
Abstract
Telomere biology disorders (TBD) are a heterogeneous group of diseases arising from germline mutations affecting genes involved in telomere maintenance. Telomeres are DNA-protein structures at chromosome ends that maintain chromosome stability; their length affects cell replicative potential and senescence. A constellation of bone marrow failure, pulmonary fibrosis, liver cirrhosis and premature greying is suggestive, however incomplete penetrance results in highly variable manifestations, with idiopathic pulmonary fibrosis as the most common presentation. Currently, the true extent of TBD burden is unknown as there is no established diagnostic criteria and the disorder often is unrecognised and underdiagnosed. There is no gold standard for measuring telomere length and not all TBD-related mutations have been identified. There is no specific cure and the only treatment is organ transplantation, which has poor outcomes. This review summarises the current literature and discusses gaps in understanding and areas of need in managing TBD.
Similar content being viewed by others
Introduction
Telomeres are noncoding repeats of the TTAGGG nucleotide sequence at chromosomal ends that provide genomic stability1,2. They shorten with each cellular division, triggering senescence and apoptosis when critically short1,3,4. Genetic mutations affecting telomere homoeostasis, thus, result in disorders of premature aging, with various manifestations like bone marrow failure, pulmonary fibrosis, liver cirrhosis, premature greying and increased cancer risk5,6,7,8,9. Various terms such as short telomere syndrome, telomeropathies and telomere biology disorders (TBD) have been employed to describe these diseases; in this review, we use the term TBD as it reflects the spectrum of the disorders and pathophysiology4,8.
The archetypal TBD is dyskeratosis congenita (DC), which was first described by Zinsser in the early twentieth century as a mucocutaneous triad of skin pigmentation, nail dystrophy and oral leukoplakia, with other systemic features identified later8. DC accounts for less than 5% of all TBD and has an estimated incidence of 1 in 1,000,0008. It is estimated that 41% of familial mixed haematologic and interstitial lung disease (ILD) and 3% of familial haematologic disorders are due to TBD, however the protean presentation makes recognition challenging and the overall incidence is unknown6.
The first DC-related mutations were described when DKC1 mutations were identified as the cause of X-linked DC10. It is now known that DC and other TBD arise from germline mutations affecting telomeres5,6,7,10,11,12,13,14,15,16,17,18,19. Inheritance can be X-linked recessive, autosomal dominant (AD) or autosomal recessive (AR) and incomplete penetrance results in manifestations of varying degrees of severity at different time points within the same pedigree5,8,17,20. Anticipation is also demonstrated with earlier, more severe presentations due to progressive telomere shortening through the generations5,20. Although there is now a greater understanding of telomere biology (Fig. 1), many mechanisms of telomere dysfunction have not been identified. This review summarises the current literature and discusses gaps in understanding and areas of need in managing TBD.

Since dyskeratosis congenita was first described in the early 1900s, there have been significant developments in the knowledge of telomere biology over the last century. It is now known that telomeres are DNA-protein structures found at the ends of chromosomes and provide stability to chromosomes and prevent deterioration during cellular replication. Telomere length attrition thus leads to cellular senescence and triggers cell death pathways. Telomere biology disorders are a group of monogenic disorders of premature aging arising due to accelerated shortening of telomere lengths. It has a highly variable presentation due to various germline mutations and incomplete penetrance. Clinical phenotypes vary from multisystem disorders presenting in childhood such as dyskeratosis congenita and single organ disorders such as idiopathic pulmonary fibrosis, which may present in late adulthood. Despite the significant advancement, the true extent of the syndrome remains unknown and further genetic mutations have yet to be identified.
Mechanisms/pathophysiology
Telomere biology
Telomeres are crucial for chromosomal stability and protect them from deterioration during mitosis or aberrant fusion with neighbouring chromosomes1,4. They address the “end-replication” problem that describes DNA loss and progressive chromosomal shortening with each cell division3. Linear DNA and broken chromosomes are unstable and have a propensity to fuse21. Critical telomere shortening or broken chromosomes, thus, result in aberrant recombination with end-to-end fusions and “breakage-fusion-bridge” cycles, which are termed “crisis”1,4,21. Crisis is characterised by replicative senescence, chromosome end-to-end fusions and extensive apoptosis, causing uneven derivative chromosomes and genomic instability21.
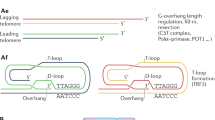
The telomere structure may result in DNA damage responses recognising their ends as double-stranded DNA breaks and various mechanisms help to maintain its integrity (Fig. 2). Telomeres have a stabilising “D-loop-T-loop” configuration; the T-loop is formed by insertion of the single-stranded G-strand terminus into the double-stranded telomere sequence, it displaces the sequence strand of the duplex telomeric DNA and forms the D-loop at the point of insertion22. Telomeres are protected by shelterin, which is a protein complex that generates and stabilises the “T-loop” structure, and facilitates and regulates telomere elongation4,23. It comprises six subunits, namely, telomere repeat-binding factor 1 (TRF1), telomere repeat-binding factor 2 (TRF2), repressor/activator protein 1 (RAP1), TRF1-interacting nuclear protein 2 (TIN2), TIN2-interacting protein 1 (TPP1) and protection of telomeres 1 (POT1)23. TRF1, TRF2 and POT1 localise the telomeric sequence, while TIN2, TPP1 and RAP1 form a complex that distinguishes telomeres from sites of DNA damage4,23. Ku is a DNA-end-binding heterodimer involved in DNA repair, but also associates with telomeres and protects them from recombination and degradation, regulates telomere addition and maintains telomere length (TL)24.

Telomeres are noncoding tandem repeats of the sequence TTAGGG, found at the ends of chromosomes in a duplex “D-loop-T-loop” configuration. RTEL1 is a helicase that disrupts T-loops for telomere replication and repair. Shelterin protects telomeres from DNA damage surveillance and comprises of six polypeptide components: TRF1, TRF2, RAP1, TIN2, TPP1 and POT1. Telomerase comprises of the essential components TERT and TERC, which synthesise telomeres and maintain telomere length. Dyskerin forms a complex with NHP2, NOP10 and GAR1. It binds to TERC to stabilise the telomerase complex. TCAB1 regulates the recruitment of telomerase to telomeres. Mutations in any of these components results telomere dysfunction, manifesting as telomere biology disorders. DC, dyskeratosis congenita; GAR1, H/ACA ribonucleoprotein complex subunit 1; HHS, Hoyeraal–Hreidarsson Syndrome; IPF, idiopathic pulmonary fibrosis; NHP2, H/ACA ribonucleoprotein complex subunit 2; NOP10, H/ACA ribonucleoprotein complex subunit 3; POT1, protection of telomeres 1; RAP1, repressor/activator protein 1; RTEL1, regulator of telomere length 1; TCAB1, Telomerase Cajal body protein 1; TERT, telomerase reverse transcriptase; TERC, telomerase RNA component; TIN2, TRF1-interacting nuclear protein 2; TPP1, TIN2-interacting protein 1; TRF1, telomere repeat-binding factor 1; TRF2, telomere repeat-binding factor 2.
Telomerase is a ribonucleoprotein complex that adds telomere repeats to chromosomal ends and maintains TL2. Its essential components are telomerase reverse transcriptase (TERT) and an RNA component (TERC), which provides the template for nucleotide addition1. Immature TERC is non-functional with short extended oligoadenylated forms25. Its maturation involves PARN, which removes oligoadenylated tails that mark nuclear RNAs for exosome-dependent degradation25. In contrast, PAPD5 oligoadenylates immature TERC and thus targets it for exosome degradation26. The nuclear exome targeting (NEXT) complex directs a subset of noncoding RNAs for exosomal degradation and is also involved in TERC maturation27. It comprises three subunits, RNA helicase Mtr4p (hMTR4), RNA recognition motif (RRM)-containing RBM7 and zinc-knuckle ZCCHC8 proteins27,28. PARN “loss-of-function” mutations and ZCCHC8 loss result in reduced mature TERC, while PAPD5 increases TERC and increases TL26,28,29.
Dyskerin, which is encoded by DKC, binds the H/ACA box of TERC to stabilise the telomerase complex4,10,30. Dyskerin forms a core complex with NHP2, NOP10 and GAR1, and associates with other proteins like NAF1 and TCAB1 for assembly, trafficking, recruitment and stabilisation4,30,31,32. There are other important components of telomere replication such as RTEL1 (regulator of telomere length 1) and CST (CTC1-STN1-TEN1). RTEL1 is a helicase that disrupts the T-loop during telomere replication, and its dysfunction results in stalling of replication and T-loop excision, shortening TL33. CST is a single-stranded DNA complex consisting of conserved telomere protection component 1 (CTC1), suppressor of cdc thirteen 1 (STN1) and telomeric pathway with STN1 (TEN1)34. CST is essential for synthesising the complementary C-strand and limiting telomerase action to prevent G-strand overhang overextension, thus preventing telomeric DNA damage signalling34,35. Germline mutations causing dysregulation of any of the components involved in telomere homoeostasis thus results in TBD (Table 1).
Telomere length influences and effects
The Hayflick limit describes the fixed number of cell replications prior to senescence and apoptosis, which is triggered by short telomeres1,3. TL thus reflects somatic cell replicative history and life span, with shorter TL correlating with increasing age36,37. At birth, the average leucocyte TL (LTL) is estimated to be 10 kb and attrition rate ranges from 27 to 41 base pairs per year37,38. The factors affecting TL and attrition are complex, but genetics has significant influence. Telomeres are longer in individuals with African ancestry and LTL attrition in Down’s syndrome is thrice that of age-matched controls36,37. Although TL and variants in non-telomeric regions related to telomere maintenance are hereditable, variable local gene expression across different cell lines results in varying TL within an individual20,36,37,39. For example, testis have longer TL than other cell types and higher TERT expression, while cells with a higher rate of turnover like blood and lung epithelial cells, have a shorter TL36.
Variation in TL is also demonstrated between twins and family members, suggesting that extrinsic factors also affect TL and attrition36,39,40 (Fig. 3). Some of these include obesity, cigarette smoking and environmental exposures36,38,40. Exposures and chronic inflammatory states like obesity and recurrent infection, result in reactive oxygen species generation and DNA damage, leading to shorter telomeres and accelerated cellular aging38,40,41. Short telomeres are implicated in the degenerative defects of aging and carcinogenesis; however the exact mechanisms have not been fully delineated yet4,8.

Schema showing how intrinsic factors such as genetic variants influence telomere length and the influence of exposures and intrinsic factors like age, on telomere attrition.
Clinical presentation
Childhood onset syndromes
DC is the best described childhood TBD, it is a syndrome of inherited bone marrow failure with 80–90% developing aplastic anaemia (AA) or bone marrow failure by age 308,15,16,17,20. Inheritance may be X-linked recessive, AD, or AR, but sporadic cases also occur8,10,13,17,20. Although patients classically develop mucocutaneous manifestations and anaemia in adolescence, the severe X-linked form may present within the first decade of life, and presentation may be at any age (Fig. 4)8,10,13,15,16,17,18,20. There may be multisystem involvement; up to 20% develop pulmonary fibrosis, and other traits include liver disease, premature greying of hair, epiphora, developmental delay, short stature, osteoporosis, avascular necrosis of the hips or shoulders, urethral stenosis and oesophageal stenosis8,10,13,15,16,17,18,20. Malignancy risk is high and patients are particularly susceptible to epithelial cancers such as head and neck squamous cell carcinoma, anorectal and skin cancers, and haematological malignancies9,42. The most common cause of death is bone marrow failure, followed by pulmonary complications and malignancy8,9,17,20,42.

Each disorder has its own spectrum of clinical features. Hoyeraal–Hreidarsson Syndrome and Revesz Syndrome present early in childhood and are considered more severe subtypes of dyskeratosis congenita. Dyskeratosis congenita presents commonly in childhood but may manifest at any age. Patients classically present with the mucocutaneous triad of abnormal skin pigmentation, nail dyskeratosis and leukoplakia, and bone marrow failure is the most common cause of death. Idiopathic pulmonary fibrosis is the most common presentation and typically presents after the age of 60. A constellation of premature greying, bone marrow failure, pulmonary fibrosis and cryptogenic liver cirrhosis is suggestive of telomere biology disorders, but due to incomplete penetrance and various inheritance patterns of germline mutations, not all features may be present. The disease demonstrates anticipation with earlier and more severe manifestations down the generations. Across all phenotypes and ages, patients are at an increased risk of cancers.
The diagnostic criteria for DC are summarised in Table 215,16,43. The severe forms of DC, Hoyeraal–Hreidarrson Syndrome (HHS) and Revesz Syndrome (RS) often present in infancy, and patients often do not survive long enough to develop all the syndromic features or mucocutaneous manifestations15,16,17. HHS presents with cerebellar hypoplasia, immunodeficiency, progressive bone marrow failure and intrauterine growth retardation8,11,14,15,17,19. RS has the additional traits of Coats bilateral exudative retinopathy, although cerebral calcifications are often present8,15,16. More recently, Coats plus, which overlaps with RS, has been described and is characterised by exudative retinopathy, gastrointestinal vascular ectasia and leukoencephalopathy12.
Pulmonary manifestations
Idiopathic pulmonary fibrosis (IPF) is the most common TBD manifestation5,44. It is a disease of progressive lung fibrosis with a deteriorating disease course and median survival of 3 years45. It has a prevalence of 1.25–23.4 per 100,000 in Europe46. IPF commonly affects male smokers over 60 and typically has a usual interstitial pneumonia (UIP) pattern on chest high-resolution computed tomography (HRCT) or lung biopsy in the appropriate clinical context45.
At least 30% of patients with sporadic or familial pulmonary fibrosis have genetic predisposing factors that are known to increase the risk of pulmonary fibrosis45,47. TERT and TERC mutations account for 8–15% of familial and 1–3% of sporadic IPF, while other telomere-related mutations like NAF1, PARN, RTEL1, DKC1, TINF2 and ZCCHC8 account for 1-3% of familial cases5,28,29,31,44,47,48,49. FIP presents earlier with a more aggressive disease course than sporadic cases, and radiological and histological heterogeneity exists even within a pedigree5,44. Non-specific interstitial pneumonia (NSIP), cryptogenic organising pneumonia (COP), respiratory bronchiolitis-interstitial lung disease (RB-ILD), unclassifiable ILD and CPFE have also been described in FIP5,29,31,44 (Fig. 5).

The heterogenous presentation of telomere biology disorders manifestations may overlap and manifest at various time points or only following certain exposures or insults (e.g., radiation and immunosuppression as preconditioning regimens prior to bone marrow transplant may lead to pulmonary fibrosis in individuals who may have presented with isolated bone marrow failure). Regardless of clinical phenotype, patients are at increased risk of malignancies and disorders of premature aging. DC, dyskeratosis congenita; HHS, Hoyeraal–Hreidarsson Syndrome; ILD, interstitial lung disease; RS, Revesz Syndrome; TBD, telomere biology disorders.
TL below the 10th percentile of age-matched controls are found in sporadic and familial IPF with no telomere-related mutations44. Other sporadic forms of ILD have also been found to have shorter telomeres and telomere-related mutations50. TL below the 10th percentile correlate with poorer outcomes in IPF patients treated with immunosuppression and are associated with faster lung function deterioration, higher mortality and poorer lung transplantation outcomes across the different ILD subtypes44,50,51,52.
More recently, pulmonary arteriovenous malformations (AVM) have been described in patients with DC and TBD even in the absence of hepatopulmonary syndrome (HPS), another known manifestation of TBD53.
Haematological manifestations
Bone marrow failure from AA or myelodysplastic syndrome (MDS) may occur as an isolated manifestation of TBD, most commonly due to TERT or TERC mutations7,20,54. TBD cases who first present with AA are younger than those who first present with IPF, suggesting that AA is a more severe phenotype5,7,54. Telomerase-related mutations account for 3–5% of AA and TERC mutations have more severe disease; they also have significantly shorter LTL compared to idiopathic and secondary cases of AA7,54. A short LTL correlates with poorer outcomes in AA and MDS, including higher rates of transformation to acute leukaemia, poorer survival and higher rates of non-relapse mortality after transplantation55,56.
Other haematological conditions described in TBD include macrocytosis, isolated cytopenias, paroxysmal nocturnal haemoglobinuria and essential thrombocythaemia5,6,8,15,20,54 (Fig. 5). Immunodeficiencies like hypogammaglobulinemia, lymphopenia and T-cell dysfunction have also been described8,14,15,16,57. It is postulated that underlying immune dysfunction and accelerated immunosenescence increased cancer risk due to failure of cancer surveillance and contribute to poor tolerance of immunosuppression15,16,52,57.
Hepatobiliary manifestations
Up to 40% of patients with TBD have liver involvement6,58,59. Manifestations include transaminitis, non-alcoholic steatohepatitis, nodular regenerative hyperplasia, cryptogenic liver cirrhosis, noncirrhotic portal hypertension and HPS (Fig. 5)6,58,59,60. Biopsy findings include inflammatory and fibrotic components, hepatic nodular regeneration, cirrhosis and idiopathic hemosiderosis58,59,60. HPS in TBD typically presents during the first four decades of life, which is earlier than that for pulmonary fibrosis; thus when younger TBD patients present with breathlessness, HPS should be evaluated for, particularly in the absence of pulmonary fibrosis60.
Shorter TL have been found in non-alcoholic steatohepatitis, which is postulated to make them vulnerable to a “second hit”58,59. Besides telomere-related germline mutations, chronic cell injury and exposures to hepatotoxic drugs and alcohol may accelerate telomere attrition, contributing to progression to liver fibrosis59. Similarly, shorter telomeres, TERC and TERT promoter mutations are linked to the pathogenesis of various hepatobiliary tumours including hepatocellular carcinoma58,59.
Diagnosis
Measuring telomere length
Most methods that are currently employed measure the average TL61. Currently, there is no common reference or gold standard method to measure and trend TL. Furthermore, TL measured using different methods do not correlate well62. These factors limit objective comparisons between different cohorts and changes in TL within an individual over time.
The earliest method used to measure TL is terminal restriction fragment (TRF) analysis by Southern blot, which measures the average TRF. TL is calculated by comparing the electrophoresis-separated TRF against known molecular weight markers63. This is time consuming and requires substantial amounts of DNA. Furthermore, it overestimates TL as subtelomeric DNA is included in the measurement61,63.
Quantitative polymerase chain reaction (qPCR) is most commonly used. It involves using two primer pairs, one targeting telomere repeats (T) and another targeting a known single-copy gene (S)64. The ratio between the T and S amplification products (T/S ratio) is calculated and this correlates with TL; the relative difference in T/S ratio between samples is proportional to the relative difference in TL. Although it does not require a large amount of starting DNA, it does not give an absolute TL value and there may be substantial variation between cohorts due to different single-copy loci used61,64.
Quantification fluorescence in-situ hybridisation (Q-FISH) has several variations that have been used. Q-FISH quantifies the fluorescence intensity after hybridisation with a nucleic acid telomeric repeat, which is then compared against a reference population for a comparative assessment of TL65. Flow-FISH is a commonly used variation that can be used for clinical purposes. It combines FISH with flow cytometry, using labelled peptide nucleic acid probes, which hybridise to telomeric repeats in cells and allows measurement of TL in specific cell subpopulations66. Q-FISH results are reproducible but it is labour intensive.
Some methods used in research settings include Single-Telomere Length Analysis (STELA), which uses a single-molecule PCR to generate highly accurate TL measurements on individual chromosomes, and Telomere Shortest Length Assay (TeSLA), which measures the shortest telomeres and their longitudinal changes67,68. These methods are labour intensive, and low throughput67,68. The methods available to measure TL are summarised in Table 3; the selection of the test would also depend on the purpose as large-scale population-based studies would have different requirements from clinical or mechanistic ones.
Diagnosing TBD
Recognising and diagnosing TBD can be challenging due to the spectrum of presentations (Fig. 4). Heterogeneity within a pedigree exists due to incomplete penetration, anticipation and the influence of TL on disease severity5,6,7,17,20. Environmental insults like smoking may result in genetically predisposed individuals developing IPF earlier in life than nonsmokers, or the use of immunosuppression may result in increased susceptibility to bone marrow failure5,15,16,52. In addition, TERC and TERT mutations may present as isolated AA or IPF and be regarded as sporadic5,6,7,54. Furthermore, not all mutations have been identified and some cases thus remain genetically uncharacterised.
Diagnostic criteria based on the various systems such as pulmonary, haematological and liver should be defined to help facilitate recognition of TBD. A thorough family history and evaluation of other involved organ systems should be considered and we propose a possible schema to guide clinicians (Fig. 6).

HRCT, high-resolution computed tomography scan; IPF, idiopathic pulmonary fibrosis; PBMC, peripheral blood mononuclear cell; US, ultrasound.
Management
Transplantation and immunosuppression
There is no specific treatment for TBD and only transplant is curative. However, immunosuppression is often poorly tolerated and outcomes are worse in TBD. Shorter TL correlate with reduced survival and a faster onset of severe chronic lung allograft dysfunction in pulmonary fibrosis patients who undergo lung transplantation51. TBD lung transplant recipients also have a shorter median survival ranging 214 days to 1.9 years compared to non-TBD recipients69. Solid organ transplant recipient with TBD are at higher risk of complications such as severe cytopenias, higher infection rates, bone marrow failure, renal complications and calcineurin inhibitor toxicity69,70,71,72,73. Furthermore, myeloablative regimens used in bone marrow transplantation for TBD are associated with poorer survival and high rates of pulmonary, liver and endothelial related complications, including unusual complications like veno-occlusive disease15,74. Although non-myeloablative fludarabine-based regimens have fewer complications and allow for successful engraftments, TBD recipients may still develop complications in organs that were unaffected prior to transplantation15,75.
To date, seven liver transplants in TBD are reported with decompensated liver cirrhosis as the most common indication60,71,72,73,76,77. A combined liver and lung transplant has also been performed for noncirrhotic hepatopulmonary syndrome in end-stage pulmonary fibrosis71. Although there is now more experience in performing transplantation in TBD, it remains high risk and poses challenges. Further experience is needed to draw conclusions longer term outcomes of transplant in TBD, particularly with regards to liver and combined organ transplantations.
Therapeutics
Androgen hormonal therapy
Androgens have been shown to improve blood counts, transfusion dependence and liver fibrosis, while also stabilising lung function in TBD patients78,79. Although androgens directly increase TERT transcription and normalise telomerase activity in mouse models, effects on TL in clinical studies is mixed, with some demonstrating improvement while others have attrition rates similar to placebo group78,80,81. Further studies are still required before danazol can be routinely used in TBD and potential adverse effects should be considered. Common complications include transaminitis, muscle cramps, lipid abnormalities and virilisation, while rare complications such as splenic peliosis and rupture have been described with concomitant granulocyte colony-stimulating factor (GSCF) use78,79,80.
Non-transplant treatment in IPF
Treatment options are very limited for patients with IPF and currently only the antifibrotics pirfenidone and nintedanib are approved. They retard the rate of forced vital capacity deterioration by 50% a year and pooled analysis suggests mortality benefit, but they are non-curative and do not improve symptoms nor quality of life82,83. They are used to treat FIP; however patients have a persistently more aggressive trajectory compared to sporadic IPF and lung transplantation should be explored early in suitable patients84.
Non-transplant treatment for bone marrow failure
In acquired severe aplastic anaemia where transplant is not suitable, immunosuppression with the use of growth factors like GCSF, erythropoietin and eltrombopag can improve treatment response and accelerate count recovery85,86. GCSF does not improve trilineage response or overall survival, and potentially increases the risk of secondary clonal disorders like AML and MDS85,87. Concomitant use of GCSF with danazol should be used cautiously or avoided if possible due to the risk of splenic peliosis and rupture79,80. Eltrombopag has been shown to restore haematopoeisis even after discontinuation, however further study is needed before this can be used as monotherapy in severe aplastic anaemia86.
MDS should be managed according to risk stratification, lower risk patients may be considered for immunosuppression, thrombopoiesis-stimulating agents or DNA-hypomethylation agents such as azacytidine88. In higher risk non-transplant candidates, DNA-hypomethylation agents may be used as tolerated88. Supportive treatments like blood transfusions alleviate symptoms such as fatigue, and antimicrobial prophylaxis reduces the risk of opportunistic infections89.
Non-transplant treatment of liver cirrhosis
Advanced liver cirrhosis is often complicated by portal hypertension with sequelae like ascites, oesophageal varices, and HPS90. This may lead to fatal gastrointestinal tract bleeding and multi-organ failure. Medications like non-selective beta-blockers to reduce portal venous pressures and endoscopic interventions such as endoscopic band ligation to reduce variceal bleeding risk are often required90. Ascites is the most common cause of decompensated liver cirrhosis and a moderate sodium-restrictive diet with diuretics should be prescribed; large volume paracentesis may be required in symptomatic or massive ascites90. Hepatotoxic drugs and alcohol should be avoided, and laxatives prescribed to prevent constipation, which may precipitate hepatic encephalopathy. Patients with recurrent hepatic encephalopathy may require rifaximin prophylaxis90.
Potential new therapies and trials
Imetelstat is a 13-mer lipid-conjugated oligonucleotide that competitively inhibits telomerase and has been shown to inhibit cellular proliferation in tumour xenografts and cancer91. A pilot study in myelofibrosis demonstrated that 21% achieved partial or complete remission, while another trial in advanced non-small cell lung cancer showed a trend towards improvement in progression-free and overall survival in patients with short telomeres92,93. The main adverse effects were severe myelosuppression and transaminitis92,93. Further trials are underway to evaluate safety profile and efficacy.
Other investigational treatments include small molecule PAPD5 inhibitors, TA-65 and telomerase gene therapy94,95,96. PAPD5 inhibitors like BCH001 and RG7834, demonstrated restoration of telomerase activity and TL in DC induced pluripotent stem cells and mouse models94. TA-65 is a small molecule activator of telomerase that extended TL compared to placebo in healthy subjects95. Telomerase gene therapy using adenovirus-associated virus (AAV)9 gene therapy vectors has been shown to lengthen telomeres, improve blood counts and survival in mice with aplastic anaemia96. However, further studies are needed to evaluate safety and outcomes before these can be translated to further trials.
Quality of life
Early palliation should be commenced in TBD patients not suitable for transplant. Common areas of need identified in patients with bone marrow failure, IPF and advanced liver disease include improving quality of life through management of symptoms such as fatigue, dyspnoea and pain, psychological support, advanced care planning, and access to resources like specialised care and hospice services89,97,98,99. The disease burden in patients with single organ disease is high and may compounded in those with multisystem involvement.
Genetic counselling and testing
TBD is rare and complex with limited treatment options. A certain level of genetic literacy is thus needed for patients to make challenging informed decisions and share information with relatives100. Furthermore, inaccurate informal sources may further hinder understanding100. Although genetic counselling and patient education initiatives improve knowledge, surveyed patients still answer less than 60% of disease-specific and general genetic knowledge questions correctly when assessed, suggesting patient education has further room for improvement100.
In suspected cases of inherited bone marrow failure syndrome, other conditions to consider besides TBD would include Diamond-Blackfan anaemia, Fanconi anaemia and Shwachman–Diamond syndrome15. IPF cases are not routinely offered genetic counselling unless they present young and have features suggestive of FIP, due to a low pretest probability44,47. Patients with short TL should be offered screening for TBD, while those with a personal or family history of early-onset lung malignancy or disease, should be counselled for surfactant-related mutation screening45 (Fig. 6). A thorough assessment should be done prior to offering genetic testing with appropriate counselling on the risks and benefits, as a confirmatory diagnosis has far-reaching implications.
Outlook
Since DC was first described, there have been significant developments in telomere biology and its implications on senescence and disease pathogenesis. TBD is a syndrome of premature aging and the prototype for degenerative diseases, as telomeres shorten with age. It is likely the most common monogenic premature aging disorder and identifying its syndromic nature has important clinical implications as seemingly sporadic diseases share a common pathways of telomere dysfunction. Its role in more complex pathogenesis such as carcinogenesis, suggests that genotype–phenotype correlations extend beyond what is currently understood. Other mechanisms of telomere dysfunction and mutations will likely be identified with further genotype–phenotype correlations.
Future research would include standardisation of TL measurement methods and benchmarking TL across different cell types and ages. This would allow for comparison of different study results and establishing a gold standard for clinical testing, where TL potentially could have utility as a biomarker of disease predisposition, prognosis and treatment response.
Establishing clinical and genetic criteria for TBD diagnosis would be invaluable for risk stratification and genetic counselling. Increased awareness and education of medical practitioners can help early recognition and facilitate diagnosis. Multidisciplinary genetics clinics with genetic counselling and support services would also help improve care. Early advanced care planning should be initiated with open discussions to explore patients’ ideas, concerns, and expectations so that appropriate supportive care is received in a timely manner.
Regarding therapeutics, androgen treatment and novel therapies targeting telomere homoeostasis pathways are areas for development. These will require further trials before they are suitable for clinical use. If successful, given the involvement of telomere dysfunction in various disease pathways, such treatments could potentially be used to treat other diseases beyond TBD that are currently incurable.
Although various aspects of care discussed in this review are regarded as standard of care in TBD, in many parts of the world, these services and enrolment into trials may not be available or easily accessible to patients. Greater awareness of TBD is needed to improve overall care of patients with this disease.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Greider, C. W. & Blackburn, E. H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337, 331–337 (1989).
Greider, C. W. & Blackburn, E. H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 43, 405–413 (1985).
Levy, M. Z., Allsopp, R. C., Futcher, A. B., Greider, C. W. & Harley, C. B. Telomere end-replication problem and cell aging. J. Mol. Biol. 225, 951–960 (1992).
Bertuch, A. A. The molecular genetics of the telomere biology disorders. RNA Biol. 13, 696–706 (2016).
Armanios, M. Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326 (2007).
Feurstein, S. et al. Telomere biology disorder prevalence and phenotypes in adults with familial hematologic and/or pulmonary presentations. Blood Adv. 4, 4883–4886 (2020).
Yamaguchi, H. et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic. Anemia. N. Engl. J. Med. 352, 1413–1424 (2005).
Niewisch, M. R. & Savage, S. A. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev. Hematol. 12, 1037–1052 (2019).
Schratz, K. E. et al. Cancer spectrum and outcomes in the Mendelian short telomere syndromes. Blood 135, 1946–1956 (2020).
Heiss, N. S. et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 19, 32–38 (1998).
Dodson, L. M. et al. From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants. Hum. Mutat. 40, 2414–2429 (2019).
Polvi, A. et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am. J. Hum. Genet. 90, 540–549 (2012).
Walne, A. J. et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 16, 1619–1629 (2007).
Henslee, G., Williams, C., Liu, P. & Bertuch, A. A. Identification and characterization of novel ACD variants: Modulation of TPP1 protein level offsets the impact of germline loss-of-function variants on telomere length. Cold Spring Harb. Mol. Case Stud. https://doi.org/10.1101/mcs.a005454 (2021).
Alter, B. P. et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 110, 1439–1447 (2007).
Walne, A. J., Vulliamy, T., Beswick, R., Kirwan, M. & Dokal, I. TINF2 mutations result in very short telomeres: Analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112, 3594–3600 (2008).
Marrone, A. et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110, 4198–4205 (2007).
Vulliamy, T. et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl Acad. Sci. USA 105, 8073–8078 (2008).
Walne, A. J., Vulliamy, T., Kirwan, M., Plagnol, V. & Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 92, 448–453 (2013).
Vulliamy, T. et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 36, 447–449 (2004).
McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 26, 234–282 (1941).
Griffith, J. D. et al. Mammalian telomeres end in a large duplex loop. Cell 97, 503–514 (1999).
De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 (2005).
Lemon, L. D., Morris, D. K. & Bertuch, A. A. Loss of Ku’s DNA end binding activity affects telomere length via destabilizing telomere-bound Est1 rather than altering TLC1 homeostasis. Sci. Rep. https://doi.org/10.1038/s41598-019-46840-2 (2019).
Moon, D. H. et al. Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat. Genet. 47, 1482–1488 (2015).
Boyraz, B. et al. Posttranscriptional manipulation of TERC reverses molecular hallmarks of telomere disease. J. Clin. Invest. 126, 3377–3382 (2016).
Lubas, M. et al. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 43, 624–637 (2011).
Gable, D. L. et al. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 33, 1381–1396 (2019).
Stuart, B. D. et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 47, 512–517 (2015).
Mitchell, J. R., Wood, E. & Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402, 551–555 (1999).
Stanley, S. E. et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aaf7837. (2016).
Zhong, F. et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 25, 11–16 (2011).
Vannier, J. B., Pavicic-Kaltenbrunner, V., Petalcorin, M. I. R., Ding, H. & Boulton, S. J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806 (2012).
Feng, X. et al. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. https://doi.org/10.1038/s41467-018-05154-z (2018).
Keller, R. B. et al. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr. Blood Cancer 59, 311–314 (2012).
Demanelis, K. et al. Determinants of telomere length across human tissues. Science https://doi.org/10.1126/science.aaz6876 (2020).
Vaziri, H. et al. Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet. 52, 661–667 (1993).
Valdes, A. M. et al. Obesity, cigarette smoking, and telomere length in women. Lancet 366, 662–664 (2005).
Slagboom, P. E., Droog, S. & Boomsma, D. I. Genetic determination of telomere size in humans: A twin study of three age groups. Am. J. Hum. Genet. 55, 876–882 (1994).
Zhang, X., Lin, S., Funk, W. E. & Hou, L. Environmental and occupational exposure to chemicals and telomere length in human studies. Occup. Environ. Med. 70, 743–749 (2013).
Risom, L., Møller, P. & Loft, S. Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 592, 119–137 (2005).
Alter, B. P., Giri, N., Savage, S. A. & Rosenberg, P. S. Cancer in the national cancer institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39 (2018).
Dokal, I., Vulliamy, T., Mason, P. & Bessler, M. Clinical utility gene card for: Dyskeratosis congenita-update 2015. Eur. J. Hum. Genet. https://doi.org/10.1038/ejhg.2014.170 (2015).
Cronkhite, J. T. et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737 (2008).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, e44–e68 (2018).
Nalysnyk, L., Cid-Ruzafa, J., Rotella, P. & Esser, D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur. Respir. Rev. 21, 355–361 (2012).
Allen, R. J. et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir. Med. 5, 869–880 (2017).
Gaysinskaya, V., Stanley, S. E., Adam, S. & Armanios, M. Synonymous mutation in DKC1 causes telomerase RNA insufficiency manifesting as familial pulmonary fibrosis. Chest 158, 2449–2457 (2020).
Alder, J. K. et al. Exome sequencing identifies mutant TINF2 in a family with pulmonary fibrosis. Chest 147, 1361–1368 (2015).
Newton, C. A. et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur. Respir. J. https://doi.org/10.1183/13993003.01641-2018 (2019).
Newton, C. A. et al. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post–lung transplantation survival. J. Hear. Lung Transplant. 36, 845–853 (2017).
Newton, C. A. et al. Telomere length and use of immunosuppressive medications in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 200, 336–347 (2019).
Khincha, P. P. et al. Pulmonary arteriovenous malformations: an uncharacterised phenotype of dyskeratosis congenita and related telomere biology disorders. Eur. Respir. J. https://doi.org/10.1183/13993003.01640-2016 (2017).
Du, H. Y. et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood 113, 309–316 (2009).
Hwang, S. M. et al. Short telomere length and its correlation with gene mutations in myelodysplastic syndrome. J. Hematol. Oncol. https://doi.org/10.1186/s13045-016-0287-9 (2016).
Myllymäki, M. et al. Short telomere length predicts nonrelapse mortality after stem cell transplantation for myelodysplastic syndrome. Blood 136, 3070–3081 (2020).
Knudson, M., Kulkarni, S., Ballas, Z. K., Bessler, M. & Goldman, F. Association of immune abnormalities with telomere shortening in autosomal-dominant dyskeratosis congenita. Blood 105, 682–688 (2005).
Kapuria, D. et al. The spectrum of hepatic involvement in patients with telomere disease. Hepatology 69, 2579–2585 (2019).
Calado, R. T. et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS ONE https://doi.org/10.1371/journal.pone.0007926 (2009).
Gorgy, A. I. et al. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest 148, 1019–1026 (2015).
Montpetit, A. J. et al. Telomere length: a review of methods for measurement. Nurs. Res. 63, 289–299 (2014).
Khincha, P. P. et al. Correlation of leukocyte telomere length measurement methods in patients with dyskeratosis congenita and in their unaffected relatives. Int. J. Mol. Sci. https://doi.org/10.3390/ijms18081765 (2017).
Kimura, M. et al. Measurement of telomere length by the southern blot analysis of terminal restriction fragment lengths. Nat. Protoc. 5, 1596–1607 (2010).
Dahlgren, P. N., Bishop, K., Dey, S., Herbert, B. S. & Tanaka, H. Development of a new monochrome multiplex qPCR method for relative telomere length measurement in cancer. Neoplasia 20, 425–431 (2018).
Canela, A., Vera, E., Klatt, P. & Blasco, M. A. High-throughput telomere length quantification by FISH and its application to human population studies. Proc. Natl Acad. Sci. USA 104, 5300–5305 (2007).
Gutierrez-Rodrigues, F., Santana-Lemos, B. A., Scheucher, P. S., Alves-Paiva, R. M. & Calado, R. T. Direct comparison of Flow-FISH and qPCR as diagnostic tests for telomere length measurement in humans. PLoS ONE https://doi.org/10.1371/journal.pone.0113747 (2014).
Lai, T. P. et al. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat. Commun. https://doi.org/10.1038/s41467-017-01291-z (2017).
Baird, D. M., Rowson, J., Wynford-Thomas, D. & Kipling, D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat. Genet. 33, 203–207 (2003).
Silhan, L. L. et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur. Respir. J. 44, 178–187 (2014).
Tokman, S. et al. Clinical outcomes of lung transplant recipients with telomerase mutations. J. Hear. Lung Transpl. 34, 1318–1324 (2015).
Moschouri, E. et al. Combined lung and liver transplantation for short telomere syndrome. Liver Transpl. 26, 840–844 (2020).
Kolb, J. M. et al. Liver transplantation for decompensated cirrhosis secondary to telomerase reverse transcriptase mutation. Hepatology 72, 356–358 (2020).
del Brío Castillo, R. et al. Successful liver transplantation in short telomere syndromes without bone marrow failure due to DKC1 mutation. Pediatr. Transplant. https://doi.org/10.1111/petr.13695 (2020).
Rocha, V. et al. Unusual complications after bone marrow transplantation for dyskeratosis congenita. Br. J. Haematol. 103, 243–248 (1998).
Dietz, A. C. et al. Disease-specific hematopoietic cell transplantation: Nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transpl. 46, 98–104 (2011).
Mahansaria, S. S., Kumar, S., Bharathy, K. G. S., Kumar, S. & Pamecha, V. Liver transplantation after bone marrow transplantation for end stage liver disease with severe hepatopulmonary syndrome in dyskeratosis congenita: a literature first. J. Clin. Exp. Hepatol. 5, 344–347 (2015).
Valenti, L. et al. Liver transplantation for hepatocellular carcinoma in a patient with a novel telomerase mutation and steatosis. J. Hepatol. 58, 399–401 (2013).
Townsley, D. M. et al. Danazol treatment for telomere diseases. N. Engl. J. Med. 374, 1922–1931 (2016).
Khincha, P. P., Wentzensen, I. M., Giri, N., Alter, B. P. & Savage, S. A. Response to androgen therapy in patients with dyskeratosis congenita. Br. J. Haematol. 165, 349–357 (2014).
Khincha, P. P. et al. Similar telomere attrition rates in androgen-treated and untreated patients with dyskeratosis congenita. Blood Adv. 2, 1243–1249 (2018).
Bär, C., Huber, N., Beier, F. & Blasco, M. A. Therapeutic effect of androgen therapy in a mouse model of aplastic anemia produced by short telomeres. Haematologica 100, 1267–1274 (2015).
Lancaster, L. et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: Pooled data from six clinical trials. BMJ Open Respir. Res. https://doi.org/10.1136/bmjresp-2018-000397 (2019).
Noble, P. W. et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 47, 243–253 (2016).
Bennett, D. et al. Pirfenidone therapy for familial pulmonary fibrosis: a real-life study. Lung 197, 147–153 (2019).
Gluckman, E. et al. Results and follow-up of a phase III randomized study of recombinant human-granulocyte stimulating factor as support for immunosuppressive therapy in patients with severe aplastic anaemia. Br. J. Haematol. 119, 1075–1082 (2002).
Desmond, R. et al. Eltrombopag restores trilineage hematopoiesis in refractory severe aplastic anemia that can be sustained on discontinuation of drug. Blood 123, 1818–1825 (2014).
Socie, G. et al. Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European group for Blood and Marrow Transplantation (EBMT). Blood 109, 2794–2796 (2007).
Steensma, D. P. Myelodysplastic syndromes current treatment algorithm 2018. Blood Cancer J. https://doi.org/10.1038/s41408-018-0085-4 (2018).
Höchsmann, B., Moicean, A., Risitano, A., Ljungman, P. & Schrezenmeier, H. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 48, 168–173 (2013).
Angeli, P. et al. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 69, 406–460 (2018).
Ouellette, M. M., Wright, W. E. & Shay, J. W. Targeting telomerase-expressing cancer cells. J. Cell. Mol. Med. 15, 1433–1442 (2011).
Tefferi, A. et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N. Engl. J. Med. 373, 908–919 (2015).
Chiappori, A. A. et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 26, 354–362 (2015).
Nagpal, N. et al. Small-molecule PAPD5 inhibitors restore telomerase activity in patient stem cells. Cell Stem Cell 26, 896–909. e8 (2020).
Salvador, L. et al. A natural product telomerase activator lengthens telomeres in humans: a randomized, double blind, and placebo controlled study. Rejuvenation Res. 19, 478–484 (2016).
Bär, C. et al. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood 127, 1770–1779 (2016).
Steensma, D. P. et al. Common troublesome symptoms and their impact on quality of life in patients with myelodysplastic syndromes (MDS): Results of a large internet-based survey. Leuk. Res. 32, 691–698 (2008).
Low, J. T. S. et al. Supportive and palliative care in people with cirrhosis: International systematic review of the perspective of patients, family members and health professionals. J. Hepatol. 69, 1260–1273 (2018).
Lindell, K. O., Kavalieratos, D., Gibson, K. F., Tycon, L. & Rosenzweig, M. The palliative care needs of patients with idiopathic pulmonary fibrosis: a qualitative study of patients and family caregivers. Hear. Lung. 46, 24–29 (2017).
Hamilton, J. G. et al. Genetic information-seeking behaviors and knowledge among family members and patients with Inherited Bone Marrow Failure Syndromes. J. Genet. Couns. 24, 760–770 (2015).
Author information
Authors and Affiliations
Contributions
M.L.W.K. and J.Y.Y.N. conceptualised the idea. M.L.W.K. prepared the manuscript. All authors contributed to the design and wrote the paper. M.L.W.K. and T.T.T.N. conceptualised and created the figures and tables. J.Y.Y.N. provided critical review of the manuscript. All authors approved the manuscript for this submission and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kam, M.L.W., Nguyen, T.T.T. & Ngeow, J.Y.Y. Telomere biology disorders. npj Genom. Med. 6, 36 (2021). https://doi.org/10.1038/s41525-021-00198-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-021-00198-5
This article is cited by
-
Telomere Biology Disorder: A Focus on Gastrointestinal and Hepatic Manifestations
Current Hematologic Malignancy Reports (2024)
-
DNA methylation variations and epigenetic aging in telomere biology disorders
Scientific Reports (2023)
-
Impacts of radiation exposure, hindlimb unloading, and recovery on murine skeletal muscle cell telomere length
npj Microgravity (2023)
-
Approach Toward Germline Predisposition Syndromes in Patients with Hematologic Malignancies
Current Hematologic Malignancy Reports (2022)
