
Abstract
Chlorine radicals are strong atmospheric oxidants known to play an important role in the depletion of surface ozone and the degradation of methane in the Arctic troposphere. Initial oxidation processes of chlorine produce chlorine oxides, and it has been speculated that the final oxidation steps lead to the formation of chloric (HClO3) and perchloric (HClO4) acids, although these two species have not been detected in the atmosphere. Here, we present atmospheric observations of gas-phase HClO3 and HClO4. Significant levels of HClO3 were observed during springtime at Greenland (Villum Research Station), Ny-Ålesund research station and over the central Arctic Ocean, on-board research vessel Polarstern during the Multidisciplinary drifting Observatory for the Study of the Arctic Climate (MOSAiC) campaign, with estimated concentrations up to 7 × 106 molecule cm−3. The increase in HClO3, concomitantly with that in HClO4, was linked to the increase in bromine levels. These observations indicated that bromine chemistry enhances the formation of OClO, which is subsequently oxidized into HClO3 and HClO4 by hydroxyl radicals. HClO3 and HClO4 are not photoactive and therefore their loss through heterogeneous uptake on aerosol and snow surfaces can function as a previously missing atmospheric sink for reactive chlorine, thereby reducing the chlorine-driven oxidation capacity in the Arctic boundary layer. Our study reveals additional chlorine species in the atmosphere, providing further insights into atmospheric chlorine cycling in the polar environment.
Similar content being viewed by others


Introduction
Active chlorine cycling has been found in the Arctic boundary layer during the springtime following polar sunrise and is acknowledged to play a key role in the depletion of surface ozone (O3) in this region1,2,3. Chlorine atoms (Cl) are also a strong oxidant in the polar troposphere, where the levels of hydroxyl radicals, another major atmospheric oxidant, are relatively low4. It is also well established that the direct reaction with Cl provides a chemical sink of methane (CH4) in the atmosphere5,6,7,8. The presence of chlorine species such as molecular chlorine (Cl2) and bromine monochloride (BrCl) has been reported in the Arctic, attributed to direct emissions from snowpacks9,10 and heterogeneous reactions of chlorine species on snow grains and airborne aerosols11. Upon photolysis, these chlorine species release reactive Cl atoms (1−2), which rapidly react with O3 to form chlorine monoxide, ClO (3)3. ClO is subsequently oxidized by bromine monoxide (BrO) or ClO, producing chlorine dioxide (OClO); reacting with HO2 to form hypochlorous acid (HOCl); or reacting with nitric oxide (NO) and nitrogen dioxide (NO2) to produce Cl atoms and chlorine nitrate (ClONO2) (as shown in reactions 4−7)12. Cl atoms can also degrade other hydrocarbons (RH) to generate hydrochloric acid (HCl; 8)13. HCl can be converted into chloride (Cl−) via hydrolysis on aerosol surfaces (9)14. Chloride can further undergo heterogeneous reactions with HOCl to produce Cl2 (10), which can, in turn, be photolyzed to recycle Cl atoms (1)15.
Despite decades of research on chlorine cycling in the atmosphere, a largely unexplored aspect entails the formation of chlorine oxyacids, such as chloric (HClO3) and perchloric (HClO4) acids. The presence of atmospheric HClO4 was first proposed to be important in the polar stratosphere and is believed to be a missing atmospheric sink process of chlorine16,17,18. Recent studies have hypothesized the potential formation of HClO3 and HClO4 in the lower atmosphere through observations of significant chlorate (ClO3−) and perchlorate (ClO4−) levels in rainwater, snow, and Arctic ice core samples19,20,21,22. Therefore, the atmospheric occurrence of chlorine oxyacids could enhance the chlorine sink, thereby affecting the oxidation capacity of the atmosphere and potentially posing environmental threats once deposited to the Earth’s surface. However, to date, there exists no direct evidence of the presence of HClO3 and HClO4 in the atmosphere, thus, limiting our full understanding of the atmospheric chlorine cycle and its associated environmental impacts.
Here, we present ambient observations of HClO3 and HClO4 in the atmosphere. Measurements were made via mass spectrometry in the Arctic at the Villum Research Station, Greenland, Ny-Ålesund, Svalbard, and over the central Arctic Ocean onboard research vessel (RV) Polarstern during the Multidisciplinary drifting Observatory for the Study of the Arctic Climate (MOSAiC) expedition. The measurements show that both chlorine oxyacids are ubiquitous and widespread during spring in the Arctic region. We find that these atmospheric species are not photoactive and therefore represent a previously unconsidered atmospheric sink of reactive chlorine in the pan-Arctic boundary layer.
Results and discussion
Observations of gas-phase HClO3 and HClO4 in the Arctic
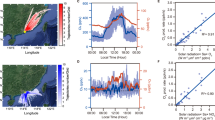
Figure 1 shows the time series of HClO3 and HClO4 measured at the Villum Research Station, Greenland, and during the MOSAiC campaign. Our observations in Greenland indicated a significant increase in the HClO3 signal measured with a nitrate-chemical ionization atmospheric pressure interface time-of-flight mass spectrometry (CI-APi-TOF; Methods), with an estimated concentration up to 1 × 106 molecules cm−3 in the spring of 2015. The HClO3 concentration began to increase when sunlight increased towards the end of February. HClO3 exhibited no diurnal pattern, but a unique feature is that a significant increase in HClO3 concentration was observed in coincidence with the depletion of O3, as shown in Fig. 1a. Typically, HClO3 peaked under relatively low O3 levels (<30 ppb). We also measured HClO3 with a nitrate CI-APi-TOF instrument during the MOSAiC expedition in 2019/2020 (Methods section). Similar to the observation in Greenland, the measurements onboard RV Polarstern in different seasons revealed a clear increment in HClO3 starting at the end of February, when solar radiation started to increase after the polar night. The estimated springtime concentration of HClO3 during the MOSAiC campaign ranged from approximately 1 × 105 to 7 × 106 molecules cm−3 (Fig. 1b). An increase in HClO3 was also observed in coincidence with the depletion of O3 over the Arctic Ocean during the MOSAiC campaign. The HClO3 levels are relatively low in the other seasons, with concentrations near detection limits of ≈104 molecule cm−3 (Supplementary Fig. S1). Further measurements at Ny-Ålesund, Svalbard also indicated the presence of HClO3 in springtime, with concentrations up to 8 × 105 molecules cm−3 (Supplementary Fig. S2). However, without direct measurement of O3 during the campaign at Svalbard, we are not able to evaluate the relationship between HClO3 and O3.

Time series of HClO3, HClO4, and O3, together with the temperature and incoming solar radiation measured at a the Villum Research Station from 1 March–15 May 2015 and b during the MOSAiC expedition from 22 February–30 April 2020. The data are displayed at a 30-min-average resolution, and any gaps in the time series are the results of instrumentation offline and maintenance periods. The dashed line represents the detection limits for HClO3 (blue) and HClO4 (pink) measurements. The uncertainty of HClO3 and HClO4 measurements was estimated to be at least a factor of two (see Methods). The map shows the location of the Villum Research Station (Nord) in Greenland, Ny-Ålesund in Svalbard, and RV Polarstern passive drifting track across the Arctic Ocean during the springtime sampling period. Note that all the time reported here is in Coordinated Universal Time (UTC). The map was created by the authors using MathWorks MATLAB (https://www.mathworks.com/products/matlab.html).
As shown in Figs. 1, 2, the increase in HClO3 was accompanied by an increase in HClO4. The HClO4 concentrations in Greenland and MOSAiC were estimated to be in the range of near detection limits (7 × 103) to 8 × 104 molecules cm−3 and near detection limits (3 × 104) to 1 × 106 molecules cm−3, respectively, during springtime, which were typically lower than the HClO3 concentration. The lower concentration of HClO4 compared with that of HClO3 is consistent with the levels of ClO4− and ClO3− measured in Arctic ice cores, where the annual depositional flux of ClO4− was reported to be several times lower than that of ClO3− (refs. 21,22).

Expanded view of HClO3 (blue solid line) tracking with the HClO4 (pink solid line) and HBr (gray shaded-area; based on the Br– normalized signal from nitrate CI-APi-TOF measurements which is most likely HBr (refer to Methods) to represent bromine chemistry), at a the Villum Research Station, Greenland, from 19 to 29 March 2015; and b onboard RV Polarstern during the MOSAiC campaign, from 15 to 25 March 2020. The dashed line represents the detection limits for HClO3 (blue) and HClO4 (pink) measurements. The uncertainty of HClO3 and HClO4 measurements was estimated to be at least a factor of two.
The results obtained during our campaigns at the different locations and time periods over the Arctic demonstrate that these chlorine oxyacids are widespread (Fig. 1 and Supplementary Fig. S2), and that their presence is a common phenomenon in the Arctic boundary layer during the springtime. The question arising here concerns the mechanisms leading to the occurrence of HClO3 and HClO4 in the Arctic.
Potential formation mechanism of atmospheric chlorine oxyacids
Although the initial steps of atmospheric chlorine oxidation are well understood (1–10)13,14, the final oxidation steps leading to chlorine oxyacid formation are not well characterized. Here, we explore the potential formation mechanisms of the observed HClO3 and HClO4 during spring in the Arctic.
As shown in Figs. 1, 2, the increase in both HClO3 and HClO4 coincides with the decrease in the O3 concentration. Springtime atmospheric surface ozone destruction is a well-known phenomenon in the Arctic and is typically linked to the shallow mixing layer and chemical reactions, including bromine and chlorine chemistry14,15,23,24. Our air mass backward trajectory analysis revealed that chlorine oxyacid-laden and O3-depleted air mass originated from the near ground surface, while the high O3 air mass originated from higher altitudes (Supplementary Fig. S3). This indicates that the ground surface, such as snowpacks in the Arctic, may play a role in the observed increases in HClO3 and HClO4 levels. It has been suggested that the heterogeneous reaction of O3 on chloride-containing aqueous and salt surfaces constitutes a potential formation mechanism of ClO3− and ClO4− (refs. 25,26,27). However, the formation of HClO3 and HClO4 via heterogeneous reactions on the aerosol surface or direct emission from the surface of snowpacks is likely not the dominant pathway in the Arctic. This assumption is justified by the remarkably low vapor pressure (6.8 mm Hg under a 70% concentration, at 298 K)28 and high Henry’s law constant (KH) of HClO4 (9.9 × 103 mol m−3 Pa−1)18. Although information for KH of HClO3 is not available, its value is very likely in between KH of HClO4 and KH of HOCl (6.5 mol m−3 Pa−1)29. These low vapor pressure and high KH suggest that the formed ClO3− and ClO4− on the aerosols or snow surface are unlikely being emitted directly as gas-phase HClO3 or HClO4 into the atmosphere. This is further supported by the detection of low HClO3 and HClO4 atmospheric concentrations in winter when the Arctic is covered by snow (see Supplementary Fig. S1). Furthermore, the observed lack of a clear pattern between HClO3 and HClO4 and the aerosol surface area during the springtime (Supplementary Fig. S4) may point to their limited partitioning from the aerosol phase. Another previously suggested potential HClO4 formation pathway via the heterogeneous reaction of ClO with sulfuric acid (H2SO4)30 may also not be important, as the results demonstrated no direct relationship between HClO3 (or HClO4) and our measured H2SO4 concentrations (coefficient of determination, R2 ≤ 0.04) during both the Greenland and MOSAiC campaigns (Supplementary Fig. S4).
Here, we propose a more likely formation mechanism of HClO3 and HClO4 over the Arctic environment during springtime, as illustrated in Fig. 3. The snowpack emissions of Cl2 and BrCl9,10,11 undergo fast photolysis, leading to the production of Cl atoms, which subsequently react with O3 to form ClO (1–3)3. In addition to photolysis, the produced ClO can then react with BrO/ClO to produce OClO, or undergo loss through reactions with OH, HO2, NO and NO2, CH3OO, and CH3COOO2. Abundant BrO and ClO must have been present during the encountered ozone-depletion events, as have been previously demonstrated by many studies2,3,31,32,33,34,35,36,37, and significant levels of BrO have been observed in spring during the MOSAiC campaign38. By using the previously reported typical ranges of BrO, ClO, and HO2 levels during Arctic ozone-depletion events11,32,33,34,35,36,37,38,39,40, we estimate that the reaction rate of ClO + BrO is much higher than that of the ClO + ClO and ClO + HO2 channels (section S1 in the Supplementary Information, SI), suggesting that the increase in BrO during ozone depletion events drives the OClO formation. The reaction of ClO + OH, ClO + CH3OO, and ClO + CH3COOO are insignificant; however, the presence of typical levels of NOx (NO and NO2) in the Arctic (i.e., 1−40 ppt) can compete with BrO for ClO (section S1 in SI). Indeed, previous ground measurements have detected significant OClO, up to 24 ppt, in the Arctic springtime35. Further observational evidence for the key involvement of bromine chemistry in the chlorine oxyacids formations comes from our observations which demonstrated that the recorded bromide signal adhered to the increase in HClO3 and HClO4 combined with a drastic decline in the O3 concentration (Fig. 2 and Supplementary Fig. S5).

Simplified diagram of the proposed potential formation mechanism of gas-phase HClO3 (blue) and HClO4 (pink) in the Arctic boundary layer during springtime after polar sunrise. The produced HClO3 and HClO4 can be taken up by the surface of aerosols and converted into ClO3− and ClO4−, respectively. The deposition of aerosols and/or the direct deposition of gas-phase HClO3 and HClO4 onto the ground surface, such as snowpacks, can function as a sink for reactive chlorine in the Arctic troposphere. The reactions are based on the literatures2,12,13,14,64. The mean boundary layer height was reported to vary between 100 and 200 m during the MOSAiC campaign74,78.
OClO can undergo further reactions, including (i) reacting with Cl to yield two ClO molecules; (ii) oxidation by OH to form HClO3; (iii) reacting with NO to recycle ClO; and (iv) oxidation by O3 to produce ClO3 (Fig. 3). Among these reactions, OClO + OH exhibits the fastest rate, with \({k}_{{{{{{\rm{OClO}}}}}}+{{{{{\rm{OH}}}}}}}[{{{{{\rm{OH}}}}}}]/{k}_{{{{{{\rm{OClO}}}}}}+{{{{{\rm{Cl}}}}}}}[{{{{{\rm{Cl}}}}}}]\) and \({k}_{{{{{{\rm{OClO}}}}}}+{{{{{\rm{OH}}}}}}}[{{{{{\rm{OH}}}}}}]/{k}_{{{{{{\rm{OClO}}}}}}+{{{{{{\rm{O}}}}}}}_{3}}[{{{{{{\rm{O}}}}}}}_{3}]\) ratios calculated to be in the range of 2 × 10−1 − 1 × 104 and 1 × 103 − 1 × 106, respectively, while the \({k}_{{{{{{\rm{OClO}}}}}}+{{{{{\rm{OH}}}}}}}[{{{{{\rm{OH}}}}}}]/{k}_{{{{{{\rm{OClO}}}}}}+{{{{{\rm{NO}}}}}}}[{{{{{\rm{NO}}}}}}]\) ratios fall in the range of 6 × 10−1 − 3 × 101 (section S2 in the SI). These results suggest that a significant fraction of OClO can be directly oxidized by OH to convert into HClO3, and produce ClO to recycle OClO. This is consistent with previous experimental studies on the OH + OClO reaction, where HClO3 was suggested to be produced at low temperatures41,42. Despite the lower O3 concentration (a source of OH), previous studies have shown that HOx (OH and HO2) chemistry is active during springtime in the Arctic with a reported OH concentration of ≈105−106 molecules cm−3 (refs. 2,4,43). This can also be indicated by our observation of significant H2SO4 concentrations (Supplementary Fig. S4), and the previously reported increase in methane sulfonic acid (MSA) levels44,45 during the Arctic springtime, both of which are products of sulfur oxidation reactions with OH and BrO. Therefore, OH is not a limiting factor of HClO3 formation in the Arctic during springtime. Given the fast reaction rate of OClO + OH, sufficient OH concentration, and enhanced OClO formation due to the increase in BrO during ozone depletion events, HClO3 production can occur efficiently. As to HClO4, the limiting factor of formation is likely the ClO3 concentration (Fig. 3), most likely due to its slow formation process via OClO + O3 (ref. 46). Therefore, the HClO4 formation is likely regulated by the O3 levels as indicated by the higher mean HClO4 concentrations observed at relatively higher O3 levels during the depletion events (Supplementary Fig. S6).
Based on these results, we conclude that the observed HClO3 and HClO4 over the Arctic atmosphere are predominantly produced through homogeneous reactions of chlorine, involving photochemical processes of HOx and bromine chemistry.
Atmospheric fate of HClO3 and HClO4
The fate of chlorine oxyacids determines their importance in the atmosphere. We first evaluate the potential removal of HClO3 and HClO4 in the troposphere through photodecomposition. Supplementary Fig. S7a shows our computed UV–Vis absorption spectra and cross-sections of HClO3 and HClO4 within the 170–340 nm wavelength range (refer to the Methods section for details). At the relevant wavelengths under tropospheric conditions (>290 nm), the estimated cross-sections of HClO3 and HClO4 are very small, suggesting that these two chlorine species are not photolabile in the troposphere. Based on these cross-sections, the loss rate constants of these oxyacids against photolysis at noon in the Arctic springtime are calculated as 4.4 × 10−12 and 2.5 × 10−18 s−1 for HClO3 and HClO4, respectively (Supplementary Fig. S7b, c).
Another possible removal pathway of HClO3 and HClO4 in the troposphere is their reactions with OH and Cl. Although there are no rate constants available for the reactions of HClO3 with Cl and OH, the reaction rate of HClO3 with either Cl or OH is expected to be low since both barriers for hydrogen abstraction from HClO3 are high47. The reactions of HClO4 with Cl and OH radicals (11 and 12, respectively) are also slow at low temperatures, with reported reaction rate coefficients of 1.00 × 10−31 and 5.8 × 10−13 cm3 molecule−1 s−1 at 253 K, respectively48. Assuming typical Cl (4 × 105 molecules cm−3)3 and OH (5 × 105 molecules cm−3)4 concentrations in the Arctic, the loss rates of kCl+HClO4[Cl] and kOH+HClO4[OH] are estimated to be 4.0 × 10−26 and 2.9 × 10−7 s−1, respectively.
Given the presence of significant aerosol particle surfaces (up to 100 µm2 cm−3) and humidity (which begins to increase in April) in the Arctic troposphere (Supplementary Fig. S8), once HClO3 and HClO4 are formed, they can undergo heterogeneous uptake on the surface of aerosol particles. Although there is no direct information available on the heterogeneous uptake coefficient (γ) of HClO3 and HClO4, previous studies have reported that the heterogeneous uptake coefficient of other chlorine acids, such as HCl, on aqueous aerosols reaches ≈0.2 at 273 K49,50. Both HClO3 and HClO4 are very strong acids with high electronegativity and can thus be easily ionized into ClO3−, ClO4−, and H3O+ in liquid water of the aerosol since HClO3 and HClO4 are highly soluble in water. The KH value of HClO4 in water was reported as 9.9 × 103 mol m−3 Pa−1 (ref. 18), while the KH value of HClO3 likely varies between the KH values of HClO4 and HOCl, with that of the latter compound reaching 6.5 mol m−3 Pa−1 (ref. 29). These higher KH values than that of HCl (KH = 0.2 mol m−3 Pa−1) may indicate that HClO3 and HClO4 could be efficiently accommodated on the surface of aerosol particles and that the fraction evaporating back into the gas phase could be small as well. By assuming that the heterogeneous uptake is accommodation limited and the γ values of HClO3 and HClO4 are similar to that of HCl (γ = 0.2)49,50, the estimated heterogeneous loss rate coefficients of HClO3 and HClO4 based on a typical aerosol surface area of 20 µm2 cm−3 during the MOSAiC campaign are 2.7 × 10−4 and 2.5 × 10−4 s−1, respectively (section S3 in SI). These rates are much (>3 orders of magnitude) higher than the rates of photodecomposition and radical attack (by OH and Cl) estimated above (<3 × 10−7 s−1). Therefore, the most relevant fate of HClO3 and HClO4 is their heterogeneous uptake by the surface of aerosol particles and subsequent deposition on the ground surface or undergo wet deposition. However, we cannot exclude the possibility of direct loss of these chlorine oxyacids to the snow surface (Fig. 3). In fact, our hypothesis is supported by previous studies in polar regions that have measured a considerable amount of ClO3− and ClO4− in ice cores21,22, snow51, and aerosols52, where atmospheric sources are strongly implicated.
Atmospheric chlorine chemistry has been regarded as a “never-ending” reaction since there is no termination process in the cycle. Indeed, the formation of HCl can serve as a sink for chlorine compounds in the troposphere, where HCl is taken up by aerosols and converted into Cl−, followed by an atmospheric deposition process14,53; although, in the presence of NOx and reactive halogens (i.e., HOI and HOBr), Cl− can be efficiently activated into reactive gas-phase chlorine again54,55,56. However, as HClO3 and HClO4 are not susceptible to photolysis and radical attack, and their conversion into ClO3− and ClO4− on aerosol surfaces or snowpacks is efficient in the Arctic boundary layer, the homogeneous formation of HClO3 and HClO4 could terminate chlorine recycling. Therefore, the formation of HClO3 and HClO4 is expected to affect the chlorine-mediated oxidation capacity in the Arctic troposphere. Furthermore, once HClO3 and HClO4 deposit on the ground surface (i.e., snowpack and sea ice), they may have environmental implications as their ions, ClO3− and ClO4−, can accumulate in the polar ice and marine sediment, and may present a toxicity risk to resident biota57,58.
In summary, our study revealed the observations of HClO3 and HClO4 in the atmosphere and their widespread occurrence over the pan-Arctic during spring. We propose a novel plausible mechanism for the formation and loss of chlorine oxyacids in the Arctic environment. The results provide evidence for chlorine oxyacids to be a previously unconsidered atmospheric sink for reactive chlorine, thereby providing further insights into chlorine chemistry in the Arctic region. We, therefore, conclude that the existence of HClO3 and HClO4 in the atmosphere should be considered when evaluating the environmental impacts of chlorine chemistry in the Arctic.
Methods
Sampling locations
This study comprises data obtained during three field measurement campaigns over the Arctic within different time ranges. We conducted a measurement campaign at the Villum Research Station (Station Nord) in high Arctic Northern Greenland (81° 36’ N 16° 39’ W). This campaign started in mid-February 2015 and continued until the end of August 2015. The measurement station is located approximately 2 km from Station Nord, and the instrumentation was set up at the station location. We also conducted measurements at the atmospheric observatory, Gruvebadet, located 2 km southeast of Ny-Ålesund, Svalbard (78° 55’ N, 11° 56’ E), from 28 March to 30 May 2017 (spring). Detailed information on the Greenland and Ny-Ålesund sampling site can be found in ref. 44. The other field study was the MOSAiC expedition, which involved RV Polarstern drifting across the central Arctic from September 2019 to October 2020. The MOSAiC expedition track and detailed campaign information can be found in ref. 59.
Detection of HClO3 and HClO4
A state-of-the-art chemical ionization atmospheric pressure interface time-of-flight mass spectrometry (CI-APi-TOF) instrument60 was employed in negative ion mode with nitrate (NO3−) ions as reagent ions to detect gas-phase HClO3 and HClO4. In Greenland, a straight, stainless steel inlet tube with a length of 1 m and an outer diameter of ¾ inch was applied at ~1.5 m above ground level (a.g.l.) to sample ambient air with a flow rate of 10 liters per minute (lpm). The inlet tube was heated to zero degrees Celsius. At Ny-Ålesund, the inlet tube length was 2 m (outer diameter of ¾ inch) and the sample was taken through the roof (height = 2 m a.g.l.), with a flow rate of 10 lpm. On RV Polarstern, the nitrate CI-APi-TOF instrument was set up in a Swiss Container on the bow deck of vessel61. The nitrate CI-APi-TOF inlet was connected to a new particle formation (NPF) inlet, through a core sampling flange system, accommodating a neutral air ion spectrometer utilized to create ~60 lpm inlet flow (height = about 15 m above sea level). The zero measurements were conducted occasionally with a high-efficiency particulate air (HEPA) filter for at least 40 min each measurement in both Greenland and MOSAiC, which cover different seasons during the measurement period.
HClO3 was detected as ClO3− (82.954 m/z), and its isotope peak was clearly observed at 84.951 m/z, with a 35Cl:37Cl ratio of approximately 3:1 (Supplementary Fig. S9). Regarding HClO4 detection, the peak of ClO4− (98.949 m/z) should be carefully identified, as it is difficult to distinguish it from the isotopic peak of deprotonated sulfuric acid, HSO4− (i.e., H34SO4− = 98.956 m/z) when the HSO4− signal is high. The m/z values of these two peaks are close to each other and may create interference with lower-mass resolution devices. Therefore, we considered the peak of 37ClO4− (100.946 m/z) to estimate the HClO4 signal in this study. We also detected the Br− signal from the peak at 78.919 m/z (together with NO3(HBr)− peak at 141.915 m/z; Supplementary Fig. S10), which is most likely attributed to hydrobromic acid (HBr)62. The raw data was pre-averaged over 10 min and processed with the MATLAB tofTools package according to the procedures described in Jokinen et al.60.
The detected ClO3− and ClO4− were the deprotonated products of HClO3 and HClO4, respectively. Quantum chemical calculations (section S4 in SI) indicated that the binding free energies of NO3− with HClO3 and HClO4 are 24.0 and 36.9 kcal mol−1, respectively (the binding free energy is simply −1 × formation free energy in Supplementary Fig. S11), and the deprotonation free energies are 7.1 and 24.8 kcal mol−1, respectively, which suggests that both HClO3 and HClO4 are efficiently deprotonated into ClO3− and ClO4−, respectively. The HClO3•NO3− cluster remains more stable against deprotonation than the HClO4•NO3− cluster, and some fraction of the former could occur, which is consistent with the detection of the HClO3•NO3− cluster (refer to Supplementary Fig. S9c,d), while the HClO4•NO3− cluster was not present in the spectrum. The isomers of HClO3 and HClO4 detected via nitrate CI-APi-TOF likely occurred in the form of HOClO2 and HOClO3, respectively, as these components are most energetically stable in the atmosphere42,63,64.
Currently, there are no available methods for HClO3 and HClO4 calibration of CI-APi-TOF instruments. More importantly, the handling of HClO3 and HClO4 is dangerous, as these substances are very corrosive and could cause violent explosions in reactions with organics, making calibration becoming very difficult. Based on quantum chemical calculations, the binding free energies of NO3− with HClO3 and HClO4, i.e., \({{{{{\mathrm{HClO}}}}}}_{3} \bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{NO}}}}}}_{3}^{-}}+{{{{{\mathrm{HClO}}}}}}_{3}\) and \({{{{{\mathrm{HClO}}}}}}_{4}\bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{NO}}}}}}_{3}^{-}}+{{{{{\mathrm{HClO}}}}}}_{4}\) (24.0 and 36.9 kcal mol−1, respectively), are similar to (the former is slightly lower than) that of \({{{{{{\mathrm{NO}}}}}}_{3}^{-}}\) with H2SO4 (\({{{{{\mathrm{H}}}}}}_{2}{{{{{\mathrm{SO}}}}}}_{4}\bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{NO}}}}}}_{3}^{-}}+{{{{{\mathrm{H}}}}}}_{2}{{{{{\mathrm{SO}}}}}}_{4}\); 34.4 kcal mol−1). However, similar to H2SO4 (\({{{{{\mathrm{H}}}}}}_{2}{{{{{\mathrm{SO}}}}}}_{4}\bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{HSO}}}}}}_{4}^{-}}+{{{{{\mathrm{HNO}}}}}}_{3}\) = 20.3 kcal mol−1), their binding free energies are higher than their deprotonation free energies (7.1 and 24.8 kcal mol−1 for \({{{{{\mathrm{HClO}}}}}}_{3}\bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{ClO}}}}}}_{3}^{-}}+{{{{{\mathrm{HNO}}}}}}_{3}\) and \({{{{{\mathrm{HClO}}}}}}_{4}\bullet {{{{{{\mathrm{NO}}}}}}_{3}^{-}}\to {{{{{{\mathrm{ClO}}}}}}_{4}^{-}}+{{{{{\mathrm{HNO}}}}}}_{3}\), respectively), which suggests that deprotonation of the formed clusters is efficient. Therefore, if HClO3 and HClO4 are dissociated during ionization, they could preferably form ClO3− and ClO4−-, respectively, and are detectable via nitrate CI-APi-TOF. This indicates that the detection of HClO3 and HClO4 is very likely as efficient as the detection of H2SO4 by NO3−, whose reaction rate is expected to occur within the kinetic limit range65. Thus, it is reasonable to assume that the instrument sensitivity to HClO3 and HClO4 is similar to the sensitivity determined for the H2SO4 measurement.
H2SO4 calibration for the nitrate CI-APi-TOF was conducted before or immediately after the campaign with a method presented in ref. 66. Regarding the Greenland and Ny-Ålesund measurements, the obtained H2SO4 calibration factor was 1.48 × 109 (ref. 44). During the MOSAiC campaign, calibration was completed twice after the campaign, and the average of two-factor values was 6 × 109. This factor includes the losses at the NPF inlet. Based on these calibration factors, the detection limits of HClO3 and HClO4 were estimated as 3 × 103 and 7 × 103 molecules cm−3 (10 min-average, 3σ), respectively, during the Greenland measurement campaign and 2 × 104 and 3 × 104 molecules cm−3 (10 min-average, 3σ), respectively, during the MOSAiC measurement campaign. The detection limit for HClO3 measurements in Ny-Ålesund was calculated to be 1 × 103 molecules cm−3 (10 min-average, 3σ).
The HClO3 and HClO4 concentrations in this study were computed with Eq. (13).
where x equals 3 or 4, and C is the calibration factor, which was assumed to be similar to the H2SO4 calibration factor. If the detected HClO3 and HClO4 clusters are not charged as efficiently as H2SO4, it could lead to underestimating the concentration of HClO3 and HClO4. The sum uncertainties of the HClO3 and HClO4 measurement from the collision limit of the target compound with its charger ions and potential inlet losses were predicted to be at least a factor of two.
Ancillary measurements
O3 was measured with a UV ozone analyser during both the Greenland and MOSAiC campaigns. More details on the O3 measurement setup in Greenland can be found in ref. 67. Regarding the MOSAiC O3 measurements, we used here an hourly merged dataset that combines cross-evaluated measurements performed by three independent instruments, as has been detailed in refs. 68,69. The particle number size distribution, 10–500 nm (9–915 nm in Greenland), was measured with a scanning mobility particle sizer (SMPS). The information on the SMPS setup in Greenland can be found in ref. 70. As for the MOSAiC campaign, the SMPS was measured from the United States Department of Energy Atmospheric Radiation Measurement (ARM) Aerosol Observation System container71,72. Water vapor was measured during the MOSAiC campaign via cavity ring-down spectroscopy (CRDS) with a commercial Picarro instrument (model G2401) also connected to the interstitial inlet of the Swiss container. Meteorological parameters (temperature and solar radiation) during the MOSAiC were obtained from the meteorological observatory Polarstern73. Details on the atmospheric and meteorological equipment during the MOSAiC campaign can be found in ref. 59 and ref. 74.
HClO3 and HClO4 photolysis rates
To obtain the photolysis rates of HClO3 and HClO4, we applied the estimated absorption cross-sections of these two compounds (refer to section S5 in SI for the cross-sections computation) in an explicit Tropospheric Halogen Chemistry Model (THAMO): a one-dimensional atmospheric chemistry model75 that has been used in many previous studies (e.g., ref. 56) to simulate the halogen chemical processes (including the photolysis) in the boundary layer. The THAMO simulations were conducted for 24 h in an Arctic environment (with a latitude of 81° 21′ N, similar to the location of Villum Research Station, Greenland) during spring (1 May) to derive the photolysis rates of HClO3 and HClO4 in the Arctic boundary layer.
Data availability
The data that support the findings of this study are available online in the following repositories. All of the data obtained from Greenland (Villum Research Station) and Ny-Ålesund observations, and absorption cross-sections and photolysis rates of HClO3 and HClO4 have been deposited in Zenodo: https://doi.org/10.5281/zenodo.7655981 and https://doi.org/10.5281/zenodo.4292239 (for H2SO4).
The MOSAiC dataset used in this study are available at https://doi.org/10.1594/PANGAEA.944393 (for merged O3), https://doi.pangaea.de/10.1594/PANGAEA.956085 (for HClO3 and HClO4), https://doi.pangaea.de/10.1594/PANGAEA.956087 (for HBr and H2SO4), https://doi.org/10.5439/1225453 (Kuang et al.72 for the particle number size distribution), https://doi.org/10.1594/PANGAEA.935265 (for meteorological data), https://doi.org/10.1594/PANGAEA.954232 (for water vapor), and https://doi.org/10.17632/bn7ytz4mfz.1 (for BrO).
References
Foster, K. L. et al. The role of Br2 and BrCl in surface ozone destruction at polar sunrise. Science 291, 471–474 (2001).
Thompson, C. R. et al. Interactions of bromine, chlorine, and iodine photochemistry during ozone depletions in Barrow, Alaska. Atmos. Chem. Phys. 15, 9651–9679 (2015).
Custard, K. D., Pratt, K. A., Wang, S. & Shepson, P. B. Constraints on Arctic atmospheric chlorine production through measurements and simulations of Cl2 and ClO. Environ. Sci. Technol. 50, 12394–12400 (2016).
Stone, D., Whalley, L. K. & Heard, D. E. Tropospheric OH and HO2 radicals: field measurements and model comparisons. Chem. Soc. Rev. 41, 6348–6404 (2012).
Hossaini, R. et al. A global model of tropospheric chlorine chemistry: Organic versus inorganic sources and impact on methane oxidation. J. Geophys. Res. Atmos. 121, 271–214,297 (2016). 14.
Strode, S. A. et al. Strong sensitivity of the isotopic composition of methane to the plausible range of tropospheric chlorine. Atmos. Chem. Phys. 20, 8405–8419 (2020).
Gromov, S., Brenninkmeijer, C. A. M. & Jöckel, P. A very limited role of tropospheric chlorine as a sink of the greenhouse gas methane. Atmos. Chem. Phys. 18, 9831–9843 (2018).
Li, Q. et al. Reactive halogens increase the global methane lifetime and radiative forcing in the 21st century. Nat. Commun. 13, 2768 (2022).
Liao, J. et al. High levels of molecular chlorine in the Arctic atmosphere. Nat. Geosci. 7, 91–94 (2014).
Custard, K. D., Raso, A. R. W., Shepson, P. B., Staebler, R. M. & Pratt, K. A. Production and release of molecular bromine and chlorine from the Arctic coastal snowpack. ACS Earth Space Chem. 1, 142–151 (2017).
McNamara, S. M. et al. Bromine chloride in the coastal Arctic: diel patterns and production mechanisms. ACS Earth Space Chem. 4, 620–630 (2020).
Atkinson, R. et al. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 7, 981–1191 (2007).
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A. & Glasow, R. Tropospheric halogen chemistry: sources, cycling, and impacts. Chem. Rev. 115, 4035–4062 (2015).
Saiz-Lopez, A. & von Glasow, R. Reactive halogen chemistry in the troposphere. Chem. Soc. Rev. 41, 6448–6472 (2012).
Simpson, W. R. et al. Halogens and their role in polar boundary-layer ozone depletion. Atmos. Chem. Phys. 7, 4375–4418 (2007).
Sander, S. P., Friedl, R. R. & Yung, Y. L. Rate of formation of the ClO dimer in the polar stratosphere: implications for ozone Loss. Science 245, 1095–1098 (1989).
Webster, C. R. et al. Chlorine chemistry on polar stratospheric cloud particles in the Arctic Winter. Science 261, 1130–1134 (1993).
Jaeglé, L., Yung, Y. L., Toon, G. C., Sen, B. & Blavier, J.-F. Balloon observations of organic and inorganic chlorine in the stratosphere: The role of HClO4 production on sulfate aerosols. Geophys. Res. Lett. 23, 1749–1752 (1996).
Dasgupta, P. K. et al. The origin of naturally occurring perchlorate: the role of atmospheric processes. Environ. Sci. Technol. 39, 1569–1575 (2005).
Rajagopalan, S. et al. Perchlorate in wet deposition across North America. Environ. Sci. Technol. 43, 616–622 (2009).
Furdui, V. I. & Tomassini, F. Trends and sources of perchlorate in Arctic snow. Environ. Sci. Technol. 44, 588–592 (2010).
Furdui, V. I., Zheng, J. & Furdui, A. Anthropogenic perchlorate increases since 1980 in the Canadian High Arctic. Environ. Sci. Technol. 52, 972–981 (2018).
Barrie, L. A., Bottenheim, J. W., Schnell, R. C., Crutzen, P. J. & Rasmussen, R. A. Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere. Nature 334, 138–141 (1988).
McConnell, J. C. et al. Photochemical bromine production implicated in Arctic boundary-layer ozone depletion. Nature 355, 150–152 (1992).
Rao, B., Anderson, T. A., Redder, A. & Jackson, W. A. Perchlorate formation by ozone oxidation of aqueous chlorine/oxy-chlorine species: Role of ClxOy radicals. Environ. Sci. Technol. 44, 2961–2967 (2010).
Rao, B. et al. Perchlorate production by photodecomposition of aqueous chlorine solutions. Environ. Sci. Technol. 46, 11635–11643 (2012).
Jackson, W. A., Wang, S., Rao, B., Anderson, T. & Estrada, N. L. Heterogeneous production of perchlorate and chlorate by ozone oxidation of chloride: Implications on the source of (per)chlorate in the solar system. ACS Earth Space Chem. 2, 87–94 (2018).
Baran, K. P. & Gad, S. Perchloric Acid, in Encyclopedia of Toxicology 3rd edn. (Wexler, P.), 796-798 (Academic Press, 2014).
Sander, R. Compilation of Henry’s law constants (version 4.0) for water as solvent. Atmos. Chem. Phys. 15, 4399–4981 (2015).
Martin, L. R., Wren, A. G. & Wun, M. Chlorine atom and ClO wall reaction products. Int. J. Chem. Kinet. 11, 543–557 (1979).
Barrie, L. & Platt, U. Arctic tropospheric chemistry: an overview. Tellus B: Chem. Phys. Meteorol. 49, 450–454 (1997).
Platt, U. & Wagner, T. Satellite mapping of enhanced BrO concentrations in the troposphere. Nature 395, 486–490 (1998).
Tuckermann, M. et al. DOAS-observation of halogen radical-catalysed arctic boundary layer ozone destruction during the ARCTOC-campaigns 1995 and 1996 in Ny-Ålesund, Spitsbergen. Tellus B: Chem. Phys. Meteorol. 49, 533–555 (1997).
Wang, S. et al. Direct detection of atmospheric atomic bromine leading to mercury and ozone depletion. Proc. Natl Acad. Sci. USA 116, 14479 (2019).
Pöhler, D., Vogel, L., Frieß, U. & Platt, U. Observation of halogen species in the Amundsen Gulf, Arctic, by active long-path differential optical absorption spectroscopy. Proc. Natl Acad. Sci. USA 107, 6582 (2010).
Martinez, M., Arnold, T. & Perner, D. The role of bromine and chlorine chemistry for arctic ozone depletion events in Ny-Ålesund and comparison with model calculations. Ann. Geophys. 17, 941–956 (1999).
Liao, J. et al. Characterization of soluble bromide measurements and a case study of BrO observations during ARCTAS. Atmos. Chem. Phys. 12, 1327–1338 (2012).
Benavent, N. et al. Substantial contribution of iodine to Arctic ozone destruction. Nat. Geosci. 15, 770–773 (2022).
Edwards, P. et al. Hydrogen oxide photochemistry in the northern Canadian spring time boundary layer. J. Geophys. Res. Atmos. 116, D22306 (2011).
Thompson, C. R. et al. Bromine atom production and chain propagation during springtime Arctic ozone depletion events in Barrow, Alaska. Atmos. Chem. Phys. 17, 3401–3421 (2017).
Poulet, G., Zagogianni, H. & Bras, G. L. Kinetics and mechanism of the OH + CIO2 reaction. Int. J. Chem. Kinet. 18, 847–859 (1986).
Francisco, J. S. & Sander, S. P. Structures, relative stabilities, and vibrational spectra of isomers of HClO3. J. Phys. Chem. 100, 573–579 (1996).
Hornbrook, R. S. et al. Arctic springtime observations of volatile organic compounds during the OASIS-2009 campaign. J. Geophys. Res. Atmos. 121, 9789–9813 (2016).
Beck, L. J. et al. Differing mechanisms of new particle formation at two Arctic sites. Geophys. Res. Lett. 48, e2020GL091334 (2021).
Jang, S. et al. Large seasonal and interannual variations of biogenic sulfur compounds in the Arctic atmosphere (Svalbard; 78.9°N, 11.9°E). Atmos. Chem. Phys. 21, 9761–9777 (2021).
Atkinson, R. et al. Evaluated kinetic, photochemical and heterogeneous data for atmospheric chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry. J. Phys. Chem. Ref. Data 26, 521–1011 (1997).
Poggi, G. & Francisco, J. S. Hydrogen atom abstraction from HOOOCl by chlorine atom and OH radical. Inorg. Chim. Acta 360, 837–841 (2007).
Zhu, R. S. & Lin, M. C. An ab initio chemical kinetic study on the reactions of H, OH, and Cl with HOClO3. Int. J. Chem. Kinet. 42, 253–261 (2010).
Ammann, M. et al. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates. Atmos. Chem. Phys. 13, 8045–8228 (2013).
Li, Y. Q. et al. Uptake of HCl(g) and HBr(g) on ethylene glycol surfaces as a function of relative humidity and temperature. J. Phys. Chem. A 106, 1220–1227 (2002).
Jiang, S., Cox, T. S., Cole-Dai, J., Peterson, K. M. & Shi, G. Trends of perchlorate in Antarctic snow: implications for atmospheric production and preservation in snow. Geophys. Res. Lett. 43, 9913–9919 (2016).
Jiang, S., Shi, G., Cole-Dai, J., An, C. & Sun, B. Occurrence, latitudinal gradient and potential sources of perchlorate in the atmosphere across the hemispheres (31°N to 80°S). Environ. Int. 156, 106611 (2021).
Faxon, C. B. & Allen, D. T. Chlorine chemistry in urban atmospheres: a review. Environ. Chem. 10, 221–233 (2013).
Finlayson-Pitts, B. J., Ezell, M. J. & Pitts, J. N. Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2. Nature 337, 241–244 (1989).
Osthoff, H. D. et al. High levels of nitryl chloride in the polluted subtropical marine boundary layer. Nat. Geosci. 1, 324–328 (2008).
Tham, Y. J. et al. Direct field evidence of autocatalytic iodine release from atmospheric aerosol. Proc. Natl Acad. Sci. USA 118, e2009951118 (2021).
Kounaves, S. P. et al. Discovery of natural perchlorate in the Antarctic dry valleys and its global implications. Environ. Sci. Technol. 44, 2360–2364 (2010).
Acevedo-Barrios, R., Rubiano-Labrador, C. & Miranda-Castro, W. Presence of perchlorate in marine sediments from Antarctica during 2017–2020. Environ. Monit. Assess. 194, 102 (2022).
Shupe, M. D. et al. Overview of the MOSAiC expedition—Atmosphere. Elem. Sci. Anth. 10, 0060 (2022).
Jokinen, T. et al. Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF. Atmos. Chem. Phys. 12, 4117–4125 (2012).
Beck, I. et al. Automated identification of local contamination in remote atmospheric composition time series. Atmos. Meas. Tech. 15, 4195–4224 (2022).
Fan, X. et al. Atmospheric gaseous hydrochloric and hydrobromic acid in urban Beijing, China: detection, source identification and potential atmospheric impacts. Atmos. Chem. Phys. 21, 11437–11452 (2021).
Rohlfing, C. M. A theoretical study of HClO3 isomers. Chem. Phys. Lett. 245, 665–670 (1995).
Zhu, R. & Lin, M.-C. Ab initio study of ammonium perchlorate combustion initiation processes: unimolecular decomposition of perchloric acid and the related OH+ ClO3 reaction. Phys. Chem. Comm. 4, 127–132 (2001).
Viggiano, A. A., Seeley, J. V., Mundis, P. L., Williamson, J. S. & Morris, R. A. Rate constants for the reactions of XO3-(H2O)n (X = C, HC, and N) and NO3-(HNO3)n with H2SO4: Implications for atmospheric detection of H2SO4. J. Phys. Chem. A 101, 8275–8278 (1997).
Kürten, A., Rondo, L., Ehrhart, S. & Curtius, J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A 116, 6375–6386 (2012).
Skov, H. et al. Variability in gaseous elemental mercury at Villum Research Station, Station Nord, in North Greenland from 1999 to 2017. Atmos. Chem. Phys. 20, 13253–13265 (2020).
Angot, H. et al. Year-round trace gas measurements in the central Arctic during the MOSAiC expedition. Sci. Data 9, 723 (2022).
Angot, H. et al. Ozone dry air mole fractions measured during MOSAiC 2019/2020 (merged dataset). PANGAEA https://doi.org/10.1594/PANGAEA.944393 (2022).
Lange, R. et al. Characterization of distinct Arctic aerosol accumulation modes and their sources. Atmos. Environ. 183, 1–10 (2018).
Uin, J. et al. Atmospheric Radiation Measurement (ARM) Aerosol Observing Systems (AOS) for surface-based in situ atmospheric aerosol and trace gas measurements. J. Atmos. Ocean. Technol. 36, 2429–2447 (2019).
Kuang, C., Singh, A. & Howie, J. Scanning mobility particle sizer (AOSSMPS). https://doi.org/10.5439/1225453 (2022).
Schmithüsen, H., Rohleder, C. & Hausen, R. Meteorological observations during POLARSTERN cruise PS122/3. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research. Bremerhaven. PANGAEA https://doi.org/10.1594/PANGAEA.935265 (2021).
Maturilli, M. et al. Initial radiosonde data from 2019-10 to 2020-09 during project MOSAiC. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven. PANGAEA https://doi.org/10.1594/PANGAEA.928660 (2021).
Saiz-Lopez, A. et al. On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime. Atmos. Chem. Phys. 8, 887–900 (2008).
Knust, R. Polar Research and Supply Vessel POLARSTERN operated by the Alfred-Wegener-Institute. 10.17815/jlsrf-3-163 3, A119 (2017).
Nixdorf, U. et al. MOSAiC extended acknowledgement. Zenodo https://doi.org/10.5281/zenodo.5541624 (2021).
Jozef, G., Cassano, J., Dahlke, S. & de Boer, G. Testing the efficacy of atmospheric boundary layer height detection algorithms using uncrewed aircraft system data from MOSAiC. Atmos. Meas. Tech. 15, 4001–4022 (2022).
Acknowledgements
This study received funding from the National Natural Science Foundation of China (42175118), European Research Council Executive Agency under the European Union’s Horizon 2020 Research and Innovation Program (Project ERC-2016-COG726349 CLIMAHAL, ERC-StG GASPARCON - grant agreement no. 714621, the EMME-CARE project – grant agreement no. 856612, and grant no. 101002728, ATM-GTP Contract No. 742206), Climate Relevant interactions and feedbacks: the key role of sea ice and Snow in the polar and global climate system (CRiceS, grant number 101003826) and Horizon Europe project Non-CO2 Forcers and their Climate, Weather, Air Quality and Health Impacts, (FOCI, project number 101056783), ACCC Flagship funded by the Academy of Finland grant no. 337549, Academy professorship funded by the Academy of Finland (grant no. 302958), Academy of Finland (project nos. 296628, 328290, 311932, 316114, 325647, 325681, 334792, 337549, 329274, 333397, 328616, 1325656, 347782, 349659, and 334514), “Quantifying carbon sink, CarbonSink+ and their interaction with air quality” INAR project funded by Jane and Aatos Erkko Foundation, University of Helsinki via ACTRIS-HY, SIOS funding (Project no: 2020_003), United States National Science Foundation (grant nos. 1807496 and 1807163), Swiss National Science Foundation (grant 200021_188478), and the Swiss Polar Institute (grant no. DIRCR-2018-004). J.S. holds the Ingvar Kamprad Chair for Extreme Environments Research, sponsored by Ferring Pharmaceuticals. J.C.-G. acknowledges “La Caixa” Foundation (ID 100010434) for the fellowship LCF/BQ/DR20/11790027. D.R.-S. is thankful to the Spanish “Ministerio de Ciencia e Innovación (MICINN)” for funding (Project ref. CTQ2017-87054-C2-2-P, Unit of Excellence María de Maeztu CEX2019-000919-M and “Ramón y Cajal” grant RYC-2015-19234. We acknowledge that the data reported in this manuscript were produced as part of the International Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition with the MOSAiC20192020 tag, with activities supported by Polarstern76 expedition AWI_PS122_00. The authors gratefully acknowledge the National Oceanic and Atmospheric Administration (NOAA) Air Resources Laboratory (ARL) for the provision of the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) transport and dispersion model and/or Real-time Environmental Applications and Display sYstem (READY) website (https://www.ready.noaa.gov) employed in this publication. The authors also acknowledge the contributions of Oskari Kausiala, Daniela Wimmer, Lauri Ahonen, and Jyri Mikkilä during the Greenland field measurement. We acknowledge Matthew Shupe for his contribution to the O3 measurements during the MOSAiC campaign. SMPS data of the MOSAiC campaign were obtained from the Atmospheric Radiation Measurement (ARM) User Facility, a U. S. Department of Energy (DOE) Office of Science User Facility Managed by the Biological and Environmental Research Program. We thank all those who contributed to MOSAiC and made this endeavor possible (ref. 77). We acknowledge Ilann Bourgeois, Jeff Peischl, Chelsea R. Thompson, and Thomas B. Ryerson for collecting and providing the ATom NOx data. This work represents a contribution to CSIC Thematic Interdisciplinary Platform PTI POLARCSIC.
Author information
Authors and Affiliations
Contributions
Y.J.T., T.J., and A.S.-L designed the research. N.S., H.A, L.L.J.Q, I.B., T.L., L.J.B., M.B., O.P., R.C.T., D.Ho., B.B., S.D.A., L.Ba., K.P., J.H., D.He. H.-W.J., H.J., A.S.M., A.M., H.S., M.Si., J.S., and T.J. performed the field measurements. Y.J.T., N.S., S.I., H.A., X.-C.H., Q.Z., and T.J. analysed the data. S.I. conducted the quantum chemical calculations of the binding energies. Q.L., J.C.-G., A.B.-S., and D.R.-S. computed the absorption spectra, cross-sections, and photolysis rates of HClO3 and HClO4. Y.J.T., N.S., S.I., M.K., J.S. F., J.S., T.J., and A.S.-L. wrote the paper with the contributions of all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks A. R. Ravishankara and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tham, Y.J., Sarnela, N., Iyer, S. et al. Widespread detection of chlorine oxyacids in the Arctic atmosphere. Nat Commun 14, 1769 (2023). https://doi.org/10.1038/s41467-023-37387-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37387-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
