
Abstract
The de novo design of antimicrobial therapeutics involves the exploration of a vast chemical repertoire to find compounds with broad-spectrum potency and low toxicity. Here, we report an efficient computational method for the generation of antimicrobials with desired attributes. The method leverages guidance from classifiers trained on an informative latent space of molecules modelled using a deep generative autoencoder, and screens the generated molecules using deep-learning classifiers as well as physicochemical features derived from high-throughput molecular dynamics simulations. Within 48 days, we identified, synthesized and experimentally tested 20 candidate antimicrobial peptides, of which two displayed high potency against diverse Gram-positive and Gram-negative pathogens (including multidrug-resistant Klebsiella pneumoniae) and a low propensity to induce drug resistance in Escherichia coli. Both peptides have low toxicity, as validated in vitro and in mice. We also show using live-cell confocal imaging that the bactericidal mode of action of the peptides involves the formation of membrane pores. The combination of deep learning and molecular dynamics may accelerate the discovery of potent and selective broad-spectrum antimicrobials.
Similar content being viewed by others


Main
De novo therapeutic molecule design remains a cost and time intensive process, typically requiring more than 10 years and US$2–3 billion for a new drug to reach the market, and the success rate could be as low as <1% (refs. 1,2). Efficient computational strategies for the targeted generation and screening of molecules with desired therapeutic properties are therefore urgently required. As a specific example, here we consider the antimicrobial peptide (AMP) design problem. AMPs are drug candidates for tackling antibiotic resistance, one of the biggest threats in global health, food security and development. Patients who are at a higher risk from drug-resistant pathogens are also more vulnerable to illness from viral lung infections, such as influenza, severe acute respiratory syndrome and coronavirus disease 2019. Drug-resistant diseases claim 700,000 lives each year globally3, which is expected to rise to 10 million deaths per year by 2050 on the basis of the present trends4. Multidrug-resistant Gram-negative bacteria are of particular concern5. AMPs that are used as the antibiotic of last resort are typically 12–50 amino acids long and produced by multiple higher-order organisms to combat invading microorganisms. Owing to their exceptional structural and functional variety6, promising activity and low tendency to induce (or even reduce) resistance, natural AMPs have been proposed as promising alternatives to traditional antibiotics and as potential next-generation antimicrobial agents7. Most reported antimicrobials are cationic and amphiphilic in nature and possess properties that are thought to be crucial for insertion into and disruption of the bacterial membrane7.
Rational methods for new therapeutic peptide design, both in the wet laboratory and in silico, rely heavily on structure–activity relationship studies8,9,10,11,12,13. Such methods struggle with the prohibitively large molecular space, complex structure–function relationships and multiple competing constraints, such as activity, toxicity, synthesis cost and stability associated with the design task. Artificial intelligence (AI) methods, in particular, statistical learning and optimization-based approaches have shown promise in designing small molecules and macromolecules, including AMPs. A comprehensive review of computational methodologies for AMP design can be found in ref. 14. A conventional approach is to build a predictive model that estimates the properties of a given molecule, which is then used for candidate screening15,16,17,18,19,20,21,22. Either a manually selected or automatically learned set of compositional, structural or physicochemical features, or a direct sequence is used to build the predictive model. A candidate is typically obtained by combinatorial enumeration of chemically plausible fragments (or subsequences) followed by random selection, from an existing molecular library, or modification thereof. Sequence optimization using genetic algorithms23,24, pattern insertion25 or sub-graph matching26 has also been used in the development of new drugs, requiring selection of initial (or template) sequence and/or defining patterns. An alternative is to develop a generative model for automated de novo design of unique molecules with user-specified properties. Deep-learning-based architectures, such as neural language models as well as deep generative neural networks, have emerged as a popular choice27,28,29,30,31,32,33,34,35. Probabilistic autoencoders36,37, a powerful class of deep generative models that learn a bidirectional mapping of the input molecules (and their attributes) to a continuous latent space, have been used for this design task.
Earlier deep generative models for targeted generation have often limited the learning to a fixed library of molecules with desired attributes to restrict the exhaustive search to a defined section of the chemical space23. Such an approach can affect the novelty as well as the validity of the generated molecules, as the fixed library represents a small portion of the combinatorial molecular space. Alternative methods include Bayesian optimization (BO) on a learned latent space, reinforcement learning38,39 (RL) or semi-supervised learning40 (SS). However, those approaches require surrogate model fitting (as in BO), optimal policy learning (as in RL) or minimizing attribute-specific loss objectives (as in SS), which suffers from additional computational complexity. As a result, controlling attribute(s) of designed molecules efficiently remains a non-trivial task.
In this Article, to address these challenges, we propose a computational framework for the targeted design and screening of molecules, which combines attribute-controlled deep generative models and physics-driven simulations. For targeted generation, we propose Conditional Latent (attribute) Space Sampling (CLaSS), which leverages guidance from attribute classifier(s) trained on the latent space of the system of interest and uses a rejection sampling scheme for generating molecules with the desired attributes. CLaSS has several advantages, as it is efficient and easily repurposable in contrast to existing machine learning algorithms for targeted generation. To encourage novelty and validity of designed sequences, we performed CLaSS on the latent space of a deep generative autoencoder that was trained on a larger dataset consisting of all known peptide sequences, instead of a limited number of known antimicrobial agents. Extensive analyses showed that the resulting latent space is informative of peptide properties. As a result, the AMPs generated from this informative space are unique, diverse, valid and optimized.
Although several AMPs are in clinical trials7, the future design of new AMP therapeutics requires minimizing the high production cost due to longer sequence length, proteolytic degradation, poor solubility and off-target toxicity. A rational path for resolving these problems is to design short peptides as a minimal physical model41,42 that captures the high selectivity of natural AMPs. That is, maximizing antimicrobial activity, while minimizing toxicity to the host.
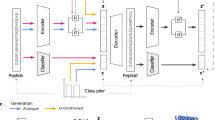
To account for these additional key requirements, such as broad-spectrum potency and low toxicity, we provide an efficient in silico screening method that uses deep-learning classifiers augmented with high-throughput physics-driven molecular simulations (Fig. 1). This computational approach for de novo antimicrobial design explicitly accounts for broad-spectrum potency and low toxicity, and we performed experimental verification of these properties. The synthesis of 20 candidate sequences (from a pool of nearly 90,000 generated sequences) that passed the screening enabled the discovery of two new and short-length peptides (YI12 and FK13) with experimentally validated strong antimicrobial activity against diverse pathogens, including a difficult-to-treat multidrug-resistant Gram-negative K. pneumoniae strain. Importantly, both sequences demonstrated low in vitro haemolysis (HC50) and in vivo lethal (LD50) toxicity. Circular dichroism (CD) experiments revealed the amphiphilic helical topology of the two new cationic AMPs. All-atom simulations of helical YI12 and FK13 show distinct modes of early lipid membrane interaction. Wet-laboratory experiments confirmed their bactericidal nature, while live-cell imaging using confocal microscopy showed bacterial membrane permeability. Both peptides displayed a low propensity to induce resistance onset in E. coli compared with imipenem, an existing antibiotic. No cross-resistance was observed for either YI12 or FK13 when tested using a polymyxin-resistant strain. Taken together, both YI12 and FK13 seem to be promising therapeutic candidates that deserve further investigation. Thus, the present strategy provides an efficient de novo approach for discovering new, broad-spectrum and low-toxic antimicrobials with therapeutic potential at 10% success rate and at a rapid (48 d) pace.

Overview and timeline of the proposed AI-driven approach for accelerated antimicrobial sequence (seq.) design.
Results
Peptide autoencoder
To model the peptide latent space, we used generative models based on a deep autoencoder36,37 composed of two neural networks, an encoder and a decoder. The encoder qϕ(z|x) parameterized with ϕ learns to map the input x to a variational distribution, and the decoder pθ(x|z) parameterized with θ aims to reconstruct the input x given the latent vector z from the learned distribution, as illustrated in Fig. 2a. Variational autoencoder (VAE), the most popular model in this family37, assumes latent variable z ~ p(z) and follows a simple prior (for example, Gaussian) distribution. And the decoder then produces a distribution over sequences given the continuous representation z. Thus, the generative process is specified as \(p\left( {\mathbf{x}} \right) = {\int} p \left( {\mathbf{z}} \right)p_\theta \left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{z}}} \right){\mathrm{d}}{\mathbf{z}}\) where we integrate out the latent variable. An alternative (and supposedly improved variant) of standard VAE is the Wasserstein Autoencoder (WAE; Supplementary Information). Within the VAE/WAE framework, the peptide generation is formulated as a density modelling problem, that is, estimating p(x) where x are short variable-length strings of amino acids. The density estimation procedure has to assign a high likelihood to known peptides. Thus, the model generalization implies that plausible new peptides can be generated from regions with a high probability density under the model. Peptide sequences are presented as text strings composed of 20 natural amino acid characters. Only sequences with a length of ≤25 were considered for model training and generation, as short AMPs are desired.

a, Training a generative autoencoder (AE) model on peptide sequences (Fig. 1, autoencoder training). b, Mapping sparse peptide attributes to the model’s latent z space and constructing the density model of the z space (Fig. 1, autoencoder evaluation). c, Sampling from the z space using our CLaSS method (Fig. 1, controlled generation).
However, instead of training a model over only known AMP sequences, one can train a model over all peptide sequences reported in the UniProt database43—an extensive database of protein/peptide sequences that may or may not have an annotation. For example, the number of annotated AMP sequences is approximately equal to 9,000, and the number of peptide sequences in UniProt is nearly 1.7 million, when a sequence length of up to 50 is considered. Here we therefore trained a density model over known peptide sequences. The fact that unsupervised representation learning by pretraining on a large corpus has recently led to impressive results for downstream tasks in text and speech44,45,46,47 as well as in protein biology48,49 also inspired this approach. Furthermore, in contrast to similar models for protein sequence generation50, we do not restrict ourselves to learning the density associated with a single protein family or a specific three-dimensional fold. Instead, we trained a global model over all known short peptide sequences expressed in different organisms. This global approach should enable meaningful density modelling across multiple families, the interpolation between them, better learning of the ‘grammar’ of plausible peptides and exploration beyond known antimicrobial templates, as shown next.
The advantage of training a WAE (instead of a plain VAE) on peptide sequences is evident from the reported evaluation metrics in Supplementary Table 1. We also observed a high reconstruction accuracy and diversity of generated sequences when the WAE was trained on all of the peptide sequences, instead of on only AMP sequences (Supplementary Table 1). We next analysed the information content of the peptide WAE, inspired by recent investigations in natural language processing. Using the ‘probing’ methods, it has been shown that encoded sentences can retain much linguistic information51. In a similar vein, we investigated whether the similarity (we used pairwise similarity, which is defined by global alignment/concordance between two sequences) between sequences52 is captured by their encoding in the latent z space, as such information is known to specify the biological function and fold of peptide sequences. Figure 3a reveals a negative correlation (Pearson correlation coefficient = −0.63) between sequence similarities and Euclidean distances in the z space of the WAE model, suggesting that WAE intrinsically captures the sequence relationship within the peptide space. The VAE latent space fails to capture such a relation.

a, The relationship between sequence similarity and Euclidean distance in latent z space when sequences were modelled using WAE. Inset: VAE. The darker points indicate similarity with itself (that is, the same exact sequence). Classifiers were trained either using WAE z-space encodings (z-) or on sequences (sequence level). Note that the class prediction accuracy values of the AMP attribute using WAE z classifiers and sequence classifiers on test data are 87.4% and 88.0%, respectively, and the corresponding values for the toxicity attribute are 68.9% and 93.7%, respectively. b, Example of a table generated using https://peptide-walk.mybluemix.net. It shows decoded sequences and their attributes during a linear interpolation (interpol) between two distant sequences in the WAE latent space. Attributes include (1) physicochemical properties, (2) sequence similarity (evo_start, evo_end) from endpoint sequences, and (3) AMP (z_amp) and toxic (z_tox) class probabilities from z classifiers. The values in red and blue are in the upper and lower quartiles, respectively. The grey rectangle indicates sequences with a low attribute similarity to endpoint sequences. The colours in the ‘peptide’ column indicate if an amino acid was added (blue letter), swapped (violet letter) or removed (red line) with respect to the previous row. The ‘mol’ column indicates that a molecular rendering is available and can be selected by clicking on it (which leads to a yellow highlight). Aro, aromaticity; Chrg, charge; HMom, hydrophobic moment; Inst, instability; MolW, molecular mass. c, As an example of further analysis, we show the relationship between AMP class probability and the instability index for all candidates in the interpolation, and a ball–stick rendering of a selected sequence in the path.
With the end-goal of a conditional generation of new peptide sequences, it is crucial to ensure that the learned encoding in the z space retains identifiable information about functional attributes of the original sequence. Specifically, we investigated whether the space is linearly separable into different attributes, such that sampling from a specific region of that space yields consistent and controlled generations. For this purpose, we trained linear classifiers for binary (yes/no) functional attribute prediction using the z encodings of sequences (Fig. 2b). Probing the z space modelled by the WAE uncovers that the space is indeed linearly separable into different functional attributes, as evident from the test accuracy of binary logistic classifiers on test data. The class prediction accuracy values of the attribute ‘AMP’ using WAE z classifiers and sequence-level classifiers on test data are 87.4% and 88.0%, respectively (Supplementary Table 2). Supplementary Table 3 shows the reported accuracy of several existing AMP classification methods, as well as of our sequence-level long short-term memory (LSTM) classifier. The reported accuracy varies widely from 66.8% for iAMPPred18 to 79% for DBAASP-SP53, which relies on local-density-based sampling using physicochemical features, to 94% for the method described in ref. 20, which uses a convolutional neural net trained directly on a large (similar to ours) corpus of peptide sequences. Our sequence-level LSTM model shows a comparable 88% accuracy. This comparison reveals a close performance of the z-level classifier compared to classifiers reported in literature16,18,19,20,21,22 or trained in-house that have access to the original sequences. We emphasize that the goal of this study is not to provide an AMP prediction method that outperforms existing machine-learning-based AMP classifiers. Rather, the goal is to have a predictor trained on latent features resulting in comparable accuracy, which can be used to automatically generate new AMP candidates by conditional sampling directly from the latent space using CLaSS. Note that comparing different AMP prediction models is non-trivial, as different methods vary widely by training AMP dataset size (for example, 712 for AMP Scanner v2 (ref. 22), 3,417 for iAMPpred18, 2,578 for CAMP19, 140 for DBAASP-SP prediction16, 1,486 for iAMP-2L21, 4,050 for ref. 20 and 6,482 in the present study), sequence length, different definition of AMP and non-AMP, and other data curation criteria. Furthermore, both the z-level and sequence-level AMP classifiers used here do not require any manually defined set of features, in contrast to many existing prediction tools16,21.
For the toxicity classification task, a much lower accuracy was found using models trained on latent features compared with similar sequence-level deep classifiers54 (Fig. 3 and Supplementary Table 2) that report accuracy values as high as 90%. These results imply that some attributes, such as toxicity, are more challenging to predict from the learned latent peptide representation; one possible reason could be that there is a higher class imbalance in training data (Supplementary Information).
We also investigated the smoothness of the latent space by analysing the sequences generated along a linear interpolation vector in the z space between two distant training sequences (Fig. 3b,c). Sequence similarity, functional attributes (AMP and toxic class probabilities), as well as several physicochemical properties, including aromaticity, charge and hydrophobic moment (indicating amphiphilicity of a helix) change smoothly during the interpolation. These results are encouraging, as the WAE latent space that was trained on the much larger amount of unlabelled data appears to carry significant structure in terms of functional, physicochemical and sequence similarity. Figure 3c also demonstrates that it is possible to identify sequence(s) during linear interpolation that are visibly different from both endpoint sequences, indicating that the trained latent space has the potential to generate new and unique sequences.
CLaSS for controlled sequence generation
For controlled generation, we aimed to control a set of binary (yes/no) attributes of interest, such as antimicrobial function and/or toxicity. We propose CLaSS for this purpose. CLaSS leverages attribute classifiers directly trained on the peptide z space, as those can capture important attribute information (Fig. 3). The goal is to sample conditionally p(x|at) for a specified target attribute combination at. This task was approached using CLaSS (Fig. 2c), which makes the assumption that attribute conditional density factors as follows: \(p\left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{a}}} \right) = {\int} {{\mathrm{d}}z} \,p\left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{a}}} \right)p\left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{z}}} \right)\). We sampled p(z|at) approximately using rejection sampling from models in the latent z space appealing to Bayes rule and p(at|z) modelled by the attribute classifiers (Fig. 2b,c and Supplementary Information). As CLaSS uses only simple attribute predictor models and rejection sampling from models of z space, it is a simple and efficient forwards-only screening method. It does not require any complex optimization over latent space compared with existing methods for controlled generation, such as BO30,55, RL38,39 or SS generative models40. CLaSS is easily repurposable and highly parallelizable at the same time and does not need a starting point in the latent space to be defined.
As the toxicity classifier trained on latent features appears to be weaker (Fig. 3), antimicrobial function (yes/no) was used as the sole condition for controlling the sampling from the latent peptide space. Generated antimicrobial candidates were then screened for toxicity using the sequence-level classifier during post-generation filtering. Notably, CLaSS does not perform a heuristic-based search (that is, genetic algorithm or node-based sampling, as reported in literature55) on the latent space, rather, it relies on a probabilistic rejection-sampling-based scheme for attribute-conditioned generation. CLaSS is also different from the local-density-based sampling approaches16, as those methods rely on clustering the labelled data and then finding the cluster assignment of the test sample by using similarity search; it is therefore suited for a forward design task. By contrast, CLaSS is formulated for the inverse design problem, and enables targeted generation by attribute-conditioned sampling from the latent space followed by decoding (Methods).
Features of CLaSS-generated AMPs
To check the homology of CLaSS-generated AMP sequences with training data, we performed a BLAST sequence similarity search. We used the expect value (E value) for the alignment score of the actual sequences to assess sequence homology, while using other alignment metrics, such as raw alignment scores, percentage identity and positive matches, gaps and coverage in alignment, to obtain an overall sense of sequence similarity. An E value indicates statistical (also known as biological) significance of the match between the query and sequences from a database of a particular size. A larger E value indicates that there is a higher chance that the similarity between the hit and the query is merely a coincidence—that is, the query is not homologous or related to the hit. We analysed the E value for the matches with the highest alignment score. Typically, when querying the UniProt non-redundant database, which contains around 220 million sequences, E values of ≤0.001 are used to infer homology56. As our training database is nearly 1,000 times smaller than UniProt, an E value of ≤10−6 can be used to indicate homology. As shown in Supplementary Table 4, about 14% of generated sequences show E values of ≥10, and another 36% have E values of >1 when considering the match with the highest alignment score, indicating that there is non-significant similarity between generated and training sequences. If only the alignments with score of >20 are considered, the average E value is found to be >2, further implying the non-homologous nature of the generated sequences. Similar criteria have also been used to detect the novelty of designed short antimicrobials8. CLaSS-generated AMPs are also more diverse, as the unique (that is, found only once in an ensemble of sequences) k-mers (k = 3–6) are more abundant compared with training sequences or their fragments (Supplementary Fig. 1). These results highlight the ability of the present approach to generate short-length AMP sequences that are, on average, unique with respect to training data, as well as diverse among themselves.
Distributions of key molecular features implicated in antimicrobial nature, such as amino acid composition, charge, hydrophobicity (H) and hydrophobic moment (μH), were compared between the training and generated AMPs, as shown in Fig. 4a–d. Additional features are reported in Supplementary Fig. 1. CLaSS-generated AMP sequences show distinct character: specifically, these sequences are richer in Arg, Leu, Ser, Gln and Cys, whereas Ala, Gly, Asp, His, Asn and Trp content is reduced, in comparison to training antimicrobial sequences (Fig. 4a). We also present the most abundant k-mers (k = 3, 4) in Supplementary Fig. 1, suggesting that the most frequent 3- and 4-mers are Lys- and Leu-rich in both generated and training AMPs, the frequency being higher in generated sequences. Generated AMPs are characterized by global net-positive charge and aromaticity somewhere between unlabelled and AMP-labelled training sequences, while the hydrophobic moment is comparable to that of known AMPs (Fig. 4b–d and Supplementary Fig. 1). These trends imply that the generated antimicrobials are still cationic and can form a putative amphiphilic α-helix, similar to the majority of known antimicrobials. Interestingly, they also exhibit a moderately higher hydrophobic ratio and aliphatic index compared with training sequences (Supplementary Fig. 1). These observations highlight the distinct physicochemical nature of the CLaSS-generated AMP sequences, as a result of the SS nature of our learning paradigm, which might help in their therapeutic application. For example, a lower aromaticity and higher aliphatic index are known to induce better oxidation susceptibility and higher heat stability in short peptides57, while lower hydrophobicity is associated with reduced toxicity58.

a–d, Comparison of amino acid composition (a), global hydrophobicity (b), hydrophobic moment (c) and charge distribution (d) of CLaSS-generated AMPs with training sequences. For a–c, data are mean ± s.d., estimated on three different sets, each consisting of 3,000 randomly chosen samples. The bars show generated AMP (Gen-amp, orange), training AMP (Train-amp, blue) and training unlabelled (Train-unlab, grey).
In silico post-generation screening
To screen the approximately 90,000 CLaSS-generated AMP sequences, we first used an independent set of binary (yes/no) sequence-level deep-neural-net-based classifiers that screens for antimicrobial function, broad-spectrum efficacy, presence of secondary structure and toxicity (Fig. 1 and Supplementary Table 2). The 163 candidates that passed this screening were then processed for coarse-grained molecular dynamics simulations of peptide–membrane interactions. The computational efficiency of these simulations makes them an attractive choice for high-throughput and physics-inspired filtering of peptide sequences.
As there exists no standardized protocol for screening antimicrobial candidates using molecular simulations, we performed a set of control simulations of known sequences with or without antimicrobial activity. From those control runs, we found that the variance in the number of contacts between positive residues and membrane lipids is predictive of antimicrobial activity (Supplementary Fig. 2). Specifically, the contact variance differentiates between high-potency AMPs and non-antimicrobial sequences with a sensitivity of 88% and a specificity of 63% (Supplementary Information). Physically, this feature can be interpreted as measuring the robust binding tendency of a peptide sequence to the model membrane. We therefore used the contact variance cut-off of 2 for further filtering of the 163 generated AMPs that passed the classifier screening.
Wet-laboratory characterization
A final set of 20 CLaSS-generated AMP sequences that passed the contact variance-based screening described above, along with their simulated and physicochemical characteristics, is reported in Supplementary Table 6. Those sequences were tested in the wet laboratory for antimicrobial activity, as measured using minimum inhibitory concentration (MIC; lower is better) against Gram-positive Staphylococcus aureus and Gram-negative E. coli (Supplementary Table 7). Eleven generated non-AMP sequences were also screened for antimicrobial activity (Supplementary Table 8). None of the designed non-AMP sequences showed MIC values that were low enough to be considered to be antimicrobials, implying that our approach is not prone to false-negative predictions. We also speculate that a domain shift between the AMP test set and generated sequences from the latent space trained on the labelled and unlabelled peptide sequences probably results in a false-positive prediction (18 out of 20).
Among the 20 AI-designed AMP candidates, two sequences, YLRLIRYMAKMI-CONH2 (YI12, 12 amino acids) and FPLTWLKWWKWKK-CONH2 (FK13, 13 amino acids), were identified to be the best with the lowest MIC values (Table 1 and Supplementary Table 7). Both peptides are positively charged and have a non-zero hydrophobic moment (Supplementary Table 6), indicating that their cationic and amphiphilic nature is consistent with known antimicrobials. These peptides were further evaluated against the more difficult-to-treat Gram-negative Pseudomonas aeruginosa and Acinetobacter baumannii, as well as against a multidrug-resistant strain of Gram-negative K. pnuemoniae. As shown in Fig. 5, both YI12 and FK13 showed potent broad-spectrum antimicrobial activity with comparable MIC values. We compared the MIC values of YI12 and FK13 with LLKKLLKKLLKK, which is an existing α-helix-forming AMP with excellent antimicrobial activity and selectivity59. The MIC values of LLKKLLKKLLKK against S. aureus, E. coli and P. aeruginosa are >500, >500 and 63, respectively. The MIC values of FK13 and FY12 are comparable to that of LLKKLLKKLLKK against P. aeruginosa. However, the MIC values of FK13 and YI12 are notably lower than those of LLKKLLKKLLKK against S. aureus and E. coli, demonstrating that the AMPs discovered in this study have a greater efficiency. We also report the results of several existing AMP prediction methods on YI12 and FK13 (Table 1). iAMPPred18 and CAMP-RF19 predict that YI12 and FK13 are both AMPs. The other methods misclassified either of the two—for example, DBAASP-SP53, which relies on local similarity search, misclassified FK13. The method described in ref. 20 predicts AMP activity against S. aureus for both sequences. The same method does not recognize that FK13 is an effective antimicrobial against E. coli.

a, Images from an all-atom simulation of YI12 (top) and FK13 (bottom). Selected residues that interact with the membrane are highlighted. b, Resistance-acquisition studies of E. coli in the presence of sub-MIC (1/2×) levels of imipenem, YI12 and FK13.
We further performed in vitro and in vivo testing for toxicity. On the basis of activity measured at 50% haemolysis (HC50) and lethal dose (LD50) toxicity values (Table 1 and Supplementary Fig. 2), both peptides seem to be biocompatible (as the HC50 and LD50 values are much higher than the MIC values); FK13 is more biocompatible than YI12. Importantly, the LD50 values of both peptides compare favourably with that of polymyxin B (20.5 mg kg−1; ref. 60), which is a clinically used antimicrobial drug for the treatment of antibiotic-resistant Gram-negative bacterial infection.
Sequence similarity analyses
To investigate the similarity of YI12 and FK13 with respect to training sequences, we analysed the alignment scores returned by the BLAST homology search in detail (Supplementary Figs. 2 and 5), in line with previous studies8,61. The scoring metrics included the raw alignment score, E value, percentage of alignment coverage, percentage of identity, percentage of positive matches or similarity, and percentage of alignment gap (indicating the presence of additional amino acids). BLAST searching with an E-value threshold of 10 against the training database did not reveal any match for YI12, suggesting that there exists no statistically significant match of YI12. We therefore searched for related sequences of YI12 in the much larger UniProt database consisting of nearly 223.5 million non-redundant sequences, only a fraction of which was included in our model training. YI12 shows an E value of 2.9 to its closest match, which is an eleven-residue segment from the bacterial EAL-domain-containing protein (Supplementary Fig. 5). This result suggests that YI12 shares low similarity, even when all protein sequences in UniProt are considered. We also performed a BLAST search of YI12 against the PATSEQ database that contains approximately 65.5 million patented peptides and still obtained a minimum E value of 1.66. The sequence nearest to YI12 from PATSEQ is an 8 amino acid segment from a 79 amino acid human protein, which has with 87.5% similarity and only 66.7% coverage, further confirming the low similarity of YI12 to known sequences.
FK13 shows less than 75% identity, a gap in the alignment and 85% query coverage compared with its closest match in the training database implying that FK13 also shares low similarity to the training sequences (Supplementary Fig. 5). However, YI12 is more unique than FK13. The closest match of FK13 in the training database is a synthetic variant of a 13 amino acid bactericidal domain (PuroA: FPVTWRWWKWWKG) of the puroindoline A protein from wheat endosperm. The antimicrobial and haemolysis activities of FK13 are close to those reported for PuroA62,63. Nevertheless, FK13 is substantially different from PuroA; FK13 is Lys rich and low in Trp content, resulting in a lower grand average of hydropathy score (−0.854 versus −0.962), a higher aliphatic index (60.0 versus 22.3) and a lower instability index (15.45 versus 58.30), which are together indicative of higher peptide stability. In fact, lower Trp content was found to be beneficial for stabilizing FK13 during wet-laboratory experiments, as Trp is susceptible to oxidation in the air. Lower Trp content has also been implicated in improving in vivo peptide stability64. Taken together, these results show that CLaSS on latent peptide space modelled by the WAE has the ability to generate unique and optimal antimicrobial sequences by efficiently learning the complicated sequence–function relationship in peptides and using that knowledge for controlled exploration. When combined with subsequent in silico screening, new and optimal lead candidates with experimentally confirmed high broad-spectrum efficacy and selectivity were identified at a success rate of 10%. The whole cycle (from database curation to wet-laboratory confirmation) took 48 d in total for a single iteration (Fig. 1).
Structural and mechanistic analyses
We performed all-atom explicit water simulations (Supplementary Information) of these two sequences in the presence of a lipid membrane starting from an α-helical structure. Different membrane-binding mechanisms were observed for the two sequences, as shown in Fig. 5a. YI12 embeds into the membrane using positively charged N-terminal Arg residues, whereas FK13 embeds either with N-terminal Phe or with C-terminal Trp and Lys residues. These results provide mechanistic insights into the different modes of action adopted by YI12 and FK13 during the early stages of membrane interaction.
Peptides were further experimentally characterized using CD spectroscopy (Supplementary Information). Both YI12 and FK13 showed a random coil-like structure in water, but formed an α-helix in 20% SDS buffer (Supplementary Fig. 2). Structure classifier predictions (Supplementary Information) and all-atom simulations were consistent with the CD results. From CD spectra, the α-helicity of YI12 seems to be stronger than that of FK13, consistent with its stronger hydrophobic moment (Supplementary Table 6). In summary, physicochemical analyses and CD spectroscopy together suggest that cationic nature and amphiphilic helical topology are the underlying factors that induce the antimicrobial nature in YI12 and FK13.
To provide insights into the mechanism of action that underlies the antimicrobial activity of YI12 and FK13, we conducted an agar plate assay and found that both peptides, YI12 and FK13, are bactericidal. There was a 99.9% reduction of colonies at 2× MIC.
As α-helical peptides such as YI12 and FK13 are known to disrupt membranes through leaky-pore formation65,66, we performed live-cell imaging using confocal fluorescence microscopy for FK13 against E. coli (Supplementary Fig. 4). The results were compared with that of polymyxin B, which is a member of a group of basic polypeptide antibiotics derived from Bacillus polymyxa. After 2 h of incubating E. coli with polymyxin B or with FK13 at 2× MIC, confocal imaging was performed. The emergence of fluorescence, as shown in Supplementary Fig. 4, confirmed that red propidium iodide (PI) had entered the bacterial cell and interacted with bacterial DNA in the presence of either FK13 or polymyxin B. This finding implies that both polymyxin and FK13 induce pore formation on the bacterial membrane and enable the PI dye to enter the bacteria. Without the pore formation, the PI dye is unable to enter the bacteria.
Resistance analyses
Finally, we performed resistance-acquisition studies of E. coli in the presence of imipenem, an intravenous β-lactam antibiotic, YI12 or FK13 at concentrations of sub-MIC levels. The results shown in Fig. 5b confirm that both YI12 and FK13 do not induce resistance after 25 passages, whereas E. coli begun to develop resistance to the antibiotic imipenem after only six passages. We also investigated the efficacy of these peptides against polymyxin-B-resistant K. pneumoniae, a strain that is resistant to polymyxin B—a beta-lactam antibiotic of last resort. Table 1 shows the MIC values of YI12, FK13 and polymyxin B, revealing no MIC increase for either of the two discovered peptides compared to the MIC against the multidrug-resistant K. pneumoniae stain (from ATCC). By contrast, the MIC of polymyxin B is 2 μg ml−1 against the same multidrug-resistant K. pneumoniae stain (from ATCC), but increases to >125 μg ml−1 once the K. pneumoniae became resistant to polymyxin B. The MIC value of YI12 and FK13 is still lower than that of polymyxin B in the polymyxin-B-resistant strain, indicating that the resistance to polymyxin B is not observed towards YI12 and FK13. Taken together, these results indicate that YI12 and FK13 hold therapeutic potential for treating resistant strains and therefore demand further investigation.
Discussion
Learning implicit interaction rules of complex molecular systems is a major goal of AI research. This direction is critical for designing new molecules/materials with specific structural and/or functional requirements, which is one of the most anticipated and acutely needed applications. The AMPs considered here represent an archetypal system for molecular discovery problems. They exhibit a near-infinite and mostly unexplored chemical repertoire, a well-defined chemical palette (natural amino acids), as well as potentially conflicting or opposing design objectives, and are of high importance owing to the global increase in antibiotic resistance and a depleted antibiotic discovery pipeline. Recent research has shown that deep learning can be used to help to screen libraries of existing chemicals for antibiotic properties67. A number of recent studies have also used AI methods to design AMPs, and provided experimental validation17,23,24,26,35,53,68. However, here we provide a fully automated computational framework that combines controllable generative modelling, deep learning and physics-driven learning for the de novo design of broad-spectrum potent and selective AMP sequences, and experimentally validate these sequences for broad-spectrum efficacy and toxicity. Furthermore, the discovered peptides show high efficacy against a strain that is resistant to a last-resort antibiotic, as well as mitigate drug-resistance onset. Wet-laboratory results confirmed the efficiency of the proposed approach for designing new and optimized sequences with a very modest number of candidate compounds synthesized and tested. The present design approach in this proof-of-concept study yielded a 10% success rate and a rapid turnaround of 48 d, highlighting the importance of combining AI-driven computational strategies with experiments to achieve more-effective drug candidates. The generative modelling approach presented here can be tuned for not only generating new candidates but also for designing unique combination therapies and antibiotic adjuvants to further advance antibiotic treatments.
As CLaSS is a generic approach, it is suitable for a variety of controlled generation tasks and can handle multiple controls simultaneously. The method is simple to implement, fast, efficient and scalable, because it does not require any optimization over the latent space. CLaSS has additional advantages regarding repurposability, as adding a new constraint requires a simple predictor training. Thus, future directions of this research will explore the effect of additional relevant constraints, such as the induced resistance, efficacy in animal models of infection and fine-grained strain specificity, on the designed AMPs using the approach presented here. Extending the application of CLaSS to other controlled molecule design tasks, such as target-specific and selective drug-like small-molecule generation is also underway69. Finally, the AI models will be further optimized in an iterative manner using the feedback from simulations and/or experiments in an active learning framework.
Methods
Generative autoencoders
To learn meaningful continuous latent representations from sequences without supervision, the VAE37 family has emerged as a principled and successful method. The data distribution p(x) over samples x is represented as the marginal of a joint distribution p(x,z) that factors out as p(z)pθ(x|z). The prior p(z) is a simple smooth distribution, whereas pθ(x|z) is the decoder that maps a point in latent z space to a distribution in x data space. The exact inference of the hidden variable z for a given input x would require integration over the full latent space: \(p\left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}} \right) = \frac{{p\left( {\mathbf{z}} \right)p_\theta \left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{z}}} \right)}}{{{\int} {\mathrm{d}} {\mathbf{z}}p\left( {\mathbf{z}} \right)p_\theta \left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{z}}} \right)}}\). To avoid this computational burden, the inference is approximated through an inference neural network or encoder qϕ(z|x). Our implementation follows ref. 70, whereby both encoder and decoder are single-layer LSTM recurrent neural networks71, and the encoder specifies a diagonal Gaussian distribution, that is, \(q_\phi \left( {{\bf{z}}{\mathrm{|}}{\bf{x}}} \right) = N\left( {{\bf{z}};\mu \left( {\bf{x}} \right),{\Sigma}\left( {\bf{x}} \right)} \right)\) (Fig. 2).
The basis for auto-encoder training is optimization of an objective consisting of the sum of a reconstruction loss and a regularization constraint loss term: \({\cal{L}}\left( {\theta ,\phi } \right) = {\cal{L}}_{{\mathrm{rec}}}\left( {\theta ,\phi } \right) + {\cal{L}}_c\left( \phi \right)\). In the standard VAE objective37, reconstruction loss \({\cal{L}}_{{\mathrm{rec}}}\left( {\theta ,\phi } \right)\) is based on the negative log-transformed likelihood of the training sample, and the constraint \({\cal{L}}_c\left( \phi \right)\) uses DKL, the Kullback–Leibler divergence
for a single sample. This exact objective is derived from a lower bound on the data likelihood; this objective is therefore called the evidence lower bound. With the standard VAE, we observed the same posterior collapse as described for natural language in the literature72, meaning q(z|x) ≈ p(z) such that no meaningful information is encoded in z space. Further extensions include β-VAE, which adds a multiplier ‘weight’ hyperparameter β onto the regularization term, and δ-VAE, which encourages the DKL term to be close to a non-zero δ to tackle the issue of posterior collapse. However, finding the right setting that serves as a workaround for the posterior collapse is tricky within these VAE variants.
Many variations within the VAE family have therefore recently been proposed, such as WAE73,74 and Adversarial Autoencoder75.
WAE factors an optimal transport plan through the encoder–decoder pair, on the constraint that the marginal posterior \(q_\phi \left( {\bf{z}} \right) = {\Bbb E}_{{\bf{x}} \sim p\left( {\bf{x}} \right)}q_\phi \left( {{\bf{z}}{\mathrm{|}}{\bf{x}}} \right)\) equals a prior distribution, that is, qϕ(z) = p(z). This is relaxed to an objective similar to \({\cal{L}}_{{\mathrm{VAE}}}\) above. However, in the WAE objective73, instead of each individual qϕ(z|x), the marginal posterior \(q_\phi \left( {\mathbf{z}} \right) = {\Bbb E}_{\mathbf{x}}\left[ {q_\phi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}} \right)} \right]\) is constrained to be close to the prior p(z). We enforce the constraint by penalizing maximum mean discrepancy76 with a random features approximation of the radial basis function77 \({\cal{L}}_c\left( \phi \right) = {\mathrm{MMD}}\left( {q_\phi \left( {\mathbf{z}} \right),p\left( {\mathbf{z}} \right)} \right)\). The total objective for WAE is \({\cal{L}} = {\cal{L}}_{{\mathrm{rec}}} + {\cal{L}}_c\) where we use the reconstruction loss \({\cal{L}}_{rec} = - {\Bbb E}_{q_\phi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}} \right)}\left[ {{\mathrm{log}}p_\theta \left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{z}}} \right)} \right]\). In WAE training with maximum mean discrepancy or with a discriminator, we found a benefit of regularizing the encoder variance as described previously74,78. For maximum mean discrepancy, we used a random features approximation of the Gaussian kernel77.
Details of autoencoder architecture and training, as well as an experimental comparison between different auto-encoder variations tested in this study are provided in Supplementary Information 3.1.1, 3.1.2 and 3.1.4. Python codes for training peptide autoencoders are available at GitHub (https://github.com/IBM/controlled-peptide-generation).
CLaSS
We propose CLaSS, which is a simple but efficient method to sample from the targeted region of the latent space from an auto-encoder that was trained in an unsupervised manner (Fig. 2).
Density modelling in latent space
We assume a latent variable model (for example, Autoencoder) that has been trained in an unsupervised manner to meet the evaluation criteria outlined in the Supplementary Information. All training data xj are then encoded in latent space: \({\mathbf{z}}_{j,k} \sim q_\phi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}_j} \right)\). These zj,k are used to fit an explicit density model Qξ(z) to approximate marginal posterior qϕ(z), and a classifier model qξ(ai|z) for attribute ai to approximate the probability p(ai|x). The motivation for fitting a Qξ(z) is to sample from Qξ rather than p(z) as, at the end of training, the discrepancy between qϕ(z) and p(z) can be substantial.
Although any explicit density estimator could be used for Qξ(z), here we consider Gaussian mixture density models and evaluate negative log-transformed likelihood on a held-out set to determine the optimal complexity. We find 100 components and untied diagonal covariance matrices to be optimal, giving a held-out log-transformed likelihood of 105.1. To fit Qξ, we used K = 10 random samples from the encoding distribution of the training data \({\mathbf{z}}_{j,k} \sim q_\phi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}_j} \right) = {\cal{N}}\left( {\mu \left( {{\mathbf{x}}_j} \right),\sigma \left( {{\mathbf{x}}_j} \right)} \right)\) with k = 1, …, K.
Independent simple linear attribute classifiers qξ(ai|z) are then fitted per attribute. For each attribute ai, the procedure consists of: (1) collecting dataset with all labelled samples for this attribute (xj, ai), (2) encoding the labelled data as before, \({\mathbf{z}}_{j,k} \sim q_\phi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{x}}_j} \right)\), (3) fitting ξ, the parameters of logistic regression classifier qξ(ai|z) with inverse regularization strength C = 1.0 and 300 limited-memory Broyden–Fletcher–Goldfarb–Shanno (lbfgs) iterations.
Rejection sampling for attribute-conditioned generation
Let us formalize that there are n different (and possibly independent) binary attributes of interest \({\mathbf{a}} \in \{ 0,1\} ^n = [a_1,a_2, \ldots ,a_n]\), and each attribute is available (labelled) for only a small and possibly disjoint subset of the dataset. As functional annotation of peptide sequences is expensive, current databases typically represent a small (nearly 100–10,000) subset of the unlabelled corpus. We posit that all plausible datapoints have those attributes, albeit mostly without label annotation. Therefore, the data distribution implicitly is generated as \(p\left( {\mathbf{x}} \right) = {\Bbb E}_{{\mathbf{a}} \sim p\left( {\mathbf{a}} \right)}\left[ {p\left( {{\mathbf{x}}{\mathrm{|}}{\mathbf{a}}} \right)} \right]\), where the distribution over the (potentially huge) discrete set of attribute combinations p(a) is integrated out and, for each attribute combination, the set of possible sequences is specified as p(x|a). As our aim is to sample new sequences x ~ p(x|a) for a desired attribute combination \({\mathbf{a}} = \left[ {a_1, \ldots ,a_n} \right]\), we are now able to approach this task through conditional sampling in latent space:
Where \(\hat p_\xi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{a}}} \right)\) will not be approximated explicitly, rather, we will use rejection sampling using the models Qξ(z) and qξ(ai|z) to approximate samples from p(z|a).
To approach this, we first use Bayes’ rule and the conditional independence of the attributes ai conditioned on z, because we assume that the latent variable captures all information to model the attributes: \(a_i \bot a_j|{\mathbf{z}}\) (that is, two attributes ai and aj are independent when conditioned on z)
This approximation is introduced to \(\hat p_\xi \left( {{\mathbf{z}}{\mathrm{|}}{\mathbf{a}}} \right)\), using the models Qξ and qξ above:
The denominator qξ(a) in equation (5) could be estimated by approximating the expectation \(q_\xi \left( {\mathbf{a}} \right) = {\Bbb E}_{Q_\xi \left( {\mathbf{z}} \right)}q_\xi \left( {{\mathbf{a}}{\mathrm{|}}{\mathbf{z}}} \right) \approx \frac{1}{N}\mathop {\sum}\nolimits_{{\mathbf{z}}_j \sim Q_\xi \left( {\mathbf{z}} \right)}^N {q_\xi } \left( {{\mathbf{a}}{\mathrm{|}}{\mathbf{z}}} \right)\). However, the denominator is not needed a priori in our rejection sampling scheme; by contrast, qξ(a) will naturally appear as the rejection rate of samples from the proposal distribution (see below).
For rejection sampling distribution with probability density function (pdf) f(z), we need a proposal distribution g(z) and a constant M, such that f(z) ≤ Mg(z) for all z, that is, Mg(z) envelopes f(z). We draw samples from g(z) and accept the sample with probability \(\frac{{f\left( {\mathbf{z}} \right)}}{{Mg\left( {\mathbf{z}} \right)}} \le 1\).
In the above, to sample from equation (5), we consider that a is constant. We perform rejection sampling through the proposal distribution \(g\left( {\mathbf{z}} \right) = Q_\xi \left( {\mathbf{z}} \right)\) that can be directly sampled. Now set \(M = \frac{1}{{q_\xi \left( {\mathbf{a}} \right)}}\) so \(Mg\left( {\mathbf{z}} \right) = \frac{{Q_\xi \left( {\bf{z}} \right)}}{{q_\xi \left( {\mathbf{a}} \right)}}\), while our pdf to sample from is \(f\left( {\mathbf{z}} \right) = \frac{{Q_\xi \left( {\mathbf{z}} \right)\mathop {\prod }\nolimits_i q_\xi \left( {a_i{\mathrm{|}}{\mathbf{z}}} \right)}}{{q_\xi \left( {\mathbf{a}} \right)}}\). We therefore accept the sample from \(Q_\xi \left( {\mathbf{z}} \right)\) with probability
The inequality trivially follows from the product of normalized probabilities. The acceptance rate is \(\frac{1}{M} = q_\xi \left( {\mathbf{a}} \right)\). Intuitively, the acceptance probability is equal to the product of the classifier’s scores, while sampling from explicit density Qξ(z). To accept any samples, we need a region in z space to exist where Qξ(z) > 0 and the classifiers assign a non-zero probability to all desired attributes, that is, the combination of attributes has to be realizable in z space.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The peptide sequence data are available at GitHub (https://github.com/IBM/controlled-peptide-generation).
Code availability
The code is available at GitHub (https://github.com/IBM/controlled-peptide-generation).
Change history
28 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41551-021-00771-4
References
DiMasi, J. A., Grabowski, H. G. & Hansen, R. W. Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ. 47, 20–33 (2016).
Desselle, M. R. et al. Institutional profile: community for open antimicrobial drug discovery—crowdsourcing new antibiotics and antifungals. Future Sci. OA 3, FSO171 (2017).
No Time to Wait: Securing the Future From Drug-Resistant Infections Technical Report (UN, 2019).
O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations Technical Report (Review on Antimicrobial Resistance, 2016).
2019 Antibacterial Agents in Clinical Development Technical Report (WHO, 2019).
Powers, J.-P. S. & Hancock, R. E. The relationship between peptide structure and antibacterial activity. Peptides 24, 1681–1691 (2003).
Mahlapuu, M., Håkansson, J., Ringstad, L. & Björn, C. Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 6, 194 (2016).
Chen, C. H. et al. Simulation-guided rational de novo design of a small pore-forming antimicrobial peptide. J. Am. Chem. Soc. 141, 4839–4848 (2019).
Torres, M. D. et al. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Biol. 1, 221 (2018).
Tucker, A. T. et al. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell 172, 618–628 (2018).
Field, D. et al. Saturation mutagenesis of selected residues of the α-peptide of the lantibiotic lacticin 3147 yields a derivative with enhanced antimicrobial activity. Microb. Biotechnol. 6, 564–575 (2013).
Fjell, C. D., Hiss, J. A., Hancock, R. E. & Schneider, G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11, 37–51 (2012).
Li, J. et al. Membrane active antimicrobial peptides: translating mechanistic insights to design. Front. Neurosci. 11, 73 (2017).
Cardoso, M. H. et al. Computer-aided design of antimicrobial peptides: are we generating effective drug candidates. Front. Microbiol. 10, 3097 (2020).
Jenssen, H., Fjell, C. D., Cherkasov, A. & Hancock, R. E. QSAR modeling and computer-aided design of antimicrobial peptides: computer-aided antimicrobial peptides design. J. Pept. Sci. 14, 110–114 (2008).
Vishnepolsky, B. et al. De novo design and in vitro testing of antimicrobial peptides against Gram-negative bacteria. Pharmaceuticals 12, 82 (2019).
Maccari, G. et al. Antimicrobial peptides design by evolutionary multiobjective optimization. PLoS Comput. Biol. 9, e1003212 (2013).
Meher, P. K., Sahu, T. K., Saini, V. & Rao, A. R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 7, 42362 (2017).
Thomas, S., Karnik, S., Barai, R. S., Jayaraman, V. K. & Idicula-Thomas, S. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 38, D774–D780 (2010).
Witten, J. & Witten, Z. Deep learning regression model for antimicrobial peptide design. Preprint at bioRxiv https://doi.org/10.1101/692681 (2019).
Xiao, X., Wang, P., Lin, W.-Z., Jia, J.-H. & Chou, K.-C. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 436, 168–177 (2013).
Veltri, D., Kamath, U. & Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018).
Porto, W. F. et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 9, 1490 (2018).
Fjell, C. D., Jenssen, H., Cheung, W. A., Hancock, R. E. & Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and cheminformatics: optimizing antibacterial peptides. Chem. Biol. Drug Des. 77, 48–56 (2011).
Porto, W. F., Fensterseifer, I. C. M., Ribeiro, S. M. & Franco, O. L. Joker: an algorithm to insert patterns into sequences for designing antimicrobial peptides. Biochim. Biophys. Acta 1862, 2043–2052 (2018).
Nagarajan, D. et al. Ω76: a designed antimicrobial peptide to combat carbapenem- and tigecycline-resistant Acinetobacter baumannii. Sci. Adv. 5, eaax1946 (2019).
Mueller, A. T., Hiss, J. A. & Schneider, G. Recurrent neural network model for constructive peptide design. J. Chem. Inf. Model. 58, 472–479 (2018).
Grisoni, F. et al. Designing anticancer peptides by constructive machine learning. ChemMedChem 13, 1300–1302 (2018).
Gupta, A. et al. Generative recurrent networks for de novo drug design. Mol. Inform. 37, 1700111 (2018).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proc. International Conference on Machine Learning 2323–2332 (2018).
Blaschke, T., Olivecrona, M., Engkvist, O., Bajorath, J. & Chen, H. Application of generative autoencoder in de novo molecular design. Mol. Inform. 37, 1700123 (2018).
Chan, H. S., Shan, H., Dahoun, T., Vogel, H. & Yuan, S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 40, 592–604 (2019).
Sanchez-Lengeling, B. & Aspuru-Guzik, A. Inverse molecular design using machine learning: generative models for matter engineering. Science 361, 360–365 (2018).
Nagarajan, D. et al. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 293, 3492–3509 (2018).
Hinton, G. E. & Salakhutdinov, R. R. Reducing the dimensionality of data with neural networks. Science 313, 504–507 (2006).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. Preprint at https://arxiv.org/abs/1312.6114 (2014).
Guimaraes, G. L., Sanchez-Lengeling, B., Outeiral, C., Farias, P. L. C. & Aspuru-Guzik, A. Objective-reinforced generative adversarial networks (ORGAN) for sequence generation models. Preprint at https://arxiv.org/abs/1705.10843 (2017).
Popova, M., Isayev, O. & Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 4, eaap7885 (2018).
Kang, S. & Cho, K. Conditional molecular design with deep generative models. J. Chem. Inf. Model. 59, 43–52 (2018).
Losasso, V., Hsiao, Y.-W., Martelli, F., Winn, M. D. & Crain, J. Modulation of antimicrobial peptide potency in stressed lipid bilayers. Phys. Rev. Lett. 122, 208103 (2019).
Cipcigan, F. et al. Accelerating molecular discovery through data and physical sciences: applications to peptide-membrane interactions. J. Chem. Phys. 148, 241744 (2018).
UniProt (EMBL-EBI, SIB, accessed August 2018); https://www.uniprot.org
Peters, M. E. et al. Deep contextualized word representations. In Proc. 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies 2227–2237 (Association for Computational Linguistics, 2018).
Radford, A. et al. Language models are unsupervised multitask learners. OpenAI Blog 1, 9 (2019).
McCann, B., Bradbury, J., Xiong, C. & Socher, R. Learned in translation: contextualized word vectors. In Proc. Advances in Neural Information Processing Systems 6297–6308 (ACM, 2017).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies 4171–4186 (Association for Computational Linguistics, 2019).
Rao, R. et al. Evaluating protein transfer learning with TAPE. In Advances in Neural Information Processing Systems 32 9689–9701 (2019).
Madani, A. et al. ProGen: language modeling for protein generation. Preprint at https://arxiv.org/abs/2004.03497 (2020).
Riesselman, A. J., Ingraham, J. B. & Marks, D. S. Deep generative models of genetic variation capture the effects of mutations. Nat. Methods 15, 816–822 (2018).
Shi, X., Padhi, I. & Knight, K. Does string-based neural MT learn source syntax? In Proc. 2016 Conference on Empirical Methods in Natural Language Processing 1526–1534 (Association for Computational Linguistics, 2016).
Yu, Y.-K., Wootton, J. C. & Altschul, S. F. The compositional adjustment of amino acid substitution matrices. Proc. Natl Acad. Sci. USA 100, 15688–15693 (2003).
Vishnepolsky, B. et al. Predictive model of linear antimicrobial peptides active against Gram-negative bacteria. J. Chem. Inf. Model. 58, 1141–1151 (2018).
Gupta, S. et al. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8, e73957 (2013).
Sattarov, B. et al. De novo molecular design by combining deep autoencoder recurrent neural networks with generative topographic mapping. J. Chem. Inf. Model. 59, 1182–1196 (2019).
Pearson, W. R. An introduction to sequence similarity (‘homology’) searching. Curr. Protoc. Bioinform. 42, 3.1.1–3.1.8 (2013).
Li, R.-F. et al. Molecular design, structural analysis and antifungal activity of derivatives of peptide CGA-N46. Interdiscip. Sci. Comput. Life Sci. 8, 319–326 (2016).
Hawrani, A., Howe, R. A., Walsh, T. R. & Dempsey, C. E. Origin of low mammalian cell toxicity in a class of highly active antimicrobial amphipathic helical peptides. J. Biol. Chem. 283, 18636–18645 (2008).
Wiradharma, N., Sng, M. Y., Khan, M., Ong, Z.-Y. & Yang, Y.-Y. Rationally designed α-helical broad-spectrum antimicrobial peptides with idealized facial amphiphilicity. Macromol. Rapid Commun. 34, 74–80 (2013).
Rifkind, D. Prevention by polymyxin B of endotoxin lethality in mice. J. Bacteriol. 93, 1463–1464 (1967).
Rončević, T. et al. Parallel identification of novel antimicrobial peptide sequences from multiple anuran species by targeted DNA sequencing. BMC Genom. 19, 827 (2018).
Jing, W., Demcoe, A. R. & Vogel, H. J. Conformation of a bactericidal domain of puroindoline a: structure and mechanism of action of a 13-residue antimicrobial peptide. J. Bacteriol. 185, 4938–4947 (2003).
Haney, E. F. et al. Mechanism of action of puroindoline derived tryptophan-rich antimicrobial peptides. Biochim. Biophys. Acta 1828, 1802–1813 (2013).
Mathur, D., Singh, S., Mehta, A., Agrawal, P. & Raghava, G. P. In silico approaches for predicting the half-life of natural and modified peptides in blood. PLoS ONE 13, e0196829 (2018).
Kumar, P., Kizhakkedathu, J. N. & Straus, S. K. Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 8, 4 (2018).
Guha, S., Ghimire, J., Wu, E. & Wimley, W. C. Mechanistic landscape of membrane-permeabilizing peptides. Chem. Rev. 119, 6040–6085 (2019).
Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 180, 688–702 (2020).
Loose, C., Jensen, K., Rigoutsos, I. & Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 443, 867–869 (2006).
Chenthamarakshan, V. et al. CogMol: target-specific and selective drug design for COVID-19 using deep generative models. In Advances in Neural Information Processing Systems 33 (eds Larochelle, H. et al.) 4320–4332 (Curran Associates, Inc., 2020).
Bowman, S. R., Angeli, G., Potts, C. & Manning, C. D. A large annotated corpus for learning natural language inference. In Proc. 2015 Conference on Empirical Methods in Natural Language Processing 632–642 (Association for Computational Linguistics, 2015).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. 9, 1735–1780 (1997).
Bowman, S. et al. Generating sentences from a continuous space. In Proc 20th SIGNLL Conference on Computational Natural Language Learning 10–21 (Association for Computational Linguistics, 2016).
Tolstikhin, I., Bousquet, O., Gelly, S. & Schölkopf, B. Wasserstein auto-encoders. In International Conference on Learning Representations (2018).
Bahuleyan, H., Mou, L., Vamaraju, K., Zhou, H. & Vechtomova, O. Stochastic Wasserstein autoencoder for probabilistic sentence generation. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies 4068–4076 (Association for Computational Linguistics, 2019).
Makhzani, A., Shlens, J., Jaitly, N., Goodfellow, I. & Frey, B. Adversarial autoencoders. In International Conference on Learning Representations(2016).
Gretton, A., Borgwardt, K. M., Rasch, M., Schölkopf, B. & Smola, A. J. A kernel method for the two-sample-problem. In Proc. Advances in Neural Information Processing Systems (eds Schölkopf, B. et al.) 513–520 (MIT Press, 2007).
Rahimi, A. & Recht, B. Random features for large-scale kernel machines. In Proc. Advances in Neural Information Processing Systems 1177–1184 (2007).
Rubenstein, P. K., Schoelkopf, B. & Tolstikhin, I. On the latent space of Wasserstein auto-encoders. Preprint at https://arxiv.org/abs/1802.03761 (2018).
Acknowledgements
We acknowledge Y. Mroueh and K. Varshney for discussions. We also thank O. Chang, E. Khabiri and M. Riemer for help with the initial phase of the work. We would like to acknowledge D. Cox, Y. Tu, P. Meyer Rojas and M. Rigotti for providing feedback on the manuscript. F.C. thanks P. Simcock for sharing knowledge. S.G. was an intern at IBM Research during this work.
Author information
Authors and Affiliations
Contributions
P.D., J.C. and A.M. conceived the project. P.D. designed and managed the project. P.D. and T.S. designed and implemented the sequence generation and screening framework and algorithm with help from K.W., I.P. and S.G. Autoencoder experiments were run and analysed by P.D., T.S., K.W., I.P., P.-Y.C., V.C. and C.d.S. Generated sequences were analysed in silico by P.D., T.S., H.S. and K.W.; F.C. with help from P.D. and J.C. designed, performed and analysed the molecular dynamics simulations. Y.Y.Y., J.P.K.T. and J.H. performed and analysed the wet-laboratory experiments. H.S. created the final figures. All of the authors contributed to writing the paper.
Corresponding author
Ethics declarations
Competing interests
The authors have filed the following patent applications related to this work: (1) application no. US2021/01025A1 on the CLaSS method for generating attribute-based samples (inventors: P.D., T.S., K.W., C.d.S., I.P. and S.G.); (2) application no. 16/880021 on the filtering of AI-designed molecules for laboratory testing (inventors: P.D., F.C., K.W., I.P., V.C., P.-Y.C., A.M., T.S. and C.d.S.); and (3) application no. 16/880280 on AI-designed antimicrobial peptides (inventors: P.D., F.C., J.H., Y.Y.Y., K.W., I.P., V.C. and J.P.K.T.).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary methods, figures, tables and references.
Rights and permissions
About this article

Cite this article
Das, P., Sercu, T., Wadhawan, K. et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat Biomed Eng 5, 613–623 (2021). https://doi.org/10.1038/s41551-021-00689-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-021-00689-x
This article is cited by
-
Antimicrobial resistance crisis: could artificial intelligence be the solution?
Military Medical Research (2024)
-
Tpgen: a language model for stable protein design with a specific topology structure
BMC Bioinformatics (2024)
-
Design of target specific peptide inhibitors using generative deep learning and molecular dynamics simulations
Nature Communications (2024)
-
Novel antimicrobial peptides against Cutibacterium acnes designed by deep learning
Scientific Reports (2024)
-
Machine learning assisted rational design of antimicrobial peptides based on human endogenous proteins and their applications for cosmetic preservative system optimization
Scientific Reports (2024)
