
Abstract
Covalent organic frameworks with abundant active sites are potential metal-free catalysts for the nitrogen reduction reaction. However, the utilization ratio of active sites is restricted in an actual reaction process due to the limited nitrogen transport. Here, we demonstrate that facilitating the N2 accessibility to boron-rich covalent organic frameworks through electrochemical excitation can achieve highly efficient nitrogen reduction activity. Simulations show that the boron sites are bonded with nitrogenous species under electrochemical condition and the resultant amorphous phase of covalent organic frameworks has much stronger affinity toward N2 to enhance the molecule collision. Combined with experimental results, the excitation process is confirmed to be a virtuous cycle of more excited sites and stronger N2 affinity, which continuously proceed until the whole system reaches the optimum reaction status. As expected, the electrochemically excited catalyst delivers significantly enhanced reaction activity, with a high Faradaic efficiency of 45.43%.
Similar content being viewed by others
Introduction
The chemical ammonia (NH3) is essential for human beings and ecological system as a fertilizer feedstock and clean energy carrier1,2,3. Yet, industrial-scale ammonia production is monopolized by traditional Haber-Bosch process under drastic operating conditions, making the energy consumption and greenhouse gas emissions unavoidable all over the world4,5,6,7. While the main precursor N2 of this process is one of the most stable substances in nature, only low N2-to-NH3 conversion efficiency (~15%) can be achieved due to the extremely high bond energy of the N≡N covalent triple bond8,9,10. Theoretical studies and previous work have confirmed that N2 reduction to NH3 is feasible under ambient conditions when suitable voltage was applied11,12. Thus, direct electrocatalytic N2 fixation powered by renewable electricity is a very promising candidate as alternative of Haber-Bosch strategy for future use13,14,15.
Acting as the cornerstone of electrochemical catalysis, heterogeneous catalyst is crucial for the chemical transformation in nitrogen reduction reaction (NRR). The extended economic challenges especially encourage the development of metal-free catalysts with highly efficient active sites to increase the output and thus reduce the overall cost16,17. Specifically, boron sites doped in carbon framework induce electron deficiency that lead to significantly improved electrocatalytic activity, for instance, toward oxygen reduction reaction18 and CO2 reduction reaction19, and they thus can be extended to NRR20,21,22. However, the boron-doping amount in carbon is rather restricted so that the reaction activity is usually far from satisfaction23. Taking this into account, covalent organic frameworks (COFs), formed by repetitive organic molecules through strong covalent bonds, provide unlimited imagination for the design of catalysts with abundant active sites24,25. This class of material offers superiorities of modularity, porosity, stability and low density that endow them great potential in catalysis. In detail, regular extension of molecular units can create plentiful active sites in COFs, and desired structure and functions can be achieved by precisely controlling the building units26,27. Among various options, COFs linked by boronate ester (C2O2B) or boroxine (B3O3) rings contain large amount of boron atoms, featuring strong Lewis acidity that greatly promote the N2 (a weak Lewis base) adsorption28. Moreover, the chemical environment of boron site in COFs is tunable compared with traditional boron-doped carbon catalysts, which holds great potential to achieve further improvement in its intrinsic activity, and thus the overall NRR performance.
In this article, we demonstrate that electrochemical excitation of boron-rich COFs could facilitate the catalyst accessibility to N2 and start up efficient NH3 synthesis under ambient conditions (Fig. 1a). Density functional theory (DFT) calculations suggest a superior electronic structure for N2 absorption and greatly reduced energy barrier of N2 dissociation over the boron atoms, and they are, therefore, easy to bond with nitrogenous species when suitable potential is applied. The formation of B–N bonds causes significant distortion of the COFs’ lattice planes, and the reconstruction greatly enhances the N2 adsorption toward the COFs networks, as illustrated by molecular dynamics (MD) simulations. The localized high N2 concentration would promote the collision probability of N2 molecules and the boron sites, so that to facilitate the whole reaction. Careful characterization of the proof-of-concept materials confirms that the COFs exhibit an evident transformation from crystalline to amorphous phase, as well as the appearance of B–N bond along with electrochemical excitation, which are also verified by in situ X-ray powder diffraction (XRD) and in situ Raman measurements. Since the linear sweep voltammetry (LSV) responses show facilitated NRR ability due to the above transformations, we ultimately conclude that the COFs undergo a virtuous cycle of more excitated sites and stronger N2 affinity until the whole system reaches the high-point ready for efficient NRR. By quantifying the NH3 produced, a high Faradaic efficiency of 45.43% is observed.

Computational studies. a Schematic illustration of the electrochemical excitation of COF. b The calculated charge distribution of COF. c Free energy diagrams and d changes of N–N bond length for ammonia synthesis. Molecular dynamics simulation snapshots of e COF and f Eex-COF. g Radial distribution function (RDF) and integrated RDF of nitrogen molecules around different models. The red, green, blue, yellow, and gray spheres represent B, C, N, O, and H atoms, respectively
Results
Theoretical calculations of the boron-rich COFs
The COFs formed by self-condensation of 1,4-benzenediboronic acid (BDBA) as monomer were chosen as the study model, and named as COF for short in this paper. The catalytic activity was preliminary investigated by DFT computations. As shown in Fig. 1b, the lowest unoccupied molecular orbital (LUMO; yellow) and the highest occupied molecular orbital (HOMO; cyan) illustrate an obviously localized electron distribution, with the positive charge mainly locates around the boron atoms, indicating that the boron in B3O3 as Lewis acid is the potential active site for N2 (a weak Lewis basic gas) fixation. Since the electron deficiency structure of boron sites provides a unique environment for N2 adsorption, we further evaluated the subsequent NRR effect of different mechanisms from the thermodynamic perspective by means of first-principles calculations (Supplementary Fig. 1). The reference potential is set to be that of the normal hydrogen electrode (NHE), and the Gibbs free energy is given at U = 0 V versus (vs.) NHE. Upon energy minimizing, the NRR reaction on boron sites prefer to follow the associative alternating pathway with only one energy hill presents at the N2 activation step (Fig. 1c). This first N–N bond cleavage process usually requires a large energy input of 4.26 eV due to the inert nature of N2, and is thus widely accepted as the rate-determining step (RDS). While, in this model, the energy barrier of RDS is greatly reduced to 0.93 eV compared with the case without the catalyst. After N2 activation, the Gibbs free energy exhibits a step-by-step downhill trend accompanied with an obvious N–N bond elongation from 1.110 to 1.459 Å (Fig. 1d). In brief, the COF could serve as efficient catalyst for NRR from the thermodynamic view. Interestingly, an unusual phenomenon was noticed in the DFT models that the lattice planes of COF were obviously distorted when the active boron sites are bonded with nitrogenous species. Considering that such transformation could be excitated by electrochemical process when sufficient voltage was applied, more efforts should be made to give a deeper investigation into its actual influences.
Since the dynamic performance is another key point for heterogeneous catalysis system, we carried out the MD simulations to explore whether the aforementioned phenomenon favors the N2 diffusion in the catalyst. According to the DFT results, the adsorbed N2 (*N2) would be the majority among the bonded nitrogenous species as it exists before the only energy barrier. Thus, the electrochemically excitated COF (Eex-COF) model was set up by symmetrically placing *N2 at specific boron sites in every B3O3, and pristine COF was also calculated for comparison. 0.1 M KOH was chosen as the electrolyte to avoid the strong HER in acidic solution. In detail, N2-saturated alkaline solution served as the initial reaction system, and was set up by randomly placing N2, H2O, K+, and OH− according to their molar concentration. The initial configuration was energy-minimized with the steepest descent algorithm, followed by short equilibration for 30 ps. Subsequently, 50 ns MD runs were performed under the NPT ensemble (298 K, 1 bar). As shown in Supplementary Fig. 2a, b, the Eex-COF exhibits an obvious distortion that in agreement with the DFT model. The snapshots (Fig. 1e, f), at the same time, illustrate a clear distinction of the two models that the N2 molecules tend to aggregate more compactly around the Eex-COF. To confirm this, radial distribution function (RDF) was calculated (Fig. 1g). Though the characterized peak of Eex-COF located at 0.48 nm from boron site is 0.06 nm farther than that in COF due to the block of *N2, the intensity is much higher, suggesting stronger N2 affinity to the structure. The integrated RDF also displays a much higher density of N2 molecules around the Eex-COF, which is in great accordance with the snapshots and RDF conclusion. The electrostatic and van der Waals (vdW) interactions of the two models and N2 molecules were further obtained by extracting the two energy groups from the MD trajectories (Supplementary Fig. 2c). Clearly, the vdW interaction is greatly enhanced after the excitation and plays a dominant part in the adsorption process. Considering the natures of N2 and the models, the facilitated N2 physisorption can be explained by the significantly increased London dispersion force (LDF)29,30,31. When significant distortion of COF happens, more atoms would interact with N2 from the same distance, resulting in much stronger LDF and thus greatly enhanced physisorption of N232,33. A localized high N2 concentration would significantly benefit the gas diffusion and supply toward the active sites, so that is a prerequisite for efficient NRR. Thus, the electrochemically excitated structure would greatly benefit the whole NRR process.
Synthesis and characterization of the Eex-COF/NC
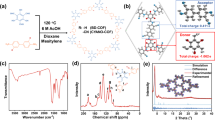
To verify the theoretical conclusions, a series of proof-of-concept experiments were carried out. Considering the poor electrical conductivity of COF, conductive nitrogen-doped carbon nanosheet (NC, Supplementary Figs. 3 and 4) was fabricated as support to avoid such disadvantage. As demonstrated by X-ray photoelectron spectroscopy (XPS, Supplementary Fig. 5), graphitic-N and pyridinic-N are the main existence forms of nitrogen in NC. With the above two nitrogen species as study models, we carried out DFT calculations to explore the catalytic activity of the NC support (Supplementary Fig. 6). Clearly, the hydrogen evolution reaction (HER) activities of graphitic-N and pyridinic-N are rather weaker than COF, and the NRR activity is even worse with significantly higher energy barriers. Thus, its performance would not cause obvious influence on the subsequent analysis of COF. The NC was preoxidized and treated with BDBA in methanol solution to create nucleation sites for the growth of COF framework, and COF was then coated on it by molecular dehydration reaction of BDBA in mesitylene-dioxane solution via solvothermal reaction (COF/NC, Supplementary Fig. 7). The eletrochemically excitated COF/NC (Eex-COF/NC) was obtained by potentiostatic electrolyzing at −0.2 V vs. reversible hydrogen electrode (RHE) in N2-saturated 0.1 M KOH for 20 min. Pure COF was also synthesized for reference. It should be pointed out that the N2 was sufficiently purified before use to avoid the possible existence of NH3 or NOx in all measurements in this work13,34 (Supplementary Figs. 8–10). The transmission electron microscopy (TEM) image of Eex-COF/NC (Fig. 2a) shows a homogeneous COF matrix over the substrate without any changes of surface morphology compared with COF/NC, with corresponding element mappings showing the uniform distribution of B, C, and N (Fig. 2b). Yet, the high-resolution TEM (HRTEM) images reveal an obvious transformation from crystalline (Fig. 2c) to amorphous phase (Fig. 2d). The XRD pattern of COF/NC shows all the characteristic peaks of COF, which is align with the previous report35, whereas the diffractions disappear after electrochemical excitation (Fig. 2e), which is consistent with the amorphous phase. This can be further confirmed by the cross-polarization magic angle spinning nuclear magnetic resonance (CP/MAS NMR) responses. The dramatical broaden and weaken of the 13C NMR spectra (Supplementary Fig. 11a) indicate a completely anisotropic chemical environment in Eex-COF/NC, and the shift of 11B NMR signal (Supplementary Fig. 11b) suggests that the bonding environment of boron might be changed. The surface chemistry of different samples was subsequently analyzed by XPS (Supplementary Fig. 12). As shown in Fig. 2f, a new peak representing B–N is captured in the high-resolution B 1s spectra of Eex-COF/NC. Simultaneously, same variation is also discovered in Fourier-transform infrared (FTIR) spectrum with the appearance of two new peaks located at 815 and 1370 cm−1 assigning to B–N36 (Fig. 2g). The strong peak appeared in the range of 620–680 cm−1 in Eex-COF/NC is ascribed to the out of plane phenyl ring deformation for benzene ring due to the significant lattice distortion, as well as C–H deformation resulting from the HER35,37,38. In the region highlighted in light green, the B–C stretching at 1023 cm−1 in COF/NC shifts to lower wavenumber in Eex-COF/NC due to the bonding between boron and nitrogen after electrochemical excitation39, and the new peak located at about 1040 cm−1 can be ascribed to benzyl ring deformations40 in COF. The results above are combined to elucidate that, the bonding of nitrogenous species on boron contributes to the distortion of the lattice, and provides direct evidence that the boron atoms serve as the NRR active sites.

Physical characterization. a Transmission electron microscopy (TEM) image and b element mappings of Eex-COF/NC. High-resolution TEM images of c COF/NC and d Eex-COF/NC. Scale bars, a 200 nm; b 100 nm; c 5 nm and d 5 nm. e X-ray powder diffraction (XRD) patterns, f high-resolution B 1s spectra, and g Fourier-transform infrared (FTIR) spectra of different samples
In situ characterizations and eletrochemical responses
To give a real-time observation of the unique conversion, in situ characterizations were employed in a tailor-made electrolytic cell (Fig. 3a). In situ XRD experiment was first implemented to examine the crystallinity change. As shown in Fig. 3b and Supplementary Fig. 13, legible characteristic peaks are detected in the pristine COF/NC electrode. While, all diffractions weakened gradually when a constant excitation voltage of –0.2 V vs. RHE was applied, and completely vanished at about 12 min. However, the characteristic peaks of COF remain almost unchanged in the same period of time when replacing N2 with Ar (Supplementary Figs. 14 and 15), suggesting that the transformation of COF from crystalline to amorphous phase is associated with the NRR process. Subsequently, we captured the in situ Raman spectra under the same operating conditions. When using N2 as gas supply (Fig. 3c), an obvious band at about 800 cm−1 attributed to B–N stretching vibration41 gradually appeared as the excitation time passed, whereas no such transformation was detected under Ar (Supplementary Fig. 16). These phenomena again imply that the lattice distortion of COF is due to the chemisorption of nitrogen or nitrogenous intermediates on the active boron sites. Now that such transformations above are proved to be beneficial for N2 aggregation around the COF by theoretical calculations, it is necessary to verify if they profit the actual NRR process. Thus, LSV curves were collected in both N2- and Ar-saturated 0.1 M KOH electrolyte (Fig. 3d and Supplementary Fig. 17). Expectedly, the LSVs exhibit a tendency that the current density gap between N2 and Ar environment increases from the pristine state to transition state, and then to the excitation state, illustrating the intensified reaction activity, accompanied by the positively shifted onset potential. That means, the COF/NC undergoes a warming-up procedure through electrochemical excitation and finally reaches an optimum status with dramatically high activity for efficient NRR. Combining the theoretical and experimental results, we can explain the mechanism and advantage for electrochemical excitation of COF as follows: the superior electronic structure and reduced energy barrier of RDS makes the boron sites in COF easy to bond with nitrogenous species under electrochemical condition; the resultant amorphous structure has stronger affinity toward the N2 molecules that could facilitate the molecular collision in the reaction to excite more boron atoms; the reconstruction induced by the enriched excitated boron sites further enhance the adsorption of N2 toward the COF networks. Such virtuous cycle continuously proceed until the whole system attains the high-point for NRR (Supplementary Fig. 18).

In situ characterizations and electrochemical responses. a Schematic illustration of the tailor-made electrolytic cell for in situ characterizations. b In situ XRD intensity map, and c in situ Raman spectra of the transformation from COF/NC to Eex-COF/NC. d Linear sweep voltammetry (LSV) curves of different states through excitation
Electrocatalytic nitrogen reduction performance of Eex-COF/NC
Quantified study of Eex-COF/NC of the NRR activity was then performed in 0.1 M KOH using a gas-tight two-compartment cell through chronoamperometry measurement (Supplementary Figs. 19 and 20). NH3 and N2H4 as possible products were both examined (Supplementary Figs. 8 and 21–23), whereas only NH3 was obtained in this work. Figure 4a, b shows the NH3 yield rate and corresponding Faradaic efficiency under different given potentials. Strikingly, the Faradaic efficiency and NH3 yield rate of Eex-COF/NC both reach the maximum value of 45.43% and 12.53 μg h−1 mg−1, respectively, at −0.2 V vs. RHE, which is a record-high activity among the metal-free catalysts to date (Supplementary Table 1). In contrast, the NC substrate exhibits an inferior NRR activity so that its contribution to the NH3 synthesized in Eex-COF/NC is almost neglectable. Compared with NC, in which the majority of electrons tends to offer up the HER, the HER is greatly reduced in the voltage range from −0.1 to −0.3 V vs. RHE after the electrochemical excitation of COF/NC (Fig. 4c), and more electrons are captured by NRR. On the other hand, the Eex-COF/NC can keep the superior performance almost unchanged under 10 successive cycles of NRR electrolysis (Fig. 4d), with the morphology and chemical construction remain constant (Supplementary Figs. 24–28), revealing the robust stability. Control experiments (Supplementary Fig. 29) exclude the probable existence of NH3 in the pristine feeding gas and the NRR ability of the carbon paper (CP), so that all the NH3 detected is confirmed to be produced by Eex-COF/NC. Furthermore, the 15N isotope labeling experiments (Supplementary Figs. 30–31) were also conducted to examine the N source of the obtained NH3. The 15N2 gas was also sufficiently purified before test (Supplementary Fig. 32). The produced 15NH3 is confirmed to be completely from the electrochemical NRR process (Supplementary Fig. 33), and the NH3 yield rate and corresponding Faradaic efficiency are in great accordance with the data obtained under 14N2 (Supplementary Fig. 34), thus making the experimental results reliable. As can be seen in Fig. 4e, when using 15N2 as feeding gas for both the electrochemical excitation and chronoamperometry measurement, only a doublet signal representing 15NH4+ is detected in the 1H NMR spectra, declaring the total N source from the feeding gas rather than any nitrogen species in NC. In addition, argon control experiments were also conducted at each given potential (Supplementary Fig. 35a). As demonstrated in ultraviolet-visible (UV-vis) spectra and 1H NMR analysis (Supplementary Figs. 35b and 36), no NH3 was detected when Ar was supplied, confirming that the nitrogen in NC substrate is not active enough to join the electrochemical conversion to NH3. Notably, we then used 14N2 for the excitation process and 15N2 for the subsequent NRR test, with both 14NH4+ and 15NH4+ observed in the evaluated products (Fig. 4f). Therefore, the pre-bonded nitrogen through electrochemical excitation are proved to be involved in the following conversion to NH3. Considering that there is no difference between the chemical constructions of Eex-COF/NC before and after the NRR test, the catalytic mechanism of Eex-COF/NC is inferred to be a continuous nitrogen substituting process, which is stable and maintains the catalyst with high nitrogen affinity that greatly favors the NRR.

Electroreduction of N2 to NH3 at ambient conditions. a NH3 yield rates, b corresponding Faradaic efficiencies, and c H2 selectivity of Eex-COF/NC and NC. The error bars correspond to the standard deviations of measurements over three separately prepared samples under the same conditions. d The NH3 production performance in durability test of Eex-COF/NC. e 1H nuclear magnetic resonance (NMR) spectra of the NRR products using the same isotope feeding gas for excitation and NRR. f 1H NMR spectrum of the NRR products using 14N2 for excitation and 15N2 for NRR. The red, green, blue, purple, yellow, and gray spheres represent B, C, 14N, 15N, O, and H atoms, respectively
Discussion
In summary, we demonstrate a seminal discovery that electrochemical excitation of COF is able to promote the catalyst accessibility to N2 and launch highly efficient NRR under ambient conditions. DFT calculations show a beneficial charge distribution and reduced energy barrier of RDS over boron sites in the COF, so that they are facile to bond with nitrogenous species under electrochemical condition. The amorphous structure caused by the fixation of nitrogenous species is confirmed to have stronger affinity to the N2 molecules by MD simulations, which could enhance the collision frequency of the N2 molecules toward the boron sites in the NRR process and thus benefit the whole reaction system. In situ XRD, in situ Raman and careful experiments are combined to verify the theories, and we finally draw a conclusion that the COF undergoes a continuous warming-up procedure until the whole system attains an optimum status with strikingly high activity for efficient NRR. As a proof of concept, the target catalyst (Eex-COF/NC) tested in an actual NRR process achieves an outstanding Faradaic efficiency as high as 45.43%, outperforming all the metal-free catalyst to date to the best of our knowledge.
Methods
Computational method and model
The first-principles calculations were conducted using Cambridge Sequential Total Energy Package (known as CASTEP). The electron–electron interaction was described using the exchange-correlation function under the generalized gradient approximation (GGA) with norm-conserving pseudopotentials and Perdew-Burke-Ernzerhof functional. In total, 750 eV was used as the energy cutoff. Several parameters were considered, including energy tolerance of 5.0 × 10−7 eV per atom, a force tolerance of 0.01 eV Å−1 and maximum displacement of 5.0 × 10−4 Å. No constraints were set for any atom in the models, which is allowed to relax to the minimum in the enthalpy. The vacuum space along the z direction was set to be 15 Å.
Adsorption energy ΔE of A group on the surface of substrates was defined as:
where *A and * denote the adsorption of A group on substrates and the bare substrates, EA denotes the energy of A group.
Gibbs free energy change (ΔG) of each chemical reaction was calculated by:
where E, ZPE, T, and S denote the calculated total energy, zero point energy, temperature, and entropy, respectively. ΔGU = −eU (U is the potential measured against NHE) and ΔGpH = −kBTln(10) × pH. Here, T = 300 K and pH = 13 are considered.
MD simulations were carried out with Gromacs (v5.0.5). One COF molecule was immersed in KOH/N2 solution, which was composed of 60 N2 molecules, 4 K+, 4 OH−, and 2000 water molecules. The Eex-COF model was constructed by placing one *N2 at specific boron site in every B3O3 ring in COF.
The Universal force field (UFF)42 was adopted to the COF model. The topology file of COF was generated by the OBGMX program43, which includes the bonded (bond stretching, angle bending, and dihedral torsion) and nonbonded (interatomic van der Waals interactions represented by Lennard Jones model) parameters from UFF, while the partial charges were obtained from Zheng et al.44. Water, N2, and KOH were represented by the SPC/E model45, TraPPE model46 and OPLS-AA47 force field, respectively. Cross interactions between unlike atom pairs were calculated with the standard Lorentz-Berthelot combination rules. Short-range interactions were truncated at 1.2 nm, while the long-range coulombic interactions were calculated with the particle mesh Ewald algorithm with a Fourier spacing of 0.12 nm. Leapfrog algorithm was used to integrate the equations of motion with a time step of 1 fs.
Energy-minimization was first conducted with the steepest descent algorithm, followed by isothermal-isobaric equilibration at 298 K and 1 bar for 50 ps. Then, 50 ns MD runs were carried out under the NPT ensemble at 298 K and 1 bar, with Nose-Hoover thermostat and Parrinello-Rahman barostat controlling the system temperature and pressure, respectively. 3-D periodic boundary conditions were applied throughout simulations.
Catalyst synthesis
NC was first prepared. Pyrrole (1 mL) was added to deionized water and treated by ultrasonic vibration to obtain a uniform suspension. Sodium chloride and zinc chloride with a molar ratio of 4:1, and 1 M HCl (1 mL) were successively dissolved into the above solution. The solution was cooled down to 0 °C in a low-temperature cooling system, followed by dropwise adding of (NH4)2S2O8 solution to trigger the pyrrole polymerization. The reaction was kept stirring for 3 h, and the obtained solution was freeze-dried for 24 h. The obtained solid was annealed for 2 h at 700 °C with argon protection. The salt template and impurities were washed away using hydrochloric acid, water, and ethanol successively. Then, the obtained products were dried under vacuum at 60 °C for 12 h. To prepare COF/NC, the NC was oxidized with piranha solution (14 mL of 98% H2SO4 and 6 mL of 30% H2O2) under vigorous stirring for 24 h, and thoroughly washed with deionized water and dried. Then 10 mg oxidized NC was sonicated in 2 mL methanol in a pyrex tube, followed by adding 30 mg BDBA. The tube was sealed under nitrogen and annealed at 90 °C for 24 h. The obtained materials were thoroughly washed with methanol, and then with mesitylene/dioxane solution (1:1 in volume). The products and 50 mg BDBA were further dispersed in 2 mL mesitylene/dioxane solution without drying. The tube was flash frozen with liquid N2, evacuated and flame sealed. The reaction was kept at 120 °C for 72 h, and the product was washed with acetone and stabilized under vacuum at 100 °C overnight to obtain COF/NC. The Eex-COF/NC was prepared by potentiostatic electrolyze the COF/NC at –0.2 V vs. RHE in N2-saturated 0.1 M KOH for 20 min. Pristine COF was synthesized using the same method as COF/NC without using the NC as substrate.
Physical characterization
The morphology was studied via field emission transmission electron microscope (FETEM, FEI Tecnai G2 F20 S-TWIN TMP) operating at 200 kV. Surface elemental analysis was performed on X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra Dld). The structure of the catalysts was characterized by X-ray powder diffraction (XRD, D8 Advance, Bruker), nuclear magnetic resonance measurements (NMR, Agilent 600 MHz) and Fourier-transform infrared spectra (FTIR, Tensor 27, Bruker).
Cathode preparation
One microgram of catalyst was dispersed in 100 μL solution consisting of 90 μL ethanol and 10 μL Nafion solution (5 wt%), and sonicated for at least 1 h to obtain a homogeneous ink. The catalyst ink was loaded on carbon paper with area of 1 × 1 cm2 to obtain a mass loading of 1 mg cm−2, and dried in N2 atmosphere at 80 °C for 1 h.
Nitrogen purification
High purity 14N2 (Messer Gas, Germany) and 15N2 (99 atom% 15N, Newradar Special Gas Co. Ltd., Wuhan, China) was purchased commercially. The nitrogen gas was flowed through acid and alkaline trap successively to remove the possible NH3 and NOx, and passed a drying tube to block the vapor before supplied into the electrochemical cell. The purified gas was continuously passed through 0.1 M KOH solution for a period of time, and the obtained solution was analyzed using spectrophotometric methods to ensure that no NH3 or NOx resides.
Detection of ammonia
NH3 was determined by the indophenol blue method48 with some modification. In detail, 2 mL of the treated electrolyte was mixed with 2 mL of 1 M NaOH solution containing sodium citrate and salicylic acid. Then, 0.2 mL of 1 wt% sodium nitroferricyanide and 1 mL of 0.05 M sodium hypochlorite solution were added into the solution. After standing in darkness for 3 h, the UV-Vis absorption spectrum was measured. The concentration of indophenol blue was determined using the absorbance at a wavelength of 655 nm. The concentration–absorbance curves were calibrated using standard ammonia chloride solution with a series of concentrations in 0.1 M KOH.
Detection of NOx
NOx was determined using N-(-1-naphthyl)-ethylenediamine dihydrochloride spectrophotometric method49 with some modification. Detailedly, 0.5 g sulfanilic acid was dissolved in 90 mL H2O and 5 mL acetic acid. Then, 5 mg n-(1-naphthyl)-ethylenediamine dihydrochloride was added and the solution was filled to 100 mL to obtain chromogenic agent. One milliliter of the treated electrolyte was mixed with 4 mL chromogenic agent and left in darkness for 15 min. The UV-Vis absorption spectrum was then measured at 540 nm. The concentration–absorbance curves were calibrated using standard sodium nitrite solution with a series of concentrations in 0.1 M KOH.
In situ characterization
A tailor-made cell was customized for electrochemical measurements. The Pt wire and Ag/AgCl (4 M KCl) were used as the counter and reference electrode, respectively. N2- or Ar-saturated 0.1 M KOH was used as the electrolyte. The catalyst ink was prepared by dispersing 10 mg COF/NC in 9 mL ethanol and 1 mL Nafion solution (5 wt%) in an ultrasonic bath for at least 1 h. The ink was coated on carbon paper to be used as the working electrode. The in situ XRD measurements were recorded at the X-ray powder diffractometer (D8 Advance, Bruker). The in situ Raman measurements were conducted at the Raman spectroscopy (HR evolution, Horiba Jobin Yvon, France). The chronoamperometric method was conducted at −0.2 V vs. RHE by CHI660E electrochemical workstation (Shanghai Chenhua Instrument Co., Ltd). The working electrode was continuously bubbled by N2 through the in situ characterization.
Electrochemical NRR measurements
The electrochemical measurements were conducted in a Nafion 211 membrane-separated two-compartment cell at room temperature. Both cathode chamber and anode chamber contained 30 mL of 0.1 M KOH electrolyte. Before measurements, the membrane was treated by hydrogen peroxide solution (5%) at 80 °C for 1 h and then deionized water at 80 °C for 1 h. Ag/AgCl (4 M KCl) was used as the reference electrode, and Pt foil was used as the counter electrode. For electrochemical NRR, potentiostatic tests were conducted in N2-saturated 0.1 M KOH, which was purged with purified N2 for 30 min before the measurement.
The potentiostatic tests were tested in 0.1 M KOH aqueous solution at different potentials including −0.1, −0.2, −0.3, −0.4, and −0.5 V vs. RHE. In order to avoid the volatilization of produced ammonia, another cell filled with 30 mL of 0.001 M H2SO4 as the gas absorption liquid was set at the end of the cathode cell. After NRR, the cathodic electrolyte and the absorption liquid were both collected and analyzed by chromogenic reactions for quantitative measurements. The total ammonia yield was the summation of that in 0.1 M KOH and 0.001 M H2SO4. Argon control electrochemical measurements were conducted using the same method with high purity argon as the gas supply.
Detection of ammonia in gas absorption cell
Concentration of the absorbed ammonia in 0.001 M H2SO4 was determined using the same method as that in 0.1 M KOH.
Detection of hydrazine
The hydrazine presented in 0.1 M KOH was estimated by the method of Watt and Chrisp50. A mixture of para-(dimethylamino) benzaldehyde (5.99 g), HCl (concentrated, 30 mL) and ethanol (300 mL) was used as a color reagent. Five microliters of the residual electrolyte after NRR potentiostatic test was removed from the electrochemical reaction vessel. Then, 5 mL of above prepared color reagent was added to the solution and stirred for 10 min at room temperature. The absorbance of the resulting solution was measured at a wavelength of 455 nm. The concentration–absorbance curves were calibrated using standard hydrazine monohydrate 0.1 M KOH solution with a series of concentrations. Concentration of produced hydrazine in 0.001 M H2SO4 was determined using the same method.
Detection of hydrogen
The hydrogen evolution analysis was performed by a gas chromatography (Agilent 7890B, USA).
Thermodynamic equilibrium potential
The standard potential of NRR at alkaline medium was calculated as below:
where F is the Faraday constant (96,485 C mol−1) and n is the number of transferred electrons (6).
As for reaction conditions, the thermodynamic equilibrium potential was determined according to the Nernst equation, assuming 1 atm of N2 in the electrolyte.
where F is the Faraday constant (96,485 C mol−1), T is the temperature in Kelvin (298.15 K), c(OH−) is the hydroxide concentration (0.1 M), R is the gas constant (8.314 J mol−1 K−1), and pH is 13. Assuming a NH4OH concentration of 10−6 M in the electrolyte, the corresponding thermodynamic equilibrium potential is calculated to be 0.207 V vs. RHE.
Ammonia yield rate and Faradaic efficiency
The yield rate and Faradaic efficiency of NH3 were calculated as below:
where m is the loading mass of the catalysts, t is the electrolysis time (1 h), V is the volume of the electrolyte, Q is the total charge passed through the electrode, F is the Faraday constant (96,485 C mol−1), and c(NH3) is the measured ammonia concentration.
The Faradaic efficiency of hydrogen evolution was calculated as below:
where itotal is the steady-state cell current, G is the gas flow rate (mL/min at room temperature and ambient pressure), v(H2) is the volume concentration of hydrogen in the exhaust gas from the electrochemical cell, p0 = 1.01 × 105 Pa, R = 8.314 J mol−1 K−1.
15N isotopic labeling experiment
In the labeling experiment, 15N2 (99 atom% 15N, Newradar Special Gas Co. Ltd., Wuhan, China) was applied as the feeding gas. The electrolyte was taken out after electrolysis at −0.2 V vs. RHE for several hours. By adding 0.5 M sulfuric acid, the pH of the electrolyte was adjusted to 3. After concentration to 1 mL, 0.9 mL solution was taken out and 0.1 mL D2O was added in as an internal standard. (15NH4)2SO4 and (14NH4)2SO4 was used as the benchmark. The quantification of the produced NH3 was performed by 1H nuclear magnetic resonance measurement (Agilent 600 MHz, USA).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Gruber, N. & Galloway, J. N. An earth-system perspective of the global nitrogen cycle. Nature 451, 293–296 (2008).
Guo, C., Ran, J., Vasileff, A. & Qiao, S.-Z. Rational design of electrocatalysts and photo(electro)catalysts for nitrogen reduction to ammonia (NH3) under ambient conditions. Energy Environ. Sci. 11, 45–56 (2018).
Chen, G.-F. et al. Ammonia electrosynthesis with high selectivity under ambient conditions via a Li+ incorporation strategy. J. Am. Chem. Soc. 139, 9771–9774 (2017).
Chen, J. G. et al. Beyond fossil fuel-driven nitrogen transformations. Science 360, eaar6611 (2018).
Licht, S. et al. Ammonia synthesis by N2 and steam electrolysis in molten hydroxide suspensions of nanoscale Fe2O3. Science 345, 637–640 (2014).
Cheng, H. et al. Molybdenum carbide nanodots enable efficient electrocatalytic nitrogen fixation under ambient conditions. Adv. Mater. 30, 1803694 (2018).
Luo, Y. et al. Efficient electrocatalytic N2 fixation with MXene under ambient conditions. Joule 3, 279–289 (2019).
van der Ham, C. J. M., Koper, M. T. M. & Hetterscheid, D. G. H. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 43, 5183–5191 (2014).
Wang, J. et al. Ambient ammonia synthesis via palladium-catalyzed electrohydrogenation of dinitrogen at low overpotential. Nat. Commun. 9, 1795 (2018).
Li, S.-J. et al. Amorphizing of Au nanoparticles by CeOx-RGO hybrid support towards highly efficient electrocatalyst for N2 reduction under ambient conditions. Adv. Mater. 29, 1700001 (2017).
Jiao, F. & Xu, B. Electrochemical ammonia synthesis and ammonia fuel cells. Adv. Mater. 30, 1805173 (2018).
Yan, D., Li, H., Chen, C., Zou, Y. & Wang, S. Defect engineering strategies for nitrogen reduction reactions under ambient conditions. Small Methods 2, 1800331 (2018).
Chen, G.-F. et al. Advances in electrocatalytic N2 reduction–strategies to tackle the selectivity challenge. Small Methods 2, 1800337 (2018).
Wang, M. et al. Over 56.55% Faradaic efficiency of ambient ammonia synthesis enabled by positively shifting the reaction potential. Nat. Commun. 10, 341 (2019).
Shi, M.-M. et al. Anchoring PdCu amorphous nanocluster on graphene for electrochemical reduction of N2 to NH3 under ambient conditions in aqueous solution. Adv. Energy Mater. 8, 1800124 (2018).
Zhao, S., Lu, X., Wang, L., Gale, J. & Amal, R. Carbon-based metal-free catalysts for electrocatalytic reduction of nitrogen for synthesis of ammonia at ambient conditions. Adv. Mater. 31, 1805367 (2019).
Zhang, L., Ding, L.-X., Chen, G.-F., Yang, X. & Wang, H. Ammonia synthesis under ambient conditions: Selective electroreduction of dinitrogen to ammonia on black phosphorus nanosheets. Angew. Chem. Int. Ed. 58, 2612–2616 (2019).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Origin of the electrocatalytic oxygen reduction activity of graphene-based catalysts: a roadmap to achieve the best performance. J. Am. Chem. Soc. 136, 4394–4403 (2014).
Sagaraa, N., Kamimuraa, S., Tsubotaa, T. & Ohnoa, T. Photoelectrochemical CO2 reduction by a p-type boron-doped g-C3N4 electrode under visible light. Appl. Catal. B: Environ. 192, 193–198 (2016).
Yu, X. et al. Boron-doped graphene for electrocatalytic N2 reduction. Joule 2, 1–13 (2018).
Ji, S., Wang, Z. & Zhao, J. A boron-interstitial doped C2N layer as a metal-free electrocatalyst for N2 fixation: a computational study. J. Mater. Chem. A 7, 2392–2399 (2019).
Chen, C. et al. B-N pairs enriched defective carbon nanosheets for ammonia synthesis with high efficiency. Small 15, 1805029 (2019).
Liu, Y. et al. Elemental superdoping of graphene and carbon nanotubes. Nat. Commun. 7, 10921 (2016).
Diercks, C. S. & Yaghi, O. M. The atom, the molecule, and the covalent organic framework. Science 355, eaal1585 (2017).
Huang, N., Wang, P. & Jiang, D. Covalent organic frameworks: a materials platform for structural and functional designs. Nat. Rev. Mater. 1, 16068 (2016).
Ding, S.-Y. & Wang, W. Covalent organic frameworks (COFs): from design to applications. Chem. Soc. Rev. 42, 548–568 (2013).
Lin, C.-Y., Zhang, L., Zhao, Z. & Xia, Z. Design principles for covalent organic frameworks as efficient electrocatalysts in clean energy conversion and green oxidizer production. Adv. Mater. 29, 1606635 (2017).
Song, Y., Sun, Q., Aguila, B. & Ma, S. Opportunities of covalent organic frameworks for advanced applications. Adv. Sci. 5, 1801410 (2018).
Hermann, J., DiStasio Jr., R. A. & Tkatchenko, A. First-principles models for van der Waals interactions in molecules and materials: concepts, theory, and applications. Chem. Rev. 117, 4714–4758 (2017).
Liptrot, D. J. & Power, P. P. London dispersion forces in sterically crowded inorganic and organometallic molecules. Nat. Rev. Chem. 1, 0004 (2017).
Wagner, J. P. & Schreiner, P. R. London dispersion in molecular chemistry–reconsidering steric effects. Angew. Chem. Int. Ed. 54, 12274–12296 (2015).
Kennedy, M. R., Burns, L. A. & Sherrill, C. D. Buckyplates and buckybowls: examining the effects of curvature on π−π interactions. J. Phys. Chem. A 116, 11920–11926 (2012).
Okamoto, Y. & Miyamoto, Y. Ab initio investigation of physisorption of molecular hydrogen on planar and curved graphenes. J. Phys. Chem. B 105, 3470–3474 (2001).
Suryanto, B. H. R. et al. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2, 290–296 (2019).
Côté, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Yu, B. et al. Thermal exfoliation of hexagonal boron nitride for effective enhancements on thermal stability, flame retardancy and smoke suppression of epoxy resin nanocomposites via sol–gel process. J. Mater. Chem. A 4, 7330–7340 (2016).
Seoudi, R., El-Bahy, G. S. & Sayed, Z. A. E. FTIR, TGA and DC electrical conductivity studies of phthalocyanine and its complexes. J. Mol. Struct. 753, 119–126 (2005).
Mohan, S., Sundaraganesan, N. & Mink, J. FTIR and Raman studies on benzimidazole. Spectrochim. Acta (A) 47, 1111–1115 (1991).
Holm, M. S. et al. Assessing the acid properties of desilicated ZSM-5 by FTIR using CO and 2,4,6-trimethylpyridine (collidine) as molecular probes. Appl. Catal. A: Gen. 356, 23–30 (2009).
Contrerasa, C. D., Ledesmaa, A. E., Zinczukb, J. & Brandán., S. A. Vibrational study of tolazoline hydrochloride by using FTIR-Raman and DFT calculations. Spectrochim. Acta (A) 79, 1710–1714 (2011).
Liu, A. & Song, Y. In situ high-pressure and low-temperature study of ammonia borane by Raman spectroscopy. J. Phys. Chem. C. 116, 2123–2131 (2012).
Rappé, A. K., Casewit, C. J., Colwell, K. S., Goddard III, W. A. & Skif, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Garberoglio, G. OBGMX: a web-based generator of GROMACS topologies for molecular and periodic systems using the universal force field. J. Comput. Chem. 33, 2204–2208 (2012).
Zheng, C. & Zhong, C. Estimation of framework charges in covalent organic frameworks using connectivity-based atom contribution method. J. Phys. Chem. C. 114, 9945–9951 (2010).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987).
Potoff, J. J., J & Siepmann, I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen. Aiche J. 47, 1676–1682 (2001).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Zhu, D., Zhang, L., Ruther, R. E. & Hamers, R. J. Photo-illuminated diamond as a solid-state source of solvated electrons in water for nitrogen reduction. Nat. Mater. 12, 836–841 (2013).
Wang, Q.-H. et al. Methods for the detection and determination of nitrite and nitrate: a review. Talanta 165, 709–720 (2017).
Watt, G. W. & Chrisp, J. D. A spectrophotometric method for the determination of hydrazine. Anal. Chem. 24, 2006–2008 (1952).
Acknowledgements
We acknowledge the support from the National Natural Science Foundation of China (Nos. 21703149, 51622208, 51872193, and 5192500409) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Contributions
C.Y. conceived and designed this work. S.L., M.W., T.Q., and J.L. prepared materials and performed the material characterizations, electrochemical measurements. H.J. conducted the theoretical calculations. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information: Nature Communications thanks the anonymous reviewers for their contributions to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, S., Wang, M., Qian, T. et al. Facilitating nitrogen accessibility to boron-rich covalent organic frameworks via electrochemical excitation for efficient nitrogen fixation. Nat Commun 10, 3898 (2019). https://doi.org/10.1038/s41467-019-11846-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-019-11846-x
This article is cited by
-
Induced dipole moments in amorphous ZnCdS catalysts facilitate photocatalytic H2 evolution
Nature Communications (2024)
-
The reformation of catalyst: From a trial-and-error synthesis to rational design
Nano Research (2024)
-
Selective Pathway of Nitrogen Reduction Controlled by Symmetry Breaking in BiErRu2O7 Pyrochlores
Catalysis Letters (2024)
-
Embedding oxophilic rare-earth single atom in platinum nanoclusters for efficient hydrogen electro-oxidation
Nature Communications (2023)
-
Cation controlled rotation in anionic pillar[5]arenes and its application for fluorescence switch
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
