing to its role in the biogenesis of Fe–S clusters and antioxidant defence75. Frataxin deficiency causes mitochondrial iron accumulation, excess production of ROS and increased lipid peroxidation, leading to the development of Friedreich ataxia76. In addition to the typical neurological symptoms, the clinical manifestations of Friedreich ataxia include severe cardiomyopathy, which is the most common cause of death among these patients77,78. The severe neurological and cardiac symptoms are to be expected, given that the heart and the brain contain a large number of mitochondria. Interestingly, mice with a cardiomyocyte-specific Fxn deletion have increased mitochondrial iron in the heart owing to pronounced changes in Tfr1 expression and alterations in iron transport from the cytosol to the mitochondria79. Co-treating patients with Friedreich ataxia with both the iron chelator deferiprone and the coenzyme Q10 analogue idebenone results in the attenuation of cardiac hypertrophy80. Other studies have also indicated that treatment with deferiprone can improve heart function, but not the neurological symptoms81,82. Nevertheless, the clinical benefits of iron chelation are currently unclear and long-term, large-scale trials are warranted.
Ineffective erythropoiesis and transfusion-induced iron overload
Iron overload is a common complication among patients with thalassaemia, a group of hereditary disorders caused by impaired haemoglobin synthesis83. Ineffective erythropoiesis, in which an increase in the production of erythroid cells is not matched by a corresponding increase in mature red blood cells, is a major cause of iron overload in organs84. In addition, repeated blood transfusions, a common treatment for thalassaemia, is another source of excess iron that can lead to iron overload85.
Iron overload can also lead to cardiomyopathy, the primary cause of morbidity and mortality in patients with thalassaemia86,87,88. Although the clinical presentation of thalassaemia-associated cardiomyopathy is both variable and complex, the majority of patients present with left-sided heart failure and reduced ejection fraction89. Another major symptom is cardiac arrhythmia, which can directly lead to sudden cardiac death89,90. To manage the deleterious effects of iron overload, chelation therapy was first used to treat patients with iron overload-related, thalassaemia-associated cardiomyopathy in the 1970s; today, iron chelation is globally accepted as the most effective treatment for this condition91,92.
Ineffective erythropoiesis and transfusion-induced iron overload can also be caused by other inherited blood disorders such as sickle cell disease (discussed below), myelodysplastic syndromes, pure red cell aplasia and leukaemia. The management of iron overload-induced cardiac conditions can be improved by monitoring tissue iron levels more precisely, developing new iron chelators and modulating hepcidin levels to reduce iron loading and cardiotoxicity93,94.
Anthracycline-induced cardiotoxicity
Since the late 1960s, anthracyclines — a class of drugs that includes doxorubicin, daunorubicin, epirubicin and idarubicin — have been widely used to treat breast cancer, leukaemia and many other types of malignancies95. Despite their powerful anticancer effects, the clinical use of anthracyclines is severely limited owing to the risk of cardiotoxicity96. Indeed, >25% of patients who received a cumulative dose of 550 mg/m2 of doxorubicin developed congestive heart failure97. Although the mechanism underlying doxorubicin-induced cardiomyopathy is unclear, a growing body of evidence suggests that several risk factors are involved, including iron overload. Systemic iron accumulation in mice mediated by a high iron diet or genetic modification significantly increased susceptibility to doxorubicin-induced cardiotoxicity98,99. Conversely, mice fed an iron-deficient diet had a lower risk of doxorubicin-induced cardiotoxicity and increased survival compared with control mice, indicating that the targeting of metabolic pathways that regulate cardiac iron levels might be a clinically effective strategy for the treatment of chemotherapy-related cardiomyopathy6.
In terms of how doxorubicin affects iron metabolism in the heart, one study found that iron accumulates specifically in the mitochondria of doxorubicin-treated cardiomyocytes owing to suppression of the mitochondrial iron exporter ABCB8 (also known as mitochondrial potassium channel ATP-binding subunit)100. Overexpression of ABCB8 or direct chelation of mitochondrial iron using dexrazoxane protects against doxorubicin-induced cardiotoxicity. In addition, doxorubicin treatment in mice has been shown to induce cardiac mitochondrial ferritin expression, and genetic deletion of mitochondrial ferritin increased the sensitivity of cardiomyocytes to doxorubicin-induced iron toxicity101.
Using RNA sequencing (RNA-seq) analysis, we showed that HO1 is upregulated via nuclear factor-erythroid 2-related factor 2 (NRF2) activation during doxorubicin-induced cardiomyopathy in mice, causing haem degradation and the release of free iron in mitochondria6. In addition, we found that treating mice with the competitive HO1 inhibitor zinc protoporphyrin IX protects against doxorubicin-induced cardiomyopathy6. A study published in 2021 found that the E3 ubiquitin protein ligase TRIM21 negatively regulates the NRF2-mediated antioxidant pathway and that Trim21-knockout mice are protected against doxorubicin-induced cardiotoxicity and death102. Additional studies examining the clinical applicability of these compounds and their molecular targets should lead to improved therapeutic strategies that are designed to reduce anthracycline-induced heart injury.
Dietary iron overload
In a typical human diet, the two forms of iron (haem iron and non-haem iron) are derived from distinct dietary sources and have different absorption mechanisms and metabolic pathways103. Haem iron, which constitutes approximately 15% of the total iron intake of a typical diet, is present exclusively in the haemoglobin and myoglobin in red meat, fish and poultry, whereas non-haem iron is present in cereals, fruits and vegetables. Given that haem iron is buried and protected within the porphyrin complex, its absorption rate is fivefold to tenfold higher than that of non-haem iron104.
To date, the population-based studies that have examined the putative association between dietary iron intake and the risk of heart disease have found inconsistent findings105,106,107,108,109,110,111,112,113,114,115,116 (Table 2). Nonetheless, meta-analyses of these prospective cohort studies suggest that a high intake of dietary haem iron, irrespective of non-haem iron intake, is significantly correlated with an increased risk of heart disease and cardiovascular death in the general population117,118. Therefore, a reduction in the consumption of foods that are high in haem iron might help to prevent heart disease.
Glutathione metabolism and ferroptosis in the heart
In addition to iron metabolism, cysteine or cysteine deficiency, glutathione depletion and inactivation of the enzyme phospholipid hydroperoxide glutathione peroxidase 4 (GPX4) have also been shown to promote ferroptosis13. Produced primarily in the liver, the master antioxidant glutathione is a tripeptide composed of the amino acids cysteine, glutamic acid and glycine. Among these three amino acids, cysteine is the rate-limiting precursor in glutathione synthesis. Although intracellular cysteine can be produced by either de novo biosynthesis or protein catabolism, most cells obtain cysteine primarily from the cystine–glutamate antiporter system xc− (also known as xCT), which consists of the solute carrier family 7A member 11 subunit (SLC7A11; also known as cystine–glutamate transporter) and the SLC3A2 subunit (also known as 4F2HC)119. The SLC7A11 subunit contains 12 transmembrane domains and primarily mediates the transporter activity of the protein complex, whereas SLC3A2 is a chaperone protein that helps to stabilize SLC7A11 and ensures the appropriate membrane localization of the complex. System xc− exports intracellular glutamate and imports extracellular cystine at a 1:1 ratio; the newly imported cystine is then converted to cysteine in the cytosol via an NADPH-consuming reduction reaction120. The structure of human system xc−, which was resolved in 2020, clearly demonstrates the interaction between the two subunits at the extracellular interface and in the transmembrane region121,122,123. In addition, a well-resolved non-protein density was found in the intracellular vestibule of SCL7A11, to which an erastin molecule can bind123. Therefore, inhibiting system xc− with erastin or its analogues can lead to the depletion of intracellular glutathione and trigger ferroptosis, suggesting a potentially viable strategy for antitumour therapies2,124.
The expression of SLC7A11 is positively regulated by the transcription factor NRF2 under conditions of cellular stress125. Moreover, both the genetic deletion of NRF2 and overexpression of KEAP1 (Kelch-like ECH-associated protein 1, which binds to and facilitates the ubiquitination and proteasomal degradation of NRF2) have been shown to promote ferroptosis in cancer cells126. Of note, the KEAP1–NRF2 axis regulates a wide range of genes involved in glutathione biosynthesis and iron metabolism, which can also affect the susceptibility of cells to ferroptosis. Conversely, the tumour suppressor protein p53 represses SLC7A11 transcription, thereby promoting ferroptosis127. The cAMP-dependent transcription factor ATF3 has also been shown to promote ferroptosis by binding to the SLC7A11 promoter and repressing its expression in a p53-independent manner128. Moreover, our research group has shown that overexpressing Slc7a11 selectively in cardiomyocytes increases cellular glutathione levels and prevents ferritin H deficiency-mediated cardiac ferroptosis, providing the first evidence that SLC7A11 has an anti-ferroptotic role in the heart27. In addition, knocking out Slc7a11 aggravates cardiac hypertrophy and dysfunction in mice, both of which can be reversed by inhibiting ferroptosis129.
The enzyme GPX4, together with glutathione as an essential cofactor, scavenge the harmful by-products of iron-dependent lipid peroxidation, thereby protecting the cell membrane from damage. Using a chemoproteomics assay, investigators have determined that overexpression of GPX4 in cancer cells can inhibit ferroptosis that is mediated by transcription factor RSL3, whereas deletion of GPX4 increases sensitivity to ferroptosis130. The synthesis and activity of GPX4, which is a selenoprotein, are affected by the concentration of the essential trace mineral selenium124,125. Selenium is required for GPX4 function, and its supplementation can increase the expression of GPX4 and protect cells against ferroptosis131,132. Interestingly, selenium deficiency in humans is thought to cause Keshan disease, an endemic cardiomyopathy present in children and pregnant women residing in the Keshan region of China where selenium levels in food are low133. Patients with Keshan disease develop dilated cardiomyopathy and heart failure. High serum levels of selenium have also been independently associated with both reduced mortality and fewer cases of new-onset heart failure in a population-based cohort study134. Therefore, well-powered interventional studies designed to further evaluate the potential benefits of high selenium levels in humans are warranted.
Given the wide range of physiological functions of glutathione, its role in the pathogenesis and development of heart disease is not surprising. In a Japanese population-based study, plasma glutathione levels were found to be significantly lower in all patients with heart disease than in healthy controls135. In a subsequent study, circulating glutathione levels were reduced by 21% and 40% in patients with asymptomatic and symptomatic heart disease, respectively136. Importantly, the investigators also measured glutathione concentrations in atrial tissue and found direct evidence that cardiac glutathione deficiency is closely associated with heart disease136. Glutathione deficiency has also been observed in patients with hypertension137.
Mice deficient in the glycoprotein apolipoprotein E (apoE), a component of all lipoproteins except LDL, are the most commonly used preclinical model of atherosclerosis138. Glutathione levels are significantly reduced in the atheroma-prone aortic arch of male apoE-deficient mice compared with age-matched, wild-type controls139. The study investigators proposed that glutathione deficiency is central to the suppression of intracellular antioxidant defences in these animals and therefore has a causal role in the pathogenesis of atherosclerosis139, an intriguing hypothesis that is supported by subsequent studies. For example, liposomal-coated glutathione was shown to significantly reduce oxidative stress and correlate with decreased levels of either lipid peroxides or oxidized LDL140. Furthermore, other studies have shown that increased glutathione levels significantly reduced liver and plasma cholesterol levels in mice fed a diet rich in saturated fats141,142.
Given that the heart has a very high energy demand and is particularly susceptible to oxidative damage, cardiomyocytes are thought to require a specialized and enhanced antioxidant system to avoid ferroptosis. Despite the lack of direct evidence that ferroptosis is involved in heart disease in humans, cardiomyocyte-specific animal disease models with genetically modified ferroptosis-related genes (such as SLC7A11 and GPX4) and studies on ferroptosis inducers or inhibitors in animal models have provided compelling preclinical evidence that the glutathione pathway has a protective role against cardiac ferroptosis.
Lipid metabolism and ferroptosis in the heart
In mammalian cells, the peroxidation of PUFA-containing phospholipids in cell membranes is an essential step in ferroptosis143. The enzyme long-chain fatty acid CoA ligase 4 (LACS4) converts PUFAs to the acylated form and is considered to be a specific driver of ferroptosis, as its upregulation increases PUFA content in phospholipids and renders the cell more susceptible to ferroptosis144,145. Although an in-depth study into the role of LACS4 in cardiac muscle has not yet been conducted, this enzyme has been shown to be a novel therapeutic target for limiting skeletal muscle cell death and preventing rhabdomyolysis146.
By contrast, LACS3 catalyses the conversion of exogenous monounsaturated fatty acids to fatty acyl-CoAs, thereby displacing PUFAs and inhibiting ferroptosis147. Conversely, the enzyme lysophosphatidylcholine acyltransferase 3 catalyses the insertion of these acylated PUFAs into membrane phospholipids; therefore, deleting this enzyme increases cellular resistance to ferroptosis145,148.
Lipoxygenases are a family of iron-containing enzymes that directly oxygenate PUFAs and PUFA-containing lipids in cellular membranes. Lipoxygenases are thought to have an important role in lipid peroxidation and ferroptosis149. In addition, the scaffold protein phosphatidylethanolamine-binding protein 1, which inhibits protein kinase cascades, has been shown to bind to and direct 15-lipoxygenase to PUFAs in the membrane, thereby promoting ferroptosis150.
The heart is highly susceptible to oxidative damage, and lipid peroxidation is an important contributor to ROS-induced heart injury. Cardiac tissue has several potential sources of endogenous ROS, including the mitochondrial electron transport chain, NADPH oxidase, xanthine oxidoreductase, nitric oxide synthases and cytochrome P450 (ref.151). In addition, several exogenous factors and chemicals can cause oxidative damage. An accumulation of lipid peroxides that originate from the oxidation of PUFA-containing membrane phospholipids suggests that ROS production cannot always be effectively balanced by the antioxidant system; as such, lipid peroxidation is one of the most pronounced manifestations of oxidative stress in the heart. Electrophilic reactive aldehydes, such as malondialdehyde, 4-hydroxyhexenal and 4-hydroxynonenal, are major end-products of PUFA oxidation, and are commonly used as markers of lipid peroxidation152. In patients with chronic heart failure, high serum levels of malondialdehyde were found to be an independent predictor of death and a combined clinical end point in a 1-year follow-up study153. These results were consistent with those of previous studies154,155, and support the hypothesis that lipid peroxidation has a role in both the development and severity of heart disease. Of note, lipid peroxidation is a driver of ferroptosis through its effects on damaging cellular membranes156. The scavenging of lipid peroxides protects against lipid peroxidation-induced damage to membranous structures in cardiomyocytes and results in inhibition of ferroptosis27,157,158.
Mitochondria and cardiac ferroptosis
As the energy powerhouse in eukaryotic cells, mitochondria coordinate essential metabolic processes such as oxidative phosphorylation. Given that the production of haem and Fe–S clusters occurs primarily in the mitochondria, this organelle contains large amounts of iron159 (Fig. 2b). Iron is taken up by mitochondria via the proteins mitoferrin 1, which is expressed in the erythroid lineage, and mitoferrin 2, which is ubiquitously expressed160. Mitochondrial iron binds to mitochondrial ferritin (a ferroxidase enzyme encoded by FTMT) to prevent ROS production, and mutations in mitochondrial ferritin can cause mitochondrial iron overload and cytoplasmic iron deficiency161. The mechanism by which iron is exported from the mitochondria is currently unknown; whether exported mitochondrial iron is conjugated to glutathione or is incorporated into Fe–S clusters or haem molecules is unclear159. Deletion of the mitochondrial protein ABCB8 selectively in the heart has been shown to cause mitochondrial iron overload, increased ROS production, defects in the cytosolic maturation of Fe–S clusters and cardiomyopathy162. Furthermore, iron–sulfur clusters transporter ABCB7, mitochondrial (ABCB7) has a role in mitochondrial iron homeostasis163, and ATP-binding cassette subfamily B member 10, mitochondrial (ABCB10) has been shown to regulate the early steps of haem synthesis in mitochondria164 and can also export biliverdin from mitochondria165.
In the mitochondria, iron is also used to synthesize haem, which functions as a cofactor in mediating catalysis and electron transfer166. The production of haem is a multistep process that requires eight different enzymes, with aminolevulinic acid synthase (ALAS; also known as ALAS-H) involved in the rate-limiting first step167. Excess haem is either exported to the cytoplasm via feline leukaemia virus subgroup C receptor-related protein 1B (FLVCR1B) or catabolized via the HO1 pathway into equimolar amounts of Fe2+, CO and biliverdin168. HO1 expression is induced ubiquitously in response to oxidative stress, whereas HO2 is constitutively expressed and not inducible169. In mammals, haem efflux is mediated primarily via the plasma membrane exporter FLVCR1A170. Conversely, FLVCR2 has been identified as a plasma membrane haem importer171. Other haem transporters such as haem transporter HRG1 and ATP-binding cassette subfamily C member 5 (ABCC5; also known as MRP5) are thought to deliver haem from various organelles, but their functions are poorly understood172.
Given that the mitochondrial respiratory chain is a major source of ROS in most mammalian cells173, mitochondria have been hypothesized to have a central role in the regulation of erastin-induced cell death since the discovery of ferroptosis in 2012. Substantial morphological changes in the mitochondria of ferroptotic cells have been observed using transmission electron microscopy, including reduced mitochondrial volume and increased mitochondrial membrane density2. In addition, certain nitroxide-based lipid peroxidation mitigators have been designed to specifically target mitochondria and are protective against erastin-induced and RSL3-induced ferroptosis174.
By contrast, evidence also exists against the link between mitochondria and ferroptosis. For example, an early report suggested that a mitochondrial DNA-depleted cancer cell line is just as sensitive to ferroptosis as its parental cell line2. A subsequent study showed that mitochondria-deficient cells can still undergo ferroptosis and can be rescued by treatment with ferrostatins and iron chelators175. Nevertheless, these findings are highly debatable. In a separate study, cysteine depletion was shown to mediate hyperpolarization of mitochondrial membranes and promote lipid peroxide accumulation, whereas inhibition of the tricarboxylic acid cycle or the electron transfer chain suppressed mitochondrial ferroptosis176. However, the investigators in this study also concluded that mitochondrial function is not required for ferroptosis induced by GPX4 inhibition, suggesting that the role of mitochondria in ferroptosis is context-dependent, consistent with observations in Gpx4−/− primary cells18.
Of note, Gpx4 knockout in mice is embryonically lethal, whereas conditional Gpx4 deletion promotes disorders in the brain, liver, endothelium, haematopoietic system and immune system177. In mammalian cells, several isoforms of GPX4, including cytosolic and mitochondrial isoforms, are encoded by a single gene. The mitochondrial isoform of GPX4 is believed to localize in mitochondria owing to the presence of a mitochondrial targeting signal at its amino terminus178,179. However, this isoform might not be expressed endogenously in the majority of cell types, with the exception of sperm cells. The cytosolic GPX4 isoform can cross the mitochondrial outer membrane and accumulate in the intermembrane space, where it helps to suppress mitochondrial lipid peroxidation178. Interestingly, overexpressing cytosolic GPX4, but not the mitochondrial isoform, prevented the death of Gpx4-knockout mouse embryonic fibroblasts180. Taken together, these findings indicate that the expression of cytosolic GPX4 is sufficient to prevent ferroptosis.
In vivo evidence also supports the notion that mitochondria have an essential role in ferroptosis. The mitochondria-targeted antioxidant MitoTEMPO has been shown to protect against ferroptosis-induced cardiac injury6. In addition, mice lacking mitochondrial ferritin develop more severe brain damage and neurological deficits after cerebral injury, as well as more typical features of ferroptosis, such as increased lipid peroxidation and disturbed glutathione antioxidative defence, whereas overexpression of mitochondrial ferritin inhibits ferroptosis and prevents these pathological changes181.
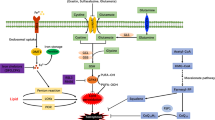
A study published in 2021 described a novel mitochondrial defence mechanism against ferroptosis182. By analysing global metabolomics data, the researchers linked the enzyme dihydroorotate dehydrogenase (quinone), mitochondrial (DHOH) to ferroptosis in vitro. DHODH is localized to the inner mitochondrial membrane, where it catalyses the rate-limiting fourth step in the de novo pyrimidine synthesis pathway, converting dihydroorotate to orotate183. Interestingly, deleting DHODH in cancer cells with low expression of GPX4 has been shown to markedly increase mitochondrial lipid peroxidation and ferroptosis182. In addition, delivering GPX4 to mitochondria rescues RSL3-induced ferroptosis in DHODH-deficient cells182. SLC25A39 has been identified as a mitochondrial glutathione importer, highlighting its potential functional role in regulating mitochondrial lipid peroxidation and ferroptotic cell death184. Nevertheless, additional studies are needed to determine the in vivo function of DHODH and identify other ferroptosis-regulating mitochondrial enzymes, particularly in the context of cardioprotection (Fig. 3).

Mitochondria host a wide range of key metabolic processes (such as the tricarboxylic acid (TCA) cycle) and are a major source of reactive oxygen species (ROS). Separate mitochondria-localized defence systems have evolved to prevent mitochondrial lipid peroxidation and ferroptosis. For example, either the mitochondrial version of phospholipid hydroperoxide glutathione peroxidase 4 (GPX4) or dihydroorotate dehydrogenase (quinone), mitochondrial (DHODH) can specifically detoxify mitochondrial lipid peroxides. Moreover, the mitochondria-specific form of ferritin (FTMT) protects mitochondria from iron overload-induced oxidative injury, and mitoNEET (also known as CISD1) suppresses ferroptosis by limiting mitochondrial iron uptake. CoQ10, coenzyme Q10; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; GSH, glutathione; GSSG, glutathione disulfide; HO1, haem oxygenase 1; LIP, labile iron pool; PL-PUFA-OOH, polyunsaturated fatty acid-containing phospholipid hydroperoxides; PLOO·, phospholipid peroxyl radical; RNF217, E3 ubiquitin protein ligase RNF217; SLC25A39, probable mitochondrial glutathione transporter SLC25A39; SLC39A14, solute carrier family 39 member 14; TF, transferrin; TFR1, transferrin receptor protein 1.
Other pathways that regulate ferroptosis
Ferroptosis suppressor protein 1 (FSP1) has been identified by two separate studies as a glutathione-independent, anti-ferroptosis factor185,186. Upon myristoylation, FSP1 is recruited to the plasma membrane, where it functions as an oxidoreductase to catalyse the production of coenzyme Q10 using NADPH185,186. Coenzyme Q10 functions as an endogenous inhibitor of ferroptosis through its antioxidative properties in the cell membrane, and depletion of coenzyme Q10 via the squalene synthase‒mevalonate pathway explains, at least in part, the mechanism by which the type 3 ferroptosis inducer Fin56 promotes ferroptosis187. Although a previous study in mice indicated that doxorubicin can activate FSP1 translocation via lipid peroxidation products in the heart188, the precise function of FSP1 in the heart remains poorly understood. Therefore, mice with a conditional Fsp1 knockout might be a potential tool to provide further mechanistic insight.
Ferroptosis in cardiovascular disease
Before the discovery in 2014 that GPX4 is a key regulator of ferroptosis18,130, numerous studies provided preliminary evidence on the role of altered GPX4 in a non-apoptotic form of cell death. For example, global Gpx4 knockout in mice is lethal by embryonic day 7.5 (refs.189,190), whereas an inducible knockout of Gpx4 in adult mice causes a rapid reduction in body weight and death within 2 weeks191. Moreover, mice with a conditional Gpx4 knockout in endothelial cells generally die within 3 weeks, owing to thromboembolic events when deprived of dietary vitamin E192. This phenotype resulting from the combination of GPX4 and vitamin E deficiency indicates an important role of GPX4 in cardiovascular physiology. However, there remains a lack of evidence to support the existence of ferroptosis in the Gpx4-knockout animal models.
Myocardial ischaemia–reperfusion injury
Ischaemia–reperfusion injury is a fairly common, life-threatening clinical complication that can occur in nearly any organ, including the heart, liver, kidneys and brain. Mitochondria-specific overexpression of GPX4 was reported to be cardioprotective after ischaemia–reperfusion injury long before the concept of ferroptosis was first established193. In 2014, two studies found that ferroptosis is a primary driver of renal and hepatic ischaemia–reperfusion injury18,194. A subsequent study demonstrated that suppressing ferroptosis by inhibiting glutaminolysis reduced ischaemia–reperfusion injury in an ex vivo heart model45. Further in vivo data provided additional evidence indicating that either inhibiting ferroptosis or chelating iron during both acute and chronic myocardial ischaemia–reperfusion injury can provide cardioprotective benefits, highlighting the potential of targeting ferroptosis as a promising novel therapeutic strategy for ischaemia–reperfusion injury6,195,196.
Ischaemia–reperfusion injury after heart transplantation can lead to serious complications such as primary graft dysfunction and increased risk of death197. In addition to the direct loss of cardiomyocytes, the release of endogenous substances during ischaemia–reperfusion induced by ferroptosis can trigger a harmful inflammatory response in the donor heart by promoting the adhesion of neutrophils to coronary vascular endothelial cells via a Toll-like receptor 4-dependent signalling pathway157. Importantly, oxidized phosphatidylethanolamine was also identified as a specific product of ferroptosis by analysing oxidative lipidomics, providing direct evidence of ferroptosis in the heart157. An accumulation of ferroptotic oxidized phosphatidylethanolamine species was also found in mitochondria isolated from hearts with ischaemia–reperfusion injury, further highlighting the role of cardiac mitochondria in the production of lipid peroxides and in ferroptotic signalling198. In addition, a new imaging protocol was developed in 2021 that can directly detect the presence and distribution of oxidized phosphatidylethanolamine in specific cells and tissues199. Application of this protocol in preclinical studies will facilitate the detection of peroxidized lipids in disease conditions, including cardiovascular disease.
Anthracycline cardiotoxicity
As discussed above, the use of doxorubicin for the treatment of malignancies is limited by its cardiotoxic effects. Our research group has examined the relative contributions of various forms of regulated cell death in doxorubicin-induced cardiotoxicity by measuring the effect of the respective inhibitors of cell death on survival in doxorubicin-treated mice and shown that inhibition of ferroptosis is cardioprotective6. In addition to inducing heart injury, doxorubicin treatment causes a robust increase in cardiac levels of iron, lipid-derived ROS and ferroptosis biomarkers. Together, these findings suggest that ferroptosis has a major role in doxorubicin-induced cardiomyopathy and death6. Furthermore, at the subcellular level, we found that mitochondria are the target of HO1-mediated release of free iron, which causes lipid peroxidation in the mitochondrial membrane.
A subsequent study published in 2020 confirmed that mitochondria-dependent ferroptosis has a major pathogenic role in doxorubicin-induced cardiotoxicity200. Specifically, the study showed that doxorubicin treatment can downregulate GPX4 expression in the heart, leading to excessive lipid peroxidation. Moreover, overexpression of GPX4 in mice prevented doxorubicin-induced cardiomyopathy, whereas knocking down Gpx4 exacerbated doxorubicin-induced cardiomyopathy200. However, the mechanism by which doxorubicin treatment can downregulate GPX4 requires further elucidation.
Finally, it is important to note that caspase-dependent apoptosis, but not ferroptosis, drives doxorubicin-induced cardiotoxicity in cultured cardiomyocytes6,200. This apparent discrepancy between in vivo and in vitro findings reflects the complexity of the mechanism of action of doxorubicin in vivo. For example, the HO1 pathway is differentially regulated in cultured cells compared with the in vivo setting, thereby affecting both iron accumulation and lipid peroxidation6,201.
Diabetic cardiomyopathy
Diabetes mellitus is a common comorbidity in patients with cardiovascular disease and can increase the susceptibility of the heart to ischaemia–reperfusion injury202. Consequently, patients with diabetes have a poorer prognosis after acute myocardial infarction than patients without diabetes202. Diabetes can aggravate myocardial ischaemia–reperfusion injury by activating the NADPH oxidase pathway in an AMPK-dependent manner, subsequently inducing various forms of programmed cell death, including ferroptosis203. Ferroptosis has been shown to have a pathogenic role in mediating myocardial ischaemia–reperfusion injury in a streptozotocin mouse model of type 1 diabetes204. Moreover, hyperglycaemia-induced endoplasmic reticulum stress seems to be involved in cardiomyocyte damage mediated by ferroptosis204.
Patients with diabetes are also at increased risk of developing myocardial dysfunction that is independent of coronary artery disease and hypertension — a phenomenon known as diabetic cardiomyopathy205. Indeed, several pathogenic factors, including oxidative stress, have been shown to contribute to the structural and functional changes that characterize the diabetic heart205. Ferroptosis was reported for the first time in the heart of diabetic mice in a study published in 2022, in which NRF2 activation was shown to prevent ferroptosis by upregulating ferritin and SLC7A11 levels158.
Sepsis‑induced cardiac injury
In patients, sepsis-induced cardiac injury and dysfunction correlate with increased mortality206. Caecal ligation and puncture is currently the most commonly used animal model for studying sepsis and involves the perforation of the caecum to allow the release of faecal material into the peritoneal cavity, which triggers an immune response mediated by polymicrobial infection207. Caecal ligation and puncture has been shown to increase cardiac iron content and lipid peroxidation levels, as well as to reduce cardiac glutathione content and GPX4 expression, suggesting that the development of sepsis-induced heart injury might involve ferroptosis207. In addition, ferroptosis has been shown to have a role in a lipopolysaccharide-induced model of septic cardiomyopathy208. A lipopolysaccharide-induced increase in sideroflexin 1 expression in the cardiac mitochondrial membrane can increase the production of mitochondrial ROS, leading to ferroptosis208.
Hypertrophic cardiomyopathy
In the heart, pressure overload (owing to pulmonary hypertension and/or systemic hypertension) can lead to cardiac hypertrophy, cardiac fibrosis and eventual heart failure. Increased cardiac NADPH oxidase 4 expression and decreased cardiac GPX4 activity in animal models after either aortic banding or isoprenaline administration suggest that ROS production and ferroptosis might participate in the progression from compensated hypertrophy to heart failure209,210. In mice, genetic deletion of the key ferroptosis regulator SLC7A11 was found to exacerbate angiotensin II-mediated cardiac fibrosis, hypertrophy and dysfunction, providing genetic evidence of the involvement of ferroptosis in hypertrophic cardiomyopathy129.
Interestingly, genetic deletion of Ncoa4 specifically in cardiomyocytes has been shown to attenuate transverse aortic constriction (TAC)-induced heart failure by suppressing ferritinophagy28. Moreover, ferroptosis inhibitors attenuated cardiac remodelling in wild-type mice that had undergone TAC, but did not provide additional protection in Ncoa4-knockout mice, implicating cardiac ferroptosis as the downstream consequence of NCOA4-mediated ferritinophagy28. In addition, in a TAC mouse model, the enzyme mixed lineage kinase 3 (also known as MAP3K11) has been shown to induce pyroptosis and ferroptosis, which are essential for the development of TAC-mediated myocardial fibrosis211. Nevertheless, the mechanisms underlying the involvement of various forms of cell death in the pathogenesis of hypertrophic cardiomyopathy remain to be determined.
Putative link between ferroptosis and arrhythmia
Cardiac arrhythmia is common among patients with heart failure212. Findings from a preclinical study suggest a possible link between ferroptosis and arrhythmia. In mice, frequent excessive alcohol consumption triggered ferroptosis and increased the inducibility of atrial fibrillation213. Ferroptosis inhibitors could partially or completely reverse most of the adverse change induced by excessive alcohol intake.
As mentioned above, ferroptosis can propagate to adjacent cells in a wave-like manner, a phenomenon that has not been reported to occur with other forms of regulated cell death15,194. However, the mechanism underlying this seemingly directed pattern of propagation remains unknown. Cardiac tissue is a functional syncytium for coordinated muscle contraction, mediated via cell-to-cell electrical and chemical coupling through gap junction channels that determine the rhythm of the heart and might contribute to the wave-like propagation of ferroptosis. Therefore, cardiac muscle might be an ideal model for studying the propagation of cell death. Interestingly, myocardial damage such as ischaemia–reperfusion injury often results in arrhythmia as well as the formation of a necrotic zone, which has been suggested to account for the cell-to-cell propagation of ferroptosis214. Further investigation of this unique feature of ferroptosis is warranted.
Other cardiovascular-related diseases
Sickle cell disease is a group of inherited red blood cell disorders characterized by the presence of haemolysis that lead to organ ischaemia and cardiovascular complications215. Increased haem levels in mice with sickle cell disease resulted in an upregulation of cardiac HO1 levels, which then promoted iron overload, lipid peroxidation and ferroptosis in the heart. Furthermore, inhibiting and inducing ferroptosis attenuated and exacerbated, respectively, the cardiomyopathy that was associated with sickle cell disease216.
Currently, the global population is in the midst of the coronavirus disease 2019 (COVID-19) pandemic, a respiratory tract infection caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)217. The cardiovascular complications associated with COVID-19 are well recognized218. Iron might also have an important role in the pathogenesis of COVID-19, given that an estimated 90% of patients admitted to hospital present with abnormal serum iron levels, and these levels correlate with disease severity219. On the basis of these observations, ferroptosis has been suggested as a potential target for the treatment of COVID-19 (ref.220). Primary pacemaker cells in the heart have been shown to develop ferroptosis-associated cardiac dysfunction after infection with SARS-CoV-2 (ref.221). Moreover, a high-throughput chemical screen showed that the tyrosine kinase inhibitor imatinib and the iron-chelating agent deferoxamine can block SARS-CoV-2 infection and associated ferroptosis221.
Ferroptosis as a promising treatment target
Given its role in the pathogenesis of heart disease, ferroptosis is a highly promising therapeutic target for the treatment and prevention of cardiovascular disease. In this section, we summarize the various small molecules that inhibit the ferroptosis pathway and discuss the use of these molecules in various models of heart disease (Table 3).
Ferrostatin 1, liproxstatin 1 and antioxidants
Ferrostatin 1 is a first-generation inhibitor of ferroptosis2. Since the anti-ferroptotic effects of this small-molecule compound were first reported, ferrostatin 1 has been tested in a wide range of diseases, including cardiovascular disease. Our group pioneered the use of ferrostatin 1 in mice with experimental cardiomyopathy and found that it can protect against doxorubicin-induced cardiac damage without affecting iron levels6. A subsequent study showed that ferrostatin 1 can also prevent doxorubicin-induced cell death in cultured cardiomyocytes200. In addition, ferrostatin 1 has been shown to improve cardiac function in animal models of acute or chronic myocardial ischaemia–reperfusion injury6,157, as well as in diabetic mice204. In a mouse model of heart transplantation, ferrostatin 1 treatment blocked the recruitment of neutrophils after cardiomyocyte cell death, suggesting a potential strategy for improving clinical outcomes in patients receiving a heart transplantation157. Moreover, ferrostatin 1 treatment has also been reported to attenuate sepsis-induced cardiomyopathy and atherosclerosis in mice208,222. Of note, some researchers have indicated that the in vivo function of ferrostatin 1 is weaker than its function in vitro, given its low stability in plasma223. To address this issue, a soluble ferrostatin analogue called UAMC-3203 was developed as a more stable and potent inhibitor of ferroptosis than ferrostatin 1. In animals, UAMC-3203 outperformed ferrostatin 1 in preventing ferroptosis-driven multiorgan dysfunction and might therefore be a suitable candidate for clinical testing224.
Liproxstatin 1, a spiroquinoxalinamine derivative, was first identified as a specific inhibitor of ferroptosis from high-throughput screening of Gpx4−/− cells18. Liproxstatin 1 is not as well studied as ferrostatin 1 in the context of cardiovascular pathophysiology, but a study in mice has shown that liproxstatin 1 can protect the myocardium against ischaemia–reperfusion injury by reducing mitochondrial ROS production and maintaining GPX4 activity225. A subsequent study found that liproxstatin 1 significantly reduces palmitic acid-induced cardiac injury, with similar protective effects exerted by ferrostatin 1 (ref.226).
Other commonly studied antioxidants in the context of cardiovascular disease are 2,2,6,6-tetramethyl-piperidinyl-1-oxyl (TEMPO) and MitoTEMPO, a mitochondria-targeted version of TEMPO that was developed to scavenge mitochondrial superoxides227. We have previously demonstrated that MitoTEMPO can potently prevent doxorubicin-induced lipid peroxidation, cardiac ferroptosis and cardiac dysfunction in mice, whereas TEMPO only mildly reduces these doxorubicin-induced effects6. These findings strongly support the notion that mitochondrial lipid peroxidation and cardiac ferroptosis work in a coordinated fashion to mediate doxorubicin-induced cardiomyopathy.
Given that ferroptosis is only one pathway that contributes to the death of cardiomyocytes228, a combination strategy targeting both ferroptosis and necroptosis might be more effective at managing heart disease. Nec-1f, a dual inhibitor that targets both receptor-interacting serine/threonine protein kinase 1 and ferroptosis primarily in kidney cells and kidney tubules, significantly increased survival in a mouse model of heart transplantation and a mouse model of renal ischaemia–reperfusion injury229.
Iron chelators and iron chelation therapy
Given that ferroptosis is an iron-dependent form of programmed cell death, it is no surprise that ferroptosis can be inhibited by iron chelation2. The iron chelator dexrazoxane is currently the only FDA-approved drug for preventing doxorubicin-induced cardiotoxicity in patients with cancer. Dexrazoxane is a cyclic derivative of ethylenediaminetetraacetic acid that readily crosses cell membranes and chelates intracellular free iron230. A study showed that inhibition of ferroptosis is the predominant mechanism by which dexrazoxane exerts its cardioprotective effect6. However, this finding raises the question of why other iron chelators do not seem to be effective against doxorubicin-induced heart injury231, and researchers have suggested that dexrazoxane can directly enter mitochondria in cardiomyocytes and reduce iron accumulation, whereas other iron chelators cannot enter the mitochondria100. This notion is further supported by the finding that Mito-FerroGreen, a novel mitochondria-specific iron chelator, provides cardioprotection in mice treated with doxorubicin200. In addition, other iron chelators such as deferiprone and deferoxamine have been shown to alleviate both myocardial ischaemia–reperfusion injury and sepsis-related cardiac damage by blocking ferroptosis45,208,232. Of note, however, the finding that an iron chelator can protect tissue from damage does not necessarily indicate that it does so by inhibiting ferroptosis.
GSH precursors
Cysteine, the reduced form of cystine imported via system xc−, is the rate-limiting precursor in glutathione biosynthesis, and the addition of cystine or cysteine to cell culture media in vitro has been shown to inhibit ferroptosis45. The antioxidant N-acetyl cysteine (NAC) was developed to improve the bioavailability of cysteine and has been shown to have beneficial effects on cardiovascular function233,234. The anti-ferroptosis effect of NAC has been demonstrated in the context of cysteine depletion and system xc− inhibition-induced ferroptosis2,235. NAC treatment has also been shown to reduce myocardial ischaemia–reperfusion injury in diabetic rats, suggesting that this compound might have clinical applications in heart disease203.
Other compounds that can affect cardiac ferroptosis
In addition to the aforementioned drugs, other compounds can also inhibit cardiac ferroptosis by acting on other targets. For example, zinc protoporphyrin IX, a competitive inhibitor of HO1, reduces doxorubicin-induced cardiac iron accumulation and ferroptosis in mice by blocking haem degradation and the resulting release of free iron6. Glutaminolysis is required for cysteine deprivation-induced ferroptosis, and compound 968, a cell-permeable, small-molecule inhibitor of glutaminolysis, has been shown attenuate myocardial ischaemia–reperfusion injury ex vivo by limiting glutamine levels45. Moreover, P22077, an inhibitor of ubiquitin-specific protease 7, has been shown to suppress ferroptosis by activating p53 and reducing TFR1 levels, providing protection against myocardial ischaemia–reperfusion injury236. Interestingly, dexmedetomidine, an α2‑adrenergic receptor agonist used clinically to sedate patients, has been shown to alleviate sepsis‑induced myocardial injury via the ferroptosis suppressor GPX4 (ref.207). In addition, puerarin, an isoflavone extracted from the kudzu root, has been shown to protect against erastin-induced and isoprenaline-mediated ferroptosis in cultured cardiomyocytes and can mitigate heart failure induced by pressure overload in rats210. Furthermore, atorvastatin has also been shown to ameliorate isoprenaline-induced cardiac dysfunction and remodelling in mice by inhibiting ferritinophagy-mediated ferroptosis, thereby providing another potential therapeutic strategy for the prevention of hypertrophic cardiomyopathy237.
Finally, commonly prescribed heart medications might have previously unidentified anti-ferroptotic activity. For example, carvedilol, which is widely used to treat hypertension and heart failure, has been shown to block ferroptosis independent of its effect on β-adrenergic receptors238,239, and the underlying mechanism might contribute to its capacity to scavenge lipid peroxides and chelate iron240.
Limitations
A growing body of evidence supports the role of ferroptosis in the initiation and progression of various cardiovascular diseases. However, a number of questions need to be addressed before the therapeutic potential of ferroptosis-targeted agents can be clinically evaluated. First, what are the crucial safeguarding mechanisms against cardiac ferroptosis? Second, can we identify reliable biomarkers for predicting ferroptosis in cardiovascular disease? The ferroptosis biomarkers currently used in preclinical studies are non-specific and present in other types of cell death and certain pathological conditions. In this fast-growing field, the lack of ferroptosis-specific biomarkers has been a long-standing bottleneck limiting the development of ferroptosis-targeted clinical applications. Third, is it possible to design effective ferroptosis-targeted strategies for preventing and treating ferroptosis-related cardiovascular disease? Finally, when and how do other forms of cell death occur together with ferroptosis in the development of cardiovascular disease? Although selective inhibition of ferroptosis has been shown to substantially improve cardiac function in a variety of animal models, no clinical trials have so far been performed using ferroptosis-specific inhibitors to treat cardiovascular disease. Additional population-based data are urgently needed to determine whether selectively blocking ferroptosis can improve cardiovascular outcomes in the clinical setting.
Conclusions
The link between iron and cardiovascular disease was first proposed nearly three decades ago, but the mechanistic pathways underlying this relationship remained elusive until the discovery of ferroptosis, an iron-dependent form of regulated cell death, just 10 years ago. Ferroptosis is induced by the activation of iron-dependent lipid peroxidation, but the key effector molecules involved in this process are unclear. Excess levels of free reactive iron can cause tissue damage, and iron chelation therapy has been widely recommended for the treatment of patients with iron overload-related cardiomyopathy. Preclinical studies are paving the way to the development of effective ferroptosis-specific antagonists for clinical testing.
References
Del Re, D. P., Amgalan, D., Linkermann, A., Liu, Q. & Kitsis, R. N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 99, 1765–1817 (2019).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). This study introduces the term ferroptosis to describe a novel form of non-apoptotic cell death and identifies the small molecule ferrostatin 1 as the first specific inhibitor of ferroptosis.
Ganz, T. Systemic iron homeostasis. Physiol. Rev. 93, 1721–1741 (2013).
Jankowska, E. A. et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur. Heart J. 35, 2468–2476 (2014).
Berdoukas, V., Coates, T. D. & Cabantchik, Z. I. Iron and oxidative stress in cardiomyopathy in thalassemia. Free Radic. Biol. Med. 88, 3–9 (2015).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019). This study draws, for the first time, a link between cardiac cell death and ferroptosis in murine models of doxorubicin cardiotoxicity or myocardial ischaemia–reperfusion.
Chen, Y. R. & Zweier, J. L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 114, 524–537 (2014).
Bajic, V. P. et al. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxid. Med. Cell. Longev. 2019, 5028181 (2019).
Eaton, J. W. & Qian, M. Molecular bases of cellular iron toxicity. Free Radic. Biol. Med. 32, 833–840 (2002).
Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514 (1955).
Murphy, T. H., Miyamoto, M., Sastre, A., Schnaar, R. L. & Coyle, J. T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 (1989).
Tan, S., Schubert, D. & Maher, P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 1, 497–506 (2001).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Green, D. R. The coming decade of cell death research: five riddles. Cell 177, 1094–1107 (2019).
Kim, S. E. et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat. Nanotechnol. 11, 977–985 (2016).
Riegman, M. et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat. Cell Biol. 22, 1042–1048 (2020).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014). This study provides direct evidence that deletion of Gpx4 causes in vivo cell death in a pathologically relevant form of ferroptosis, and identifies liproxstatin 1 as an inhibitor of ferroptosis.
Koleini, N., Shapiro, J. S., Geier, J. & Ardehali, H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J. Clin. Invest. 131, e148671 (2021).
Luck, A. N. & Mason, A. B. Transferrin-mediated cellular iron delivery. Curr. Top. Membr. 69, 3–35 (2012).
Xu, W. et al. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 13, 533–545 (2015).
Liu, Q., Azucenas, C., Mackenzie, B. & Knutson, M. Metal-ion transporter SLC39A14 Is required for cardiac iron loading in the Hjv mouse model of iron overload. Blood 138 (Suppl. 1), 758 (2021).
Sukhbaatar, N. & Weichhart, T. Iron regulation: macrophages in control. Pharmaceuticals 11, 137 (2018).
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W. & Kimmelman, A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Quiles Del Rey, M. & Mancias, J. D. NCOA4-mediated ferritinophagy: a potential link to neurodegeneration. Front. Neurosci. 13, 238 (2019).
Yambire, K. F. et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. eLife 8, e51031 (2019).
Fang, X. et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 127, 486–501 (2020). This study demonstrates that ferritin has a major role in protecting against cardiac ferroptosis and suggests SLC7A11 as a potential new therapeutic target for cardiomyopathy and heart failure.
Ito, J. et al. Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. eLife 10, e62174 (2021).
Lakhal-Littleton, S. et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl Acad. Sci. USA 112, 3164–3169 (2015).
Fang, X., Wang, H., An, P., Min, J. & Wang, F. Cardiomyocyte-specific deletion of ferroportin using MCK-Cre has no apparent effect on cardiac iron homeostasis. Int. J. Cardiol. 201, 90–92 (2015).
Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090–2093 (2004).
Jiang, L. et al. RNF217 regulates iron homeostasis through its E3 ubiquitin ligase activity by modulating ferroportin degradation. Blood 138, 689–705 (2021).
Lesbordes-Brion, J. C. et al. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood 108, 1402–1405 (2006).
Roetto, A. et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat. Genet. 33, 21–22 (2003).
Lakhal-Littleton, S. et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 5, e19804 (2016).
Rouault, T. A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 (2006).
Bayeva, M., Chang, H. C., Wu, R. & Ardehali, H. When less is more: novel mechanisms of iron conservation. Trends Endocrinol. Metab. 24, 569–577 (2013).
Bayeva, M. et al. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. 16, 645–657 (2012).
Sandler, H., Kreth, J., Timmers, H. T. & Stoecklin, G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 39, 4373–4386 (2011).
Sato, T. et al. mRNA-binding protein tristetraprolin is essential for cardiac response to iron deficiency by regulating mitochondrial function. Proc. Natl Acad. Sci. USA 115, E6291–E6300 (2018).
Wang, H. et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66, 449–465 (2017).
Yu, Y. et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood 136, 726–739 (2020).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Doll, S. & Conrad, M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life 69, 423–434 (2017).
Gao, M., Monian, P., Quadri, N., Ramasamy, R. & Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015). This study shows that inhibition of ferroptosis by targeting glutaminolysis might be a potential therapy for ischaemic heart disease.
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Song, Y. et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 37, 51–64 (2021).
Mumbauer, S., Pascual, J., Kolotuev, I. & Hamaratoglu, F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 15, e1008396 (2019).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Protchenko, O. et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology 73, 1176–1193 (2021).
Brown, C. W. et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell 51, 575–586.e4 (2019).
Geng, N. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 22, 3826–3836 (2018).
Tuo, Q. Z. et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530 (2017).
Shang, Y. et al. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell. Signal. 72, 109633 (2020).
Paradkar, P. N., Zumbrennen, K. B., Paw, B. H., Ward, D. M. & Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 29, 1007–1016 (2009).
Zhang, Z. et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 36, 101619 (2020).
Kwon, M. Y., Park, E., Lee, S. J. & Chung, S. W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 6, 24393–24403 (2015).
Chang, L. C. et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137 (2018).
Adedoyin, O. et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 314, F702–F714 (2018).
Wang, Y. Q. et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308 (2016).
Alvarez, S. W. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Yuan, H., Li, X., Zhang, X., Kang, R. & Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 478, 838–844 (2016).
Kim, E. H., Shin, D., Lee, J., Jung, A. R. & Roh, J. L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 432, 180–190 (2018).
Kremastinos, D. T. & Farmakis, D. Iron overload cardiomyopathy in clinical practice. Circulation 124, 2253–2263 (2011).
Fleming, R. E. & Ponka, P. Iron overload in human disease. N. Engl. J. Med. 366, 348–359 (2012).
Powell, L. W., Seckington, R. C. & Deugnier, Y. Haemochromatosis. Lancet 388, 706–716 (2016).
Gulati, V. et al. Cardiac involvement in hemochromatosis. Cardiol. Rev. 22, 56–68 (2014).
Roest, M. et al. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation 100, 1268–1273 (1999).
Tuomainen, T. P. et al. Increased risk of acute myocardial infarction in carriers of the hemochromatosis gene Cys282Tyr mutation: a prospective cohort study in men in eastern Finland. Circulation 100, 1274–1279 (1999).
Gaenzer, H. et al. Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J. Am. Coll. Cardiol. 40, 2189–2194 (2002).
Lodi, R., Tonon, C., Calabrese, V. & Schapira, A. H. Friedreich’s ataxia: from disease mechanisms to therapeutic interventions. Antioxid. Redox Signal. 8, 438–443 (2006).
Campuzano, V. et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 (1996).
Stemmler, T. L., Lesuisse, E., Pain, D. & Dancis, A. Frataxin and mitochondrial FeS cluster biogenesis. J. Biol. Chem. 285, 26737–26743 (2010).
Koutnikova, H. et al. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 16, 345–351 (1997).
Hewer, R. L. Study of fatal cases of Friedreich’s ataxia. Br. Med. J. 3, 649–652 (1968).
Tsou, A. Y. et al. Mortality in Friedreich ataxia. J. Neurol. Sci. 307, 46–49 (2011).
Whitnall, M. et al. The MCK mouse heart model of Friedreich’s ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl Acad. Sci. USA 105, 9757–9762 (2008).
Velasco-Sanchez, D. et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 10, 1–8 (2011).
Elincx-Benizri, S. et al. Clinical experience with deferiprone treatment for Friedreich ataxia. J. Child Neurol. 31, 1036–1040 (2016).
Pandolfo, M. & Hausmann, L. Deferiprone for the treatment of Friedreich’s ataxia. J. Neurochem. 126 (Suppl. 1), 142–146 (2013).
Taher, A. T. & Saliba, A. N. Iron overload in thalassemia: different organs at different rates. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 265–271 (2017).
Rivella, S. Iron metabolism under conditions of ineffective erythropoiesis in β-thalassemia. Blood 133, 51–58 (2019).
Saliba, A. & Taher, A. Iron overload in transfusion-dependent thalassemia. Hematology 20, 311–312 (2015).
Lekawanvijit, S. & Chattipakorn, N. Iron overload thalassemic cardiomyopathy: iron status assessment and mechanisms of mechanical and electrical disturbance due to iron toxicity. Can. J. Cardiol. 25, 213–218 (2009).
Zurlo, M. G. et al. Survival and causes of death in thalassaemia major. Lancet 2, 27–30 (1989).
Borgna-Pignatti, C. et al. Survival and complications in thalassemia. Ann. NY Acad. Sci. 1054, 40–47 (2005).
Kremastinos, D. T. et al. Heart failure in beta thalassemia: a 5-year follow-up study. Am. J. Med. 111, 349–354 (2001).
Russo, V., Rago, A., Papa, A. A. & Nigro, G. Electrocardiographic presentation, cardiac arrhythmias, and their management in β-thalassemia major patients. Ann. Noninvasive Electrocardiol. 21, 335–342 (2016).
Piga, A., Gaglioti, C., Fogliacco, E. & Tricta, F. Comparative effects of deferiprone and deferoxamine on survival and cardiac disease in patients with thalassemia major: a retrospective analysis. Haematologica 88, 489–496 (2003).
Pennell, D. J. et al. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 107, 3738–3744 (2006).
Sheth, S. Strategies for managing transfusional iron overload: conventional treatments and novel strategies. Curr. Opin. Hematol. 26, 139–144 (2019).
Casu, C., Nemeth, E. & Rivella, S. Hepcidin agonists as therapeutic tools. Blood 131, 1790–1794 (2018).
Young, R. C., Ozols, R. F. & Myers, C. E. The anthracycline antineoplastic drugs. N. Engl. J. Med. 305, 139–153 (1981).
Singal, P. K. & Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 339, 900–905 (1998).
Swain, S. M., Whaley, F. S. & Ewer, M. S. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97, 2869–2879 (2003).
Panjrath, G. S. et al. Potentiation of doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll. Cardiol. 49, 2457–2464 (2007).
Miranda, C. J. et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood 102, 2574–2580 (2003).
Ichikawa, Y. et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630 (2014). This study showed that cardiotoxicity develops via mitochondrial iron accumulation after doxorubicin treatment and can be prevented by reducing mitochondrial iron levels.
Maccarinelli, F. et al. Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. J. Mol. Med. 92, 859–869 (2014).
Hou, K. et al. Loss of TRIM21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. EBioMedicine 69, 103456 (2021).
Hallberg, L. Advantages and disadvantages of an iron-rich diet. Eur. J. Clin. Nutr. 56 (Suppl. 1), S12–S18 (2002).
Hurrell, R. & Egli, I. Iron bioavailability and dietary reference values. Am. J. Clin. Nutr. 91, 1461S–1467S (2010).
Ascherio, A., Willett, W. C., Rimm, E. B., Giovannucci, E. L. & Stampfer, M. J. Dietary iron intake and risk of coronary disease among men. Circulation 89, 969–974 (1994).
Liao, Y., Cooper, R. S. & McGee, D. L. Iron status and coronary heart disease: negative findings from the NHANES I epidemiologic follow-up study. Am. J. Epidemiol. 139, 704–712 (1994).
Klipstein-Grobusch, K. et al. Dietary iron and risk of myocardial infarction in the Rotterdam Study. Am. J. Epidemiol. 149, 421–428 (1999).
Malaviarachchi, D., Veugelers, P. J., Yip, A. M. & MacLean, D. R. Dietary iron as a risk factor for myocardial infarction. Public health considerations for Nova Scotia. Can. J. Public Health 93, 267–270 (2002).
van der A, D. L., Peeters, P. H., Grobbee, D. E., Marx, J. J. & van der Schouw, Y. T. Dietary haem iron and coronary heart disease in women. Eur. Heart J. 26, 257–262 (2005).
Lee, D. H., Folsom, A. R. & Jacobs, D. R. Jr Iron, zinc, and alcohol consumption and mortality from cardiovascular diseases: the Iowa Women’s Health Study. Am. J. Clin. Nutr. 81, 787–791 (2005).
Qi, L., van Dam, R. M., Rexrode, K. & Hu, F. B. Heme iron from diet as a risk factor for coronary heart disease in women with type 2 diabetes. Diabetes Care 30, 101–106 (2007).
Casiglia, E. et al. Dietary iron intake and cardiovascular outcome in Italian women: 10-year follow-up. J. Women’s Health 20, 1565–1571 (2011).
de Oliveira Otto, M. C. et al. Dietary intakes of zinc and heme iron from red meat, but not from other sources, are associated with greater risk of metabolic syndrome and cardiovascular disease. J. Nutr. 142, 526–533 (2012).
Zhang, W. et al. Associations of dietary iron intake with mortality from cardiovascular disease: the JACC study. J. Epidemiol. 22, 484–493 (2012).
Kaluza, J., Larsson, S. C., Hakansson, N. & Wolk, A. Heme iron intake and acute myocardial infarction: a prospective study of men. Int. J. Cardiol. 172, 155–160 (2014).
Wang, W. et al. Dietary iron and vitamins in association with mortality. Clin. Nutr. 40, 2401–2409 (2021).
Fang, X. et al. Dietary intake of heme iron and risk of cardiovascular disease: a dose-response meta-analysis of prospective cohort studies. Nutr. Metab. Cardiovasc. Dis. 25, 24–35 (2015).
Han, M. et al. Dietary iron intake and risk of death due to cardiovascular diseases: a systematic review and dose-response meta-analysis of prospective cohort studies. Asia Pac. J. Clin. Nutr. 29, 309–321 (2020).
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620 (2021).
Bannai, S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J. Biol. Chem. 261, 2256–2263 (1986).
Oda, K. et al. Consensus mutagenesis approach improves the thermal stability of system xc− transporter, xCT, and enables cryo-EM analyses. Protein Sci. 29, 2398–2407 (2020).
Parker, J. L. et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc. Nat. Commun. 12, 7147 (2021).
Yan, R. et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. https://doi.org/10.1038/s41422-022-00642-w (2022).
Zhang, Y. et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e9 (2019).
Rojo de la Vega, M., Chapman, E. & Zhang, D. D. NRF2 and the hallmarks of cancer. Cancer Cell 34, 21–43 (2018).
Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371 (2017).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Wang, L. et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc−. Cell Death Differ. 27, 662–675 (2020).
Zhang, X. et al. SLC7A11/xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc. Drugs Ther. 36, 437–447 (2021).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422.e21 (2018).
Alim, I. et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279.e25 (2019).
Loscalzo, J. Keshan disease, selenium deficiency, and the selenoproteome. N. Engl. J. Med. 370, 1756–1760 (2014).
Al-Mubarak, A. A. et al. High selenium levels associate with reduced risk of mortality and new onset heart failure: data from PREVEND. Eur. J. Heart Fail. 24, 299–307 (2022).
Shimizu, H. et al. Relationship between plasma glutathione levels and cardiovascular disease in a defined population: the Hisayama study. Stroke 35, 2072–2077 (2004).
Damy, T. et al. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 4, e4871 (2009).
Redon, J. et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension 41, 1096–1101 (2003).
Lo Sasso, G. et al. The Apoe(−/−) mouse model: a suitable model to study cardiovascular and respiratory diseases in the context of cigarette smoke exposure and harm reduction. J. Transl. Med. 14, 146 (2016).
Biswas, S. K., Newby, D. E., Rahman, I. & Megson, I. L. Depressed glutathione synthesis precedes oxidative stress and atherogenesis in Apo-E(−/−) mice. Biochem. Biophys. Res. Commun. 338, 1368–1373 (2005).
Rosenblat, M., Volkova, N., Coleman, R. & Aviram, M. Anti-oxidant and anti-atherogenic properties of liposomal glutathione: studies in vitro, and in the atherosclerotic apolipoprotein E-deficient mice. Atherosclerosis 195, e61–e68 (2007).
Lin, C. C., Yin, M. C., Hsu, C. C. & Lin, M. P. Effect of five cysteine-containing compounds on three lipogenic enzymes in Balb/cA mice consuming a high saturated fat diet. Lipids 39, 843–848 (2004).
Chaves, F. J. et al. Inadequate cytoplasmic antioxidant enzymes response contributes to the oxidative stress in human hypertension. Am. J. Hypertens. 20, 62–69 (2007).
Hassannia, B., Van Coillie, S. & Vanden Berghe, T. Ferroptosis: biological rust of lipid membranes. Antioxid. Redox Signal. 35, 487–509 (2021).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
He, S. et al. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J. Cachexia Sarcopenia Muscle 13, 1717–1730 (2022).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432.e9 (2019).
Dixon, S. J. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Kuhn, H., Banthiya, S. & van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 1851, 308–330 (2015).
Wenzel, S. E. et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641.e26 (2017).
D’Oria, R. et al. The role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid. Med. Cell. Longev. 2020, 5732956 (2020).
Esterbauer, H., Schaur, R. J. & Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 (1991).
Romuk, E. et al. Malondialdehyde and uric acid as predictors of adverse outcome in patients with chronic heart failure. Oxid. Med. Cell Longev. 2019, 9246138 (2019).
Radovanovic, S. et al. Markers of oxidative damage and antioxidant enzyme activities as predictors of morbidity and mortality in patients with chronic heart failure. J. Card. Fail. 18, 493–501 (2012).
Walter, M. F. et al. Serum levels of thiobarbituric acid reactive substances predict cardiovascular events in patients with stable coronary artery disease: a longitudinal analysis of the PREVENT study. J. Am. Coll. Cardiol. 44, 1996–2002 (2004).
Gianazza, E. et al. Lipid peroxidation in atherosclerotic cardiovascular diseases. Antioxid. Redox Signal. 34, 49–98 (2021).
Li, W. et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J. Clin. Invest. 129, 2293–2304 (2019). This study shows that neutrophil recruitment and other inflammatory responses that occur after heart transplantation are initiated through ferroptotic cell death.
Wang, X. et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm. Sin. B 12, 708–722 (2022).
Richardson, D. R. et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl Acad. Sci. USA 107, 10775–10782 (2010).
Shaw, G. C. et al. Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 (2006).
Corsi, B. et al. Human mitochondrial ferritin expressed in HeLa cells incorporates iron and affects cellular iron metabolism. J. Biol. Chem. 277, 22430–22437 (2002).
Ichikawa, Y. et al. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl Acad. Sci. USA 109, 4152–4157 (2012).
Pearson, S. A. & Cowan, J. A. Evolution of the human mitochondrial ABCB7 [2Fe-2S](GS)(4) cluster exporter and the molecular mechanism of an E433K disease-causing mutation. Arch. Biochem. Biophys. 697, 108661 (2021).
Bayeva, M. et al. ATP-binding cassette B10 regulates early steps of heme synthesis. Circ. Res. 113, 279–287 (2013).
Shum, M. et al. ABCB10 exports mitochondrial biliverdin, driving metabolic maladaptation in obesity. Sci. Transl Med. 13, eabd1869 (2021).
Poulos, T. L. Heme enzyme structure and function. Chem. Rev. 114, 3919–3962 (2014).
Stojanovski, B. M. et al. 5-Aminolevulinate synthase catalysis: the catcher in heme biosynthesis. Mol. Genet. Metab. 128, 178–189 (2019).
Chiabrando, D. et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122, 4569–4579 (2012).
Maines, M. D. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 2, 2557–2568 (1988).
Vinchi, F. et al. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 146, 1325–1338 (2014).
Duffy, S. P. et al. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell Biol. 30, 5318–5324 (2010).
Chambers, I. G., Willoughby, M. M., Hamza, I. & Reddi, A. R. One ring to bring them all and in the darkness bind them: the trafficking of heme without deliverers. Biochim. Biophys. Acta Mol. Cell Res. 1868, 118881 (2021).
Balaban, R. S., Nemoto, S. & Finkel, T. Mitochondria, oxidants, and aging. Cell 120, 483–495 (2005).
Krainz, T. et al. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent. Sci. 2, 653–659 (2016).
Gaschler, M. M. et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020 (2018).
Gao, M. et al. Role of mitochondria in ferroptosis. Mol. Cell 73, 354–363.e3 (2019).
Maiorino, M., Conrad, M. & Ursini, F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid. Redox Signal. 29, 61–74 (2018).
Liang, H. et al. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 284, 30836–30844 (2009).
Schneider, M. et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 23, 3233–3242 (2009).
Imai, H. et al. Depletion of selenoprotein GPx4 in spermatocytes causes male infertility in mice. J. Biol. Chem. 284, 32522–32532 (2009).
Wang, P. et al. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 12, 447 (2021).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021). This study describes a DHODH-mediated ferroptosis defence mechanism in mitochondria, which operates in parallel to GPX4 or FSP1.
Loffler, M., Jockel, J., Schuster, G. & Becker, C. Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell Biochem. 174, 125–129 (1997).
Wang, Y. et al. SLC25A39 is necessary for mitochondrial glutathione import in mammalian cells. Nature 599, 136–140 (2021).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019). This study identifies FSP1 as a previously unrecognized anti-ferroptotic protein, which acts independently of GPX4 to suppress ferroptosis on the plasma membrane.
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Miriyala, S. et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 91, 68–80 (2016).
Imai, H. et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 305, 278–286 (2003).
Yant, L. J. et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 34, 496–502 (2003).
Yoo, S. E. et al. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic. Biol. Med. 52, 1820–1827 (2012).
Wortmann, M. et al. Combined deficiency in glutathione peroxidase 4 and vitamin E causes multiorgan thrombus formation and early death in mice. Circ. Res. 113, 408–417 (2013). This is a pioneering study on a GPX4-dependent antioxidant pathway that regulates endothelial cell death and thrombus formation.
Dabkowski, E. R., Williamson, C. L. & Hollander, J. M. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic. Biol. Med. 45, 855–865 (2008).
Linkermann, A. et al. Synchronized renal tubular cell death involves ferroptosis. Proc. Natl Acad. Sci. USA 111, 16836–16841 (2014). This study showed that ferroptosis is of functional relevance in a mouse model of acute kidney injury, representing the first in vivo evidence of ferroptosis.
Conrad, M. & Proneth, B. Broken hearts: iron overload, ferroptosis and cardiomyopathy. Cell Res. 29, 263–264 (2019).
Hampton, T. New target to protect against cardiomyopathy. Circulation 139, 2278–2279 (2019).
Kobashigawa, J. et al. Report from a consensus conference on primary graft dysfunction after cardiac transplantation. J. Heart Lung Transpl. 33, 327–340 (2014).
Jang, S. et al. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 45, 102021 (2021).
Sparvero, L. J. et al. Direct mapping of phospholipid ferroptotic death signals in cells and tissues by gas cluster ion beam secondary ion mass spectrometry (GCIB-SIMS). Angew. Chem. Int. Ed. 60, 11784–11788 (2021).
Tadokoro, T. et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747 (2020).
Bernuzzi, F., Recalcati, S., Alberghini, A. & Cairo, G. Reactive oxygen species-independent apoptosis in doxorubicin-treated H9c2 cardiomyocytes: role for heme oxygenase-1 down-modulation. Chem. Biol. Interact. 177, 12–20 (2009).
Lejay, A. et al. Ischemia reperfusion injury, ischemic conditioning and diabetes mellitus. J. Mol. Cell Cardiol. 91, 11–22 (2016).
Wang, C. et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell Mol. Med. 24, 6670–6679 (2020).
Li, W., Li, W., Leng, Y., Xiong, Y. & Xia, Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 39, 210–225 (2020).
Dillmann, W. H. Diabetic cardiomyopathy. Circ. Res. 124, 1160–1162 (2019).
Martin, L. et al. The septic heart: current understanding of molecular mechanisms and clinical implications. Chest 155, 427–437 (2019).
Wang, C. et al. Dexmedetomidine alleviated sepsisinduced myocardial ferroptosis and septic heart injury. Mol. Med. Rep. 22, 175–184 (2020).
Li, N. et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic. Biol. Med. 160, 303–318 (2020).
Chen, X., Xu, S., Zhao, C. & Liu, B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem. Biophys. Res. Commun. 516, 37–43 (2019).
Liu, B. et al. Puerarin protects against heart failure induced by pressure overload through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 497, 233–240 (2018).
Wang, J. et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 11, 574 (2020).
Hynes, B. J. et al. Atrial fibrillation in patients with heart failure. Curr. Opin. Cardiol. 18, 32–38 (2003).
Dai, C. et al. Inhibition of ferroptosis reduces susceptibility to frequent excessive alcohol consumption-induced atrial fibrillation. Toxicology 465, 153055 (2022).
Riegman, M., Bradbury, M. S. & Overholtzer, M. Population dynamics in cell death: mechanisms of propagation. Trends Cancer 5, 558–568 (2019).
Gladwin, M. T. Cardiovascular complications and risk of death in sickle-cell disease. Lancet 387, 2565–2574 (2016).
Menon, A. V. et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood 139, 936–941 (2022).
Lin, P., Wang, M., Wei, Y., Kim, T. & Wei, X. Coronavirus in human diseases: mechanisms and advances in clinical treatment. MedComm 1, 270–301 (2020).
Wang, X. et al. Comorbid chronic diseases and acute organ injuries are strongly correlated with disease severity and mortality among COVID-19 patients: a systemic review and meta-analysis. Research 2020, 2402961 (2020).
Zhao, K. et al. Serum iron level as a potential predictor of coronavirus disease 2019 severity and mortality: a retrospective study. Open Forum Infect. Dis. 7, ofaa250 (2020).
Yang, M. & Lai, C. L. SARS-CoV-2 infection: can ferroptosis be a potential treatment target for multiple organ involvement? Cell Death Discov. 6, 130 (2020).
Han, Y. et al. SARS-CoV-2 infection induces ferroptosis of sinoatrial node pacemaker cells. Circ. Res. 130, 963–977 (2022).
Bai, T., Li, M., Liu, Y., Qiao, Z. & Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 160, 92–102 (2020).
Devisscher, L. et al. Discovery of novel, drug-like ferroptosis inhibitors with in vivo efficacy. J. Med. Chem. 61, 10126–10140 (2018).
Van Coillie, S. et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat. Commun. 13, 1046 (2022).
Feng, Y., Madungwe, N. B., Imam Aliagan, A. D., Tombo, N. & Bopassa, J. C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 520, 606–611 (2019).
Wang, N. et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J. Mol. Cell Cardiol. 150, 65–76 (2021).
Dikalova, A. E. et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 107, 106–116 (2010).
Zhang, T. et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 22, 175–182 (2016).
Tonnus, W. et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 12, 4402 (2021).
Gammella, E., Maccarinelli, F., Buratti, P., Recalcati, S. & Cairo, G. The role of iron in anthracycline cardiotoxicity. Front. Pharmacol. 5, 25 (2014).
Simunek, T. et al. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 61, 154–171 (2009).
Tang, L. J. et al. Ferroptosis occurs in phase of reperfusion but not ischemia in rat heart following ischemia or ischemia/reperfusion. Naunyn Schmiedebergs Arch. Pharmacol. 394, 401–410 (2021).
Dludla, P. V. et al. A systematic review on the protective effect of N-acetyl cysteine against diabetes-associated cardiovascular complications. Am. J. Cardiovasc. Drugs 18, 283–298 (2018).
Bartekova, M., Barancik, M., Ferenczyova, K. & Dhalla, N. S. Beneficial effects of N-acetylcysteine and N-mercaptopropionylglycine on ischemia reperfusion injury in the heart. Curr. Med. Chem. 25, 355–366 (2018).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Tang, L. J. et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TfR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic. Biol. Med. 162, 339–352 (2021).
Ning, D. et al. Atorvastatin treatment ameliorates cardiac function and remodeling induced by isoproterenol attack through mitigation of ferroptosis. Biochem. Biophys. Res. Commun. 574, 39–47 (2021).
Mishima, E. et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J. Am. Soc. Nephrol. 31, 280–296 (2020).
Conlon, M. et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat. Chem. Biol. 17, 665–674 (2021).
Oettl, K. et al. Radical-scavenging and iron-chelating properties of carvedilol, an antihypertensive drug with antioxidative activity. Biochem. Pharmacol. 62, 241–248 (2001).
Acknowledgements
The authors receive support from the National Natural Science Foundation of China (82170644 and 31900835 to X.F., 31970689 to J.M. and 31930057 to F.W.), the National Key R&D Program (2018YFA0507801 to J.M. and 2018YFA0507802 to F.W.), the Zhejiang Provincial Natural Science Foundation of China (Y22C115257 to X.F.), the NIH (grants NHLBI HL127646, HL140973, R01 HL140927 and HL138982 to H.A.) and the Leducq Foundation (to H.A.). The authors thank L. Luo (Guangdong Medical University, China) for helping to draft the figures before submission. The authors thank X. Bi, X. Yang and Y. Yu (Zhejiang University School of Medicine, China) for their contribution to the discussion of this Review and apologize to their colleagues whose valuable contributions are not cited in this manuscript owing to space limitations.
Author information
Authors and Affiliations
Contributions
All the authors contributed substantially to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Cardiology thanks Sabzali Javadov, Andreas Linkermann and Eikan Mishima for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fang, X., Ardehali, H., Min, J. et al. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol 20, 7–23 (2023). https://doi.org/10.1038/s41569-022-00735-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-022-00735-4
This article is cited by
-
Relationship between trajectories of dietary iron intake and risk of type 2 diabetes mellitus: evidence from a prospective cohort study
Nutrition Journal (2024)
-
Cordycepin alleviates osteoarthritis by inhibiting chondrocyte ferroptosis via Keap1/Nrf2 axis
Future Journal of Pharmaceutical Sciences (2024)
-
Inhibition of ferroptosis reverses heart failure with preserved ejection fraction in mice
Journal of Translational Medicine (2024)
-
Selenoprotein P expression in glioblastoma as a regulator of ferroptosis sensitivity: preservation of GPX4 via the cycling-selenium storage
Scientific Reports (2024)
-
Calycosin Induces Ferroptosis by SLC7A11 Through the PI3K/Akt Pathway in Acute Myelocytic Leukemia
Revista Brasileira de Farmacognosia (2024)
