
Abstract
Primary coenzyme Q10 deficiency-7 (COQ10D7) is a rare mitochondrial disease caused by biallelic mutations in COQ4. Here we report the largest cohort of COQ10D7 to date, with 11 southern Chinese patients confirmed with biallelic COQ4 mutations. Five of them have the classical neonatal-onset encephalo-cardiomyopathy, while the others have infantile onset with more heterogeneous clinical presentations. We also identify a founder mutation COQ4 (NM_016035.5): c.370G>A, p.(Gly124Ser) for COQ10D7, suggesting a higher chance of occurrence in the southern Chinese. This study helps improve understanding of the clinical spectrum of this disorder.
Similar content being viewed by others

Introduction
Coenzyme Q10 (CoQ), also known as ubiquinone, is crucial for the function of mitochondrial respiratory chain complexes. Currently, the pathway of CoQ biosynthesis is known to involve at least 18 proteins. Primary CoQ deficiencies in humans involve genetic mutations in COQ2, PDSS1, PDSS2, COQ8A, COQ9, COQ6, COQ4, or COQ7.1
The primary CoQ10 deficiency-7 (COQ10D7, MIM: 616276) is caused by autosomal-recessive mutations in COQ4. COQ4 is hypothesized to take part in stabilizing the CoQ complex.2 To date, biallelic COQ4 mutations have been described in 16 patients from 11 unrelated families. They were reported in two case series (with five to six cases each) and three case reports. The patients described had common features of cardiomyopathy, encephalopathy, lactic acidosis often with a neonatal onset, and death in the neonatal or infantile period.2,3,4 Two siblings of childhood onset presenting with spinocerebellar ataxia and stroke-like episodes5 and two Chinese siblings with neonatal onset of dystonia, seizures, lactic acidosis, and cerebellar atrophy were described in recent case reports.6
In this report, we have 11 patients (4 males and 7 females) from 9 unrelated families who were managed by the Medical Genetics division of the Department of Paediatrics and Adolescent Medicine at the University of Hong Kong and the Department of Medical Genetics and Pediatrics at the National Taiwan University Hospital in the period of 2014–2018. They presented with two overlapping phenotypes: the classical neonatal-onset encephalo-cardiomyopathy and infantile-onset encephalopathy with or without cardiomyopathy. They were diagnosed as COQ10D7 due to homozygous or compound heterozygous COQ4 mutations. All genetic diagnoses were made by whole-exome sequencing (WES) except Patient 4, Patient 5, and Patient 11 due to their known family history and recognizable features of COQ10D7. More importantly, we have identified a Chinese-specific COQ4 founder mutation in 10 subjects, 5 of whom are homozygous for that mutation.
Case reports
The study was approved by the institutional review board of the University of Hong Kong/Hospital Authority Hong Kong West Cluster (UW12-211) and the National Taiwan University Hospital (201703073RINB). Written informed consent was obtained from subjects or their parents. A summary of the clinical, biochemical, and radiological characteristics of the 11 patients with COQ4 mutations identified is presented in Table 1. Patients 1–5 had the classical neonatal-onset phenotype described by Brea-Calvo et al. and Chung et al.,2,3 whereas Patients 6–11 had later onset and more heterogeneous features. The frequency of distinct phenotypes compared with previous studies is summarized in Table 2. The corresponding MRI (magnetic resonance imaging) images are presented in Fig. 1. The pedigrees of the nine families are presented in Fig. 2.

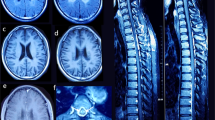
Cerebral magnetic resonance (MR) findings. a Axial T1W, b axial FLAIR, c DWI from Patient 1 at neonatal stage, and d axial T2W at follow-up; e axial T1W and f MR spectroscopy (MRS) at basal ganglia at neonate and g axial T1W at infant stages from Patient 4; h coronal T2W, i sagittal T1W, j axial T2W, k axial T1W from Patient 11. MR features include cerebellar atrophy (white arrows) (h, i) with progression (e, g); cerebral atrophy with frontal and anterior temporal lobar predominance (i–k); thinning of the corpus callosum (i); white matter loss and cystic change with frontal predominance (asterisks) (j, k); basal ganglia involvement with restricted diffusion and cystic change on follow-up (curved arrows) (a–d); lactate peak at around 1.3 ppm on MRS (arrowheads) (f)

Pedigrees of 9 families with 11 subjects described in our study
Patient 1
Patient 1 was a male with an antenatal history of oligohydramnios and intrauterine growth restriction (IUGR). He was born at 38 weeks. At 7 days old, he developed recurrent episodes of apnea, decreased activity, and mild lactic acidemia. On day 14, he developed circulatory collapse with severe metabolic acidosis and lactic acidosis up to 28.36 mmol/L (reference range 0.5–2.20), hypotension requiring multiple inotropes, an episode of pulseless ventricular tachycardia, and respiratory failure requiring intubation and ventilator support. Thereafter, he developed recurrent convulsions controlled by phenobarbitone and levetiracetam. MRI of the brain at 3 weeks of age showed symmetrical T1 and T2 hyperintensity with restricted diffusion at bilateral lentiform nuclei. Foci of restricted diffusion were also detected at bilateral frontal white matter (Fig. 1a–c). Magnetic resonance spectroscopy (MRS) showed raised lactate peaks at bilateral basal ganglia and cerebral white matter. Subsequent follow-up MRI showed established infarcts with cystic changes at bilateral lentiform nuclei (Fig. 1d). Mild cerebellar hypoplasia was also noted. Serial echocardiography in the following months showed progressive septal and ventricular myocardial hypertrophy. WES identified compound heterozygous mutations in COQ4, a missense c.370G>A, p.(Gly124Ser) and a splicing mutation c.402+1G>C. Functional analysis of the skin fibroblasts showed ETC complex II+III deficiency due to low CoQ concentration. At 5 months of age, CoQ10 supplement up to 40 mg/kg/day. At 8 months, in view of poor neurological prognosis and poor response to CoQ10 treatment, he was diverted to comfort care and extubated. He passed away shortly from respiratory failure.
Patient 2
Patient 2 was a term newborn male. He developed respiratory distress, hypotension, and progressive metabolic acidosis with a lactate level of 2.6 mmol/L and hyperalaninemia requiring intubation and inotropic support on day 1 of life. Echocardiogram revealed hypertrophic cardiomyopathy. Plasma amino acids revealed high alanine, proline, and tyrosine, and acylcarnitine profile was unremarkable. CoQ10 supplement at 15 mg/kg/day and carnitine at 100 mg/kg/day were tried on his second day of life but the patient succumbed. WES revealed compound heterozygous mutations in COQ4: a missense c.370G>A, p.(Gly124Ser) and a splicing mutation of c.402+1G>C.
Patient 3
Patient 3 was born full term at 37 weeks. She developed transient respiratory distress shortly after birth. On day 22 of life, she had cardiogenic shock. Echocardiogram showed poor contractility with a left ventricular ejection fraction of 20% and a moderate pericardial effusion. There was associated lactic acidemia (24 mmol/L; reference range 0.5–2.2) and hyperammonemia (139 µmol/L; reference range <100). She was empirically given CoQ10 supplementation and intravenous immunoglobulin. Her cardiac function improved gradually and normalized by day 32 of life. She developed seizures at 4 months of age requiring multiple anticonvulsants. She is severely delayed developmentally. WES revealed a homozygous COQ4 mutation c.370G>A, p.(Gly124Ser).
Patient 4 and Patient 5
Patient 4 was the younger sister of Patient 5. She had antenatal history of IUGR and was born at 38 weeks. Immediately postnatal, she developed respiratory distress with intermittent apnea and lactic acidemia (up to 10 mmol/L; reference range 0.5–2.2). MRI brain showed symmetrical T1 hyperintensity at bilateral basal ganglia, with mild cerebellar hypoplasia (Fig. 1e). MRS showed raised lactate peaks at bilateral basal ganglia (Fig. 1f). She developed treatment-resistant seizures at 2 months of age. Subsequent MRI at 9 months of age showed generalized progressive cerebellar and cerebral atrophy, with diffuse white matter loss including thinning of the corpus callosum. Cystic changes were seen within the cerebral white matter, bilateral basal ganglia, and thalami (Fig. 1g). Serial echocardiogram showed progressive moderate left ventricular hypertrophy. Owing to the recognizable clinical presentation, Sanger sequencing was performed and revealed compound heterozygous COQ4 mutation: c.370G>A, p.(Gly124Ser) and c.402+1G>C. CoQ10 supplement has been started since age of 4 years 5 months.
Patient 5 was the elder sister of Patient 4. She was born at 39 weeks with an antenatal history of IUGR. She developed seizures from 2 months of age with associated lactic acidosis and respiratory failure requiring home ventilation. Chest radiograph showed cardiomegaly. CoQ10 supplementation was tried at 1 year of age but passed way from respiratory failure 1 month after. After the genetic diagnosis of her younger sister, Sanger sequencing was performed retrospectively and revealed the same compound heterozygous COQ4 mutation as her sister, c.370G>A, p.(Gly124Ser) and c.402+1G>C.
Patient 6
Patient 6 presented at 8 months of life with severe global developmental delay, microcephaly, generalized dystonia, cortical visual impairment, and oromotor dysfunction. Metabolic workup revealed lactic acidemia of 2.5–5.9 mmol/L and hyperalanemia (626 µmol/L; reference range 143–439). WES revealed compound heterozygous mutations in the COQ4 gene: c.550T>C, p.(Trp184Arg) and c.402+1G>A. Functional analysis of the skin fibroblasts showed ETC complex II+III deficiency with low CoQ concentration. There was no further follow-up because of overseas adoption.
Patient 7
Patient 7 was a girl, born full term. She had bilateral cortical visual impairment since birth and progressive oromotor dysfunction requiring gastrostomy feeding. She had severe global developmental delay. She developed generalized dystonia and spasticity around 5 months of age. Lactic acidemia of 2.4–3.2 mmol/L was present. WES revealed a homozygous variant in the COQ4 gene: c.370G>A, p.(Gly124Ser). Functional analysis of the skin fibroblast showed ETC complex II+III deficiency and low CoQ concentration. She had been on CoQ10 supplement since 2 years old. There was no clinical improvement and the patient died at 3 years and 6 months of age.
Patient 8
Patient 8 is a girl, born full term. She developed infantile spasms at 6 months of age. Metabolic workup showed lactic acidemia at 2.2–4.2 mmol/L and hyperalanemia (487 µmol/L; reference range 143–439). WES revealed compound heterozygous variants of the COQ4 gene c.371G>T, p.(Gly124Val) inherited from the mother and c.370G>A, p.(Gly124Ser) inherited from the father. Interestingly, respiratory chain enzymology of the skeletal muscle activities was normal but skin fibroblast functional analysis showed ETC complex II+III deficiency and low CoQ10 concentration. CoQ10 supplement has been given since 9 months of age, with subjective improvement in responsiveness. She is alive and has achieved fair seizure control with levetiracetam and global developmental delay.
Patient 9
Patient 9 is a boy, born full term at 40 weeks. He presented with infantile spasms at 2 months of age. CoQ10 supplementation started at 7 years of age and has remained stable. WES revealed a homozygous COQ4 mutation, c.370G>A, p.(Gly124Ser). Skin fibroblast functional analysis showed ETC complex II+III deficiency and low CoQ concentration.
Patient 10 and Patient 11
Patient 10 is the younger sister of Patient 11. She was born at 36 weeks. She developed transient respiratory distress after birth. She was asymptomatic until 2 months of age when she developed progressive hypotonia, cortical visual impairment, severe developmental delay, and seizures requiring multiple anticonvulsants. Her echocardiogram showed progressive dilated cardiomyopathy and mitral regurgitation. WES revealed a homozygous COQ4 mutation: c.370G>A, p. (Gly124Ser). CoQ10 supplement at 30 mg/kg/day was started at 11 months of age, and her seizure control improved.
Patient 11 is the elder sister of Patient 10. She was born full term. At 4 months of age, she presented with seizures, hypotonia, spasticity, oromotor dysfunction, and severe developmental delay. She also developed an episode of acute myocarditis during which her echocardiogram showed diastolic dysfunction. Brain MRI at 14 months showed mild cerebellar atrophy and cerebral atrophy, white matter cystic changes with bilateral frontal and anterior temporal predominance, and thinning of the corpus callosum. Basal ganglia and brainstem appeared preserved (Fig. 1h–k). No lactic acidosis was detected. Owing to the exome findings of her sister, Sanger sequencing was performed and revealed a homozygous COQ4 mutation: c.370G>A, p.(Gly124Ser). She was not on CoQ10 supplement and passed away at 20 months due to an episode of sepsis.
Results
Pathogenicity of the COQ4 variants
We analyzed the COQ4 variants identified in our cohort by previously reported literatures, ClinVar, population frequency in gnomAD,7 conversation score by Combined Annotation-Dependent Depletion,8 in silico prediction by Rare Exome Variant Ensemble Learner,9 and protein stability change prediction by STRUM10 (Table 3). All variants demonstrated a deleterious effect. Together with the reduced level of CoQ10 of the patients, the pathogenicity of these variants is strongly supported by the biochemical findings of the patients.
Founder mutation analysis
Among these 11 patients, we identified the same missense mutation c.370G>A, p.(Gly124Ser) in 10 of them. This missense mutation is a rare variant with a population frequency of 0.001118 and it is exclusively found in South East Asians in the gnomAD database.7 Further analysis of the DNA of the five homozygous patients using Infinium OmniZhongHua-8 BeadChip SNP array showed a common haplotype of 0.464–3.290 cM implying that the mutation was inherited from a common ancestor 27 generations ago (Fig. 3). In addition, by principal component analysis), our SNP array data are clustered with Chinese and Japanese of HapMap Phase II (n = 270) and a local southern Chinese database (n = 612). Furthermore, the pathogenicity of this mutation has been established via the reduced CoQ level in the skin fibroblasts in Patients 7 and 9 who have homozygous c.370G>A mutation (Table 4). Altogether our study confirms that the missense mutation c.370G>A, p.(Gly124Ser) represents a pathogenic founder mutation in the southern Chinese population.

Founder mutation analysis. Shared haplotypes among homozygous COQ4:c.370G>A. Red square indicates the location of homozygous COQ4:c.370G>A, while black square indicates the nearest heterozygous single-nucleotide polymorphism. The length of the haplotype for each subject is at the right panel. The maximum shared length is approximately 577 kb
Discussion
To our knowledge, this is the largest case series of primary COQ10D7 reported. In the literature, primary COQ10D7 cases have been described predominantly with a neonatal onset, with only two cases of childhood onset.2,3,4,5,8 In this study, we have expanded the phenotypic spectrum of primary COQ10D7 from neonatal to infantile onset.
We have five patients exhibiting the well-described neonatal presentation of COQ10D7 as in the literature, characterized by respiratory distress, encephalopathy, seizures, hypotonia, and cardiomyopathy. Previously, it was believed that phenotypes from affected males with COQ4 mutation would be more severe and highly likely lethal.3,6 However, in our study the male-to-female death ratio was 2:3.
We have six patients with infantile-onset phenotypes. Unlike those of neonatal onset, MRI brain for those infantile-onset patients did not show characteristic basal ganglia lesions. Dystonia was observed in two out of the six patients with infantile-onset presentation in our cohort, and it was also observed in the two neonatal-onset cases reported by Lu et al.8 but not reported in non-Chinese patient.
A summary of the predominant phenotypes in the spectrum of neonatal, infantile, and childhood onset of COQ10D7 is shown in Fig. 4. The variegated symptoms and disease onsets explain the frequent delay of diagnoses of COQ10D7. This also highlights the importance of the complementarity of biochemical screening for children with unexplained neurological disturbances and the prompt application of WES in order to reach a genetic diagnosis that has an impact on patient management.

Phenotypic spectrum of neonatal, infantile, and childhood onset of COQ10D7 from all the reported cases including this study. †Lactic acidosis is not mentioned in the childhood-onset cases. (The color intensity representing the percentage of patients having that clinical presentation in arbitrary form)
Functional analysis to demonstrate the deficiency of CoQ should be carefully examined because mitochondrial enzymology can be tissue specific. In this study, among the five patients (Patients 1, 6, 7, 8, 9) with ETC chain analysis in the skin fibroblast, all of them shown a reduced level of succinate:cytochrome c oxidoreductase (complex II+III). Measurement of CoQ level was also found significantly decreased. For Patients 7 and 8, complex II+III analysis and CoQ level measurement were also performed in the muscle. Interestingly, the CoQ level from the muscle is normal but that from skin fibroblasts was reduced. From the Genotype-Tissue Expression (GTex) data, the COQ4 median expression in the muscle is 7.58 transcripts per million (TPM) while in the skin it is 44.14 TPM, demonstrating a 6-fold lower expression in the muscle.
More significantly, we have identified a common founder pathogenic COQ4 mutation associated with COD10D7. In this study, 10 out of the 11 patients carry the COQ4: c.370G>A, p.(Gly124Ser) allele. This mutation fulfills the criteria of a founder mutation: (1) all patients with the mutant alleles share a haplotype associated with the mutation; (2) the haplotype is shared among affected families with a genetic distance >1 cM; (3) the mutant allele is rare and specific to the population; and (4) all carriers are delineated to the same geographic region. It is likely that this founder mutation causes a relatively higher rate of COQ10D7 in southern Chinese individuals, and that may explain why we can present a larger cohort as compared to past studies in this field.
CoQ10 oral supplementation was previously reported effective in COQ4 mutation cases.2,3,4,6
Among the 10 patients who received CoQ10 supplement and with continuous follow-up, those shown with stabilized cardiac condition or seizure control are those of genotype of homozygous missense variant c.370G>A (Patients 3, 9, 10). Another patient on CoQ supplement with improved clinical condition is Patient 8 with genotype of compound heterozygous missense variants c.370G>A/c.371G>T. For those without improvement are patients with genotype in the presence of a splicing mutation c.402+1G>A (Patients 1, 2, 4, 5, 6). Among these five patients, three of them (Patients 1, 2, and 5) died from the disease. Retrospectively, Patients 1, 4, and 5 were documented IUGR antenatally. This may suggest that the presence of c.402+1G>A, a loss-of-function mutation, would cause more severe neonatal onset of phenotypes and less responsive to CoQ10 supplement.
In this study, we have expanded the phenotypic spectrum of COQ4 mutation. Now COQ10D7 can range from neonatal, infantile to childhood onset. We have also identified a pathogenic COQ4 founder mutation in the southern Chinese population. The importance of complementarity of biochemical screening and prompt application of WES on patients with unexplained neurological symptoms is highlighted.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The data that support the findings in this study are available on request from the corresponding authors (N.-C.L., C.-W.F., B.H.-Y.C.). The data are not publicly available as they contain information that could compromise research participant privacy or consent.
References
Desbats, M. A., Lunardi, G., Doimo, M., Trevisson, E. & Salviati, L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J. Inherit. Metab. Dis. 38, 145–156 (2015).
Brea-Calvo, G. et al. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am. J. Hum. Genet. 96, 309–317 (2015).
Chung, W. K. et al. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J. Med. Genet. 52, 627–635 (2015).
Sondheimer, N. et al. Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ10 deficiency. Mol. Genet. Metab. Rep. 12, 23–27 (2017).
Bosch, A. M. et al. Coenzyme Q10 deficiency due to a COQ4 gene defect causes childhood-onset spinocerebellar ataxia and stroke-like episodes. Mol. Genet. Metab. Rep. 17, 19–21 (2018).
Lu, M. et al. Clinical phenotype, in silico and biomedical analyses, and intervention for an East Asian population-specific c.370G>A (p.G124S) COQ4 mutation in a Chinese family with CoQ10 deficiency-associated Leigh syndrome. J. Hum. Genet. 64, 297–304 (2019).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47(D1), D886–D894 (2019).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Quan, L., Lv, Q. & Zhang, Y. STRUM: structure-based stability change prediction upon single-point mutation. Bioinformatics 32, 2911–2919 (2016).
Acknowledgements
This work was supported by the Ministry of Science and Technology, R.O.C., under grant number MOST 106-3114-B-002-009, the Society for Relief of Disabled Children, and Joshua Hellmann Foundation for Orphan Disease. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 13/01/2019.
Author information
Authors and Affiliations
Contributions
M.H.-C.Y., M.H.-Y.T., S.L., M.S.-P.H. and B.W. drafted the manuscript. D.M.L.T. and W.L. interpreted the MRI images. R.J.T.R. and J.S. provided functional analysis. M.H.-C.Y., M.H.-Y.T., A.K.-Y.K., C.C.-Y.M., K.-S.Y. and J.L.-F.F. performed the data analysis. Y.-Y.C., S.-P.L., C.M.Q., W.-L.H., Y.-H.C., P.-L.K., V.C.-M.C., C.T., S.-C.C., J.H., N.-C.L., C.-W.F. and B.H.-Y.C recruited and managed the patients. N-.C.L., C-.W.F. and B.H-.Y.C. conceived and supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, M.HC., Tsang, M.HY., Lai, S. et al. Primary coenzyme Q10 deficiency-7: expanded phenotypic spectrum and a founder mutation in southern Chinese. npj Genom. Med. 4, 18 (2019). https://doi.org/10.1038/s41525-019-0091-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-019-0091-x
This article is cited by
-
Neuromitochondrial Disorders
Clinical Neuroradiology (2021)
-
New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts
Journal of Neurology (2021)
-
Delineation of molecular findings by whole-exome sequencing for suspected cases of paediatric-onset mitochondrial diseases in the Southern Chinese population
Human Genomics (2020)
