
Abstract
Non-alcoholic fatty liver disease (NAFLD) is currently considered an important component of metabolic syndrome (MetS). The spectrum of NAFLD includes conditions that range from simple hepatic steatosis to non-alcoholic steatohepatitis. NAFLD is correlated with liver-related death and is predicted to be the most frequent indication for liver transplantation by 2030. Insulin resistance is directly correlated to the central mechanisms of hepatic steatosis in NAFLD patients, which is strongly correlated to the imbalance of the renin–angiotensin system, that is involved in lipid and glucose metabolism. Among the emerging treatment approaches for NAFLD is the anti-hypertensive agent telmisartan, which has positive effects on liver, lipid, and glucose metabolism, especially through its action on the renin–angiotensin system, by blocking the ACE/AngII/AT1 axis and increasing ACE2/Ang(1–7)/Mas axis activation. However, treatment with this drug is only recommended for patients with an established indication for anti-hypertensive therapy. Thus, there is an increased need for large randomized controlled trials with the aim of elucidating the effects of telmisartan on liver disease, especially NAFLD. From this perspective, the present review aims to provide a brief examination of the pathogenesis of NAFLD/NASH and the role of telmisartan on preventing liver disorders and thus to improve the discussion on potential therapies.
Similar content being viewed by others
Introduction
Diet and lifestyle changes have led to a worldwide increase in the prevalence of metabolic syndrome (MetS), a complex disorder in which obesity, glucose intolerance, insulin resistance (IR), dyslipidemia, and hypertension are associated [1,2,3]. Non-alcoholic fatty liver disease (NAFLD) is now considered a hepatic component of MetS because of the close association between the two conditions, which share the same risk factors [3,4,5,6].
NAFLD is currently the most common liver disorder [7,8,9], with a prediction to be the most frequent indication for liver transplantation by 2030 [4]. The global estimated prevalence of NAFLD ranges from 6.3 to 33% in the general population with a median prevalence of 20%, the median prevalence is higher in developed countries [8,9,10]. NAFLD is defined as a lipodystrophy with lipid-deposit accumulation in the hepatocytes accounting for more than 5–10% of the total hepatic weight, which is not due to excessive alcohol use (women ≤ 20 g/d, men ≤ 30 g/d). An accurate diagnosis requires imaging or histological techniques, and secondary causes of hepatic steatosis should be excluded, such as drugs, hepatitis C virus, surgical procedures, total parenteral nutrition, and various innate metabolism disorders [5, 9].
NAFLD is a term that encompasses the entire spectrum of this disease, ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which can lead to life-threatening hepatic cirrhosis and hepatocellular carcinoma in its most severe form [4, 6, 9, 11]. Histologically, NASH is characterized by hepatic steatosis and signs of intralobular inflammation with ballooning degeneration of the hepatocytes. The estimated prevalence of NASH is much lower than NAFLD and ranges from 3 to 5% [9, 10]. Twenty percent of NASH patients are reported to develop cirrhosis, and 30–40% of patients with NASH cirrhosis experience a liver-related death [12].
Among the emerging treatments for NAFLD, the anti-hypertensive drug telmisartan is of particular interest, as it seems to have positive hepatic and extra-hepatic impacts, according to preliminary studies [13,14,15,16].
From this perspective, the present review aims to provide a brief examination of the pathogenesis of NAFLD/NASH and the role of telmisartan in preventing liver disorders, and thus improving the discussion on potential therapies.
NAFLD pathogenesis and insulin resistance
Although the pathogenesis of NAFLD development and progression toward NASH is somewhat unclear, insulin resistance (IR), with or without fully developed MetS, is reportedly related as the central mechanism of hepatic steatosis in patients with NAFLD, which in turn develops in the setting of an inappropriate diet, sedentary lifestyle, obesity, and advancing age. Additionally, imbalances in pro-oxidant and anti-oxidant mechanisms and pro- and anti-inflammatory cytokines are important components in NAFLD pathogenesis. The role of genetic variations in the predisposition to steatosis development by affecting various steps in the normal fat and carbohydrate metabolism is also under investigation [1, 5, 14].
Normally, insulin acts on skeletal muscles, adipocytes, and liver for maintaining glucose and lipid homeostasis. Insulin performs in the following ways: increases the uptake of free fatty acid (FFA) conversion to triglycerides and the storage along with decreased lipolysis in adipose tissue; stimulates glucose uptake by skeletal muscles; promotes the storage of glucose as glycogen in the liver, with inhibition of glycogenolysis and gluconeogenesis; and reduces oxidation of fatty acids in the liver. The net result of all these actions is the utilization of glucose, reduction in the lipolysis of FFAs, and promotion of the storage of fats as triglycerides in adipose tissue [5].
Insulin resistance has traditionally been defined as a condition in which cells fail to respond to the normal actions of insulin. The body produces insulin, but the cells in adipose tissue, muscle, and liver become resistant and are unable to use it as effectively. In skeletal muscles, IR causes reduced glucose uptake leading to hyperglycemia. In adipose tissue, IR impairs the anti-lipolytic action of insulin leading to an increased release of FFAs [5, 17].
The basic defect in the development of hepatic steatosis is the fat imbalance between import and export to and from the liver, secondary to IR [5]. IR and subsequent hyperinsulinemia seem to be the major factors behind the alterations in the hepatic pathways of uptake, synthesis, degradation, and secretion of FFAs, which ultimately leads to the accumulation of lipids in the hepatocytes [18, 19]. Elevated plasma concentrations of insulin, glucose, and fatty acids promote hepatic fatty acid and triglyceride uptake, de-novo lipid synthesis and impaired β-oxidation of fatty acids by negative feedback. The result is an inappropriate shifting of FFAs to non-adipose tissue, such as the liver; together with inflammatory cytokines and specific adipokines associated with mitochondrial dysfunction, a vicious cycle is initiated that contributes to impaired insulin signaling and hepatic IR [5, 15, 20, 21].
The “two-hit” and the “multiple parallel hits” hypothesis
Traditionally, the two-hit hypothesis was established to explain the pathogenesis of NAFLD [22], that is, the initial insult occurs with the accumulation of lipids in the hepatocytes, which thus promotes IR. These changes seem to make the liver susceptible to a second insult, resulting in an inflammatory response and progression to liver damage. The second hit occurs due to increased hepatic oxidative stress, which is associated with increased FFA metabolism, diminished anti-oxidant activity, increased pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and endotoxin levels, and especially mitochondrial dysfunction and/or endoplasmic reticulum stress in the liver [18, 19].
The “two-hit” hypothesis has been challenged by the recognition that numerous complex pathways are responsible for the pathogenesis of NASH and progression to fibrosis, leading to a heterogeneous patient cohort with diverse clinical presentations [23]. With this insight, a new and more realistic model was proposed, namely, the “multi-parallel hits” hypothesis [24]. The main concept of this theory is that different events occur that in turn determine liver damage, but these events take place in parallel, not consecutively. We can consider the following elements as the main pathological protagonists: IR, oxidative stress, adipose tissue and pancreas toxicity, altered lipid metabolism, bile acids, gut microbiota, bacterial endotoxins, serum and liver iron overload, innate and adaptive immunity, and finally the polymorphisms of the genes involved in lipid accumulation, oxidative stress and hepatic fibrosis [5, 14, 25,26,27]. It is important to underline that all the mechanisms of damage that are involved in the pathogenesis of NAFLD enhance two conditions that are central in the progression of NASH, that is, IR and systemic chronic inflammation [13].
NAFLD treatment
While much progress has been made in elucidating the epidemiology, natural history, and pathogenesis of NAFLD/NASH, there remains no effective therapy, with limited options of evidence-based clinical guidelines for patient management. Pharmacological treatment of patients with NAFLD is still evolving, with no single therapy that has clearly been proven effective, especially, in modifying the course of the disease [5, 13,14,15, 19, 28].
Since cardiovascular and metabolic risk factors are highly prevalent among NAFLD/NASH patients, the backbone of the treatment regimens for these patients is general lifestyle interventions, including dietary changes and increased physical activity, which should be recommended for all patients [1, 5, 14,15,16, 19]. However, most patients may experience problems regarding long-term adherence to lifestyle interventions due to their attitudes or physical inability; therefore, pharmacotherapy becomes indispensable in this context [13, 15, 19].
The priority for NAFLD pharmacotherapy is to prevent the transformation of NAFLD into NASH and to improve the pathophysiology of the disease [9]. Therefore, in theory, insulin resistance should be the main target, together with anti-oxidative, anti-inflammatory, and anti-fibrotic agents. Currently, the first-line pharmacological approaches for NASH are the antioxidant vitamin E and the insulin sensitizer, pioglitazone. Both agents are unique, in that they have provided a sufficient degree of evidence in terms of efficacy, showing a positive effect on transaminases, fat accumulation, and inflammation. However, vitamin E has no proven effect on fibrosis and on long-term morbidity and mortality, and pioglitazone has a negative impact on weight. In addition, the safety of pioglitazone remains uncertain as data about its long-term use are not available [5, 13,14,15,16]. Other drugs have been studied, such as metformin, ursodeoxycholic acid, statins, pentoxifylline, and orlistat, but with only partially positive results. Among the emerging treatments, telmisartan is particularly interesting, as it seems to have an impact on insulin resistance, liver steatosis, inflammation, and fibrosis, according to preliminary studies [13,14,15,16]. However, the lack of large randomized clinical trials precludes the use of this drug as a consolidated option treatment, being formally recommended only in NALFD patients with an established indication for anti-hypertensive therapy [13, 19].
In the present review, we have focused on the available data of the anti-hypertensive drug telmisartan for treating and preventing fatty liver disease.
Renin–angiotensin system: the current view
Telmisartan is an angiotensin (Ang) type I receptor blocker (ARB) that has been widely used for the treatment of hypertension and hypertension-related cardiovascular end-organ damage [29]; it is considered a first-line drug in mild-to-moderate hypertension with an excellent safety profile [30]. Among the ARBs, telmisartan is emerging as the most promising drug for the treatment of NAFLD, in terms of both safety and efficacy [13]. The importance of the renin–angiotensin system (RAS) in MetS and particularly in NAFLD should be considered for a better comprehension of the role of telmisartan in the treatment of this condition.
The RAS is classically conceived as a single hormonal cascade responsible for controlling cardiovascular, renal, and adrenal functions [31]. It is primarily initiated by the expression of angiotensinogen (AGT), which is a protein that is produced by several cell types, including hepatocytes, adipocytes, and kidney cells. However, the liver is considered the primary source of circulating AGT in normal physiology [32, 33]. The kidney-derived renin converts angiotensinogen into angiotensin I (Ang I), a biologically inactive peptide that is rapidly hydrolyzed by angiotensin-converting enzyme (ACE) to octapeptide angiotensin II (Ang II) [34]. Ang II mediates the biological responses through two G-protein-coupled receptors, Ang II receptor type 1 (AT1R) and Ang II receptor type 2 (AT2R); however, the main described effects are mediated by AT1R [35, 36]. The AT1R is in abundance in adult tissues, whereas AT2R is mainly expressed during fetal development and is upregulated in pathologic conditions. The activation of ACE–Ang II–AT1R frequently contributes to physiopathological changes, such as excessive renal sodium reabsorption, abnormal vascular smooth muscle cell contraction, disproportionately high aldosterone secretion, and inappropriate cardiovascular responses. In addition, several pro-inflammatory, pro-oxidant, pro-thrombotic, and pro-fibrotic pathways are stimulated by AT1R activation [37, 38]. Individuals with increased FFA levels caused by lipid/heparin infusion, which mimic the lipid profile of patients with visceral fat obesity and insulin resistance, have significantly higher Ang II production in mononuclear and polymorphonuclear cells and also Ang-dependent leukocyte activation, which leads to endothelial dysfunction [37, 38]. AT2R is generally reported to mediate effects that are opposing and counterbalancing those mediated by AT1R in vitro as well as in vivo [29].
Advances in cellular and molecular biology, as well as physiological and pharmacological approaches, have generated a substantial change in our understanding of the RAS [31]. It is now clear that the circulating and tissue RASs are far more complex than previously anticipated. In addition to the classical components, the modern concept of the RAS includes novel enzymes, peptides, receptors, and biological actions. Additionally, the tissue RAS has been characterized in different organs and systems, in which significant interactions between receptors, mediators, and metabolic pathways have been discovered [21, 31, 39, 40].
One of the most significant conceptual changes of this hormonal system is the characterization of Ang-(1–7) through the G protein-coupled receptor Mas, it has opposing actions to Ang II [41, 42]. Several enzymatic routes may be involved in Ang-(1–7) formation, but it is mainly formed directly from Ang II by a new enzyme, a homolog to ACE, called ACE2 [31, 34, 43,44,45,46]. The ACE2–angiotensin-(1–7)/Mas axis can produce nitric oxide-dependent vasodilation as well as anti-arrhythmic, anti-proliferative, anti-inflammatory, anti-fibrotic, and anti-thrombotic effects [28, 41, 42, 47,48,49,50]. In humans, evidence shows that Ang(1–7) not only potentiates the vasodilating effect of bradykinin, possibly through a mechanism(s) involving nitric oxide release, but also antagonizes the vasoconstrictor effect of angiotensin II [51, 52].
Considering the opposite role of the two main mediators of RAS, Ang II, and Ang-(1–7), research groups have proposed a new view of RAS, adding a counter-regulation in the system. In this model, the RAS can be envisioned as a dual function system in which the vasoconstrictor/proliferative or vasodilator/anti-proliferative actions are primarily driven by the balance between both arms of the RAS, ACE–Ang II–AT1R, and ACE2–Ang-(1–7)–Mas. Thus, it is now accepted that the ACE2/Ang-(1–7)/Mas axis is able to counteract most of the deleterious actions of the ACE/AngII/AT1R axis, especially in pathological conditions when the renin–angiotensin system is activated [32, 42,43,44, 46, 53,54,55,56].
Renin–angiotensin system and NAFLD
The function of the RAS in metabolic homeostasis and disease regulation has been the subject of considerable interest in the last decade [39, 57, 58]. There is an increasing body of evidence showing the RAS involvement in metabolic regulation, playing an important role in lipid and glucose metabolism [42, 49, 55, 59, 60]. Recent studies have noted the importance of local balance between the ACE/Ang-II/AT1R and ACE2/Ang-(1–7)–Mas arms to avoid liver metabolic diseases [61].
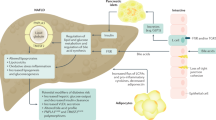
Results from experimental animals and humans suggest that obesity activates the RAS arm, composed of ACE/Ang II/AT1R [49, 62, 63] (Fig. 1). Ang II has been implicated as a major player in the altered hepatic lipid metabolism observed in NAFLD [64], influencing intracellular insulin signaling by several mechanisms, which may result in worsening insulin resistance, the main pathophysiological element of NAFLD [39, 65,66,67]. Ang II also induces the generation of reactive oxygen species (ROS), initiating and propagating the production of pro-inflammatory mediators, including TNF-α, interleukin-6 (IL-6), and plasminogen activator inhibitor-1 (PAI-1), resulting in inflammation and additional impairment of insulin signaling. Kanno et al. and Wei et al. showed that an increased expression of Ang II induces NAFLD and modulates inflammatory cell recruitment into the liver during liver injury [68, 69]. Accordingly, liver injuries in AT1-knockout mice present reduced inflammation and fibrosis [70, 71]. Paizis et al. demonstrated that AT1R genes are upregulated in areas of active hepatic fibrogenesis, meaning that chronic injury upregulates RAS in local tissues, which seems to contribute to the vicious cycle of steatosis–necroinflammation–fibrosis [13, 72].

Ang II is a major player in NAFLD. Ang II impairs intracellular insulin signaling resulting in worsening of insulin resistance, the main pathophysiological element of NAFLD. Ang II also induces generation of reactive oxygen species (ROS), initiating and propagating the production of pro-inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and plasminogen activator inhibitor-1 (PAI-1). These results in inflammation, additional impairment of insulin signaling, and upregulation of the AT1R genes, contributing to the vicious cycle of steatosis–necroinflammation–fibrosis
Moreover, increasing evidence has shown the beneficial effects of the Ang-(1–7)/Mas axis on liver pathology and metabolic disorders, exerting an important anti-obesity role by improving insulin sensitivity, glucose tolerance and type 2 diabetes, reducing body fat, increasing adiponectin production, and reverting hyperleptinemia [32, 42, 49,50,51, 59, 61]. For the first time, Feltenberger et al. evaluated the effects of a mouse model of oral Ang-(1–7) administration in high-fat-induced steatosis, liver metabolism, and inflammation. A reduction in fat mass, liver weight, and hepatic steatosis was observed and associated with decreased circulating total cholesterol, triglycerides, and alanine transaminase enzyme, improved lipid metabolism, and decreased expression of pro-inflammatory cytokines [64]. These effects were associated with the beneficial regulation of the RAS gene expression. Other reports indicated an important role of the ACE2/Ang-(1–7)/Mas axis in the liver, suggesting that oral treatment with Ang-(1–7) improves the status of steatohepatitis, as well as reduces adipogenesis-related markers [46, 60, 64]. Lastly, in humans with liver disease, both ACE2 gene expression and plasma Ang-(1–7) are increased, compared to that in healthy livers, confirming that the key regulator of Ang-(1–7) production in the alternative axis of the RAS is upregulated in response to hepatic injury [73]. All these observations confirmed the protective role of ACE2/Ang(1–7)/Mas axis activation, and might represent a promising strategy for the treatment targeting hepatic disorders [61].
NAFLD and RAS blockers
The established role of both circulating and local RAS in the pathogenesis of NAFLD and NASH produced considerable interest in the effect of RAS inhibitors because they are widely used, reasonably inexpensive, and have an excellent safety profile [28]. ACE inhibitors (ACEi) and angiotensin receptor blockers (ARBs) are used to inhibit the ACE/Ang II/AT1 arm and stimulate the activity of the ACE2/Ang-(1–7)/Mas axis [66, 74]. Both drug classes have been broadly used in congestive heart failure, hypertension, proteinuria, and chronic kidney disease [75, 76] and present significant effects in improving lipid and glucose metabolism [21, 49, 59, 64, 77]. Because the AT1R has a well-established role in mediating most of the deleterious actions of Ang II in the liver, the AT1R antagonism is considered more specific than ACE inhibition [32, 78]. Evidence indicates that treatment with ARBs results in greater improvement in insulin sensitivity and larger reduction in the risk of new onset diabetes mellitus. One possible explanation could be the inhibitory action of ACEi on both the AT1R and AT2R, resulting in suppression of the counterbalancing effects of AT2R on the actions of AT1R [28].
Additionally, there is substantial evidence to suggest that Ang-(1–7) is involved in the beneficial actions of RAS inhibitors [79]. It is important to mention that after chronic administration of AT1R blockers, Ang-(1–7) levels increase several times [80, 81] suggesting that this heptapeptide may contribute to the metabolic effects of the RAS [31, 66, 74].
The evidence is accumulating to show that ARBs can restore impaired intracellular insulin signaling and promote the redistribution of excess fat from the ectopic sites to mature adipocytes, resulting in improved insulin sensitivity [3]. Additionally, ARBs may improve transaminases, hepatic steatosis, and inflammation, in the NAFLD setting [19, 28]. The effect of ARBs on hepatic fibrosis in different animal models is well-described in the literature. ARBs inhibit stellate cell activity in obese mice, leading to a decrease in hepatic fibrosis [61, 82]. The evidence in humans shows that subjects with NAFLD who have been submitted to ARB treatment commonly have decreased liver fibrosis markers [71, 83]. These results suggest that liver fibrosis is mediated via the renin–angiotensin system, ACE/Ang II/AT1R axis, which demonstrates a beneficial role of angiotensin receptor blockers [61].
The benefits for each type of ARB deservers further evaluation, since it appears that not all ARB types have the same effects concerning receptor selectivity, binding mode, and metabolism [3]. In this context, the ARB telmisartan has a prominent role in treating and preventing NAFLD.
Telmisartan: the most promising ARB
New evidence indicates that telmisartan has beneficial actions, such as limiting MS development and improving diabetes, insulin resistance, and protecting patients with hypertension [66, 84,85,86,87,88]. Indeed, metabolic parameters, such as body weight, fat accumulation, fat cell size, and insulin resistance have been consistently ameliorated with telmisartan treatment in animal studies [3, 66, 87, 89,90,91,92,93,94] frequently with a superior performance, compared to that of other ARBs or to other RAS blockers [90, 93, 95, 96].
Kudo et al. showed for the first time that telmisartan decreases the adipocyte size and upregulates the adiponectin secretion without affecting food intake in the murine NASH model, thereby reducing the accumulation of visceral fat [97]. Moreover, telmisartan, but not ARB valsartan, increased the expression of both nuclear-encoded and mitochondrial-encoded genes in skeletal muscles that are known to play important roles in mitochondrial energy metabolism. Thus, in addition to a class effect of ARBs in modulating adipocyte size, these findings raise the possibility that certain molecules, such as telmisartan, may have a particularly strong impact on fat-cell volume and fat accumulation, as well as distinctive effects on energy metabolism, which may help to protect against dietary-induced visceral obesity and weight gain [90]. The efficacy of telmisartan in reducing visceral-fat mass may be relevant for patients, because an increase in visceral fat is related to hypertension, dyslipidemia, and an impaired metabolic pattern. Additionally, visceral fat serves as an independent predictor of mortality in men [98].
In the liver, telmisartan has been proven to positively affect the hepatic fibrosis markers in different pathological contexts [61]. The anti-oxidative effects of telmisartan have been studied [99], and its beneficial effects on hepatocytes can be partially based on the drug’s anti-oxidative properties [61]. Telmisartan reduces hepatic injuries resulting from type I diabetes mellitus [85], and when associated with propranolol, reduces several liver fibrosis signals, such as hydroxyproline, bile duct proliferation procollagen-α1, endothelin-1, and metalloproteinases in a primary sclerosing cholangitis (PSC)-like mouse model [100]. Telmisartan also prevented liver fibrosis in rat bile duct ligation model [101]. In mice on a high-fat diet that had received a low dose of streptozotocin (STZ) two days after birth, treatment with telmisartan reduced hepatic inflammation and fibrosis [102]. Similarly, telmisartan reduced liver fibrosis, that had been induced in rats by a short- or long-term methionine-deficient and choline-deficient diet [103], and also prevented the occurrence of hepatocellular carcinoma [104].
As mentioned earlier in this article, pathological changes in NAFLD may at least partly result from the activation of the inflammatory arm of RAS [94], especially in the context of insulin resistance. Telmisartan not only blocks the RAS, but also has been consistently proven to reduce insulin resistance [66, 84,85,86,87,88], thus, its effects on liver histology should be expected. Indeed, morphological evidence of the amelioration of NASH was found in several animal studies using telmisartan [91, 94, 97, 105], thus reducing inflammation and fibrosis by suppressing macrophage infiltration into the liver [94, 97]. Telmisartan treatment attenuated liver steatosis with decreased hepatic triglycerides, attenuated liver fibrogenesis with decreased type I collagen, and transforming growth factor-β1 (TGF-β1) mRNA expressions [97]. Clinical studies have resulted in similar outcomes. Hypertensive patients with MetS treated with low doses of the ARB telmisartan (20 mg/day) for 4 weeks showed a significant insulin-sensitizing effect, compared with that of hypertensive patients treated with valsartan (40 mg/day). It was the first time that a lower dose of telmisartan was proven to be sufficient to significantly improve glucose metabolism, whereas such an effect was not observed in valsartan-treated subjects [106]. Similarly, in a randomized control trial on hypertension-associated NASH patients, telmisartan (20 mg/day)-improved steatosis, necroinflammation, fibrosis, insulin resistance, and the lipid profile more consistent than valsartan (80 mg/day) in a 20-month treatment period, despite a similar blood pressure reduction [107]. Enjoji et al. evaluated the therapeutic efficacy of ARB telmisartan (40 mg/day) and olmesartan (20 mg/day) in NAFLD patients in a 6-month treatment period and concluded that both drugs significantly improved insulin resistance and liver injury, but the effect tended to be more distinct with telmisartan. This result suggests that these two ARBs may be used as liver-protecting agents in NAFLD patients. Although they did not investigate the effect of other ARBs (losartan, valsartan, candesartan), patients already using these drugs for hypertension treatment had the medication changed to olmesartan or telmisartan with a resultant significant improvement in insulin resistance and transaminases levels [3]. Another randomized trial in which telmisartan and losartan were administered did not display statistically significant differences between these two drugs on improvement in liver enzyme levels, suggesting that telmisartan may exert more potential effects in improving fatty liver, which is an important evaluated outcome [108]. In a recent 1-year randomized control trial, Alam et al. observed that telmisartan (40 mg/day or 80 mg/day if the patient was still hypertensive after taking 40 mg/day) significantly improved the overall histology of NASH patients, reducing inflammation and fibrosis markers independent of weight reduction [30]. Additionally, telmisartan was similarly effective in hypertensive and non-hypertensive NASH patients and had very minimal side effects during this period of treatment. The fact that the adverse effects encountered with telmisartan were less frequent than with other ARBs or ACE inhibitors is noteworthy [109].
Given the superiority of telmisartan in treating and reversing metabolic and hepatic parameters of Mets, the possibility that its effects may go beyond just blockade of the type 1 angiotensin II receptor has been raised [90]. To test this theory, Rong et al. demonstrated for the first time that telmisartan ameliorated diet-induced obesity, insulin resistance, and fatty liver in AT1R knock-out mice on a high-fat diet, suggesting that this drug may exert additional AT1R-independent beneficial effects on metabolism [87].
Concerning AT1R-independent effects, it is important to emphasize the relationship between telmisartan and peroxisome proliferator-activated receptor gamma (PPAR-γ) activation. Telmisartan works as a partial agonist of PPAR-γ [61, 87, 91, 97, 110], a property that does not appear to be shared by other ARBs [94, 111], except for losartan, which has already been reported to exert agonistic effects on this gene [112]. The nuclear PPARγ receptor is a transcription factor regulating many genes related to adipogenesis, lipid metabolism, and insulin sensitivity [91]. Telmisartan influences the expression of PPAR-γ target genes involved in carbohydrate and lipid metabolism. It has been shown to reduce glucose, insulin, and triglyceride levels in rats fed on a high-fat and high-carbohydrate diet [89]. PPAR-γ increases the insulin sensitivity, high-density lipoprotein levels, and decreases inflammation, oxidative stress, cell proliferation, migration, and fatty acid and triglyceride levels, but without causing the fluid collection associated with full agonists of PPAR-γ, such as pioglitazone or rosiglitazone [35, 61, 110, 113, 114]. Figure 2 illustrates these two mechanisms of action of telmisartan in the treatment of NAFLD.

The liver-protecting actions of telmisartan in NAFLD are shown. Telmisartan blocks the AT1R, inhibits the ACE/Ang II/AT1R axis and stimulates the Ang-(1–7)/Mas axis of RAS, improves insulin sensitivity, lipid metabolism, and decreases the expression of proinflammatory cytokines, with the suppression of macrophage infiltration into the liver. The result is morphological improvement in hepatic steatosis and in fibrogenic markers. Telmisartan also has anti-oxidative properties and works as a partial agonist of the nuclear receptor PPAR-γ, an action that contributes to increase insulin sensitivity and decrease inflammation, oxidative stress, cell proliferation, and fatty acid/triglyceride levels
The concept of an agent with dual PPAR-γ agonist and AT1 receptor antagonist actions is indeed promising, particularly from the standpoint of synergistic metabolic actions. However, some limitations should be noted [115]. PPAR-γ effects are elicited only by micromolar concentrations of telmisartan, whereas low nanomolar concentrations are sufficient to block AT1 receptors [116]. Thus, telmisartan is >1000-fold more potent as an AT1 receptor antagonist than as a PPAR-γ agonist [115]. According to this fact, most of the effect of telmisartan on metabolism is likely to be mediated by the blockade of AT1 receptors, together with the compensatory overstimulation of AT2 receptors and activation of the ECA2/Ang-(1–7)/Mas axis, as angiotensin II levels rise during chronic AT1 blockade.
Furthermore, structural differences between ARBs result in differences in their pharmacological and pharmacokinetic properties, and subsequently in their binding affinity to the Ang II receptor. Among the ARBs, the effectiveness of telmisartan might be adequately explained by its greatest affinity for AT1R, a longer half-life and greater lipophilicity, resulting in a greater in vivo blockade of AT1R [13, 28, 90, 115]. However, the synergy between PPAR-γ activation and angiotensin receptor blockade is clear from the preclinical data [115].
As another possible PPARg-independent mechanism, Miesel et al. investigated the hypothalamic–pituitary–adrenal (HPA) axis activity after AT1R blockade [93]. The HPA hyper-reactivity has been verified in rats and patients with diabetes [117], and the AT1R has been identified as a regulator of stress reactions [118]. Moreover, the Ang II-stimulated hyper-reactivity in the HPA axis was found to account for the reduction in glucose utilization in obese Zucker rats [119], revealing a functionally relevant crosstalk between Ang II, the HPA axis, and metabolic functions. It was demonstrated that HPA reactivity was reduced after AT1R blockade in rats with diet-induced metabolic syndrome. The simultaneous decrease in adrenocorticotropic hormone (ACTH) indicates a pituitary mechanism, requiring that peripherally administered TEL penetrates the blood–brain barrier. This was verified by measuring TEL concentrations in the cerebrospinal fluid and indirectly by the ability of TEL to antagonize the central effects of Ang II [120]. All these mechanisms could explain the differences between ARBs on their effects on insulin resistance, transaminase levels and liver histology, and the superiority of telmisartan in treating NAFLD and in preventing its progression to hepatic cirrhosis.
Conclusion
In conclusion, blocking RAS to inhibit the ACE/Ang II/AT1R axis and increase ACE2/Ang-(1–7)/Mas axis activation is an important strategy in treating NAFLD. Of all the RAS blockers, telmisartan is the most promising drug, not only because of its favorable pharmacokinetic proprieties and safety, but also because it seems to exert additional AT1-independent benefits on metabolism via partial PPAR-γ agonism and/or a positive central action on the HPA axis. However, large multicenter randomized-controlled trials are needed to consolidate these findings. Currently, treatment with telmisartan and with other RAS blockers can only be formally recommended in NALFD patients with an established indication of anti-hypertensive therapy.
References
Alberti KG, Zimmet P, Shaw J. Metabolic syndrome--a new world-wide definition. A consensus statement from the International Diabetes Federation. Diabet Med. 2006;23:469–80.
Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–28.
Enjoji M, Kotoh K, Kato M, Higuchi N, Kohjima M, Nakashima M, Nakamuta M. Therapeutic effect of ARBs on insulin resistance and liver injury in patients with NAFLD and chronic hepatitis C: a pilot study. Int J Mol Med. 2008;22:521–7.
Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1 Suppl):47–64.
Duseja A, Singh SP, Saraswat VA, Acharya SK, Chawla YK, Chowdhury S, Dhiman RK, Jayakumar RV, Madan K, Misra SP, Mishra H, Modi SK, Muruganathan A, Saboo B, Sahay R, Upadhyay R. Non-alcoholic fatty liver disease and metabolic syndrome-position paper of the Indian National Association for the Study of the Liver, Endocrine Society of India, Indian College of Cardiology and Indian Society of Gastroenterology. J Clin Exp Hepatol. 2015;5:51–68.
Koplay M, Sivri M, Erdogan H, Nayman A. Importance of imaging and recent developments in diagnosis of nonalcoholic fatty liver disease. World J Hepatol. 2015;7:769–76.
Law K, Brunt EM. Nonalcoholic fatty liver disease. Clin Liver Dis. 2010;14:591–604.
Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–31.
Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–23.
Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–85.
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43(2 Suppl 1):99–112.
McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40(Suppl 1):17–29.
Gitto S, Vitale G, Villa E, Andreone P. Treatment of nonalcoholic steatohepatitis in adults: present and future. Gastroenterol Res Pract. 2015;2015:732870.
Milic S, Mikolasevic I, Krznaric-Zrnic I, Stanic M, Poropat G, Stimac D, Vlahovic-Palcevski V, Orlic L. Nonalcoholic steatohepatitis: emerging targeted therapies to optimize treatment options. Drug Des Dev Ther. 2015;9:4835–45.
Takahashi Y, Sugimoto K, Inui H, Fukusato T. Current pharmacological therapies for nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2015;21:3777–85.
Nakajima K. Multidisciplinary pharmacotherapeutic options for nonalcoholic fatty liver disease. Int J Hepatol. 2012;2012:950693.
Gallagher EJ, Leroith D, Karnieli E. Insulin resistance in obesity as the underlying cause for the metabolic syndrome. Mt Sinai J Med. 2010;77:511–23.
Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–92.
Baran B, Akyuz F. Non-alcoholic fatty liver disease: what has changed in the treatment since the beginning? World J Gastroenterol. 2014;20:14219–29.
Chaldakov GN, Stankulov IS, Hristova M, Ghenev PI. Adipobiology of disease: adipokines and adipokine-targeted pharmacology. Curr Pharm Des. 2003;9:1023–31.
Santos SH, Andrade JM. Angiotensin 1-7: a peptide for preventing and treating metabolic syndrome. Peptides. 2014;59:34–41.
Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–5.
Younossi ZM, Reyes MJ, Mishra A, Mehta R, Henry L. Systematic review with meta-analysis: non-alcoholic steatohepatitis - a case for personalised treatment based on pathogenic targets. Aliment Pharmacol Ther. 2014;39:3–14.
Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–46.
Baskol G, Baskol M, Kocer D. Oxidative stress and antioxidant defenses in serum of patients with non-alcoholic steatohepatitis. Clin Biochem. 2007;40:776–80.
Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, Kowdley KV, Nonalcoholic Steatohepatitis Clinical Research N. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology. 2011;53:448–57.
Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–24.
Paschos P, Tziomalos K. Nonalcoholic fatty liver disease and the renin-angiotensin system: Implications for treatment. World J Hepatol. 2012;4:327–31.
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–72.
Alam S, Kabir J, Mustafa G, Gupta U, Hasan SK, Alam AK. Effect of telmisartan on histological activity and fibrosis of non-alcoholic steatohepatitis: a 1-year randomized control trial. Saudi J Gastroenterol. 2016;22:69–76.
Pereira RM, dos Santos RA, da Costa Dias FL, Teixeira MM, Simoes e Silva AC. Renin-angiotensin system in the pathogenesis of liver fibrosis. World J Gastroenterol. 2009;15:2579–86.
Santos SH, Simoes e Silva AC. The therapeutic role of renin-angiotensin system blockers in obesity- related renal disorders. Curr Clin Pharmacol. 2014;9:2–9.
Santos RA, Ferreira AJ. Angiotensin-(1-7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2007;16:122–8.
Giacchetti G, Sechi LA, Rilli S, Carey RM. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol Metab. 2005;16:120–6.
Kurtz TW, Pravenec M. Antidiabetic mechanisms of angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists: beyond the renin-angiotensin system. J Hypertens. 2004;22:2253–61.
Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther. 2010;128:119–28.
Tamura K, Tanaka Y, Tsurumi Y, Azuma K, Shigenaga A, Wakui H, Masuda S, Matsuda M. The role of angiotensin AT1 receptor-associated protein in renin-angiotensin system regulation and function. Curr Hypertens Rep. 2007;9:121–7.
Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res. 2009;32:533–6.
Putnam K, Shoemaker R, Yiannikouris F, Cassis LA. The renin-angiotensin system: a target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2012;302:H1219–1230.
Herichova I, Szantoova K. Renin-angiotensin system: upgrade of recent knowledge and perspectives. Endocr Regul. 2013;47:39–52.
Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–63.
Santos RA, Ferreira AJ, Simoes ESAC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol. 2008;93:519–27.
Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9.
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32.
Yoshiji H, Noguchi R, Ikenaka Y, Namisaki T, Kitade M, Kaji K, Shirai Y, Yoshii J, Yanase K, Yamazaki M, Tsujimoto T, Kawaratani H, Akahane T, Aihara Y, Fukui H. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Res Notes. 2009;2:70
Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–R17.
Ferreira AJ, Santos RA. Cardiovascular actions of angiotensin-(1-7). Braz J Med Biol Res. 2005;38:499–507.
Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Intern Med. 2008;264:224–36.
Santos SH, Fernandes LR, Pereira CS, Guimaraes AL, de Paula AM, Campagnole-Santos MJ, Alvarez-Leite JI, Bader M, Santos RA. Increased circulating angiotensin-(1-7) protects white adipose tissue against development of a proinflammatory state stimulated by a high-fat diet. Regul Pept. 2012;178:64–70.
Mario EG, Santos SH, Ferreira AV, Bader M, Santos RA, Botion LM. Angiotensin-(1-7) Mas-receptor deficiency decreases peroxisome proliferator-activated receptor gamma expression in adipocytes. Peptides. 2012;33:174–7.
Ueda S, Masumori-Maemoto S, Ashino K, Nagahara T, Gotoh E, Umemura S, Ishii M. Angiotensin-(1-7) attenuates vasoconstriction evoked by angiotensin II but not by noradrenaline in man. Hypertension. 2000;35:998–1001.
Ueda S, Masumori-Maemoto S, Wada A, Ishii M, Brosnihan KB, Umemura S. Angiotensin(1-7) potentiates bradykinin-induced vasodilatation in man. J Hypertens. 2001;19:2001–9.
Wei HS, Li DG, Lu HM, Zhan YT, Wang ZR, Huang X, Zhang J, Cheng JL, Xu QF. Effects of AT1 receptor antagonist, losartan, on rat hepatic fibrosis induced by CCl(4). World J Gastroenterol. 2000;6:540–5.
Seki E, Park E, Fujimoto J. Toll-like receptor signaling in liver regeneration, fibrosis and carcinogenesis. Hepatol Res. 2011;41:597–610.
Silva AR, Aguilar EC, Alvarez-Leite JI, da Silva RF, Arantes RM, Bader M, Alenina N, Pelli G, Lenglet S, Galan K, Montecucco F, Mach F, Santos SH, Santos RA. Mas receptor deficiency is associated with worsening of lipid profile and severe hepatic steatosis in ApoE-knockout mice. Am J Physiol Regul Integr Comp Physiol. 2013;305:R1323–1330.
Villela DC, Passos-Silva DG, Santos RA. Alamandine: a new member of the angiotensin family. Curr Opin Nephrol Hypertens. 2014;23:130–4.
de Kloet AD, Krause EG, Woods SC. The renin angiotensin system and the metabolic syndrome. Physiol Behav. 2010;100:525–34.
Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev. 2012;13:136–49.
Santos SH, Braga JF, Mario EG, Porto LC, Rodrigues-Machado Mda G, Murari A, Botion LM, Alenina N, Bader M, Santos RA. Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1-7). Arterioscler Thromb Vasc Biol. 2010;30:953–61.
Bilman V, Mares-Guia L, Nadu AP, Bader M, Campagnole-Santos MJ, Santos RA, Santos SH. Decreased hepatic gluconeogenesis in transgenic rats with increased circulating angiotensin-(1-7). Peptides. 2012;37:247–51.
Moreira de Macedo S, Guimaraes TA, Feltenberger JD, Sousa Santos SH. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides. 2014;62:189–96.
Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, Cassis LA. Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol. 2004;287:R943–949.
Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep. 2008;10:93–98.
Feltenberger JD, Andrade JM, Paraiso A, Barros LO, Filho AB, Sinisterra RD, Sousa FB, Guimaraes AL, de Paula AM, Campagnole-Santos MJ, Qureshi M, dos Santos RA, Santos SH. Oral formulation of angiotensin-(1-7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension. 2013;62:324–30.
Marrero MB, Fulton D, Stepp D, Stern DM. Angiotensin II-induced insulin resistance and protein tyrosine phosphatases. Arterioscler Thromb Vasc Biol. 2004;24:2009–13.
Prasad A, Quyyumi AA. Renin-angiotensin system and angiotensin receptor blockers in the metabolic syndrome. Circulation. 2004;110:1507–12.
Wei Y, Sowers JR, Nistala R, Gong H, Uptergrove GM, Clark SE, Morris EM, Szary N, Manrique C, Stump CS. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J Biol Chem. 2006;281:35137–46.
Kanno K, Tazuma S, Nishioka T, Hyogo H, Chayama K. Angiotensin II participates in hepatic inflammation and fibrosis through MCP-1 expression. Dig Dis Sci. 2005;50:942–8.
Wei Y, Clark SE, Morris EM, Thyfault JP, Uptergrove GM, Whaley-Connell AT, Ferrario CM, Sowers JR, Ibdah JA. Angiotensin II-induced non-alcoholic fatty liver disease is mediated by oxidative stress in transgenic TG(mRen2)27(Ren2) rats. J Hepatol. 2008;49:417–28.
Kanno K, Tazuma S, Chayama K. AT1A-deficient mice show less severe progression of liver fibrosis induced by CCl(4). Biochem Biophys Res Commun. 2003;308:177–83.
Yokohama S, Tokusashi Y, Nakamura K, Tamaki Y, Okamoto S, Okada M, Aso K, Hasegawa T, Aoshima M, Miyokawa N, Haneda M, Yoneda M. Inhibitory effect of angiotensin II receptor antagonist on hepatic stellate cell activation in non-alcoholic steatohepatitis. World J Gastroenterol. 2006;12:322–6.
Paizis G, Gilbert RE, Cooper ME, Murthi P, Schembri JM, Wu LL, Rumble JR, Kelly DJ, Tikellis C, Cox A, Smallwood RA, Angus PW. Effect of angiotensin II type 1 receptor blockade on experimental hepatic fibrogenesis. J Hepatol. 2001;35:376–85.
Liu J, Gong H, Zhang ZT, Wang Y. Effect of angiotensin II and angiotensin II type 1 receptor antagonist on the proliferation, contraction and collagen synthesis in rat hepatic stellate cells. Chin Med J. 2008;121:161–5.
Kyvelou SM, Vyssoulis GP, Karpanou EA, Adamopoulos DN, Zervoudaki AI, Pietri PG, Stefanadis CI. Effects of antihypertensive treatment with angiotensin II receptor blockers on lipid profile: an open multi-drug comparison trial. Hellenic J Cardiol. 2006;47:21–28.
Alfie J, Aparicio LS, Waisman GD. Current strategies to achieve further cardiac and renal protection through enhanced renin-angiotensin-aldosterone system inhibition. Rev Recent Clin Trials. 2011;6:134–46.
Meune C, Wahbi K, Duboc D, Weber S. Meta-analysis of renin-angiotensin-aldosterone blockade for heart failure in presence of preserved left ventricular function. J Cardiovasc Pharmacol Ther. 2011;16:368–75.
Santos RA, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP, Campagnole-Santos MJ, Baltatu O, Iliescu R, Reudelhuber TL, Bader M. Expression of an angiotensin-(1-7)-producing fusion protein produces cardioprotective effects in rats. Physiol Genom. 2004;17:292–9.
Carey RM, Padia SH. Angiotensin AT2 receptors: control of renal sodium excretion and blood pressure. Trends Endocrinol Metab. 2008;19:84–87.
Ferrario CM, Averill DB, Brosnihan KB, Chappell MC, Iskandar SS, Dean RH, Diz DI. Vasopeptidase inhibition and Ang-(1-7) in the spontaneously hypertensive rat. Kidney Int. 2002;62:1349–57.
Campbell DJ. The renin-angiotensin and the kallikrein-kinin systems. Int J Biochem Cell Biol. 2003;35:784–91.
Simoes e Silva AC, Diniz JS, Pereira RM, Pinheiro SV, Santos RA. Circulating renin angiotensin system in childhood chronic renal failure: marked increase of angiotensin-(1-7) in end-stage renal disease. Pediatr Res. 2006;60:734–9.
Hirose A, Ono M, Saibara T, Nozaki Y, Masuda K, Yoshioka A, Takahashi M, Akisawa N, Iwasaki S, Oben JA, Onishi S. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology. 2007;45:1375–81.
Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–5.
Investigators O, Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–59.
Halici Z, Bilen H, Albayrak F, Uyanik A, Cetinkaya R, Suleyman H, Keles ON, Unal B. Does telmisartan prevent hepatic fibrosis in rats with alloxan-induced diabetes? Eur J Pharmacol. 2009;614:146–52.
Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes Tissue Repair. 2010;3:21.
Rong X, Li Y, Ebihara K, Zhao M, Naowaboot J, Kusakabe T, Kuwahara K, Murray M, Nakao K. Angiotensin II type 1 receptor-independent beneficial effects of telmisartan on dietary-induced obesity, insulin resistance and fatty liver in mice. Diabetologia. 2010;53:1727–31.
Guimaraes GG, Santos SH, Oliveira ML, Pimenta-Velloso EP, Motta DF, Martins AS, Alenina N, Bader M, Santos RA, Campagnole-Santos MJ. Exercise induces renin-angiotensin system unbalance and high collagen expression in the heart of Mas-deficient mice. Peptides. 2012;38:54–61.
Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA, Kurtz TW. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002.
Sugimoto K, Qi NR, Kazdova L, Pravenec M, Ogihara T, Kurtz TW. Telmisartan but not valsartan increases caloric expenditure and protects against weight gain and hepatic steatosis. Hypertension. 2006;47:1003–9.
Clemenz M, Frost N, Schupp M, Caron S, Foryst-Ludwig A, Bohm C, Hartge M, Gust R, Staels B, Unger T, Kintscher U. Liver-specific peroxisome proliferator-activated receptor alpha target gene regulation by the angiotensin type 1 receptor blocker telmisartan. Diabetes. 2008;57:1405–13.
Souza-Mello V, Gregorio BM, Cardoso-de-Lemos FS, de Carvalho L, Aguila MB, Mandarim-de-Lacerda CA. Comparative effects of telmisartan, sitagliptin and metformin alone or in combination on obesity, insulin resistance, and liver and pancreas remodelling in C57BL/6 mice fed on a very high-fat diet. Clin Sci. 2010;119:239–50.
Miesel A, Muller-Fielitz H, Johren O, Vogt FM, Raasch W. Double blockade of angiotensin II (AT(1))-receptors and ACE does not improve weight gain and glucose homeostasis better than single-drug treatments in obese rats. Br J Pharmacol. 2012;165:2721–35.
Michel MC, Brunner HR, Foster C, Huo Y. Angiotensin II type 1 receptor antagonists in animal models of vascular, cardiac, metabolic and renal disease. Pharmacol Ther. 2016;164:1–81.
Miura Y, Yamamoto N, Tsunekawa S, Taguchi S, Eguchi Y, Ozaki N, Oiso Y. Replacement of valsartan and candesartan by telmisartan in hypertensive patients with type 2 diabetes: metabolic and antiatherogenic consequences. Diabetes Care. 2005;28:757–8.
Vitale C, Mercuro G, Castiglioni C, Cornoldi A, Tulli A, Fini M, Volterrani M, Rosano GM. Metabolic effect of telmisartan and losartan in hypertensive patients with metabolic syndrome. Cardiovasc Diabetol. 2005;4:6.
Kudo H, Yata Y, Takahara T, Kawai K, Nakayama Y, Kanayama M, Oya T, Morita S, Sasahara M, Mann DA, Sugiyama T. Telmisartan attenuates progression of steatohepatitis in mice: role of hepatic macrophage infiltration and effects on adipose tissue. Liver Int. 2009;29:988–96.
Kuk JL, Katzmarzyk PT, Nichaman MZ, Church TS, Blair SN, Ross R. Visceral fat is an independent predictor of all-cause mortality in men. Obesity. 2006;14:336–41.
Cianchetti S, Del Fiorentino A, Colognato R, Di Stefano R, Franzoni F, Pedrinelli R. Anti-inflammatory and anti-oxidant properties of telmisartan in cultured human umbilical vein endothelial cells. Atherosclerosis. 2008;198:22–28.
Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92:1866–74.
Yi ET, Liu RX, Wen Y, Yin CH. Telmisartan attenuates hepatic fibrosis in bile duct-ligated rats. Acta Pharmacol Sin. 2012;33:1518–24.
Cynis H, Kehlen A, Haegele M, Hoffmann T, Heiser U, Fujii M, Shibazaki Y, Yoneyama H, Schilling S, Demuth HU. Inhibition of glutaminyl cyclases alleviates CCL2-mediated inflammation of non-alcoholic fatty liver disease in mice. Int J Exp Pathol. 2013;94:217–25.
Nakagami H, Kiomy Osako M, Nakagami F, Shimosato T, Minobe N, Moritani T, Shimamura M, Miyake T, Shimizu H, Takeya Y, Morishita R. Prevention and regression of non-alcoholic steatohepatitis (NASH) in a rat model by metabosartan, telmisartan. Int J Mol Med. 2010;26:477–81.
Tamaki Y, Nakade Y, Yamauchi T, Makino Y, Yokohama S, Okada M, Aso K, Kanamori H, Ohashi T, Sato K, Nakao H, Haneda M, Yoneda M. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J Gastroenterol. 2013;48:491–503.
Rosselli MS, Burgueno AL, Carabelli J, Schuman M, Pirola CJ, Sookoian S. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis. 2009;206:119–26.
Ichikawa Y. Comparative effects of telmisartan and valsartan on insulin resistance in hypertensive patients with metabolic syndrome. Intern Med. 2007;46:1331–6.
Georgescu EF, Ionescu R, Niculescu M, Mogoanta L, Vancica L. Angiotensin-receptor blockers as therapy for mild-to-moderate hypertension-associated non-alcoholic steatohepatitis. World J Gastroenterol. 2009;15:942–54.
Hirata T, Tomita K, Kawai T, Yokoyama H, Shimada A, Kikuchi M, Hirose H, Ebinuma H, Irie J, Ojiro K, Oikawa Y, Saito H, Itoh H, Hibi T. Effect of Telmisartan or Losartan for treatment of nonalcoholic fatty liver disease: fatty liver protection trial by Telmisartan or Losartan study (FANTASY). Int J Endocrinol. 2013;2013:587140.
Karlberg BE, Lins LE, Hermansson K. Efficacy and safety of telmisartan, a selective AT1 receptor antagonist, compared with enalapril in elderly patients with primary hypertension. TEES study group. J Hypertens. 1999;17:293–302.
Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation. 2004;109:2054–7.
Kurtz TW. Treating the metabolic syndrome: telmisartan as a peroxisome proliferator-activated receptor-gamma activator. Acta Diabetol. 2005;42(Suppl 1):9–16.
Schupp M, Lee LD, Frost N, Umbreen S, Schmidt B, Unger T, Kintscher U. Regulation of peroxisome proliferator-activated receptor gamma activity by losartan metabolites. Hypertension. 2006;47:586–9.
Clasen R, Schupp M, Foryst-Ludwig A, Sprang C, Clemenz M, Krikov M, Thone-Reineke C, Unger T, Kintscher U. PPARgamma-activating angiotensin type-1 receptor blockers induce adiponectin. Hypertension. 2005;46:137–43.
Pershadsingh HA. Treating the metabolic syndrome using angiotensin receptor antagonists that selectively modulate peroxisome proliferator-activated receptor-gamma. Int J Biochem Cell Biol. 2006;38:766–81.
Ernsberger P, Koletsky RJ. Metabolic actions of angiotensin receptor antagonists: PPAR-gamma agonist actions or a class effect? Curr Opin Pharmacol. 2007;7:140–5.
Kakuta H, Sudoh K, Sasamata M, Yamagishi S. Telmisartan has the strongest binding affinity to angiotensin II type 1 receptor: comparison with other angiotensin II type 1 receptor blockers. Int J Clin Pharmacol Res. 2005;25:41–46.
Johren O, Dendorfer A, Dominiak P, Raasch W. Gene expression of mineralocorticoid and glucocorticoid receptors in the limbic system is related to type-2 like diabetes in leptin-resistant rats. Brain Res. 2007;1184:160–7.
Aguilera G, Kiss A, Luo X. Increased expression of type 1 angiotensin II receptors in the hypothalamic paraventricular nucleus following stress and glucocorticoid administration. J Neuroendocrinol. 1995;7:775–83.
Muller-Fielitz H, Hubel N, Mildner M, Vogt FM, Barkhausen J, Raasch W. Chronic blockade of angiotensin AT(1) receptors improves cardinal symptoms of metabolic syndrome in diet-induced obesity in rats. Br J Pharmacol. 2014;171:746–60.
Gohlke P, Weiss S, Jansen A, Wienen W, Stangier J, Rascher W, Culman J, Unger T. AT1 receptor antagonist telmisartan administered peripherally inhibits central responses to angiotensin II in conscious rats. J Pharmacol Exp Ther. 2001;298:62–70.
Acknowledgements
The authors acknowledge the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG—Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq—Brazil) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES—Brazil).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Borém, L.M.A., Neto, J.F.R., Brandi, I.V. et al. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: a brief review. Hypertens Res 41, 394–405 (2018). https://doi.org/10.1038/s41440-018-0040-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-018-0040-6
This article is cited by
-
Interaction of telmisartan and related sartans with the programmed cell death-ligand 1 (PD-L1) protein dimer: a molecular docking analysis
Future Journal of Pharmaceutical Sciences (2023)
-
Chronic angiotensin receptor activation promotes hepatic triacylglycerol accumulation during an acute glucose challenge in obese-insulin-resistant OLETF rats
Endocrine (2022)
-
The spike of SARS-CoV-2 promotes metabolic rewiring in hepatocytes
Communications Biology (2022)
-
S-adenosylmethionine upregulates the angiotensin receptor-binding protein ATRAP via the methylation of HuR in NAFLD
Cell Death & Disease (2021)
