
Abstract
Production of syngas with tunable CO/H2 ratio from renewable resources is an ideal way to provide a carbon-neutral feedstock for liquid fuel production. Ag is a benchmark electrocatalysts for CO2-to-CO conversion but high overpotential limits the efficiency. We synthesize AgP2 nanocrystals (NCs) with a greater than 3-fold reduction in overpotential for electrochemical CO2-to-CO reduction compared to Ag and greatly enhanced stability. Density functional theory calculations reveal a significant energy barrier decrease in the formate intermediate formation step. In situ X-ray absorption spectroscopy (XAS) shows that a maximum Faradaic efficiency is achieved at an average silver valence state of +1.08 in AgP2 NCs. A photocathode consisting of a n+p-Si wafer coated with ultrathin Al2O3 and AgP2 NCs achieves an onset potential of 0.2 V vs. RHE for CO production and a partial photocurrent density for CO at −0.11 V vs. RHE (j−0.11, CO) of −3.2 mA cm−2.
Similar content being viewed by others

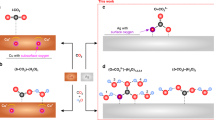

Introduction
Syngas with a tunable CO/H2 ratio is a particularly attractive commodity because it can subsequently be upgraded to synthetic liquid fuels by industrial Fischer–Tropsch reactions1. Currently, non-renewable fossil fuels are the predominant sources for syngas production, all of which contribute to environmental pollution2. The conversion of CO2 into value-added chemicals by use of renewable energy (e.g. solar energy) is an ideal approach to achieving a carbon-neutral energy cycle3, and the development of an efficient and selective catalyst is critical to make this technology feasible.
For most metallic CO2 reduction catalysts, both theory and experiment have shown that only an H2-rich mixture is achievable because the HER process dominates in aqueous solution4. Au and Ag have emerged as benchmark electrocatalysts for highly selective conversion of CO2 to CO5. Compared to the Au electrocatalyst, Ag is considerably cheaper and shows comparable activity. Various strategies, such as morphology-nanostructuring6,7, size-tuning8, defect-engineering9, surface ligand functionalization10, and bimetal alloying11, have been utilized to decrease the required overpotential (η) for high selectivity of CO2-to-CO on Ag-based electrocatalysts. However, Ag still suffers from a high overpotential and the current density of syngas at low overpotential (η < 500 mV) is usually below a metric value of 10 mA/cm2. Moreover, the ratio of CO:H2 is generally limited to the range of 1:2 to 1:1, severely restricting the flexibility for integration with existing chemical infrastructure.
Incorporation of non-metal elements such as phosphorus, sulfur, and boron into metals to form multicomponent alloys is an effective way to tailor the electronic structure and surface properties of metal electrocatalysts12,13,14. Our previous work studying the hydrogen evolution reaction (HER) on CoP found that phosphorus was able to regulate the binding strength of reaction intermediates adsorbed on cobalt active sites to improve electrocatalytic activity15. While silver diphosphide (AgP2) is an attractive material for electrocatalysis given the success of other metal phosphide systems, it is rarely reported due to a lack of synthetic methods to control the nanostructured morphology and purity16,17. Colloidal synthesis has been shown to yield high monodispersity with controllable morphology but only limited work has been done on noble metal phosphide (e.g. AgP2) NCs and they have not been employed for CO2 reduction18. It is highly desirable to understand how the incorporation of P into Ag affects the catalytic efficiency, stability, and selectivity for CO2 reduction applications.
One attractive method for converting solar energy into chemical fuels is via photoelectrochemical (PEC) conversion. However, overall PEC device performance still suffers from the limitations of high overpotential, poor selectivity, and instability in the long-term operation19. Rational design is required to optimize the interface between semiconductor and cocatalyst for efficient charge transfer, and a catalyst that exhibits a fast reaction rate at low overpotential while maintaining high selectivity. Recently, a wide range of semiconductors, such as p-Si20,21, CuFeO2/CuO22, ZnTe23,24, and GaN25 have been studied in PEC CO2 reduction to produce CO or formate. These semiconductors are typically paired with an effective and selective metal cocatalyst such as Au, Ag, and Cu. For efficient PEC CO2 reduction, selective cocatalysts are generally integrated with a p-type semiconductor. P-type silicon (p-Si) is one of the most promising candidates for PEC CO2 reduction due to its narrow band gap (1.1 eV) and abundance26. Incorporation of a high level of surface n-dopant can decouple the band bending in the p-Si from the semiconductor-liquid junction to form an n+p built-in depletion region, which increases the photovoltage of the p-Si photocathode27. However, direct contact between n+p-Si and a metal cocatalyst can lead to severe surface recombination due to Fermi level pinning, lowering the built-in photovoltage28. Inserting an ultrathin insulator layer to form a buried heterojunction can eliminate Fermi level pinning and provide a higher photovoltage to drive the chemical reaction, while also protecting the semiconductor from the corrosive electrolyte. Ultrathin and pin-hole free metal oxides have been fabricated by atomic layer deposition (ALD) and used as tunnel layers for efficiently mediating charge transfer from semiconductor to cocatalyst29,30,31. In the past few years, photoelectrodes with buried junction have been investigated in PEC water splitting32,33,34,35 but have not been realized in PEC CO2 reduction for syngas production due to the lack of efficient and selective metal cocatalysts.
Results
Synthesis and characterization of AgP2 nanocrystals
To synthesize monodisperse and crystalline silver phosphide NCs, a reactive phosphorus precursor is key for achieving the necessary balance of Ag+ reduction and P incorporation. The use of trioctylphosphine (TOP) requires a high temperature (>280 °C) to achieve TOP decomposition to generate the PH3 intermediate36. For silver phosphide NCs synthesis, the reduction rate for Ag+ to Ag is too fast at high temperature and severe aggregation and precipitation occurs37. Tris(trimethylsilyl)phosphine (P(SiMe3)3) is an alternative to in situ PH3 generation, opening a wider temperature window. High-quality silver phosphide NCs are obtained by hot-injection at 180 °C, maintaining the temperature for 10 min, then fast cooling to 80 °C where the temperature is held for 60 min Inductively coupled plasma mass spectroscopy (ICP-MS) gives an atomic ratio of silver to phosphorus of 0.505, consistent with AgP2 (Supplementary Table 1). Energy-disperse X-ray spectroscopy (EDS) of AgP2 shows a similar Ag:P ratio of 0.517 (Supplementary Fig. 1). The X-ray diffraction (XRD) pattern of AgP2 NCs is highly matched with the monoclinic structure (ICDD PDF: 01-018-1185) (Supplementary Fig. 2), and notably does not show the existence of a crystalline Ag impurity in the silver phosphide NCs. The central Ag atom is octahedrally coordinated by P atoms, while P atoms are tetrahedrally coordinated by P and Ag atoms (Supplementary Fig. 3). The P-rich composition distorts the Ag cubic structure and potentially leads to exposure of abundant uncoordinated sites to be exploited for catalysis. The transmission electron microscopy (TEM) image of the as-synthesized AgP2 NCs shows a relatively uniform spherical morphology with the average size of 3.5 ± 0.5 nm (Fig. 1a). Two sets of lattice fringes are observed, showing interplane distances of 0.41 and 0.39 nm corresponding to the (011) and (\(\bar 1\)02) planes of the monoclinic AgP2 (Fig. 1b). The intersected angle between the (011) and (\(\bar 1\)02) planes is 63°, very close to the monoclinic theoretical value of 65°. The two lattice fringes are also found in the selected-area electron diffraction (SAED) image and fast Fourier transform (FFT) image (Supplementary Fig. 4). To better understand the effects of ultrasmall NC size, AgP2 NCs with a larger size of around 10 nm were obtained by extending the reaction time at 180 °C to 25 min. For the 10 nm AgP2 NCs, only a general (111) plane is presented on nanocrystal surface (Supplementary Fig. 5a, b), indicating the critical role of controlling nanocrystal size on enhancing the exposure of high-index planes which may exhibit higher catalytic activity8. High-angle annular dark-field scanning TEM (HAADF-STEM) image shows a single AgP2 NC that possesses well-defined spherical geometry (Fig. 1c). Figure 1d–f show the elemental mapping of an individual AgP2 NC obtained by HAADF-STEM-EDS, in which the Ag and P elements are evenly distributed throughout the whole NC.

Materials characterization of AgP2 NCs. a TEM image, scale bar 10 nm. b HRTEM image, c HAADF-STEM, d–f elemental mapping images of AgP2 NCs. Scale bars 2 nm. g X-ray absorption near-edge structure (XANES) spectra of 4 nm AgP2 NCs, 10 nm AgP2 NCs, 10 nm Ag NCs, and Ag foil. h Corresponding extended X-ray absorption fine structure (EXAFS) spectra.
X-ray absorption spectroscopy further reveals the effect of phosphorus incorporation on the local geometric and electronic structures of silver in AgP2 NCs. Figure 1g shows the Ag K-edge X-ray absorption near-edge structure (XANES) spectra of 4 nm AgP2 NCs, 10 nm AgP2 NCs, 10 nm Ag NCs, and Ag foil. The rising edge of AgP2 NCs shows a positive shift compared with that of Ag NCs due to the increased Ag valence oxidation state after incorporation of P into Ag. This can be ascribed to an electron density shift from Ag to P-rich regions with high electronegativity38. The X-ray photoelectron spectroscopy (XPS) binding energy (Supplementary Fig. 6) of Ag 3d5/2 for AgP2 NCs is positively shifted (0.7 eV) from that of Ag 3d5/2 for Ag NCs, while there is negative shift for the binding energy of P 2p3/2 (128.6 eV) in AgP2 NCs when compared to that of elemental P (130.0 eV)15. The white line intensity for AgP2 NCs is stronger than that for Ag NCs, which indicates a decrease in the number of electrons in the Ag frontier orbitals39. Moreover, decreasing the size of AgP2 NCs leads to increases of both rising edge intensity and white line intensity, which are ascribed to the changes of coordination environment and surface states40. The Fourier transforms of the k3-weighted extended Ag K-edge X-ray absorption fine structure (EXAFS) spectra were shown in Fig. 1h. The EXAFS fitting results are summarized in Supplementary Fig. 7 and Table 2. The Ag–Ag bond length for AgP2 NCs is larger than that for Ag NCs and Ag foil (2.65 Å), which is ascribed to the negatively charged P-rich clusters strongly binding the surrounding Ag atoms and thus weakening the Ag–Ag bonding strength. The P incorporation into Ag also results in a decrease of coordination numbers of Ag–Ag and structural disorder (Supplementary Table 2).
Electrochemical CO2 reduction
The CO2 electroreduction performance of 4 nm AgP2 NCs was measured in a Nafion membrane-separated H-type cell. For comparison, the performance of 10 nm AgP2 NCs, 4 nm Ag NCs, and 10 nm Ag NCs were also measured. The gaseous and liquid products were determined by a gas chromatography (GC) and H1 NMR, respectively, and only CO and H2 were observed. The geometric current density, CO and H2 Faradaic efficiency, and CO partial current density as a function of potential for 4 nm AgP2 NCs are summarized in Supplementary Table 3. Figure 2a shows the polarization curves acquired in a CO2-saturated 0.5 M KHCO3. The 4 nm AgP2 NCs achieve a current density as high as −15.2 mA cm−2 at −1.0 V vs. RHE, over twice that of 4 nm Ag NCs, and 3.6 times higher than that of 10 nm Ag NCs. The enhanced overall electrochemical activity of the 4 nm AgP2 NCs is potentially related to increased electrochemical active surface area (EASA) and interfacial charge transfer rate (Supplementary Figs. 8a and 9). The onset potential (Eonset, CO) for initial CO generation (defined as the potential required for jCO = −0.01 mA cm−2) on 4 nm AgP2 NCs is −0.22 V vs. RHE, which is only 110 mV lower than the thermodynamic potential of CO2-to-CO reaction. In comparison, the Eonset, CO for Ag NCs is −0.59 V vs. RHE (η = 480 mV). The 4 nm AgP2 NCs exhibit an overpotential for the same partial density of CO (jCO) that is consistently on the order of 0.3 V lower than 10 nm Ag NCs (Supplementary Fig. 10). The source of the anodic shift of onset potential and decreased overpotential for CO2-to-CO will be discussed below in detail. The performance of 10 nm AgP2 NCs is also provided as a same-size comparison to 10 nm Ag NCs, and also exhibit uniformly superior performance compared to Ag NCs. Notably, the 4 nm AgP2 NCs exhibit a remarkable geometric jCO of −0.27 and −8.7 mA cm−2 at −0.5 and −1.0 V vs. RHE, respectively, which is the one of the highest values reported for Ag-based electrocatalysts for CO2 reduction (Supplementary Table 4). A maximum FECO of 82% and minimum FEH2 of 15% is simultaneously achieved at −0.8 V vs. RHE (Fig. 2b), indicating CO2-to-CO reduction is more favorable than the competitive HER on 4 nm AgP2 NCs. The CO:H2 ratio in the syngas product varies from 1:3 to 5:1 in the potential range of −0.4 to −1.0 V vs. RHE. This is the widest range of syngas proportions for all Ag-based electrocatalysts (Supplementary Table 4), and even superior or comparable to recently reported electrocatalysts with impressive syngas efficiency, such as Au–Cu (1:2 to 4:1)41, Pd/C (1:4 to 1:1)42, Co3O4-CDots-C3N4 (1:4 to 14:1)43, MoSeS (1:3 to 1:1)44, and CdSxSe1−x (1:4 to 4:1)45. Notably, the 4 nm AgP2 NCs achieve all the key syngas ratios with nearly 100% Faradaic efficiency within an overpotential of 500 mV at stability for 12 h (Supplementary Table 3, Supplementary Fig. 11). The contribution to the H2 from catalyst substrates (Ketjet carbon or glassy carbon) was characterized and is minimal below −0.8 V vs. RHE (Supplementary Fig. 12). For comparison, a much lower FECO,max and narrower CO:H2 range is observed for Ag NCs (Supplementary Tables 6 and 7). To check that the NC surface is exposed, the effects of removing surface capping ligands on the electrochemical surface area and FECO are shown in Supplementary Fig. 13a–c. The Tafel slope for 4 nm AgP2 NCs, 10 nm AgP2 NCs, 4 nm Ag NCs, and 10 nm Ag NCs is 105, 117, 168, and 184 mV/dec, respectively (Supplementary Fig. 14). The smallest Tafel slope for 4 nm AgP2 NCs indicates that phosphorus plays a critical role in stabilizing the CO2- intermediate and accelerating the first electron transfer rate-determining step6. Additionally, the durability of 4 nm AgP2 NCs, 10 nm AgP2 NCs, and 10 nm Ag NCs was tested at a high constant potential of −0.8 V vs. RHE (Fig. 2c). A stable jCO with negligible decay within 12 h is observed on 4 nm AgP2 NCs, compared to 13.2% and 21.8% activity decay for 10 nm AgP2 NCs and Ag NCs, respectively. ICP-MS, TEM, and XPS confirm that the 4 nm AgP2 NCs remain almost the same after 12 h CO2 reduction (Supplementary Figs. 15–17).

Electrochemical measurements in CO2-saturated 0.5 M KHCO3 solution. a Linear sweep voltammetric curves; b Faradaic efficiency of CO and H2 and CO/H2 ratio; c stability of partial CO current density (jCO) measured at −0.8 V vs. RHE for 12 h. d In situ silver K-edge XANES and e EXAFS spectra of 4 nm AgP2 NCs after 30 min at the specified potential. Pristine AgP2 NCs, AgO, Ag2O, and Ag foil are included as references. f Faradaic efficiency of the CO and CO:H2 ratio as a function of Agδ+ in AgP2 and Ag.
To gain insight into the electronic structure and relationship between the chemical state of the 4 nm AgP2 NCs and the catalytic selectivity during CO2 electroreduction, operando XANES and EXAFS measurements were carried out under various applied potentials for 30 min in aqueous CO2-saturated 0.1 M KHCO3 (Fig. 2d, e). Commercial AgO, Ag2O, and Ag foil were used as references and a linear combination of XANES spectra was fitted to the in situ Ag K-edge spectra (Supplementary Fig. 18). The absorption edge (E0) and corresponding oxidation state of the silver species in AgP2 under various potentials are summarized in Supplementary Table 8. For the as-prepared AgP2 NCs, the absorption edge and radial distance of Ag–Ag are close to that of the AgO sample, and the initial oxidation state of the silver species is calculated to be +1.96. Under open-circuit potential (OCP), the oxidation state remains the same. When the applied potential increases to −0.4 V vs. RHE, the absorption edge shows a negative shift and a small Ag–Ag peak appears at the radial distance of 2.74 Å, suggesting the AgP2 is partially reduced and the average oxidation state decreases to +1.72. At the potential of −0.8 V vs. RHE, which corresponds to the peak in selectivity for CO2-to-CO, the oxidation state of the silver species is further reduced to +1.08. The evolution of the silver oxidation state in AgP2 from +1.96 to +1.08 occurs rapidly within 2 min under applied potential of −0.8 V vs. RHE and remains stable for 2 h. In comparison, AgO and Ag2O are reduced to Ag0 in less than 50 s at −0.8 V vs. RHE (Supplementary Fig. 19), which suggests that the rich-P content is beneficial to stabilize Ag+1.08. Maintaining this positive oxidation state under a highly reductive potential may be the key to the lowered the activation barrier of the initial CO2 reduction step and the stable and selective production of CO2-to-CO at −0.8 V. If the applied potential goes further to −1.2 V vs. RHE, the spectrum is matched with the Ag foil, indicating a complete transition from Ag+1.08 to Ag0. The operando measurements suggest phosphorus can stabilize a relatively high silver oxidation state which is critical to high FECO and wide CO:H2 range within a low overpotential window (Fig. 2f).
Theoretical investigation of intermediates
The schematic diagram of CRR and HER processes for tunable syngas production is summarized in Fig. 3a, where the ratio of syngas can be tuned over a wide range by changing applied potentials to meet input ratios necessary for the Fischer-Tropsch synthesis of hydrocarbons and alcohols. Silver-based CO2 reduction electrocatalysts generally follow two hydrogenation steps plus one nonelectrochemical CO desorption, i.e., \({\mathrm{CO}}_2 \to \ast {\mathrm{COOH}} \to \ast {\mathrm{CO}} \to {\mathrm{CO}}\). Density functional theory (DFT) was used to understand the key role of phosphorus alloying on the low overpotential and highly tunable syngas ratio observed on AgP2 NCs relative to Ag NCs. Low index (111) and high-index (211) facets were selected as representative surfaces for AgP246. Since HER is a competitive process with CO2 reduction reaction (CRR) in aqueous solution, DFT calculation was initially used to determine the free energy for atomic hydrogen adsorption (ΔGH*) on AgP2 and Ag electrocatalysts. The calculated ΔGH* on AgP2 (211), AgP2 (111), and Ag (111) surfaces are shown in Fig. 3b and corresponding complete structures and energies are provided in Supplementary Fig. 21 and Table 9. The closed packed Ag (111) surface exhibits a large positive ΔGH* of 0.510 eV, which implies a high-energy barrier for hydrogen adsorption process, consistent with previously reported Ag-based HER catalysts47. For AgP2 (211) and AgP2 (111), weak absorption of atomic H* on silver is still observed, with an average ΔGH* of 0.806 and 0.443 eV for AgP2 (211) and AgP2 (111), respectively. However, the HER process can proceed via P sites, and the AgP2 (111) surface has P sites (denoted P1 and P2) with a ΔGH* of 0.036 and −0.039 eV, favorable for hydrogen production. Interestingly, for the high-index surface AgP2 (211), all three P HER active sites (denoted as P1, P2, and P3) have much lower ΔGH* values (−0.360, −0.436, and −0.486 eV, respectively). Not only will strong adsorption of H* on phosphorus sites suppress the HER rate, but the adsorbed H* may also be delivered to the adsorbed carbon species on adjunct silver sites to promote the hydrogenation reaction.

Density functional theory calculations. a Schematic diagram of selective CO2-to-syngas on AgP2 (211). Free energy change (ΔG) calculated for the b HER and c selective CO2-to-CO reaction. d Optimized *COOH adsorption configuration and e corresponding PDOS on AgP2 (211) and Ag (111).
The optimized geometrics of *COOH and *CO intermediates on AgP2 (211), AgP2 (111), and Ag (111) surfaces and corresponding adsorption energy are included in Supplementary Fig. 22 and Table 10. Figure 3b depicts the free energy diagram for the lowest free energy path for the selective CO2-to-CO pathway at an equilibrium potential of −0.11 V vs. RHE. For all three surfaces, the initial CO2 hydrogenation is the rate-determined step for the CO2-to-CO process; however ΔG*COOH for both AgP2 surfaces is over threefold lower than that for Ag. This is in excellent agreement with the experimentally observed decrease in the overpotential for CO2 reduction. The minimal applied potential (Ump) for CO2-to-CO reduction on AgP2 (211), AgP2 (111), and Ag (111) is determined as −0.355, −0.434, and −1.134 V vs. RHE, respectively (Supplementary Fig. 23). This theoretical Ump is of greater magnitude than the experimental Eonset,CO on 4 nm AgP2 NCs of −0.22 V vs. RHE, indicating non-thermodynamic factors might be involved in lowering the activation energy barrier of the first hydrogenation process. At the same time, CO remains only weakly adsorbed on AgP2 with a binding free energy of around 0.085 eV. While CO is bound more tightly for AgP2 than for Ag, it is far from the strong adsorption regime that causes catalyst poisoning for Pt-based catalysts where binding strength on the order of −1 eV48. Comparing the 4 nm AgP2 NCs versus 10 nm AgP2 NCs, the higher electrocatalytic performance and ability to achieve high CO content syngas for the 4 nm AgP2 is consistent with the enhanced presence of high-index planes such as (211) on the ultrasmall NCs8.
Analysis of the partial density of states (PDOS) and Bader charge is used to understand the role of P in increasing the binding to Ag. PDOS shows that in AgP2 the valence Ag4d orbital is more delocalized with higher density d-valence band states compared to Ag at the Fermi level (Fig. 3e). This leads to a higher overlap between Ag4d and C2p, consistent with stronger *COOH adsorption49. The Ag–C bond lengths of 2.15 Å for AgP2 is shorter than that for Ag (Fig. 3d), suggesting tighter binding of *COOH on AgP2. Bader charge analysis shows that the bond strength increases with the partial positive charge on Ag, with the most stable *COOH adsorption site on AgP2 (211) having a Bader charge of Ag0.2+ compared to metallic Ag0 sites50.
Hybrid photocathode for PEC
Inspired by the efficient activity, selectivity, and stability of 4 nm AgP2 NCs toward syngas from CO2 electroreduction, we integrated 4 nm AgP2 NCs onto a surface-protected n+p-Si semiconductor to build a hybrid photocathode of n+p-Si/Al2O3/AgP2 based on the buried junction design. Briefly, an ultrathin 2 nm Al2O3 layer was deposited by ALD to protect the underlying silicon semiconductor from electrolyte corrosion and eliminate the potential for additional interfacial resistance due to in situ growth of silicon oxide. Diluted 4 nm AgP2 NCs were deposited using spin coating without any aggregation (Supplementary Fig. 24). The mechanism for PEC CO2 reduction is depicted in Fig. 4a and begins with the photogenerated minor carrier electrons in n+p-Si accumulating at the n+p-Si/Al2O3 interface. The Al2O3 is kept thin enough to allow efficient electron tunneling into the AgP2 cocatalyst, and selective CO2 electroreduction occurs at the AgP2 surface. The work function of AgP2 NCs (4.2 eV) is smaller than that of Ag NCs (4.8 eV) and AgP2 NCs are metallic. (Supplementary Fig. 25 and Fig. 26). Figure 4b shows a cross-sectional high-resolution transmission electron microscopy (HRTEM) image of the n+p-Si/Al2O3/AgP2 interface. The corresponding elemental distribution from STEM-EDS line scan across the n+p-Si/Al2O3/AgP2 interface is shown in Fig. 4c. The Al signal is evident at the interface between the Si and Ag signal. The Si semiconductor and AgP2 cocatalyst are well separated by the Al2O3 layer, which is expected to help passivate and protect the Si surface28. The n+p-Si/Al2O3/AgP2 shows similar absorption (Supplementary Fig. 27) compared with n+p-Si and n+p-Si/Al2O3, indicating the negligible light absorption and reflection loss from the ultrathin ALD oxide and sparse AgP2 nanocrystal layer.

Photoelectrochemical device and performance. a Schematic energy level alignment. b Cross-sectional HRTEM image of n+p-Si/Al2O3/AgP2 hybrid photocathode and c corresponding line scan elemental EDS spectra. Scale bar 5 nm. d j–E curves of as-prepared n+p-Si, n+p-Si/AgP2, n+p-Si/Al2O3, and n+p-Si/Al2O3/AgP2 photocathodes under simulated solar illumination (AM-1.5G, 100 mW cm−2) in CO2-saturated 0.5 M KHCO3 solution. e Faradaic efficiency of CO and H2 at different potentials for n+p-Si/Al2O3/AgP2.
Figure 4d shows the current density vs. potential (j–E) curves of the as-prepared n+p-Si/Al2O3/AgP2 photocathode in a CO2-saturated 0.5 M KHCO3 solution under simulated AM-1.5G solar illumination. Under dark condition, the n+p-Si/Al2O3/AgP2 shows a very negative onset potential of −0.16 V vs. RHE (defined as the potential to attain a total current of −0.1 mA cm−2) and negligible current in the potential window. Under illumination, n+p-Si/Al2O3/AgP2 exhibits the most positive onset potential (Eonset = 0.42 V vs. RHE) compared to control devices in which select layers are missing: n+p-Si/Al2O3 (0.28 V vs. RHE), n+p-Si/AgP2 (0.31 V vs. RHE), and n+p-Si (0.12 V. RHE). The photovoltage for n+p-Si/Al2O3/AgP2 is around 580 mV, as estimated by the difference in potential required to achieve −0.1 mA cm−2 under dark versus illumination. This high output photovoltage is superior or comparable to state-of-the-art p-Si photocathodes, such as Ni-Mo/n+p-Si NW (560 mV)51, CoP-n+p Si (580 mV)52, and p-Si/NiCoSex53 (500 mV). The high photovoltage is attributed to (1) the ultrathin Al2O3 overlayer passivating the surface states of n+p-Si and increasing the output photovoltage34, and (2) the AgP2 acting as a cocatalyst for lowering the reaction barrier and providing a fast interfacial reaction rate. However, the saturated photocurrent density (jsat) of n+p-Si/Al2O3/AgP2 is lower than that (~−30 mA cm−2) of an ideal state-of-the-art Si-based photocathode, which is ascribed to the non-ideal p–n homojunction and the Si/Al2O3 interface (Supplementary Fig. 28). At the CO2-to-CO reduction potential of −0.11 V vs. RHE, n+p-Si/Al2O3/AgP2 achieves a total current density (j−0.11, total) of −5.2 mA cm−2. The remarkable j−0.11, total is higher than that of the other photocathodes towards CO2 reduction, such as Au3Cu NP/Si NW (j−0.11,total = −2.2 mA cm−2)20, RA-Au/n+p-Si (j−0.11,total = −4.0 mA cm−2)21, N:C/N:ZnTe (j−0.11,total = −1.2 mA cm−2)54, and Cu-ZnO/GaN/n+p-Si (j−0.11,total = −1.2 mA cm−2)55. The Faradaic efficiency for H2 and CO in the potential range from −0.6 to 0.2 V vs. RHE is summarized in Fig. 4e and Supplementary Table 11. Detectable CO (FECO = 18%) starts to be observed at 0.2 V vs. RHE (Eonset, CO) for n+p-Si/Al2O3/AgP2, while the Eonset, CO negatively shifts to 0.09 V vs. RHE for n+p-Si/Al2O3/Ag, indicating that AgP2 is more suitable as a cocatalyst for CO2-to-CO than Ag, consistent with the CRR results presented above. The n+p-Si/Al2O3/AgP2 simultaneously shows a maximum FECO of 67% and minimum FEH2 of 31% at −0.2 V vs. RHE. When further increasing the applied potential, the FECO is decreased due to the limitation of CO2 mass transport and hydrogen evolution becomes the dominant reaction. In the potential window from −0.6 to 0.2 V vs. RHE, the CO/H2 ratio in the PEC syngas product can be tuned in a large range of 1:5 to 2:1. For the hybrid n+p-Si/Al2O3/AgP2 photocathode, it is challenging to determine the fraction of H2 generated from the AgP2 catalytic sites rather than the bare n+p-Si/Al2O3 photoelectrode; however, comparing the activity of different surfaces suggests the majority of H2 is from AgP2 NCs (Supplementary Figs. 29 and 30, Table 13). Notably, the impressive Eonset, CO and partial photocurrent density for CO at −0.11 V vs. RHE (j−0.11, CO = −3.2 mA cm−2) are the best values among all the reported photocathodes for CO2 reduction (detailed comparison is in Supplementary Table 14). The stability for PEC CO2 reduction was tested at a constant potential of −0.2 V vs. RHE for 12 h (Supplementary Fig. 31). Only a minimal change is observed in both total photocurrent density and FECO during long-term measurement. The incident photon-to-current conversion efficiency (IPCE) of n+p-Si/Al2O3/AgP2 reaches a maximum of 67% at 680 nm, while the absorbed photon-to-current conversion efficiency, calculated using the absorption, reflection, and transmission data from Supplementary Fig. 27, reaches a maximum of 85% (Supplementary Fig. 32). The addition of 1-butyl-3-methylimidazolium (BMIM-BF4) ionic liquid into the 0.5 M KHCO3 solution, an enhanced FECO of 89% at −0.2 V vs. RHE is reached (Supplementary Table 14). The adsorbed BMIM-BF4 on the catalysts surface lowers the energy barrier for reducing CO2 to CO2˙ˉ, enhancing CO production while simultaneously suppressing hydrogen evolution56,57.
The critical roles of each layer in the buried junction structure are revealed by comparison to structures with a component missing. While n+p-Si/Al2O3 is expected to be active for HER, no CO is detected in gas products for both n+p-Si and n+p-Si/Al2O3 within the overpotential of 400 mV because hydrogen evolution is more favorable than CO2 reduction without the selective cocatalyst (Supplementary Table 12). For the n+p-Si/AgP2 photocathode without the protective ALD layer, a more negative Eonset,CO is observed compared to n+p-Si/Al2O3/AgP2 (Supplementary Table 14), confirming the role of Al2O3 in ensuring high photovoltage output. Further, a 75% photocurrent decay is observed during the stability test of n+p-Si/AgP2 (Supplementary Fig. 31), which is attributed to either etching or an in situ increase in the thickness of the silicon oxide insulator.
Interfacial charge transport
Cyclic voltammograms (CVs) in ferri/ferrocyanide redox solution elucidate charge transfer in the as-prepared photocathodes as shown in Fig. 5a. Under dark conditions, only an oxidation peak is present in the CV curve of n+p-Si/Al2O3/AgP2. The lack of a reduction peak can be explained by the absence of electrons at the solid electrode/electrolyte interface for Fe (III) reduction or water reduction32,58. In contrast, a symmetric CV curve with narrow peak-to-peak splitting (61 mV) and a positive shift (380 mV) in the anodic oxidation peak is observed under illumination, indicating the photogenerated electrons are driven to the solid/electrolyte interface and participate in Fe(III) reduction. Without AgP2, n+p-Si/Al2O3 exhibits less peak current and wider peak-to-peak splitting (590 mV), suggesting the role of AgP2 in accelerating the interfacial reaction rate. Increasing the thickness of Al2O3 beyond the 2 nm optimal thickness results in a decrease in the reduction and oxidation peaks due to reduced tunneling through the passivation layer (Supplementary Table 15)28. For 0.5- and 1-nm- thick Al2O3 layers there is a large reduction in peak current after 10 CV cycles due to incomplete protection of the n+p-Si (Supplementary Fig. 35).

Detailed characterization of photoelectrochemical response. a Cyclic voltammogram of n+p-Si/2 nm Al2O3, n+p-Si/2 nm Al2O3/AgP2, and n+p-Si/5 nm Al2O3/AgP2 in ferri/ferrocyanide redox solution. All measurements were conducted in the simulated light illumination except as marked. b In situ transient absorption spectra of as-prepared samples measured in CO2-saturated 0.5 M KHCO3 solution under open-circuit potential. The samples were excited by 450 nm laser with a power of 50 µW.
In situ transient absorption (TA) spectroscopy is a useful tool to understand interfacial charge transfer processes for PEC water oxidation and reduction59. However, there are few TA studies of photocathodes for PEC CO2 reduction due to the timescale mismatch between charge transfer and the surface catalysis process60. Figure 5b shows the in situ TA spectra of as-prepared photocathodes immersed in CO2-saturated 0.5 M KHCO3 solution at OCP. Most notable is the difference in timescales between the n+p-Si sample and the samples passivated with Al2O3. For bare n+p-Si, between 5 and 50 ps the photogenerated charge carriers are trapped reducing the absorption signal. The wide absorption peak at 580 nm blueshifts to 550 nm and narrows, which is ascribed to electrons relaxing into deep lying localized states61. For the samples coated by ALD, only a slight change in OD is observed between 5 and 50 ps and so slow TA spectra (Supplementary Fig. 36) were measured. The normalized decay trace for bare n+p-Si yields a representative lifetime (t1/2) of 3.5 ps, which is over five orders of magnitude shorter than that of n+p-Si/Al2O3 (3.2 µs) and n+p-Si/Al2O3/AgP2 (6.3 µs). In TA absorption spectra (at 1 ms) of n+p-Si and n+p-Si/Al2O3/AgP2 at an applied potential of −0.2 V vs. RHE (Supplementary Fig. 37), unmodified n+p-Si/Al2O3 exhibits a TA spectrum peaking at 610 nm. After AgP2 decoration, the amplitude at 610 nm is lost, and a new peak appears at 690 nm, which suggests efficient electron transfer from the n+p-Si/Al2O3 surface to AgP262. Transient photocurrent (TPC) measurements were carried out at 0 V vs. RHE for the n+p-Si/Al2O3/AgP2 photocathode (Supplementary Fig. 38). With the addition of AgP2 cocatalyst, the lifetime of the decay (t1/2) is increased from 8.7 to 64.5 µs due to capture of the electrons by AgP2.
In summary, AgP2 NCs show a remarkable 0.3 V decrease in overpotential for CO generation compared to the benchmark Ag catalyst. The AgP2 NCs achieve a maximum Faradaic efficiency of 82% for CO production and a wide CO:H2 ratio from 1:3 to 5:1 within an overpotential window of 500 mV. DFT calculations reveal a threefold energy barrier decrease for the formate intermediate formation step compared to Ag, resulting in a high CO:H2 ratio at low overpotential. A n+p-Si/Al2O3/AgP2 photocathode exhibits an impressive onset potential of 0.2 V vs. RHE for PEC CO2-to-CO reduction and a maximum FECO of 67% at −0.2 V vs. RHE.
Methods
Chemicals and materials
Silver acetate (97%), oleylamine (OAm, tech. 70%), oleic acid (OA, tech. 90%), 1-octadecene (ODE, tech. 90%), and Nafion 117 solution were purchased from Sigma-Aldrich. Tris(trimethylsilyl)phosphine ((Me3Si)3P, 98%) dissolved in hexane (10 wt%) was purchased from Stream Chemicals. Commercial carbon black was obtained from FuelCellStore. Si wafers with a resistivity of 1–100 Ω cm (p-type, B-doped, (100) orientation, prime grade, 500 μm thickness, 100 mm diameter) were obtained from University Wafer.
Synthesis of sub-4 nm silver phosphide (AgP2) NCs
Silver acetate (0.5 mmol, 0.0835 g), OAm (6 mL), OA (1 mL), and ODE (6 mL) were added into a 50-mL three-neck, round bottom flask containing a stirring bar. This solution was placed under vacuum at 60 and 120 °C to degas for 30 min, respectively, to remove the low boiling point impurities and oxygen. The solution was placed under nitrogen and heated to 180 °C. Meanwhile, a second precursor solution comprising 2 mL (Me3Si)3P (10 wt% in hexane) and 1 mL ODE was degassed under vacuum and room temperature for 10 min in a 10 mL vial with a septum cap to remove hexane. The (Me3Si)3P solution was rapidly injected into the reaction flask at 180 °C and kept for 10 min at this temperature. Finally, the solution was maintained at 80 °C for 1 h to complete the Ostwald ripening and then cooled to room temperature. The reaction mixture was centrifuged at 3300 × g for 6 min The isolated precipitate was re-suspended in hexane, then acetone and methanol were added to create a 1:6:1 (v:v:v) hexanes:acetone:methanol solution to cause aggregation of the NCs. The resulting cloudy solution was centrifuged. This isolated solid was redissolved in hexane and the process repeated for an additional three times. Finally, the product was dispersed in hexane (2 mg/mL).
For comparison, 10 nm AgP2 NCs were synthesized similarly but the reaction time at 180 °C was 25 min instead of 10 min, 4 and 10 nm Ag NCs were synthesized by co-heating the precursors of silver acetate (0.5 mmol), 8 mL OAm, 0.5 mL OA at 180 °C for 20 min and 2 h, respectively.
Materials characterizations
Elemental analysis was performed using inductively coupled plasma mass spectroscopy (ICP-MS; Teledyne Leeman Labs). XRD patterns were obtained on an X-ray diffractometer (Bruker D2 Phaser) using Cu Kα radiation. HRTEM was carried out on a Tecnai G2 F20 microscope at 200 or 300 kV. STEM and EDS were performed on a JEOL JEM 2200FS STEM/TEM microscope at an acceleration voltage of 200 kV equipped with a CEOS probe corrector (Heidelberg, Germany) to provide a nominal resolution of ~0.07 nm. A Bruker-AXS silicon drift detector (SDD) was used for all EDS elemental analysis. All colloidal samples for TEM measurements were prepared by ultrasonic dispersion in hexane and were dropcast onto copper grids covered with a carbon film. For the hybrid photocathode, the corresponding cross-sectional TEM sample was prepared by mechanical sectioning and polishing to a thickness of 20 µm. The polished film was further thinned until perforation by focused ion beam and measured by HRTEM (Tecnai G2 F20 microscope at 300 kV). XPS (Kratos Axis Ultra DLD, UK) was used to detect the chemical composition of the as-prepared samples. The binding energy was calibrated based on the C 1s peak at 284.8 eV. Ultraviolet photoelectron spectroscopy (UPS) was performed using the same XPS instrument with a 165 mm hemispherical sector analyser. UPS was measured with a He I (21.2 eV) radiation source and a total energy resolution of 200 meV. Ag K-edge X-ray absorption fine structure (XAFS) measurements were made at the beamline 14W1 in Shanghai Synchrotron Radiation Facility (SSRF), China. The storage ring of SSRF was operated at 3.5 GeV with a current of 300 mA. The extended XAFS (EXAFS) data were processed according to the standard procedures using the WinXAS3.1 program. Theoretical amplitudes and phase-shift functions were calculated with the FEFF8.2 code using the crystal structural parameters of the Ag foil, Ag2O, and AgO. The parameters describing the electronic properties (e.g., correction to the photoelectron energy origin, E0) and local structure environment including CN, bond distance (R), and Debye Waller (DW) factor around the absorbing atoms were allowed to vary during the fit process. The fitted ranges for k and R spaces were selected to be k = 3.0–11.5 Å−1 and R = 1–3 Å (k3 weighted), respectively. XANES calculation was based on self-consistent multiple-scattering (MS) methods, and was carried out using the FEFF8.2 code. The experimental absorption coefficients as a function of energies μ(E) were processed by background subtraction and normalization procedures, and reported as normalized absorption. Optical transmission measurements were performed using a Shimadzu 3600 UV–Vis–NIR spectrophotometer. To perform the optical absorption measurements, the sample was placed inside an integrating sphere and illuminated with white light from a 300 W xenon lamp. All light that was not absorbed was collected and measured using an Ocean Optics Jaz EL 200-XR1 spectrometer. Reflection data were calculated assuming that the absorbed, transmitted, and reflected light fractions sum to one. The femtosecond TA spectroscopy was carried out using PHAROS laser operating at 10 kHz coupled to an ORPHEUS optical parametric amplifier in tandem with a LYRA harmonic generator to produce the desirable wavelength for sample excitation. Relatively low pump intensity of 50 µW and 400 µJ cm−2 was used for femtosecond and microsecond TA experiment. A portion of laser output was split off to pump a sapphire crystal to generate a white light continuum for the probe beam in the region of 500–900 nm. Diffuse reflectance mode was adopted due to the non-transparent silicon photocathodes. The TA intensity is displayed as % absorption = (1−R/R0) × 100, where R and R0 are the probe pulse intensities with and without excitation, respectively.
Catalyst inks preparation
For the electrochemical measurements, the as-synthesized NC solution (5 mL at 2 mg/mL) was mixed with an equal amount by weight of Ketjen carbon (C) support (10 mg) in a 20 mL vial. This colloidal mixture was sonicated for 1 h to ensure complete distribution of NCs onto the carbon support. After evaporation of the volatile hexane, 5 mL of acetic acid was added to the NC/C solid and stirred for 12 h at 60 °C. The dispersion was cooled down to room temperature. Ethanol (8 mL) was added and the mixture was centrifuged at 3300 × g for 6 min This procedure was repeated three times. Finally, the solid NCs/C was then re-dispersed in a mixture of deionized water, isopropanol, and Nafion 117 solution (v:v:v = 4:2:0.05) to form a catalyst ink (2 mg/mL), 50 μL of which was deposited on the glassy carbon working electrode (D = 5 mm) to form a catalyst film with a loading amount of about 0.5 mg/cm2 and dried at ambient conditions. Pure Ketject carbon and AgP2/Ketject carbon mixtures with different ratios were also prepared using the same procedures for comparison.
For the spin coating of the NC cocatalyst on the semiconductor, the concentration of ink solution was diluted to 0.5 mg/mL.
Electrochemical measurements
CO2 electroreduction was carried out in a customized H-type cell separated by a Nafion 117 membrane with 0.5 M KHCO3. Each compartment contained 20 mL electrolyte and 10 mL headspace. A conventional three-electrode system, namely a disk shaped glassy carbon electrode (0.20 cm2) coated with a catalyst was used as the working electrode, Ag/AgCl as the reference electrode, and graphite rod as the counter electrode, was employed for CO2 reduction with a Reference 600 potentiostat (Gamry Instrument Inc.). The electrolyte solution was purged with high purity CO2 gas for at least 1 h until a pH of 6.8 was reached before the electrochemical measurements. Before CO2 electrolysis, the headspace was also purged for 20 min. The polarization curves were obtained with a scanning rate of 5 mV/s. Chronoamperometry was performed at a fixed potential for 2 h by maintaining a constant CO2 flow rate. Gas-phase products were sampled every 30 min using a 50 µL gas-tight syringe (Hamilton). A gas chromatograph (GC, Agilent 6890) with a thermal conductivity detector was used for quantification. Liquid products were detected and quantified using a Bruker Avance 400 MHz NMR spectrometer. Typically, a 500 µL electrolyte was sampled after electrolysis and mixed with 100 µL D2O and 1.67 ppm DMSO. The electrode stability was tested by chronoamperometry under a constant potential of −0.8 V vs. RHE for 12 h. Electrochemical impedance spectroscopy under dark conditions was performed on a Reference 600 (Gamry Instrument Inc.) with the working electrode biased at a constant potential of −0.1 V vs. RHE, while sweeping the frequency from 105 to 0.1 Hz with a 10 mV AC dither. The EASA values of the as-prepared electrodes were evaluated by CV using the ferri-/ferrocyanide redox couple ([Fe(CN)6]3−/4−) as a probe. Cyclic voltammetry was carried out in a nitrogen-purged 5 mM K3Fe(CN)6/0.1 M KCl solution with platinum foil as the counter electrode. EASA values were calculated using the Randles-Sevcik equation (Eq. (1)),
where Ip is peak current (A), n = 1, D = 4.34 × 10–6 cm2 s−1, A is the EASA (cm2), C is the concentration of potassium ferricyanide (5 × 10–6 mol cm−2), and υ is the scan rate (5 mV s−1). All potentials reported in this paper were converted from vs. Ag/AgCl to vs. RHE by adding a value of 0.197 + 0.059 × pH.
Fabrication of hybrid photocathode
Degenerately boron-doped Si wafers with a resistivity of 0.002 Ω cm−1 (p-type, 500 µm) were cut into 2 × 1.5 cm2 pieces. The p-Si wafers were cleaned and etched in hydrofluoric acid to remove the native oxide and trace contaminants. The surface n-type doping of the p-Si wafers was treated with phosphorus from POCl3 gas for 10 min at 900 °C55. We also prepared the control samples with different doping time (20 and 30 min). The backside of each p-Si-based photocathode was scratched and painted with a gallium indium eutectic for good electrical contact. The Al2O3 surface protective layers were deposited on the n+p-Si by a GEMSTAR-6 ALD system. Trimethylaluminum (TMA) and distilled water (H2O) were used as precursors of Al and O, respectively. The ALD of Al2O3 was carried out at a substrate temperature of 175 °C. Each precursor was held in the chamber for 2.2 s, followed by a 28 s nitrogen purge. The growth rate per cycle (GPC) was identified by X-ray reflectivity on films deposited on optically polished silicon wafers with a native surface oxide. The GPC was 1.2 Å for Al2O3. Next, the AgP2 NC cocatalyst layer was deposited by layer-by-layer spin coating using 200 μL of the AgP2 NCs solution (0.2 mg/mL) for each cycle. The spin coating was carried out at 1467 × g for 45 s. The optimized spin cycle for AgP2 deposition is identified as one cycle. Different catalyst coverage was monitored by controlling the spin cycles of diluted AgP2 ink. Finally, the hybrid photocathode was treated by annealing at 200 °C for 60 min in a tube furnace under a flow of N2.
PEC measurements
The photoelectrochemical activity of the photocathodes was measured in a home-made PEC reactor56 with a three-electrode system using a Reference 600 potentiostat (Gamry Instrument Inc.). The prepared n+p-Si/Al2O3/AgP2 served as the working electrode, a graphite rod as the counter electrode, and Ag/AgCl as the reference electrode. A Nafion 117 membrane was inserted into the PEC reactor to separate the working and counter electrodes. The electrolyte was a CO2-saturated 0.5 M KHCO3 solution. A 300 W Xenon arc lamp (Newport) equipped with an AM-1.5G filter was used to simulate the solar spectrum. The light intensity was calibrated to 100 mW/cm2 through a Newport 843-R power meter. The geometric area of the photocathode under light illumination was measured and maintained at nearly 1 cm2. The incident photoelectron conversion efficiency was measured by a specially designed IPCE system (Zolix Solar Cell Scan 100) with a 150 W Xe lamp and a monochromator (Oriel Cornerstone 130). For the interfacial charge transport characterization, CVs were recorded under dark or light illumination in a ferri/ferrocyanide solution which was made of 10 mM of both K3Fe(CN)6 and K4Fe(CN)6 in 1 M aqueous KCl.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request
Change history
27 October 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41467-022-33833-5
References
James, O. O., Mesubi, A. M., Ako, T. C. & Maity, S. Increasing carbon utilization in Fischer–Tropsch synthesis using H2-deficient or CO2-rich syngas feeds. Fuel Process. Technol. 91, 136–144 (2010).
Goeppert, A. et al. Recycling of carbon dioxide to methanol and derived products—closing the loop. Chem. Soc. Rev. 43, 7995–8048 (2014).
Li, F. F., Lau, J. & Licht, S. Sungas instead of syngas: efficient coproduction of CO and H2 with a single beam of sunlight. Adv. Sci. 2, 1500260 (2015).
Feaster, J. T. et al. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 7, 4822–4827 (2017).
Wang, Y., Liu, J., Wang, Y., Al-Enizi, A. M. & Zheng, G. Tuning of CO2 reduction selectivity on metal electrocatalysts. Small 13, 1701809 (2017).
Lu, Q. et al. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 5, 3242 (2014).
Liu, S. et al. Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates. J. Am. Chem. Soc. 139, 2160–2163 (2017).
Kim, C. et al. Achieving selective and efficient electrocatalytic activity for CO2 reduction using immobilized silver nanoparticles. J. Am. Chem. Soc. 137, 13844–13850 (2015).
Mistry, H. et al. Enhanced carbon dioxide electroreduction to carbon monoxide over defect-rich plasma-activated silver catalysts. Angew. Chem. Int. Ed. 56, 11394–11398 (2017).
Kim, C. et al. Insight into electrochemical CO2 reduction on surface-molecule-mediated Ag nanoparticles. ACS Catal. 7, 779–785 (2017).
Luc, W. et al. Ag–Sn bimetallic catalyst with a core–shell structure for CO2 reduction. J. Am. Chem. Soc. 139, 1885–1893 (2017).
Anantharaj, S. et al. Recent trends and perspectives in electrochemical water splitting with an emphasis on sulfide, selenide, and phosphide catalysts of Fe, Co, and Ni: A review. ACS Catal. 6, 8069–8097 (2016).
Zheng, X. et al. Role of non-metallic atoms in enhancing the catalytic activity of nickel-based compounds for hydrogen evolution reaction. Chem. Sci. 9, 1822–1830 (2018).
Li, H. et al. Earth-abundant iron diboride (FeB2) nanoparticles as highly active bifunctional electrocatalysts for overall water splitting. Adv. Energy Mater. 7, 1700513 (2017).
Li, H. et al. Colloidal cobalt phosphide nanocrystals as trifunctional electrocatalysts for overall water splitting powered by a zinc–air battery. Adv. Mater. 30, 1705796 (2018).
Weizer, V. G. & Fatemi, N. S. Low resistance silver contacts to indium phosphide: electrical and metallurgical considerations. J. Appl. Phys. 73, 2353 (1993).
Massey, A. G., Thompson, N. R., Johnson, B. F. G. & Davis R. The Chemistry of Copper, Silver and Gold in Comprehensive Inorganic Chemistry (ed. Bailar J.C., Emeleus H. J., Nyholm R., Trotman-Dickenson, A.F.) 101–102 (Elsevier, 1973).
Callejas, J. F., Read, C. G., Roske, C. W., Lewis, N. S. & Schaak, R. E. Synthesis, characterization, and properties of metal phosphide catalysts for the hydrogen-evolution reaction. Chem. Mater. 28, 6017–6044 (2016).
Zhang, L., Zhao, Z. J., Wang, T. & Gong, J. Nano-designed semiconductors for electro- and photoelectro-catalytic conversion of carbon dioxide. Chem. Soc. Rev. 47, 5423–5433 (2018).
Kong, Q. et al. Directed assembly of nanoparticle catalysts on nanowire photoelectrodes for photoelectrochemical CO2 reduction. Nano Lett. 16, 5675–5680 (2016).
Song, J. T. et al. Nanoporous Au thin films on Si photoelectrodes for selective and efficient photoelectrochemical CO2 reduction. Adv. Energy Mater. 7, 1601103 (2017).
Kang, U. et al. Photosynthesis of formate from CO2 and water at 1% energy efficiency via copper iron oxide catalysis. Energy Environ. Sci. 8, 2638–2643 (2015).
Jang, J. W. et al. Aqueous-solution route to zinc telluride films for application to CO2 reduction. Angew. Chem. Int. Ed. 53, 5852–5857 (2014).
Jang, Y. J. et al. Selective CO production by Au coupled ZnTe/ZnO in the photoelectrochemical CO2 reduction system. Energy Environ. Sci. 8, 3597–3604 (2015).
DuChene, J. S., Tagliabue, G., Welch, A. J., Cheng, W. H. & Atwater, H. A. Hot hole collection and photoelectrochemical CO2 reduction with plasmonic Au/p-GaN photocathodes. Nano Lett. 18, 2545–2550 (2018).
Sun, K. et al. Enabling silicon for solar-fuel production. Chem. Rev. 114, 8662–8719 (2014).
Boettcher, S. W. et al. Photoelectrochemical hydrogen evolution using Si microwire arrays. J. Am. Chem. Soc. 133, 1216–1219 (2011).
Zhu, T. & Chong, M. N. Prospects of metal–insulator–semiconductor (MIS) nanojunction structures for enhanced hydrogen evolution in photoelectrochemical cells: a review. Nano Today 12, 347–373 (2015).
Hisatomi, T. et al. Cathodic shift in onset potential of solar oxygen evolution on hematite by 13-group oxide overlayers. Energy Environ. Sci. 4, 2512–2515 (2011).
Hu, S. et al. Amorphous TiO2 coatings stabilize Si, GaAs, and GaP photoanodes for efficient water oxidation. Science 344, 1005–1009 (2014).
Ji, L. et al. Localized dielectric breakdown and antireflection coating in metal–oxide–semiconductor photoelectrodes. Nat. Mater. 16, 127–131 (2017).
Chen, Y. W. et al. Atomic layer-deposited tunnel oxide stabilizes silicon photoanodes for water oxidation. Nat. Mater. 10, 539–544 (2011).
Esposito, D. V., Levin, I., Moffat, T. P. & Talin, A. A. H2 evolution at Si-based metal–insulator–semiconductor photoelectrodes enhanced by inversion channel charge collection and H spillover. Nat. Mater. 12, 562–568 (2013).
Scheuermann, A. G. et al. Design principles for maximizing photovoltage in metal-oxide-protected water-splitting photoanodes. Nat. Mater. 15, 99–105 (2016).
Digdaya, I. A., Adhyaksa, G. W. P., Trzesniewski, B. J., Garnett, E. C. & Smith, W. A. Interfacial engineering of metal-insulator-semiconductor junctions for efficient and stable photoelectrochemical water oxidation. Nat. Commun. 8, 15968 (2017).
Henkes, A. E., Vasquez, Y. & Schaak, R. E. Converting metals into phosphides: A general strategy for the synthesis of metal phosphide nanocrystals. J. Am. Chem. Soc. 129, 1896–1897 (2007).
Carenco, S., Portehault, D., Boissiere, C., Mezailles, N. & Sanchez, C. Nanoscaled metal borides and phosphides: recent developments and perspectives. Chem. Rev. 113, 7981–8065 (2013).
Mikhlin, Y. L. et al. XPS and Ag L3-edge XANES characterization of silver and silver–gold sulfoselenides. J. Phys. Chem. Solids 116, 292–298 (2018).
Becknell, N. et al. Atomic structure of Pt3Ni nanoframe electrocatalysts by in situ X-ray absorption spectroscopy. J. Am. Chem. Soc. 137, 15817–15824 (2015).
Dai, Y. et al. Inherent size effects on XANES of nanometer metal clusters: size-selected platinum clusters on silica. J. Phys. Chem. C. 121, 361–374 (2017).
Ross, M. B. et al. Tunable Cu enrichment enables designer syngas electrosynthesis from CO2. J. Am. Chem. Soc. 139, 9359–9363 (2017).
Sheng, W. et al. Electrochemical reduction of CO2 to synthesis gas with controlled CO/H2 ratios. Energy Environ. Sci. 10, 1180–1185 (2017).
Guo, S. et al. A Co3O4-CDots-C3N4 three component electrocatalyst design concept for efficient and tunable CO2 reduction to syngas. Nat. Commun. 8, 1828 (2017).
Xu, J. et al. Carbon dioxide electroreduction into syngas boosted by a partially delocalized charge in molybdenum sulfide selenide alloy monolayers. Angew. Chem. Int. Ed. 56, 9121–9125 (2017).
He, R. et al. Achieving the widest range of syngas proportions at high current density over cadmium sulfoselenide nanorods in CO2 electroreduction. Adv. Mater. 30, 1705872 (2018).
Qi., Z. et al. Sub-4 nm PtZn intermetallic nanoparticles for enhanced mass and specific activities in catalytic electrooxidation reaction. J. Am. Chem. Soc. 139, 4762–4768 (2017).
Zhao, Z. et al. Computational screening of near-surface alloys for CO2 electroreduction. ACS Catal. 8, 3885–3894 (2018).
Kortlever, R. et al. Catalysts and reaction pathways for the electrochemical reduction of carbon dioxide. J. Phys. Chem. Lett. 6, 4073–4082 (2015).
Zhou, Y. et al. Dopant-induced electron localization drives CO2 reduction to C2 hydrocarbons. Nat. Chem. 10, 974–980 (2018).
Xiao, H. et al. Cu metal embedded in oxidized matrix catalyst to promote CO2 activation and CO dimerization for electrochemical reduction of CO2. Proc. Natl. Acad. Sci. USA 114, 6685–6688 (2017).
Warren, E. L., McKone, J. R., Atwater, H. A., Gray, H. B. & Lewis, N. S. Hydrogen-evolution characteristics of Ni–Mo-coated, radial junction, n+p-silicon microwire array photocathodes. Energy Environ. Sci. 5, 9653–9661 (2012).
Hellstern, T. R., Benck, J. D., Kibsgaard, J., Hahn, C. & Jaramillo, T. F. Engineering cobalt phosphide (CoP) thin film catalysts for enhanced hydrogen evolution activity on silicon photocathodes. Adv. Energy Mater. 6, 1501758 (2016).
Zhang, H. et al. A p-Si/NiCoSex core/shell nanopillar array photocathode for enhanced photoelectrochemical hydrogen production. Energy Environ. Sci. 9, 3113–3119 (2016).
Jang, Y. J. et al. Metal-free artificial photosynthesis of carbon monoxide using N-doped ZnTe nanorod photocathode decorated with N-doped carbon electrocatalyst layer. Adv. Energy Mater. https://doi.org/10.1002/aenm.201702636 (2018).
Chu, S. et al. Tunable syngas production from CO2 and H2O in an aqueous photoelectrochemical cell. Angew. Chem. Int. Ed. 55, 14262–14266 (2016).
Matsubara, Y., Grills, D. C. & Kuwahara, Y. Thermodynamic aspects of electrocatalytic CO2 reduction in acetonitrile and with an ionic liquid as solvent or electrolyte. ACS Catal. 5, 6440–6452 (2015).
Asadi, M. et al. Nanostructured transition metal dichalcogenide electrocatalysts for CO2 reduction in ionic liquid. Science 353, 467–470 (2016).
Scheuermann, A. G., Prange, J. D., Gunji, M., Chidsey, C. E. D. & McIntyre, P. C. Effects of catalyst material and atomic layer deposited TiO2 oxide thickness on the water oxidation performance of metal-insulator-silicon anodes. Energy Environ. Sci. 6, 2487–2496 (2013).
Godin, R. et al. Electron transfer dynamics in fuel producing photosystems. Curr. Opin. Electrochem. 2, 136–143 (2017).
Xiong, X. et al. Time-resolved spectroscopy of ZnTe photocathodes for solar fuel production. J. Phys. Chem. C 121, 22073–22080 (2017).
Cherepy, N. J. et al. Ultrafast studies of photoexcited electron dynamics in γ and α-Fe2O3 semiconductor nanoparticles. J. Phys. Chem. B 102, 770–776 (1998).
Moss, B. et al. Unraveling charge transfer in CoFe Prussian blue modified BiVO4 photoanodes. ACS Energy Lett. 4, 337–342 (2019).
Acknowledgements
This work was financially supported by Wake Forest University, and Shenzhen Bureau of Science, Technology and Innovation Commission (JCYJ 20160525163956782). A portion of this research was conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. This work benefited from the Shanghai Synchrotron Radiation Facility (SSRF).
Author information
Authors and Affiliations
Contributions
H.L., X.M., M.K. and C.L. synthesized the NCs and fabricated the photocathodes. P.W., S.A., C.D., Z.H. and M.C. performed the physical characterizations. H.L. performed all the electrochemical and photoelectrochemical measurements. D.I. performed the DFT calculation. H.L., Y.Q. and S.M.G designed the experiments and prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Jae Sung Lee, and the other, anonymous, reviewer for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41467-022-33833-5
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, H., Wen, P., Itanze, D.S. et al. RETRACTED ARTICLE: Colloidal silver diphosphide (AgP2) nanocrystals as low overpotential catalysts for CO2 reduction to tunable syngas. Nat Commun 10, 5724 (2019). https://doi.org/10.1038/s41467-019-13388-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-13388-8
This article is cited by
-
Combining Fe nanoparticles and pyrrole-type Fe-N4 sites on less-oxygenated carbon supports for electrochemical CO2 reduction
Nature Communications (2023)
-
Room-temperature liquid metal synthesis of nanoporous copper-indium heterostructures for efficient carbon dioxide reduction to syngas
Science China Materials (2022)
-
Photoelectrochemical technology for solar fuel generation, from single photoelectrodes to unassisted cells: a review
Environmental Chemistry Letters (2022)
-
Fast operando spectroscopy tracking in situ generation of rich defects in silver nanocrystals for highly selective electrochemical CO2 reduction
Nature Communications (2021)
-
Atomic-level-designed copper atoms on hierarchically porous gold architectures for high-efficiency electrochemical CO2 reduction
Science China Materials (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
