
Abstract
Non-ideal thermodynamics of solid solutions can greatly impact materials degradation behavior. We have investigated an actinide silicate solid solution system (USiO4–ThSiO4), demonstrating that thermodynamic non-ideality follows a distinctive, atomic-scale disordering process, which is usually considered as a random distribution. Neutron total scattering implemented by pair distribution function analysis confirmed a random distribution model for U and Th in first three coordination shells; however, a machine-learning algorithm suggested heterogeneous U and Th clusters at nanoscale (~2 nm). The local disorder and nanosized heterogeneous is an example of the non-ideality of mixing that has an electronic origin. Partial covalency from the U/Th 5f–O 2p hybridization promotes electron transfer during mixing and leads to local polyhedral distortions. The electronic origin accounts for the strong non-ideality in thermodynamic parameters that extends the stability field of the actinide silicates in nature and under typical nuclear waste repository conditions.
Similar content being viewed by others

Introduction
Uranothorite (UxTh1−xSiO4) is the only known natural silicate system where actinides, U and Th, form a continuous solid solution between thorite (ThSiO4) and coffinite (USiO4)1,2,3,4,5,6,7,8,9,10,11,12. Uranothorite is of geological interest because of its natural occurrence in uranium deposits4,5,7,13,14,15,16, suggesting an enhanced stability field for the coffinite endmember. Furthermore, because zircon (ZrSiO4) has been proposed as a possible phase for the incorporation and immobilization of Pu17,18,19,20,21,22, uranothorite, as an actinide solid solution system based on the zircon structure (I41/amd), can provide critical insights into coexisting actinides in this crystalline matrix. How the mixing of various actinides impacts the structural and thermodynamic stabilities is essential to the understanding of behaviors of Pu in the zircon structure type. On the other hand, coffinite is known to be one of the key alternation phases of uraninite (UO2 in nature) or UO2 in the spent nuclear fuel (SNF) in a geologic repository23,24,25,26,27,28,29,30 due to the contact with groundwater under anoxic and silica-enriched conditions31,32. However, whether uranothorite or other actinide silicate solid solutions can be more important sinks for U, Pu, and other minor actinides that control the dissolution and distribution of radionuclides is unknown and requires a fundamental thermodynamic assessment. Furthermore, the degradation or formation processes may also be influenced by material morphology. Particularly, the presence of nanophases and surface defects may be catalytic and initialize intragranular cracking or intragranular corrosion, expediting the degradation process33. Therefore, to demonstrate the feasibility of using zircon as a ceramic waste form for the immobilization of multiple actinides (e.g., U and Pu) under geological repository conditions and predict the fate of this actinide silicate solid solution under relevant mineralization and weathering processes, a better understanding of the thermodynamics and structures of uranothorite are essential.
Uranothorite, coffinite, and thorite all crystallize in the zircon structure (I41/amd), MSiO4, isostructural with zircon (Fig. 1), where the large MO8 polyhedra can be occupied by many tetravalent cations, such as transition metals (Zr, Hf), lanthanides (Ce), and actinides (Th, U, Np, Pu)1,2,3,5,8,9,10,11,12,34,35,36,37,38,39,40,41,42,43,44,45. This is due to the high oxygen coordination of the MO8 site that favors the presence of large cations, following Pauling’s rules46. MO8 triangular dodecahedra share edges with neighboring polyhedra forming a zigzag chain parallel to the a (and b)-axis, and alternate with SiO4 tetrahedra along the c-axis2,6,12. Although isostructural, coffinite and thorite have significant differences in many aspects, such as geological conditions of formation, thermodynamic stability, and ease of synthesis7,8,10,35,40,47,48,49,50, which also lead to different material degradation behaviors in nature. Thorite is an important accessory mineral of Th in igneous and metamorphic rocks7,35. This phase can also be easily obtained by solid state reaction between thorianite (ThO2) and quartz (SiO2) under laboratory conditions34. This observation is consistent with its negative standard enthalpy of formation (ΔH°f = −2143.5 ± 6.8 kJ/mol) and enthalpy of formation from oxides (ΔHf,ox = −6.4 ± 5.7 kJ/mol)12. Coffinite, on the other hand, is difficult to synthesize in the laboratory, unless by hydrothermal reactions followed by sequential purification processes8,10, or more recently by alteration of UO223,29. Direct heating of several kinds of mixtures (e.g., uraninite (UO2) and quartz) has not been found to lead to the formation of coffinite. This is also reflected by the natural occurrence of coffinite as nanosized grains (second most abundant U(IV) containing phase on Earth, after uraninite), indicating that hydrothermal formation mediated by uranium hydroxo-silicate colloids may have occurred40,48. Its metastability was confirmed recently by independent thermodynamic studies40. Guo et al.40 determined the enthalpy of formation of coffinite from oxides to be 25.6 ± 3.9 kJ/mol (ΔH°f = −1970.0 ± 4.2 kJ/mol) using high temperature oxide melt solution calorimetry. Szenknect et al.49 determined the Gibbs free energy associated with the formation of coffinite from oxides to be −5.25 ± 0.05 kJ/mol (ΔG°f = −1867.6 ± 3.2 kJ/mol)49 by performing solubility experiments of coffinite at 25 °C under 1 bar.

a The projection of a representative uranothorite phase (U0.11Th0.89SiO4) possessing the zircon structure down to the b–c plane with the c-axis pointing up. The blue and cyan dodecahedra are the UO8 and ThO8, respectively; the yellow tetrahedra are SiO4, b AO8 dodecahedra are coordinated with two sets of oxygen atoms O1 and O2.
These important differences between the two isostructural endmembers raise the key question of whether their intermediate compositions (uranothorite) carry transitional structural and energetic characteristics and whether those differences control the thermodynamically and kinetically driven degradation or transformation processes. Due to the similar chemical characters of actinide cations and close ionic radii of 8-coordinated Th4+, U4+, and Pu4+ (1.19, 1.14, and 0.96 Å, respectively)51, an ideal mixing behavior in the zircon structure was initially expected. However, based on recent experimental work, as well as the present study, UxTh1−xSiO4 compounds have both structures and thermodynamic parameters that deviate negatively from the linear Vegard’s law, suggesting non-ideality behaviors12. Specifically, a regular solution model was proposed for uranothorite given the negative enthalpy of mixing, which favors the formation of uranothorites by lowering their formation energies that facilitates accommodating U in the zircon structure12. By contrast, Ferriss et al. proposed different solid solution models using the density functional theory (DFT) and Monte Carlo methods by taking into account the four nearest homo- or hetero-adjacent atom interaction energies, which suggests positive mixing enthalpies and phase exsolution52. The disagreement between experimental and computational results motivated this comprehensive structural study on uranothorite, coffinite, and thorite to investigate (i) the origins of the thermodynamic non-ideality by investigating the atomic-scale arrangements and the electronic features in the local atomic-scale, (ii) the entropic term which has contributed significantly to the thermodynamic stability at elevated temperature (80–300 °C generated from the decay heat)53, as is described by the mixing model of U and Th, and (iii) geochemical implication if a multi-element solid solution system, such as uranothorite, can extend the stability field of the immobilized actinides (e.g., U and Pu) in the waste matrix when stored in a geologic repository.
With the structural insights, we further revisited the previously published calorimetric data12 and refined them with the updated subregular solid solution model associated with a negative enthalpy of mixing. The subregular model describes the excess Gibbs free energy or the enthalpy of mixing by Ωx(1−x), where Ω is concentration-dependent and controlled by two interaction parameters, Ω = w12(1−x) + w21x. Though high temperature calorimetry has been demonstrated to be sensitive to bond strength, it does not reveal the origin of the enthalpic stabilization for intermediate compositions. Generally speaking, such a negative deviation from ideal mixing can be attributed to (i) short-range ordering, (ii) local distortions of coordination polyhedra, (iii) electronic features, or any combination of these three possibilities54. All three aspects were examined in this work by neutron total scattering data of thorite, coffinite, and three uranothorite samples (U0.11Th0.89SiO4, U0.35Th0.65SiO4, and U0.84Th0.16SiO4).
The diffraction data were analyzed by the Rietveld method for long-range structural information, and the diffuse scattering was subject to pair distribution function (PDF) analysis using real-space fitting that yields short-range structural features, such as the extent of cation ordering (or clustering) and polyhedral distortion. PDF analysis provides a direct determination of the atomic correlations. Fitting a PDF of a crystalline material may be performed using real-space Rietveld refinement or reverse Monte Carlo (RMC) modeling methods, both of which remove symmetry operations (unless introduced as constraints to the fit). In this work, both fitting techniques were used to explore the atomic-scale arrangement (ordering, clustering, or disordering) of U and Th in the zircon structure, which was further assessed by unsupervised machine learning (ML) (K-means clustering) to reveal the mid-range (~2 nm) arrangement of cations in their sublattices as belonging to various clusters. Additionally, DFT calculations were performed on coffinite and thorite to reveal their electronic structures, particularly actinide covalency within the metal polyhedra, the difference of which may cause the thermodynamic non-ideality when U and Th are mixed.
Lastly, we performed multi-component system thermodynamic calculations using the refined subregular solid solution model from this work and the reported or extrapolated thermodynamic properties (enthalpy, Gibbs free energy, heat capacity and entropy) to evaluate the phase equilibria of uranothorite under various oxygen fugacities and moderately elevated temperatures55,56,57. The outcome of the thermodynamic calculations allows the prediction of whether uranothorite will be stabilized or be subject to degradation during possible mineralization and weathering processes in nature or in a geological repository.
Results and discussion
Neutron diffraction and scattering data analyses
The long-range crystalline structures of coffinite, uranothorite, and thorite were refined by performing Rietveld analysis of the neutron diffraction (ND) data. Compared to X-ray diffraction, ND is more sensitive to oxygen and more suited to distinguish neighboring elements (e.g., U and Th) which have similar electron densities. The experimental diffraction data and fitted patterns can be found in Supplementary Fig. 1. From the final convergence of the refinements, the fitted unit-cell parameters, volumes and Rwp values (ranging from 3.4% to 7.6%) are shown in Table 1. The obtained atomic coordinates of oxygen and atomic displacement parameters are listed in Supplementary Table 1. The real-space Rietveld refinement of the neutron total scattering data was then performed using PDFgui with the experimental G(r) data and fits shown in Supplementary Fig. 2, and fitted values listed in Table 1. Because there is no superstructure in the model defined in PDFgui, the fitted result represents a random atom configuration, in which U and Th atoms are considered to be fully disordered in the crystallographic sites. The fitted unit-cell parameters, atomic coordinates, displacement parameters, and Rwp values (ranging from 17.4% to 28.9%) are summarized in Table 1 and Supplementary Table 2. These two obtained sets of lattice parameters are general in agreement with previous values determined from X-ray diffraction12,50. The unit-cell volume exhibits a slight increase with increasing Th addition (Fig. 2). This trend is consistent with the previous findings12 and is supported by the substitution of U4+ (1.00 Å, Shannon ionic radius in eightfold coordinated) by Th4+ (1.05 Å)51.

a Unit-cell volume (Vcell) fitted by Vcell = 321.2−1.0x − 65.1x2 + 53.3x3 Å3 (adjusted R2 = 0.9998) shown as the dashed red line. Open green circles are from nPDF analysis this work, solid black circles from neutron diffraction with Rietveld refinement in this work, and open blue triangles from ref. 12. b Volume of mixing (ΔVmix) modeled by ΔVmix = [11.6(1−x) + (−41.7)x]x(1−x) Å3 (adjusted R2 = 0.9998) shown as the dashed red line. c Revised enthalpy of mixing (ΔΗmix)12 modeled by ΔHmix = [(−88.6)(1–x) + (−155.2)x]x(1– x) kJ/mol (adjusted R2 = 0.9888) shown as the dashed red line.
To explore the mixing geometries of U and Th atoms beyond short-range distances (i.e., first three nearest-neighbor metal cation) and the extent of their ordering or disordering in the matrix, RMC modeling was performed on the scattering data of the three uranothorite samples (U11, U35, and U84). Compared to the real-space Rietveld method (PDFgui) that fits the PDF from a starting crystallographic model assuming infinitely large crystallographic planes, the RMC fitting is performed on the normalized reciprocal space scattering pattern, F(Q), and the PDF by varying the positions of atoms in a predefined atom “box” following a set of constraints, and then periodically comparing the model to data, which allows for the investigation of the arrangements of U and Th atoms in a 10 × 10 × 10 supercell (see the “Methods” section and Supplementary Information for details). Figure 3 provides the fitting results of RMC modeling. The starting mixing configurations for the three uranothorites are locally ordered structures (Fig. 4). The divergence of U and Th from the initial ordered models was achieved by atom swapping and translation per refinement step, which was constrained by distance window (Supplementary Table 3) and bond valence sum (BVS) constraints (Supplementary Table 4). Here we examine the atomic positions relaxed after RMC modeling as the “true” configuration to evaluate the mixing behavior of Th and U, in comparison with their “ordered” ones in the supercells (Fig. 4). Previously, the variation of the a lattice parameter through X-ray diffraction suggested that chemical ordering may occur in a direction parallel to the a-axis12. However, in our current assessment from the neutron scattering data, there seems to be no strong indication of ordering occurred in either the a- or the c-axis, which was also reflected in Fig. 4. There is also no preference of U–Th clustering along the (010) or (001) planes. The representations of the final atom models perpendicular to the c-axis may be slightly misleading to visual inspection with respect to the extent of clustering in the systems analyzed. This is because U and Th atoms are closer-packed along the (001) plane; however, the (010) plane is actually closer-packed with 2 U/Th atoms per plane as compared to 1 U/Th atom per plane for the (001) plane. In order to understand how the arrangements of U and Th atoms propagate to nanoscale, a further detailed categorization of the distribution of all U and Th atoms in the 10 × 10 × 10 supercell was quantified by an unsupervised ML algorithm using the atomic arrangement from the RMC-relaxed superlattice “box” before and after refinement (Fig. 5).

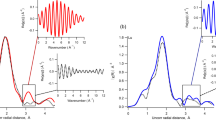
Measured F(Q) and GRMC(r) data are provided in black and fitted F(Q) and GRMC(r) curves are overlaid in red and cyan, respectively. The differences between the measured data and the RMC models are provided underneath the overlaid plots.

The blue and cyan circles represent U and Th atoms, respectively. Atomistic models are compared to the initial configurations along both the c-axis (left column of each pair) and b-axis (right column of each pair).

The insets on the inertia-number diagrams show the positions of the U/Th clusters (denoted with different colors) for the case when 50 clusters were defined (K = 50).
Further, the U–O, Th–O, and U–Th partial PDFs, g(r), for the three uranothorites obtained from RMC modeling and PDFgui fitting are shown in Supplementary Fig. 3. The integration of g(r) over distance r yields the coordination number distribution function N(r) which is shown for each pair, U–O, Th–O, and U–Th in Supplementary Figs. 4 and 6. Three coordination shells are highlighted in red, yellow, and blue for the U–Th pairs in their nearest neighbors in the a–b planes, the second nearest, and the third in the a–b planes, respectively.

The coordination number distribution functions N(r) obtained through RMCProfile are shown as solid black lines (referred to left axis). The statistically calculated Warren–Cowley parameters, α (referred to right axis, values from −0.3 to 1.0), for RMC fits and fully ordered models are depicted in black and red open circles, respectively, for RMC fits and fully ordered models. The coordinates covered by red, yellow, and blue correspond to the U–Th pairs in their nearest neighbors in the a–b planes, the second nearest, and the third in the a–b planes, respectively. The dashed black line indicates the value of α in the case of random distribution of U and Th atoms.
Solid solution model of uranothorite
By using the refined ND data, the unit-cell volume (Vcell) in terms of USiO4 mole fraction x is fitted by the following polynomial function:
where a = 321.2 ± 0.1 Å3, b = −1.0 ± 1.2 Å3, c = −65.1 ± 2.9 Å3, d = 53.3 ± 1.9 Å3. The excess volume (ΔVmix) can then be modeled by a subregular volumetric model based on Eq. (1),
where ωG1 = 11.6 Å3 and ωG2 = −41.7 Å3 are the asymmetric volumetric interaction parameters. Such a relation follows a subregular model, which is similar to the previous finding that ΔVmix has a negative deviation from Vegard’s law (ideal mixing) behavior. This updated model suggests more complicated mixing behavior in uranothorite which is asymmetric with the minimum volume of mixing located around 70 mol% of U (Fig. 2b). This updated model also explains our recent work on the high-temperature behavior of uranothorite, where U0.90Th0.10SiO4 was found to decompose above 800 °C to phases firstly with a decreasing trend of unit-cell volume, presumably having U content move from x = 0.9–0.8, and then a reversed increasing trend of unit-cell volume at higher temperatures, corresponding to the formation of U-depleted uranothorite (x < 0.8)50. The continuing shifts in chemical composition are driven by thermodynamics due to the mixing-induced enthalpic stabilization, evidenced by a previous calorimetric study where negative enthalpies of mixing (ΔHmix) were reported12. Inspired by the subregular volumetric model better describing the uranothorite solid solution, we revisited the published ΔHmix data12, and refined them with a subregular model,
where w12 = −88.6 kJ/mol and w21 = −155.2 kJ/mol, with adjusted R2 = 0.9888 (Fig. 2c). The negative interaction parameters w12 and w21 indicate that the uranothorite solid solution is expected to be stabilized by the enthalpic effect.
Such negative deviations from Vegard’s law in both formation enthalpy and unit-cell volume data could originate from (i) a favorable mixing between Th and U in the local range, (ii) the distortion of U or Th occupied polyhedra, and/or (iii) the difference in bonding characters (5f0 configuration for Th4+ and 5f2 configuration for U4+)58,59. First, based on the results from PDF analysis, we compared the coordination number distribution function N(r) of U–O, Th–O, and U–Th pairs determined from the RMC-derived models with the same quantities from a hypothetical random distribution model and show the comparison across three coordination shells in Fig. 6. No noticeable difference is observed, or there is no particular feature in the data that would lead to the locally ordered model. This is consistent with the PDFgui refinements which use the disordered model that produces good fits with acceptable Rwp values.
In order to further quantitatively evaluate the degree of cationic ordering, we calculated the Warren–Cowley (WC) parameters60,61 of U and Th based on the atomistic models from RMC fitting, and compared with the ordered and random configurations. Here the WC parameter, αi,j(r), is defined as follows:
where \(P_{i,j}\left( r \right)\) is the probability of finding an atom of type j at r given an atom of type i at the origin, and cj is the average concentration of j-type atoms. Thus, the U-centered WC parameter αU,Th has a value ranging from −NTh/NU to 1 (and a similar definition for Th-centered WC parameter αTh,U). For an assumed ideal random distribution of U and Th atoms in the sublattice sites, αU,Th (or αTh,U) is zero and depicted as the dashed lines in Fig. 6. A positive value indicates a tendency for like pairing (i.e., clustering or formation of domains) and the positiveness with the upper limit of 1 represents the “strength” of such a tendency. A negative value indicates the tendency to form unlike pairs (U–Th pairs). Thus, on each U/Th ratio, we compared their WC parameters calculated from RMC-determined atomistic models and those from ordered and random configurations. In Fig. 6, the first three polyhedral coordination shells were color coded, with the coordinates in red, yellow, and blue corresponding to the U–Th pairs in their nearest neighbors in the a–b planes, the second nearest, and the third in the a–b planes, respectively, which are defined by the coordination number distribution function NTh–U(r). The calculated α, superimposed as open circles, are in almost-zero or very weakly negative values in the region for all three solid-solution compositions within the first three coordination shells, again suggesting the disordering of U and Th atoms. Despite the fact that a random mixing model is more probable, the ordered model cannot be completely rejected. Potential ordering may have also been underestimated due to the randomization of atoms which is intrinsically built into the RMC algorithm62. A low level of short-range ordering may still be possible, as suggested by the slight negativity of WC parameters within a certain coordination shell (e.g., the first one in U11 or the third one in U35), because \(P_{i,j}\left( r \right)\) is obtained by statistically averaging all same-type atoms in the supercell and thus less sensitive to some finer local information.
Thus, ND in this work, previous XRD analysis12, and EXAFS analysis (performed on similar uranothorite compositions in a study by Labs et al.)11 are all consistent with the above RMC-derived interpretation and WC parameters. Particularly, Labs et al.11 found no evidence for the formation of secondary phases and no significant degrees of atom-clustering through EXAFS and powder diffraction of hydrothermally-grown uranothorites. Lastly, a previous DFT calculation on uranothorite predicted a tendency to form local clusters with a positive enthalpy of mixing52, which contradicts the present experimental evidence. This is not too surprising when the first contribution to non-ideality is mostly considered that is usually important only when mixing cations have large size differences, thus less probable in uranothorite due to the close cation sizes of U4+ (1.00 Å) and Th4+ (1.05 Å).
To further examine the atomistic models and how the local disordering propagates to the outer coordination shells and further to macroscopic-scale structures, the unsupervised ML algorithm (K-means clustering, details provided in the “Methods” section) was implemented on the large dataset of coordinates of U and Th atoms in uranothorite supercells. Figure 5 provides the inertia-cluster number (used to determine the appropriate number of clusters per cell) and cluster sizes of U or Th atoms in uranothorite supercells (end member compositions yielded non-physical values and have been omitted). Noted that here the “cluster” does not represent non-repeating local U–U or Th–Th clusters in the conventional structural sense that is opposite of short-range ordering. Instead, in K-means clustering algorithm, it is a group of U or Th atoms that are found by ML to be of similar types. Thus, the distribution of such “clusters” can shed light on the local patterns of U or Th from the impact of cation mixing. Gaussian functions were used to fit the distributions (Supplementary Table 5). The K-means clustering results on RMC fits are compared with those of the “ordered configuration” (used as the “initial” model, exhibited in Fig. 4, for RMC modeling). The RMC modeling leads to a slightly broader distribution than the “ordered” configurations (Supplementary Table 5). For instance, the ordered model of U11 has σ = 1.9 compared to the RMC model which has σ = 3.3, where σ is the variance in the number of atoms per cluster. Therefore, the cluster sizes (i.e., the number of atoms per cluster) of RMC models overall resemble those of the ordered models in the med-range scale.
This is an interesting finding, suggesting that although U and Th are mixed randomly in the cationic sublattice, such disordering could locally differ, forming nanosized domains similar to what the ordered arrangement may introduce, despite the drastic change in the atom positions (Fig. 5). Specifically, the statistical results of K-means clustering on RMC-derived models determined the size of U “clusters” in U11 or Th “clusters” in U84 to be approximately 5–20 atoms (mean value 9.8 atoms), suggesting that U or Th cations form similar local environments in the size of the second cation coordination shells (color coded in yellow in Fig. 6). Such a mixing-induced feature is more pronounced near the equimolar composition. U35 has approximately 20–40 atoms for U “clusters” (mean value 27 atoms), generally equivalent to the size of the third cation coordination shell. Such locally contained disordering could cause the formation of nanodomains and hence nanosized heterogeneity. Similar disorder heterogeneity was recently highlighted in pyrochlore solid solution systems63. Nanoscale disorder heterogeneities may change the interpretation of the thermodynamic properties of uranothorite, possibly affecting chemical processes such as the selective dissolution of U and Th.
Structural and electronic origins for the thermodynamic non-ideality
With a cationic disordering model, instead of a short-range ordering model, suggested by RMC for uranothorites, the local distortion in U- and Th-occupied polyhedra becomes a more probable origin for the thermodynamic non-ideality. Note that the MO8 polyhedra (M = U, Th) are triangular dodecahedra that can be considered as two intersecting tetrahedra with two sets of O4 atoms (Fig. 1b), M(O1)4 and M(O2)46,64. The first set consist of four oxygen atoms (O1) forming longer bonds with U (2.421 Å)11 or Th (2.467 Å)2, described as the extended tetrahedra, sharing the edges with SiO4 tetrahedra aligning along the c-axis. Such <U–O1> or <Th–O1> bonds are not very sensitive to U and Th composition, which is the reason for having a near linear trend of lattice parameter c when varying the U content12,65. On the other hand, four other oxygen atoms (O2) have shorter bonds with U (2.292 Å)11 or Th (2.384 Å)65, described as the compressed tetrahedra, sharing corners with other compressed tetrahedra along the a–b planes, which are more sensitive to substitution12,65. Corner-sharing also make the disphenoidal M(O2)4 easier to be relaxed to distorted geometry under chemical strain (e.g., cationic substitution) or physical strain (e.g., compression). The different susceptibility of the two types of tetrahedra to substitution is confirmed by RMC’s results that <U–O2> expands by 2.4% while <U–O1> only expands by 1.4% when U content decreases to 11 mol% in uranothorite. Similarly, in Th sublattices, <Th–O2> shrinks by 3.2% compared to 2.2% for <Th–O1> (Table 2). Such local features have impacts on the long range which leads to a greater magnitude in unit-cell variance with an anisotropic behavior. Mixing introduces greater changes along the a (and b)-axis due to the flexibility of the second-type oxygen (O2), while encountering more resistance along the c-axis because of the insensitivity of the first-type oxygen (O1) and the more rigid edge-sharing M(O1)4–Si(O1)4 chains.
Furthermore, coffinite and thorite have different bond-length ratios (<M–O1>/<M–O2>) of 1.057 and 1.035, respectively, which suggests that the corresponding dodecahedra have slightly different local environments. This could be due to the distinct covalent tendency of U coupled with crystal field splitting similar to the Jahn–Teller distortion54, so that U 5f can strongly overlap with O 2p orbitals to form hybridization66,67. In order to investigate the bonding structure and its impacts on local geometry, we performed DFT + U calculations on coffinite and thorite. The U covalency is more directional along a–b planes in the compressed tetrahedra resulted in a shorter bond length of <U–O2>. The structure optimized by DFT + U method is in an antiferromagnetic (AFM) ordering, in agreement with the experimental results and previous calculations68 (Supplementary Tables 6 and 7). As shown in the projected density of states (pDOS, Fig. 7a, d), the overlap between the U 5f orbital and O 2p orbital density of states from −8 to 0 eV demonstrates a strong hybridization of these two orbitals, which provides a covalency signature for the <U–O> bonding. On the contrary, a limited overlap of Th 6d orbital and O 2p orbital density of states from −8 to 0 eV proved a marginal hybridization and poor covalency of <Th–O> bonding. Furthermore, more hybridization between Th and O emerges above the Fermi level, which can enhance anti-bonding and decrease the <Th–O> covalent bonding strength.

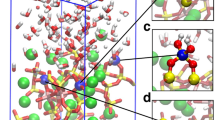
a Projected density of states (pDOS) diagram of USiO4 in an AFM magnetic order. b Representation of the half plane bisecting the <U–O1> and <U–O2> bonding, where the gray spheres are U, the blue spheres are Si, and red spheres are O. c Charge density in units of e Å−3 corresponding to the half plane presented in b. d Projected density of states (pDOS) diagram of ThSiO4. e Representation of the half plane bisecting the <Th-O1> and <Th-O2> bonding, where the green spheres are Th, the blue spheres are Si, and red spheres are O. f Charge density in unit of e Å−3 corresponding to the half plane presented in e.
To further understand the local bonding structure, charge density and electron localization function (ELF) were calculated and depicted in Fig. 7 and Supplementary Fig. 469. Figure 7c, f shows that although most charge density is localized near U or Th nuclei, indicating a strong ionic bond signature, a small proportion of electron density is distributed in the interstices of <U–O> in coffinite, which is consistent with a previous DFT result that U 5f was found to contribute to <U–O> bonds and exhibit a mixture of covalent/ionic characteristics68. More importantly, contour plotting reveals that electron density of U overlaps more with O2 than O1, hence the <U–O2> bonding has more covalent character. The excess covalency in <U–O2> was observed to be more than that in <Th–O2> due to the excess density overlapping in <U–O2> bond (Fig. 7c, f). Specifically, in the USiO4 matrix, oxygen attractors are in the shape of a meniscus heading to nearby U attractors, which indicates strong polarization between U and O attractors (Supplementary Fig. 5). While in ThSiO4, this polarization level is smaller and hence the local electron density is nearly spherical (Supplementary Fig. 5). Lastly, ThSiO4 was found to have higher ELF values, again indicating stronger ionic bonding of <Th–O> bonds. In general, ionic bonding is stronger than covalent bonding, and the difference in bonding nature may also explain why ThSiO4 is more thermodynamically stable than USiO4.
Such a difference in <U/Th–O> bonding also makes the M(O2)4 tetrahedra more “compressed” in coffinite than in thorite (Table 2 and Fig. 8). Interestingly, when U and Th mix in uranothorite with comparable amounts, such local polyhedral differences become “smeared” (Fig. 8), with UO8 dodecahedra having more ThO8-like features and vice versa. Additionally, another shorter metal–oxygen bonds <M–O3> emerge upon mixing, further distorting the local disphenoidal geometries and forming an irregular triangular dodecahedron (Table 2). The UO8 polyhedral distortion could be a result of an initial distortion introduced by substituted Th that breaks the local symmetry and then attenuates the crystal field splitting, leading to the weakening of the covalency of <U–O2> and its elongation. Thus, the distortion in UO8 is more pronounced in the U(O2)4 tetrahedra, which makes it resembles its counterpart in ThO8. While in the U- or Th-rich uranothorite, the introduction of different type cation starts to distort the two disphenoidal M(O)4 differently. We proposed an electronic interaction mechanism between UO8 and ThO8 dodecahedra to explain the variance in <M–O1>/<M–O2> bond ratio. Considering the excess covalent bonding property in <U–O2> bond could result in additional electron density accumulation in O2 atom, when U starts to enter the Th-rich matrix or vice versa, more electron density can transfer from <U–O2> bond to <Th–O1> bond through the bridging O2 atom (the same oxygen labeled as O1 atom for Th, Fig. 8b). Electron depletion from <U–O2> results in a less stable and hence longer <U–O2> bond while leaving <U–O1> untouched since it bridges to Th as O2 atom and remains mostly ionic. This results in a reduction of <U–O1>/<U–O2> bond ratio (Fig. 8a). On the other hand, electrons density flows into the <Th–O1> bond and could fill in the anti-bonding orbitals according to the pDOS and destabilize the <Th–O1> bond with an extended bond length. This also explains the increase of <Th–O1>/<Th–O2>. Such electron transfer interaction may be the main origin of the non-ideal thermodynamic behavior of uranothorite.

a The bond-length ratio (<M–O1>/<M–O2>) for M = U or Th, as a function of the U concentration x. Bond lengths of <M–O1> and <M–O2> were obtained from RMC-fitted atomistic models. b Conceptual representation of electronic transfer interaction between Th- and adjacent U-centered dodecahedra, where the gray spheres are U, the green spheres are Th, the blue spheres are Si, and red spheres are O.
Thermodynamic modeling of uranothorite and implications for material degradation
As a conclusion of RMC analyses and DFT calculations, the above proposed geometrical distortion coupled with the alteration in electronic structures is responsible for stabilizing intermediate U/Th composition in uranothorites with negative enthalpies of mixing and volume of mixing54,70. Another important implication for thermodynamics of uranothorite is that the configurational entropy may be determined by using the Boltzmann entropy formula (\({\Delta}S_{{\mathrm{Boltzmann}}} = - 4R\left[ {x\ln x + \left( {1 - x} \right)\ln \left( {1 - x} \right)} \right]\))71,72, where R is the ideal gas constant and x is the U concentration in the dodecahedral site. The configurational entropy due to mixing for uranium concentration of 0.11, 0.35, and 0.84 were calculated to be 2.88, 5.38, and 3.65 J·mol−1·K−1 (Supplementary Table 8). Such determination is usually not an easy case for other non-ideal solid-solution systems, because only random distribution of mixed ions will make the configurational entropy possible to be evaluated by the Boltzmann entropy ΔSBoltzmann, and certain favorable atomic arrangements in the short range will lead to the reduction from the theoretical maximum value70,73,74,75,76.
For a full thermodynamic assessment, vibration entropy and heat capacity (Cp) are also needed. However, due to the lack of experimentally determined Cp and entropy data, we estimated them for USiO4, ThSiO4, and uranothorite based on empirical methods, cross-checked by DFT phonon calculations in this work. The vibration entropy was estimated based on an empirical equation between the unit volume and the standard entropy values optimized for silicate minerals, So = (1.262 ± 0.028)·Vcell/4 (Å3) + (13 ± 5)77, which yielded 110 ± 15 and 114 ± 15 J·mol−1·K−1 for USiO4 and ThSiO4, respectively (Supplementary Table 9). The vibrational entropy was also estimated from the integration of Cp. Although there is even no experimental data for endmembers, USiO4 and ThSiO4, we applied different theoretical methods to obtain heat capacities, including density functional perturbation theory-based calculations by Phonopy78, Neumann–Kopp’s rule79, and n-dimensional Debye model80. A comparison of Cp derived from the three methods and from previous work is shown in Supplementary Figs. 6–8, with their integrated entropy values summarized in Supplementary Table 9. All values are in general consistent within the error range, except for the n-dimensional Debye model, due to its limited application on multi-atom ceramic materials. By accepting the volume–entropy empirical method, the vibrational mixing entropies of uranothorite were further determined to be 0.20, −0.26, and −0.87 J·mol−1·K−1, respectively for U11, U35, and U84 samples (Supplementary Table 8). Thus, configurational entropy dominates the entropy contribution based on these two estimations. Then total entropy (2.78–5.12 J·mol−1·K−1, Supplementary Table 8) can contribute positively to the thermodynamic stability of uranothorite at 300 °C (peak temperature for the nuclear repository backfill barrier materials)53 by −6 to −12 kJ/mol, and at 1000 °C (ceramic waste processing temperature)22,47,71 by −14 to −27 kJ/mol, respectively.
The availability of Cp, ΔH, ΔS, and ΔG of this solid solution system from this work and from literature12,40,49,81 now enables evaluation of changes in mineral’s stability fields triggered by the presence of uranothorite in complex systems, which in this work were simplified by including the following solid phases with their thermodynamic parameters: coesite82, cristobalite82, quartz82, Si83, soddyite84, stishovite82, thorianite83, Th83, tridymite82, uraninite85, U83, chalcedony86, γ-UO385, β-UO385, α-UO385, U3O885, U3O785, β-U4O985, U4O985, β-UO2(OH)285, coffinite40,49,81, and thorite12,49. This evaluation was performed through thermodynamic modeling using the Hch/Unitherm code87,88. The data for uranothorite, coffinite, and thorite used in these calculations are summarized in Table 3. Calculations of equilibrium compositions were performed on three model systems by analyzing limited numbers of factors (temperature and redox condition) that can affect the formation of (i) coffinite in the first, (ii) uranothorite described by the ideal mixing model in the second, and (iii) uranothorite described by the revised subregular solid solution model (from section “Solid solution model of uranothorite”) in the third calculation. Here the redox parameter was set in the calculations through fixed fugacity of O2, gas, the thermodynamic properties of which were taken from refs. 83,89. The range of log(fO2) was chosen from −40 to −8 to simulate a wide range of natural redox environments. Temperature effects were evaluated from 25 to 60 °C, which, although lower than the average nuclear waste repository temperature, is relevant to the formation, weathering, or material degradation processes in nature. An additional reason for selecting such a narrow temperature interval is also in limited applicability of the mixing model developed for uranothorite: this model was developed for ambient conditions and its applicability to higher temperatures is not evident. However, we believe that the effects observed even for such a narrow temperature interval can serve as a good illustration of how accounting for the presence of solid solutions can change the predicted phase stability fields. These effects can potentially be manifested to much higher degree at higher temperatures.
These calculations can reveal the impacts of the uranothorite solid solution system and mixing models describing the system on the stability of U-bearing zircon-type phases under various log(fO2)–temperature conditions. The starting composition of the model system was selected to be 1 mol of thorite and 1 mole of coffinite for the solid solution system for simplification. Significant differences were identified for the three models (Fig. 9 and Supplementary Table 9). Without the presence of uranothorite, coffinite only starts to be stabilized at 45 °C but under very reducing conditions, log(fO2) < −36 (Fig. 9a). This result is consistent with the natural observation that coffinite precipitation was found no greater than 50 °C in the uranium diagenetic unconformity-related deposits in the Athabasca basin, where Th is present in only trace concentrations and uranothorite is not formed90,91. When uranothorite is possible to form (model 2), the formation temperature is lowered down to 40 °C under very reducing conditions (Fig. 9b). At 55 °C and above, due to the very high stability of thorite, the formation of uranothorite also becomes possible even under very oxidizing conditions. This comparison illustrated the positive impact of the solid solution system on the stabilization of coffinite endmember. In model 3, we improved the mixing behavior of U and Th in uranothorite by using the subregular solid solution model (Eq. 3). The formation of uranothorite was found to move down in temperature even at 25 °C and log(fO2) < −30 (Fig. 9c). The stability field of uranothorite with relatively high U content expands as temperature increases (Fig. 9c, f), which eventually covers the whole oxygen fugacity range at 50 °C, suggesting the favorable stabilization of U-bearing uranothorites. These calculations illustrate that thorite overall can stabilize coffinite by absorbing U into the matrix to form uranothorite solid solutions under the conditions where pure coffinite is unstable (Fig. 9d, e). Furthermore, this work also demonstrates the significant contribution of the non-ideal mixing effect to the formation and stabilization of high U content uranothorites in much wider environmental conditions, which suggest the weathering or material degradation processes of uranothorite to be thermodynamically unfavorable.

a The phase fraction of coffinite in model 1 where no uranothorite is allowed for form. b The phase fraction of uranothorite in model 2 where an ideal mixing model is used. c The phase fraction of uranothorite in model 3 where a subregular solid solution model (Eq. 3) is used. d The difference in phase fraction of uranothorite obtained from models 3 and 2. e The difference in U content in uranothorite phase obtained from models 3 and 2. f The distribution of U content in uranothorite phase calculated from model 3. Noted that the distributions of fraction and U content have been curve-smoothed, and the raw data can be found in Supplementary Table 9.
Although the thermodynamics favors the stabilization of uranothorite over a wide range of oxygen fugacity and temperature ranges, uranothorite may still undergo material degradation due to kinetic or chemical processes. According to the discussion in section “Solid solution model of uranothorite”, uranothorite exhibits nanoscale heterogeneity, which controls chemical reactions and processes occurring at the interfaces or surfaces that may promote selective dissolution of U and Th. For instance, U can be involved in redox reactions leading to the more mobile uranyl-based species, but Th remains tetravalent, so the dissolution rate of uranothorite could be enhanced due to the nanoscale heterogeneity in the material. Plus, these small sized heterogeneities could also yield a significant amount of highly active sites in the interfaces or surfaces, which can act as catalysts for reactions with the aqueous solution to promote dissolution92,93,94. As a consequence, the heterogeneity can potentially introduce the incongruent corrosion process and induce intragranular stress in the bulk structure due to dislocation activity. This may also partially explain that why in nature, ~35 mol% U were mostly documented for uranothorite minerals that are also fine-grained, which could be in fact the degraded product of more U-rich uranothorites after selective dissolution of U upon the contact of hydrothermal fluids and/or the sequential recrystallization95. Such conclusion may also be more general for other actinide solid solutions, particularly for those when Pu is mixed with U or with multi-elements in the zircon structure. Although Pu, due to its even stronger covalent character96,97,98, may be more strongly stabilized in zircon due to potential thermodynamic non-ideality when mixed with U, Th, and/or minor actinides, the possibility of selective dissolution of Pu due to nanoscale disorder heterogeneous should not be overlooked.
In summary, we provide an updated insight of the atomic-scale arrangement of U and Th cations in uranothorite. Despite being a non-ideal mixture, mixing of U and Th in uranothorite is suggested, based on nPDF and ML algorithm, to follow a disordering model over the short range while being heterogeneous over the nanosized range. Local geometric distortions are driven by the electron transfer due to actinide covalency explain the thermodynamic non-ideality, specifically the negative deviations in volume and enthalpy from the Vegard’s law. This discovery emphasizes the role of 5f electrons in local geometric distortion and the resultant thermodynamic anomalies when actinides (Th, U, Np, and Pu) mix with other elements (transition metals or lanthanides) in natural minerals or actinide nuclear waste forms. Lastly, the solid solution model describing uranothorite was revised according to the structural findings to a subregular solid solution model, which was further used in the multi-component system thermodynamic modeling to investigate the phase equilibria of uranothorite under various redox–temperature conditions. Due to the non-ideal mixing effect, the solid solution system was found to have an enhanced and extended stability field for forming uranothorite with higher U content where pure coffinite is unstable, suggesting its weathering or material degradation to be thermodynamically unfavorable. Thus, covalency-driven nanosized disorder heterogeneity and thermodynamic non-ideality were demonstrated to have important implications for actinide-containing solid solution systems in general and their potential applications for immobilizing Pu and minor actinides.
Methods
Sample preparation
All the uranothorite samples were prepared through hydrothermal treatment, according to the procedure already described in literature8,10,99. Mixtures of U(IV) and Th(IV) concentrated solutions prepared in hydrochloric acid were added dropwise to a slightly off-stoichiometric amount of Na2SiO3 (≈5%) previously dissolved in water. The pH of each solution was adjusted to 11.0–11.5 using 8 M NaOH, then NaHCO3 was finally added to buffer the solutions at pH = 8.7. The final mixtures were transferred into 23 mL Teflon containers placed in autoclaves (Parr-type instruments). Hydrothermal treatments were performed at 250 °C for 1 week. The obtained precipitates were separated from the solutions by centrifugation at 4000 r.p.m., washed with water then ethanol, and finally dried overnight in air at room temperature. The final uranothorite samples contained amorphous silica and thorium–uranium dioxides as impurities. In order to proceed to their purification, a procedure involving successive washing steps in static and dynamic conditions was successfully developed. The complete elimination of impurities was reached in a reasonable time with respect to the chemical durability of the uranothorite solid solutions10. Chemical compositions of the samples were checked by scanning electron microscopy–wavelength dispersive spectroscopy (SEM-WDS), and those results were published previously12. Table 1 provides the naming convention for the sample compositions which were analyzed as part of this work.
ND and scattering
Samples were measured at the Nanoscale Ordered Materials Diffractometer (NOMAD), Spallation Neutron Source (SNS), and Oak Ridge National laboratory (ORNL)100. Prior to the measurements, the samples were annealed at 500 °C for 12 h according to the thermogravimetric studies on these samples12. Samples were then contained in boron free silica NMR tubes (Wilmad) with an inner diameter of 4.2065 ± 0.0065 mm and a wall-thickness of 0.38 mm. A diamond standard was used to calibrate the conversion of time-of-flight to Q for each pixel100. A vanadium rod with diameter 5.85 mm was measured for data normalization, taking into account effects such as the incident neutron spectrum and the spectral efficiency of the detectors. An identical empty NMR tube is measured as a background for the sample measurements. All sample measurements were collected for an accelerator proton charge of 4 C, which at the time of the experiment corresponded to roughly 75 min. The PDFs have been calculated with Qmax = 31 Å−1 leading to a real-space resolution of 0.02 Å.
GSAS refinement
The obtained ND data were analyzed by the Rietveld method using General Structure Analysis System (GSAS) software101. The background was fitted by a Chebyshev function, and the peak profiles were fitted to pseudo-Voigt convolution functions with a peak asymmetry correction102. The starting structure parameters for the uranothorite refinement were taken from work done by Fuchs et al.103. Data from all detector banks were simultaneously refined for the lattice parameters, atomic coordinates, and isotropic atomic displacement factors of U, Th, Si, and O. The detailed Rietveld refinement procedures were also stated previously elsewhere104,105,106,107.
Real-space fitting
A PDF is the probability that an atomic correlation exists at a corresponding radial distance. The neutron PDF (nPDF) was fitted using a combination of PDFgui and RMCProfile software tools108,109. The first silicon–oxygen bond distance was truncated in order avoid fitting the contribution of the silica NMR tube due to the limited amounts of samples. Real-space Rietveld refinement was performed on neutron total scattering data by implementing PDFgui109. GPDF(r) data from the Fourier transformation is fitted in PDFgui, and its detailed mathematical form can be found in Supplementary Information. The higher weighted R in Table 1 when compared with real-space Rietveld refinements to GSAS Rietveld refinements is likely a function of the background intensity which can significantly influence the weighted R110.
RMC modeling
RMC modeling was performed on the same neutron total scattering data. The software implemented for RMC modeling was RMCProfile108. GRMC(r) and F(Q) data were used for RMC modeling. For RMC modeling through RMCProfile, the nPDF was normalized according to the single atom scattering definition and will be denoted as GRMC(r) in this work, which differs from GPDF(r) used in PDFgui by a mathematical relationship showed in Supplementary Information. The starting atomic configurations for RMC fitting were obtained by creating a unit cell comprised of U, Si, and O using the atomic positions and lattice parameters obtained through PDFgui fitting (thereby avoiding the need to estimate a mass density), arbitrarily substituting Th for U to match the desired composition, then extending a unit cell comprised of U, Th, Si, and O to a 10 × 10 × 10 supercell. The swapping of U and Th during RMC fitting was implemented to relax the structure from the ordering introduced into the initial configuration. RMC modeling of samples U11 and U35 was carried out using distance window constraints and BVS constraints applied in order to confine silicon, uranium, and thorium atoms within oxygen polyhedra. This helps avoid the formation of interstitial atoms of Si, U, or Th and oxygen atoms without charge compensation. During the RMC modeling of samples U35 and U11, 12,600,000 attempted moves with 10,100,000 attempted swaps of U and Th (swap rate of 0.8) were carried out. For sample U84, a reasonable fit could only be obtained by significantly reducing the duration of the fit to 8,800,000 attempted moves with 7,080,000 attempted swaps. It is worth noting for reference that a swap rate of 0.5 indicates that the number of attempted atom swaps will equal the number of accepted atom displacements. The r-grid spacing was 0.02 Å and the maximum atomic moves were restricted 0.02 Å. The weighing ratio [F(Q): GRMC(r)] assigned to data during RMC fitting was 100:7.
Evaluation of cluster sizes through unsupervised ML
A K-means cluster algorithm was used to group the atoms in the atom boxes. An empirical relationship commonly referred to as the “inertia” parameter was used to ascertain the appropriate number of clusters of varying atom populations111,112. K-means clustering is one of the unsupervised learning algorithms for cluster analysis, and the distribution from Sci-kit was used for this work111, where more information can be found in the source literature. In K-means clustering, the algorithm partitions a set of available data (in this case the Euclidean atomic coordinates) into a predefined number of non-overlapping groups (where K is the predefined number). The clusters defined by the algorithm are selected such that they reduce an “inertia” parameter which is ∑(xi−cj)2, where xi is the position of the ith atom and cj is the position of the jth centroid. Empirically, the number of K clusters that best describes the dataset can be approximated by the “shoulder method” where the eponymous “shoulder” is the point at which the slope of the curve approaches zero on the graph of inertia vs. number of clusters. Selecting too few clusters results in high inertia values, suggesting the dataset is under-fit while selecting too many clusters does not significantly affect the inertia, indicating the dataset is over-fit. For this study, the value corresponding to the shoulder was K = 50 clusters. The clustering algorithm was performed using U atoms from the RMCProfile atom models of samples U11 and U35, and Th atoms for sample U84. Clustering calculations were performed for each RMC atom model, where the starting partitions were selected from 40 configurations prior to refinement. Histograms were generated based on the averages of five clustering trials.
Computational methods
DFT + U calculations were performed by using Vienna Ab initio Simulation Package (VASP) 5.4.4 (ref. 113). Generalized gradient approximation (GGA) approximation was used for the exchange-correlation function, and projector-augment wave (PAW) was used to orient the effect of core electrons114,115. DFT-D3 correction with Becke–Johnson damping was added to interpret van de Waals interaction in the system116. Effective U parameters for 5f electrons in uranium and thorium was set as 4.0 eV to correctly describe the highly localized f electrons117,118. The valence electrons of uranium, thorium, silicon, and oxygen were respectively set as 5f36s26p66d17s2, 6s26p66d27s2, 3s23p2, and 2s22p4. In ionic and electronic optimization steps, total energy was converged to 1 × 10−5 eV and force between ions was converged to 1 × 10−4 eV/Å. Based on the convergence study, integration of the Brillion zones of coffinite and thorite were done in a Gamma grid of 5 × 5 × 5 and cutoff energy was set as 550 eV. Considering the magnetic structure of USiO4, both ferromagnetic (FM) and colinear 1−k AFM phases have been tested in this study.
Multi-component system thermodynamic calculation
The calculations were performed using the Hch/Unitherm code87,88. The models used here were designed to highlight the stability field of uranothorite as a function of oxygen fugacity and temperature when used the ideal mixing model vs. the subregular solid solution model. Thus, in the highly simplified hypothetical geochemical scenarios, we have the following solid phases to be considered: coesite, cristobalite, quartz, Si, soddyite, stishovite, thorianite, Th, tridymite, uraninite, U, chalcedony, γ-UO3, β-UO3, α-UO3, U3O8, U3O7, β-U4O9, U4O9, β-UO2(OH)2, coffinite, thorite, and uranothorite. All calculations reported in this work are based on the thermodynamic dataset for the above solids and relevant aqueous species12,40,49,81,82,83,84,85,86,119,120,121,122.
Data availability
The data that support the findings of this study are available from the corresponding author upon request or can be accessed at https://doi.org/10.17632/wp87pcpx6h.1.
References
Stieff, L. R., Stern, T. & Sherwood, A. Preliminary description of coffinite—a new uranium mineral. Science 121, 608–609 (1955).
Fuchs, L. H. & Gebert, E. X-Ray studies of synthetic coffinite, thorite and uranothorites. Am. Mineral. 43, 243–248 (1958).
Fuchs, L. H. & Hoekstra, H. R. The preparation and properties of uranium(IV) silicate. Am. Mineral. 44, 1057–1063 (1959).
Pointer, C. M., Ashworth, J. R. & Ixer, R. A. The zircon-thorite mineral group in metasomatized granite, Ririwai, Nigeria. 1. Geochemistry and metastable solid-solution of thorite and coffinite. Mineral. Petrol. 38, 245–262 (1988).
Pointer, C. M., Ashworth, J. R. & Ixer, R. A. The zircon-thorite mineral group in metasomatized Granite, Ririwai, Nigeria. 2. Zoning, alteration and exsolution in zircon. Mineral. Petrol. 39, 21–37 (1988).
Finch, R. J. & Hanchar, J. M. Structure and chemistry of zircon and zircon-group minerals. Rev. Mineral. Geochem. 53, 1–25 (2003).
Forster, H. J. Composition and origin of intermediate solid solutions in the system thorite-xenotime-zircon-coffinite. Lithos 88, 35–55 (2006).
Costin, D. T. et al. How to explain the difficulties in the coffinite synthesis from the study of uranothorite? Inorg. Chem. 50, 11117–11126 (2011).
Szenknect, S. et al. From uranothorites to coffinite: a solid solution route to the thermodynamic properties of USiO4. Inorg. Chem. 52, 6957–6968 (2013).
Clavier, N. et al. Purification of uranothorite solid solutions from polyphase systems. J. Nucl. Mater. 441, 73–83 (2013).
Labs, S. et al. Synthesis of coffinite, USiO4, and structural investigations of UxTh(1-x)SiO4 solid solutions. Environ. Sci. Technol. 48, 854–860 (2014).
Guo, X. et al. Energetics of a uranothorite (Th1−xUxSiO4) solid solution. Chem. Mater. 28, 7117–7124 (2016).
Ifill, R. O., Cooper, W. C. & Clark, A. H. Mineralogical controls on the oxidative acid leaching of radioactive phases in Elliot Lake Ores—brannerite, uraninite and uranothorite. CIM Bull. 80, 72–72 (1987).
Smits, G. Uranium-Thorium Silicates, with Specific Reference to the Species in the Witwatersrand Reefs (Council for Mineral Technology, 1987).
Lira, R. & Ripley, E. M. Hydrothermal alteration and REE-Th mineralization at the Rodeo-De-Los-Molles Deposit, Las-Chacras-Batholith, Central Argentina. Contrib. Mineral. Petrol. 110, 370–386 (1992).
Sharma, G. S., Purohit, R. K., Roy, M., Sengupta, B. & Singh, J. Geochemistry and petrography of U-Th-Y mineralisation in alkali feldspar granite (Alaskite) dykes around Dhanota, Mahendragarh District, Haryana, India. J. Geol. Soc. India 55, 189–196 (2000).
Lutze, W. & Ewing, R. C. Radioactive Waste Forms for the Future (Elsevier Science Pub. Co., Inc, 1988).
Ewing, R. C., Lutze, W. & Weber, W. J. Zircon—a host-phase for the disposal of weapons plutonium. J. Mater. Res. 10, 243–246 (1995).
Ewing, R. C. Nuclear waste forms for actinides. Proc. Natl Acad. Sci. USA 96, 3432–3439 (1999).
Yudintsev, S. V., Stefanovsky, S. & Ewing, R. C. in Structural Chemistry of Inorganic Actinide Compounds (eds. Krivovichev, S. V. & Tananaev, I.) 457–490 (Elsevier, 2007).
Ewing, R. & Weber, W. in The Chemistry of the Actinide and Transactinide Elements 3813–3887 (Springer, 2010).
Weber, W. J. & Ewing, R. C. in Uranium—Cradle to Grave Vol. Short Course (eds. Burns, P. C. & Sigmon, G. E.) Ch. 10, 317–336 (Mineralogical Association of Canada, 2013).
Janeczek, J. & Ewing, R. Coffinitization—a mechanism for the alteration of UO2 under reducing conditions. In Scientific Basis for Nuclear Waste Management Xv Vol. 257, 497–504 (Cambridge University Press, 1992).
Bruno, J., Duro, L. & Grivé, M. The applicability and limitations of thermodynamic geochemical models to simulate trace element behaviour in natural waters. Lessons learned from natural analogue studies. Chem. Geol. 190, 371–393 (2002).
Pomiès, C., Hamelin, B., Lancelot, J. & Blomqvist, R. 207Pb/206Pb and 238U/230Th dating of uranium migration in carbonate fractures from the Palmottu uranium ore (southern Finland). Appl. Geochem. 19, 273–288 (2004).
Deditius, A. P., Utsunomiya, S. & Ewing, R. C. The chemical stability of coffinite, USiO4·nH2O; 0 < n < 2, associated with organic matter: a case study from Grants uranium region, New Mexico, USA. Chem. Geol. 251, 33–49 (2008).
Amme, M., Wiss, T., Thiele, H., Boulet, P. & Lang, H. Uranium secondary phase formation during anoxic hydrothermal leaching processes of UO2 nuclear fuel. J. Nucl. Mater. 341, 209–223 (2005).
Macmillan, E., Cook, N. J., Ehrig, K., Pring, A. & Krivovichev, S. Chemical and textural interpretation of late-stage coffinite and brannerite from the Olympic Dam IOCG-Ag-U deposit. Mineral. Mag. 81, 1323–1366 (2017).
Szenknect, S. et al. Coffinite formation from UO2+x. Sci. Rep. 10, 12168 (2020).
Ding, H. et al. Safely probing the chemistry of Chernobyl nuclear fuel using micro-focus X-ray analysis. J. Mater. Chem. A 9, 12612–12622 (2021).
Gaucher, É. C. et al. Modelling the porewater chemistry of the Callovian–Oxfordian formation at a regional scale. C. R. Rendus Geosci. 338, 917–930 (2006).
Gaucher, E. C. et al. A robust model for pore-water chemistry of clayrock. Geochim. Cosmochim. Acta 73, 6470–6487 (2009).
Hall, D. S., Behazin, M., Binns, W. J. & Keech, P. G. An evaluation of corrosion processes affecting copper-coated nuclear waste containers in a deep geological repository. Prog. Mater. Sci. 118, 100766 (2020).
Keller, C. Untersuchungen über die germanate und silikate des typs ABO4 der vierwertigen elemente thorium bis americium. Nukleonik 5, 41–48 (1963).
Speer, J. The actinide orthosilicates. Rev. Mineral. Geochem. 5, 113–135 (1980).
Burakov, B. E. et al. Synthesis and study of (239)Pu-doped ceramics based on zircon, (Zr,Pu)SiO4, and hafnon, (Hf,Pu)SiO4. In Scientific Basis for Nuclear Waste Management Xxiv, Vol. 663, 307–313 (Cambridge University Press, 2000).
Skakle, J., Dickson, C. & Glasser, F. The crystal structures of CeSiO4 and Ca2Ce8(SiO4)6O2. Powder Diffr. 15, 234–238 (2000).
Gibb, F. G. F., Taylor, K. J. & Burakov, B. E. The ‘granite encapsulation’ route to the safe disposal of Pu and other actinides. J. Nucl. Mater. 374, 364–369 (2008).
Schlüter, J., Malcherek, T. & Husdal, T. A. The new mineral stetindite, CeSiO4, a cerium end-member of the zircon group. Neues Jahrb. Mineral. Abhandlungen: J. Mineral. Geochem. 186, 195–200 (2009).
Guo, X. et al. Thermodynamics of formation of coffinite, USiO4. Proc. Natl Acad. Sci. USA 112, 6551–6555 (2015).
Estevenon, P. et al. Preparation of synthetic CeSiO4 from aqueous precursors under soft hydrothermal conditions. Inorg. Chem. 48, 7551–7559 (2019).
Estevenon, P. et al. Formation of CeSiO4 from cerium (III) silicate precursors. Dalton Trans. 48, 10455–10463 (2019).
Estevenon, P. et al. The formation of PuSiO4 under hydrothermal conditions. Dalton Trans. 49, 6434–6445 (2020).
Strzelecki, A. C. et al. Thermodynamics of CeSiO4: implications for actinide orthosilicates. Inorg. Chem. 59, 13174–13183 (2020).
Strzelecki, A. C. et al. High temperature thermodynamics of cerium silicates, A-Ce2Si2O7 and Ce4.67(SiO4)3O. ACS Earth Space Chem. 4, 2129–2143 (2020).
Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 51, 1010–1026 (1929).
Navrotsky, A., Shvareva, T. & Guo, X. in Uranium—Cradle to Grave Vol. Short Course (eds. Burns, P.C. & Sigmon, G. E.) Ch. 5, 147–164 (Mineralogical Association of Canada, 2013).
Mesbah, A. et al. Coffinite, USiO4, is abundant in nature: so why is it so difficult to synthesize? Inorg. Chem. 54, 6687–6696 (2015).
Szenknect, S. et al. First experimental determination of the solubility constant of coffinite. Geochim. Cosmochim. Acta 181, 36–53 (2016).
Strzelecki, A. C. et al. Role of water and hydroxyl groups in the structures of stetindite and coffinite, MSiO4 (M = Ce, U). Inorg. Chem. 60, 718–735 (2020).
Shannon, R. D. Revised Effective ionic-radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 32, 751–767 (1976).
Ferriss, E. D. A., Ewing, R. C. & Becker, U. Simulation of thermodynamic mixing properties of actinide-containing zircon solid solutions. Am. Mineral. 95, 229–241 (2010).
Greenburg, H. & Wen, J. Repository layout and host rock thermal gradient trade study for large waste packages in clay/shale: using the DSEF thermal analytical model. LLNL-TR-639869-38 (2013).
Davies, P. K. & Navrotsky, A. Thermodynamics of solid solution formation in NiO-MgO and NiO-ZnO. J. Solid State Chem. 38, 264–276 (1981).
Nisbet, H. et al. An experimental study of the solubility and speciation of thorium in chloride-bearing aqueous solutions at temperatures up to 250 C. Geochim. Cosmochim. Acta 239, 363–373 (2018).
Timofeev, A. et al. Uranium transport in acidic brines under reducing conditions. Nat. Commun. 9, 1469 (2018).
Migdisov, A. A., Guo, X., Nisbet, H., Xu, H. & Williams-Jones, A. E. Fractionation of REE, U, and Th in natural ore-forming hydrothermal systems: thermodynamic modeling. J. Chem. Thermodyn. 128, 305–319 (2019).
Goldschmidt, V. M. The principles of distribution of chemical elements in minerals and rocks. The Seventh Hugo Müller Lecture, delivered before the Chemical Society on March 17th, 1937. J. Chem. Soc. (Resumed), 655–673 (1937).
Ringwood, A. The principles governing trace element distribution during magmatic crystallization. Part I: The influence of electronegativity. Geochim. Cosmochim. Acta 7, 189–202 (1955).
Cowley, J. An approximate theory of order in alloys. Phys. Rev. 77, 669 (1950).
de Fontaine, D. The number of independent pair-correlation functions in multicomponent systems. J. Appl. Crystallogr. 4, 15–19 (1971).
Owen, L., Playford, H., Stone, H. & Tucker, M. Analysis of short-range order in Cu3Au using X-ray pair distribution functions. Acta Mater. 125, 15–26 (2017).
O’Quinn, E. C. et al. Predicting short-range order and correlated phenomena in disordered crystalline materials. Sci. Adv. 6, eabc2758 (2020).
Mursic, Z., Vogt, T. & Frey, F. High-temperature neutron powder diffraction study of ZrSiO4 up to 1900 K. Acta Crystallogr. B 48, 584–590 (1992).
Mondal, S. K., Das, P. K., Mandal, N. & Arya, A. A novel approach to the structural distortions of U/Th snub-disphenoids and their control on zircon→reidite type phase transitions of U1−xThxSiO4. J. Phys. Condens. Matter 32, 145401 (2020).
Neidig, M. L., Clark, D. L. & Martin, R. L. Covalency in f-element complexes. Coord. Chem. Rev. 257, 394–406 (2013).
Vitova, T. et al. The role of the 5f valence orbitals of early actinides in chemical bonding. Nat. Commun. 8, 16053 (2017).
Su, L., Wan, L., Gao, T. & Ao, B. First-principles calculations of the electronic structure, chemical bonding, and thermodynamic properties of USiO4. AIP Adv. 10, 075018 (2020).
Savin, A., Silvi, B. & Colonna, F. Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem. 74, 1088–1096 (1996).
Davies, P. K. & Navrotsky, A. Quantitative correlations of deviations from ideality in binary and pseudobinary solid-solutions. J. Solid State Chem. 46, 1–22 (1983).
Guo, X. et al. A. charge-coupled substituted garnets (Y3−xCa0.5xM0.5x)Fe5O12 (M = Ce, Th): structure and stability as crystalline nuclear waste forms. Inorg. Chem. 54, 4156–4166 (2015).
Guo, X. et al. Structure and thermodynamics of uranium containing iron garnets. Geochim. Cosmochim. Acta 189, 269–281 (2016).
O’Neil, H. S. C. & Navrotsky, A. Cation distributions and thermodynamic properties of binary spinel solid solutions. Am. Mineral. 69, 733–753 (1984).
Fei, Y., Saxena, S. & Eriksson, G. Some binary and ternary silicate solution models. Contrib. Mineral. Petrol. 94, 221–229 (1986).
Holland, T. & Powell, R. Thermodynamics of order-disorder in minerals: II. Symmetric formalism applied to solid solutions. Am. Mineral. 81, 1425–1437 (1996).
Ganguly, J. Thermodynamic modelling of solid solutions. EMU Notes Mineral. 3, 37 (2001).
Jenkins, H. D. B. & Glasser, L. Standard absolute entropy, values from volume or density. 1. inorganic materials. Inorg. Chem. 42, 8702–8708 (2003).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Leitner, J., Sedmidubsky, D., Růžička, K. T. & Svobo, P. in Applications of Calorimetry in a Wide Context-Differential Scanning Calorimetry, Isothermal Titration Calorimetry and Microcalorimetry, Elkordy A. A. (ed) 385–406 (IntechOpen, 2013).
Eser, E., Koc, H., Gokbulut, M. & Gursoy, G. Estimations of heat capacities for actinide dioxide: UO2, NpO2, ThO2, and PuO2. Nucl. Eng. Technol. 46, 863–868 (2014).
Szenknect, S., Dacheux, N., Ewing, R. & Navrotsky, A. Reply to comment by Konings and Plyasunov on “First experimental determination of the solubility constant of coffinite” [Geochim. Cosmochim. Acta 181 (2016) 36–53]. Geochim. Cosmochim. 212, 374–376 (2017).
Holland, T. & Powell, R. An internally consistent thermodynamic data set for phases of petrological interest. J. Metamorph. Geol. 16, 309–343 (1998).
Robie, R. A. & Hemingway, B. S. Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar pressure and at higher temperatures. U.S. Geol. Surv. Bull. 2131 (1995).
Shvareva, T. Y., Fein, J. B. & Navrotsky, A. Thermodynamic properties of uranyl minerals: constraints from calorimetry and solubility measurements. Ind. Eng. Chem. Res. 51, 607–613 (2011).
Guillaumont, R. et al. Chemical Thermodynamics Vol. 5. Update on the Chemical Thermodynamics of Uranium, Neptunium, Plutonium, Americium and Technetium (ELSEVIER, 2003).
Johnson, J. W., Oelkers, E. H. & Helgeson, H. C. SUPCRT92: a software package for calculating the standard molal thermodynamic properties of minerals, gases, aqueous species, and reactions from 1 to 5000 bar and 0 to 1000 C. Comput. Geosci. 18, 899–947 (1992).
Shvarov, Y. V. & Bastrakov, E. HCh: a software package for geochemical equilibrium modelling. User’s Guide 25, 60 (1999).
Shvarov, Y. V. OptimA: A Program for the Calculation of the Free Energies of Dissolved Aqueous Species from the Results of Chemical Experiments. http://www.geol.msu.ru/deps/geochems/soft/ (2010).
Secretariat, C. CODATA recommended key values for thermodynamics 1977. CODATA Bull. 28, 903–906 (1978).
Mercadier, J., Cuney, M., Cathelineau, M. & Lacorde, M. U redox fronts and kaolinisation in basement-hosted unconformity-related U ores of the Athabasca Basin (Canada): late U remobilisation by meteoric fluids. Miner. Deposita 46, 105–135 (2011).
Chi, G. et al. Uranium-rich diagenetic fluids provide the key to unconformity-related uranium mineralization in the Athabasca Basin. Sci. Rep. 9, 1–10 (2019).
Wang, G., Batista, E. R. & Yang, P. Excess electrons on reduced AnO2 (111) surfaces (An= Th, U, Pu) and their impacts on catalytic water splitting. J. Phys. Chem. C 123, 30245–30251 (2019).
Guo, X. et al. Energetics of hydration on uranium oxide and peroxide surfaces. J. Mater. Res. 34, 3319–3325 (2019).
Wang, G., Batista, E. R. & Yang, P. Water on actinide dioxide surfaces: a review of recent progress. Appl. Sci. 10, 4655 (2020).
Finch, R. & Murakami, T. Systematics and paragenesis of uranium minerals. Rev. Mineral. 38, 152–154 (1999).
Prodan, I. D., Scuseria, G. E. & Martin, R. L. Covalency in the actinide dioxides: systematic study of the electronic properties using screened hybrid density functional theory. Phys. Rev. B 76, 033101 (2007).
Wen, X.-D., Martin, R. L., Henderson, T. M. & Scuseria, G. E. Density functional theory studies of the electronic structure of solid state actinide oxides. Chem. Rev. 113, 1063–1096 (2013).
Butorin, S. M., Kvashnina, K. O., Vegelius, J. R., Meyer, D. & Shuh, D. K. High-resolution X-ray absorption spectroscopy as a probe of crystal-field and covalency effects in actinide compounds. Proc. Natl Acad. Sci. USA 113, 8093–8097 (2016).
Costin, D. T. et al. Preparation and characterization of synthetic Th0.5U0.5SiO4 uranothorite. Prog. Nucl. Energy 57, 155–160 (2012).
Neuefeind, J., Feygenson, M., Carruth, J., Hoffman, R. & Chipley, K. K. The Nanoscale Ordered MAterials Diffractometer NOMAD at the Spallation Neutron Source SNS. Nucl. Instrum. Methods Phys. Res. B 287, 68–75 (2012).
Toby, B. H. & Dreele, Von R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Thompson, P., Cox, D. & Hastings, J. Rietveld refinement of Debye–Scherrer synchrotron X-ray data from Al2O3. J. Appl. Crystallogr. 20, 79–83 (1987).
Fuchs, L. H. & Gerbert, E. X-ray studies of synthetic coffinite, thorite, and uranothorites. Am. Mineral. 43, 243–248 (1958).
Xu, H. et al. Crystal structure and thermodynamic stability of Ba/Ti-substituted pollucites for radioactive Cs/Ba immobilization. J. Am. Ceram. Soc. 98, 2634–2640 (2015).
Xu, H., Guo, X. & Bai, J. M. Thermal behavior of polyhalite: a high-temperature synchrotron XRD study. Phys. Chem. Miner. 44, 125–135 (2017).
Guo, X. et al. H. Bulk moduli and high pressure crystal structure of U3Si2. J. Nucl. Mater. 523, 135–142 (2019).
Lü, X. et al. Regulating off-centering distortion maximizes photoluminescence in halide perovskites. Natl. Sci. Rev. nwaa288 (2020).
Tucker, M. G., Keen, D. A., Dove, M. T., Goodwin, A. L. & Hui, Q. RMCProfile: reverse Monte Carlo for polycrystalline materials. J. Phys. Condens. Matter 19, 1–16 (2007).
Farrow, C. L. et al. PDFfit2 and PDFgui: computer programs for studying nanostructure in crystals. J. Phys. Condens. Matter 19, 335219 (2007).
Toby, B. H. R factors in Rietveld analysis: How good is good enough? Powder Diffr. 21, 67–70 (2006).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Syakur, M. A., Khotimah, B. K., Rochman, E. M. S. & Satoto, B. D. Integration K-means clustering method and elbow method for identification of the best customer profile cluster. IOP Conf. Ser. Mater. Sci. 336, 1–6 (2017).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Guo, X. et al. U(V) in metal uranates: a combined experimental and theoretical study of MgUO4, CrUO4 and FeUO4. Dalton Trans. 45, 4622–4632 (2016).
Emery, A. A. & Wolverton, C. High-throughput dft calculations of formation energy, stability and oxygen vacancy formation energy of abo 3 perovskites. Sci. Data 4, 170153 (2017).
Glushko, V. P. & Gurvich, L. V. Termodinamicheskie svoistva individualnykh veshchestv (Akademiia nauk SSSR. Leningrad, 1962).
Robie, R. A. Thermodynamic properties of minerals and related substances at 298.15° K and 1 bar (105 pascals) pressure and at higher temperatures. (U.S. Geological Survey, 1978).
Oelkers, E. H. & Helgeson, H. C. Calculation of activity coefficients and degrees of formation of neutral ion pairs in supercritical electrolyte solutions. Geochim. Cosmochim. Acta 55, 1235–1251 (1991).
Helgeson, H. C., Kirkham, D. H. & Flowers, G. C. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes by high pressures and temperatures; IV, Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600 degrees C and 5kb. Am. J. Sci. 281, 1249–1516 (1981).
Acknowledgements
This work was supported by the institutional funds from the Department of Chemistry and New Faculty Seed Grant from ORAP at Washington State University. Funding for E.T.N. and J.M. was provided by the Department of Energy (DOE) Waste Treatment & Immobilization Plant (WTP) Federal Project Office under the direction of Dr. Albert A. Kruger, contract numbers DE-EM0002904 and DE-EM0003207. Funding for J.S.M.C. was provided by DOE Office of Nuclear Energy, Nuclear Energy University Programs, award # DE-NE0008689. Research presented in this article was also supported by the Laboratory Directed Research and Development (LDRD) program of Los Alamos National Laboratory (LANL) under project number 20180007 DR (to H.X.). LANL, an affirmative action/equal opportunity employer, is managed by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE under contract 89233218CNA000001. A portion of this research used resources at the Spallation Neutron Source, the Oak Ridge National Laboratory, was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract No. DE-AC05- 00OR22725. We also acknowledge the support from Alexandra Navrotsky Institute for Experimental Thermodynamics and the WSU-PNNL Nuclear Science and Technology Institute. We thank Dr. Francisco Espinosa for discussion and Mr. Ke (Edward) Zhao for help with visualization.
Author information
Authors and Affiliations
Contributions
X.G. conceived the research. X.G., H.X., and J.C.N. performed neutron scattering experiments. A.M., S.S., and N.D. synthesized all phases. J.M., Y.Z., and X.G. analyzed and refined all nPDF data. H.X. analyzed all ND data. X.Z. and L.Q. performed DFT calculations. X.G and A.A.M. performed multi-component system thermodynamic modeling. R.C.E. and J.S.M. advised on the nuclear waste form application. All authors, J.M., Y.Z., X.Z., H.X., A.M., E.T.N., S.S., J.C.N., J.L., L.Q., A.A.M., R.C.E., N.D., J.S.M., and X.G., participated in discussions, interpretation of the data, writing and editing of the manuscript. J.M., Y.Z., and X.Z. have equal contribution and are considered as co-first authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marcial, J., Zhang, Y., Zhao, X. et al. Thermodynamic non-ideality and disorder heterogeneity in actinide silicate solid solutions. npj Mater Degrad 5, 34 (2021). https://doi.org/10.1038/s41529-021-00179-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00179-0
