
Abstract
The MCM2-7 helicase is a heterohexameric complex with essential roles as part of both the pre-replication and pre-initiation complexes in the early stages of DNA replication. Meier-Gorlin syndrome, a rare primordial dwarfism, is strongly associated with disruption to the pre-replication complex, including a single case described with variants in MCM5. Conversely, a biallelic pathogenic variant in MCM4 underlies immune deficiency with growth retardation, features also seen in individuals with pathogenic variants in other pre-initiation complex encoding genes such as GINS1, MCM10, and POLE. Through exome and chromium genome sequencing, supported by functional studies, we identify biallelic pathogenic variants in MCM7 and a strong candidate biallelic pathogenic variant in MCM3. We confirm variants in MCM7 are deleterious and through interfering with MCM complex formation, impact efficiency of S phase progression. The associated phenotypes are striking; one patient has typical Meier-Gorlin syndrome, whereas the second case has a multi-system disorder with neonatal progeroid appearance, lipodystrophy and adrenal insufficiency. We provide further insight into the developmental complexity of disrupted MCM function, highlighted by two patients with a similar variant profile in MCM7 but disparate clinical features. Our results build on other genetic findings linked to disruption of the pre-replication and pre-initiation complexes, and the replisome, and expand the complex clinical genetics landscape emerging due to disruption of DNA replication.
Similar content being viewed by others


Introduction
DNA replication is a highly coordinated process, with complex regulation to enable efficient progression through S phase in proliferating cells whilst controlling initiation mechanisms to prevent re-replication, a source of replication stress and genome instability. A new round of DNA replication starts during late mitosis and early G1 of the cell cycle, when the prereplication complex (preRC) is loaded onto chromatin, licensing genomic origins. The origin recognition complex (ORC), composed of ORC1-6 subunits, binds to DNA along with CDC6, and then via interaction with the cofactor CDT1, loads the inactive MCM helicase complex, formed by MCM2-7 subunits. At the onset of S-phase, the multiprotein preinitiation complex (preIC) acts on the MCM helicase and through the binding of the GINS1-4 complex and CDC45, forms and activates CMG helicase to unwind DNA, permitting establishment of a functional replisome, formed by DNA polymerase and associated proteins. The MCM2-7 helicase has multiple functions during this process, in both being loaded onto chromatin as a double hexamer as part of the preRC [1, 2], and then once activated as part of the CMG helicase, sliding along the DNA as a single hexamer to unwind DNA. Additional proteins including DNA polymerase subunits bind to the preIC to form the replisome, which together facilitate efficient polymerase movement and DNA replication [3, 4].
There have been several disorders associated with dysfunction of initiation of DNA replication. The most strongly associated is Meier-Gorlin syndrome (MGORS, MIM #224690), a generally autosomal recessive condition caused by variants in ORC1, ORC4, ORC6, CDT1, CDC6, GMNN, MCM5, CDC45, and most recently DONSON [5,6,7,8,9,10]. All biallelic variants act either as hypomorphic loss-of-function alleles (or in a de novo gain-of-function manner for GMNN), reducing levels of active protein on chromatin and slowing S-phase progression [6, 10, 11]. While MGORS is a form of primordial dwarfism, the reduction in proportionate growth is more variable than other subtypes [12]. Intrauterine growth delay is observed, however childhood growth is normal [12], suggesting there is a specific period of development especially sensitive to reduced activity of MGORS-associated proteins. Other characteristic features include facial dysmorphism with microtia, a/hypo-plasia of patella, external genital anomalies, and in females, absent breast development [13]. Only one individual has been described harboring biallelic variants in MCM5 [10]. This individual presented with the cardinal features of MGORS, including a mild growth delay, typical facial features and absent patellae. Patient-derived cells confirmed a delay in S phase progression and increased sensitivity to replication stress [10].
In contrast to other preRC components, biallelic variants in MCM4 cause a slightly different phenotype, where in addition to short stature and microcephaly individuals have adrenal failure and NK cell deficiency [14,15,16]. All cases reported to date are homozygous for the same variant and show common ancestry to Traveler communities. This splice site variant causes a frameshift and premature stop early in the transcript, resulting in the increased use of an alternative downstream methionine for protein translation. Total MCM4 levels remain relatively normal in patient-derived cells despite the N-terminal deletion, but absence of the full-length MCM4 leads to cell cycle defects and genome instability [16]. The amino-terminal region of MCM4 is required for CDK and DDK phosphorylation, and MCM helicase activity, suggesting disruption of this may underlie at least some of the observed phenotypes in affected families [17, 18]. Interestingly, adrenal failure and NK cell deficiency is also associated with pathogenic variants in genes encoding downstream steps in replication initiation, such as DNA polymerase subunits, POLE, POLE2, and POLA1, as well as preIC encoding genes GINS1, and more recently, MCM10 [19,20,21,22,23,24,25]. Together with the MGORS studies, insights from human genetic studies have revealed several common clinical themes such as growth retardation, with some tissue-specific differences, caused by the disruption of DNA replication.
Through exome and genome sequencing, here we report biallelic variants in MCM3 and MCM7 in three patients either clinically diagnosed with Meier-Gorlin syndrome or displaying a multisystem phenotype with progeroid appearance at birth and development of lipodystrophy and adrenal insufficiency later in life.
Methods
Ethics declaration
Ethical approval for this study was provided by the New Zealand Health and Disability Ethics Committee (16/STH/3) and the Ethics Committee of the Hamburg Medical Chamber (PV3802). Informed consent was obtained for involvement in the study, and separate consent obtained for the publication of photos.
Sequencing methodologies
10× Genomics Chromium genome sequencing was undertaken on samples from P1 and P2 to prioritize biallelic variants of interest. Processing and prioritization of variants was undertaken as previously described [9]. Exome sequencing was performed on genomic DNA extracted from leukocytes of P3 and his healthy parents by CeGaT (Tübingen, Germany). Enrichment was carried out using the SureSelect Human All Exon V6 kit (Agilent). Each captured library was then loaded and sequenced on the HiSeq platform (Illumina). Variant analysis was performed as previously described [26].
All variants were confirmed in patients and parents by PCR and bidirectional Sanger sequencing. Oligonucleotides are listed in Supplementary Table 1. Variants have been uploaded to gene-specific LOVD databases (IDs: Individuals #00324523, #00324524, and #00324525).
Protein alignment
Clustal Omega was used to align protein sequences of Homo sapiens (MCM3: NP_002379.3, MCM7: NP_005907.3), Macaca mulatta (MCM3: XP_001106966.1, MCM7: XP_001101053.2), Mus musculus (MCM3: NP_032589.1, MCM7: NP_032594.1), Danio rerio (MCM3: NP_997732.1, MCM7: NP_997734.1), Drosophila melanogaster (MCM3: NP_511048.2, MCM7: NP_523984.1), Saccharomyces cerevisiae (MCM3: NP_010882.1, MCM7:NP_009761.4), Schizosaccharomyces pombe (MCM3: NP_587795.1, MCM7:NP_596545.1).
Protein modeling
Human MCM3 and MCM7 structural models were generated with SWISS-MODEL [27], and were superimposed on the S. cerevisiae ORC-Cdc6-Cdt1 bound MCM2-7 structure (PDB: 5V8F) [28] using Pymol (Version 2.1, Schrödinger, LLC). To assess the likely molecular interactions affected by the MCM3 and MCM7 missense variants, the human ORC1-5 structure (PDB: 5UJM) [29] along with structural models for human CDC6 and remaining MCM subunits, generated with SWISS-MODEL, were also superimposed on the S. cerevisiae structure.
Cell culture
HEK293FT and patient-derived primary fibroblasts were maintained in DMEM with 10% FBS and penicillin/streptomycin antibiotics, except for cells derived from P3, which were maintained in DMEM with 20% FBS and penicillin/streptomycin antibiotics, all in 5% CO2 and normoxic conditions.
Plasmid DNA
Plasmids encoding individual MCM subunits fused to either 2×Strep-V5 or 3×FLAG C-terminal tags were generated by PCR amplification of human MCM2-7 subunits, either from cDNA extracted from a HEK293 cell line or from IMAGE clone containing-plasmids (Source BioScience), and insertion by Gibson assembly into a SacI/XbaI cut p3XFLAG vector. Site-directed mutagenesis was carried out to introduce the MCM3 and MCM7 variants, and Sanger sequencing was used to confirm the coding sequences of the plasmids.
Immunoblot analysis and antibodies
Plasmid transfections were undertaken using Lipofectamine 2000 (Thermo Scientific) in Optimem reagent, as per manufacturer’s instructions. Whole cell extracts were prepared by lysis and sonication of fibroblasts in RIPA buffer (Thermo Scientific) containing protease inhibitor cocktail (Roche), or HEK293FT cells using lysis buffer (10 mM Tris-HCl pH 7.4, 140 mM NaCl, 2 mM EDTA, 1% Triton X-100, 2 mM CaCl2 plus protease inhibitor cocktail and phosphatase inhibitor). Proteins were separated by 6–10% SDS-PAGE and transferred onto nitrocellulose membranes using standard procedures, and blocked with 5% (w/v) milk powder in PBS. Antibodies: MCM3: mouse anti-MCM3 (Sigma, SAB1412669; 1:500), MCM7 (combination of SCBT, sc-56324:1:200, SCBT, sc-9966:1:200, Abcam, Ab2360:1:200), HDAC (Abcam, Ab19845:1:1000), FLAG (Sigma, F1804-50UG:1:7000), V5 (Thermo Scientific, R960-25:1:3000), goat anti-mouse secondary (IRDye 800CW, LI-COR), goat anti-rabbit secondary antibody (IRDye 680RD, LI-COR). Images were obtained using a LI-COR Odyssey CLx instrument and bands quantified using Image Studio software.
Co-immunoprecipitation
HEK293FT cells were co-transfected with pMCM2/3/4/5/6-3×FLAG and pMCM7-2×Strep-V5 (encoding either canonical protein sequence or with patient substitutions). After 24 h, lysates were extracted with lysis buffer and MCM7-protein complexes enriched using 5% (v/v) MagStrep XT Bead solution (IBA), and mixed at 4 °C overnight. Proteins were then detected by western immunoblotting.
EdU pulse chase
Primary fibroblasts were seeded at 75% confluency, and incubated at 10 μM EdU in DMEM plus 10% FBS for 30 min. Cells were then harvested at time zero, or media replaced with DMEM plus 10% FBS until the appropriate timepoint. Cells were trypsinized, then fixed and permeabilized with ice-cold 70% ethanol. EdU was detected by using the Click-iT™ Plus EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit (Thermo Scientific), and following washing, cells were stained by DAPI 1 μg/mL)with 250 μg/mL RNase A in 1% BSA in PBS. Samples were analysed on a Gallios flow cytometer and analysed using Kaluza (Beckman Coulter). Single cells were selected by gating based on forward scatter and side scatter populations, and also by a doublet discriminating plot resulting from DAPI staining. At least 10,000 single cell events were collected per sample, with six independent experiments performed for each sample. EdU vs DAPI plots were analysed using FlowJo (FlowJo, TreeStar Software, Ashland, OR), from which the percentage of cells in G1, S (early, mid, and late) and G2/M phases were calculated. Statistical analysis was carried out by GraphPad Prism.
Immunocytochemistry
Fibroblasts were grown on coverslips, then fixed and permeabilized with ice-cold methanol and incubated at −20 °C for 10 min. Cells were blocked with 2% goat serum in 0.2% Triton X-100 and 2% BSA in PBS. Cells were stained with either mouse anti-MCM3 (Sigma, SAB1412669; 1:500) or mouse anti-MCM7 (SCBT, sc-9966:1:50) at 4 °C overnight. These antibodies were then detected using goat anti-mouse Alexa Fluor 568 (Thermo Scientific, A-11004:1:1000), and following staining with DAPI, coverslips mounted using Vectashield Mounting Medium. Images were taken using an Olympus BX53 upright microscope, using 100× oil immersion lens, at the same exposure.
qRT-PCR
RNA was extracted from fibroblasts using the RNeasy kit (Qiagen) according to manufacturer’s instructions. cDNA was synthesized with SuperScript IV VILO Master Mix with ezDNase. qRT-PCR was undertaken using PowerUP SYBR Green Master Mix (Thermo Fisher), oligonucleotide sequences are listed in Supplementary Table 1.
Results
Identification of candidate disease genes underlying MGORS
To identify novel candidate variants in individuals with MGORS, we undertook 10× Chromium whole genome sequencing, which uses microfluidic barcoding to link sequencing reads derived from the same chromosome and thereby enable phasing of variants. In two patients, we identified biallelic variants in MCM subunit encoding genes, MCM3 and MCM7 (Table 1). P1 is homozygous for c.2417A>T, p.(Gln806Leu) in MCM3 (NM_002388.4) and parents were confirmed as heterozygous, whereas P2 is compound heterozygous for c.[415C>T];[1616A>G]/p.[(Gln139*)];[(Tyr539Cys)] in MCM7 (NM_005916.4). Sanger sequencing confirmed appropriate heterozygous segregation in the parental samples of P2. A further family was identified via Genematcher [30], where trio exome sequencing identified biallelic variants in MCM7 in the affected child (Table 1), with unaffected parents and an unaffected sibling all heterozygous for only variant (Supplementary Fig. 1). This individual, P3, is compound heterozygous for c.[776G>C];[1579C>T]/p.[(Gly259Ala)];[(Arg527*)]—a similar variant profile to P2. All variants identified are either very rare or absent from gnomAD.
All missense variants demonstrated high CADD scores (Table 1) and the affected amino acids lie within conserved domains of their respective proteins (Fig. 1A). In the MCM3 protein, the substitution sites are highly conserved through to Drosophila melanogaster, while for MCM7 both sites are conserved as distantly as Saccharomyces cerevisiae (Fig. 1B). Structurally, all affected residues are located in functionally important sites (Fig. 1C, Supplementary Fig. 2). In MCM3, the conserved p.Gln806 residue is located at the surface of the winged-helix domain within the C-terminal domain and forms part of the binding site for ORC2 and CDC6. Substitution of this residue with a leucine would likely disrupt binding of the MCM2-7 helicase to the ORC1-6/CDC6 complex. The MCM7 p.Gly259 residue is located within the core of the N-terminal domain where substitution with an alanine may impact upon MCM7 protein structure. Tyr539 is located at the surface of the AAA + ATPase domain, within a region identified as being required for MCM3 and ATP binding [28].

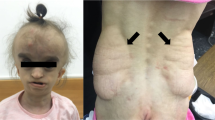
A Schematic indicating amino acid positions of identified variants, numbering according to RefSeq NM_002388.4 (MCM3) and NM_005916.4 (MCM7). NTD, N-terminal domain; AAA + , AAA ATPase domain; WD, winged-helix domain; red bar, nuclear localization signal. B Clustal Omega alignment demonstrating strong conservation of substituted residues in MCM3 and MCM7. C Substitution of MCM3 Gln806 (yellow) with a Leu may impact upon binding of MCM3 (green) to the ORC-CDC6 complex, where interactions with ORC2 (red) and CDC6 (blue) would likely be impacted. Substitution of MCM7 Gly259 (yellow) with an Ala may distort the MCM7 protein core (blue), as the side-chain would clash with that of Glu135. Substitution of MCM7 Tyr539 (yellow) with a Cys may distort the alpha helix and surrounding structures of MCM7 (blue), thereby impacting MCM3 (green) and ATP binding. ASG (yellow) represents the likely binding mode of ATP. D Photographs of patients. P1 and P2 demonstrate classical features of Meier-Gorlin syndrome, with microtia, thick lower lip vermillion and a pointy jaw. P2 and P3 share similar features of narrow face, prominent nose with a high nasal bridge and prominent teeth. P3 displayed a progeroid appearance at age 4 months (top). Facial photographs at the age of 14 yrs are shown in the bottom panel.
Clinical variability in patients with MCM3 or MCM7 variants
The three affected individuals demonstrate a broad set of developmental features (Table 2, Fig. 1D, and Supplementary Note). Individuals P1 and P2 were clinically diagnosed with MGORS. In addition to proportionate growth retardation, both individuals demonstrated microtia and facial dysmorphism commonly observed in individuals with MGORS, such as thick lower lip vermillion and micrognathia. While patella in P1 were of normal size, P1 had congenital lung emphysema, another commonly observed feature of MGORS [13]. Individual P2 (aged 27 years) had absent patella as well as absent breast development—an almost invariant feature in females of pubertal age with MGORS [13].
P3 presented with some overlapping features, particularly facial dysmorphism with micrognathia and small ears. While small at birth, he did not demonstrate reduced stature or microcephaly at age 14.5 years, which is intriguing given the similar variant profile as P2. P3 showed a neonatal progeroid appearance (see Fig. 1D at 4 months of age). At 4 months of age he had an Addisonian crisis and a primary adrenal insufficiency was diagnosed and is managed with continuing cortisol therapy. Hypertriglyceridemia was noticed in early childhood making dietary treatment necessary. Intermittent gastrostomy was necessary. He developed lipodystrophy and had dystrophy of unknown origin (Fig. 1D). Additionally, hypothyroidism was noticed and treated accordingly. Development of puberty was normal and timely. Low cholesterol levels, especially LDL-cholesterol, were also repeatedly observed. Reanalysis of the exome sequencing data did not suggest any clear additional candidates to explain his complex phenotype.
MCM7 variants reduce protein levels or complex interactions
To investigate the cellular and functional consequences of these variants, we used a combination of patient-derived fibroblasts and overexpression analyses.
We first wanted to understand the consequence of the variants on transcript and protein levels. To measure effects on transcripts, we undertook qRT-PCR on RNA extracted from patient-derived fibroblasts. The missense variant in MCM3 in P1, p.(Gln806Leu), did not reduce MCM3 transcripts in P1 fibroblasts (Fig. 2A), and similarly had no effect on protein levels at the cellular level, with comparable MCM3 levels as control fibroblasts detected by immunoblotting (Fig. 2A). For MCM7, both P2 and P3 were compound heterozygous for a missense variant in trans with a protein-truncating variant, which through introducing a premature stop codon, is predicted to cause nonsense-mediated decay of those transcripts. As therefore expected, MCM7 transcript levels were reduced in both patient-derived cell lines (overall mean ± SEM, P2: 67 ± 4%, P3: 61 ± 5%) (Fig. 2B). In analysing protein lysates from fibroblasts derived from P2 and P3, we observed a significant reduction in MCM7 levels (Fig. 2B) (mean ± SD, 42 ± 3% in P2 cells, p = 0.0040 and 35 ± 7% in P3 cells, p = 0.0028), aligning with the variants identified and the measured transcript levels. Immunocytochemistry of patient fibroblasts demonstrated no effects of MCM3 or MCM7 variants on protein localization, when compared to a control cell line (Supplementary Fig. 3).

Ai, A Quantification of MCM3 in cells from P1. (left) qRT-PCR of MCM3 from Control and P1 fibroblasts, quantified by 2(−Δ ΔCT) method, normalized to RPS18. (Three primer sets located across the transcript were used, n = 3 independent experiments). (right) Representative blot and quantification of MCM3 in P1 vs. control fibroblasts, normalized to HDAC (student’s t-test, mean ± SD, n = 3 independent experiments). B Quantification of MCM7 in cells from P2 and P3. (left) qRT-PCR of MCM7 from Control, P2, and P3 fibroblasts, quantified by 2(−Δ ΔCT) method, normalized to RPS18. (Three primer sets located across the transcript were used, n = 3 independent experiments). ****p < 0.0001, one-way ANOVA, treating each cell line as one group). (right) Representative blot and quantification of MCM7 in control fibroblasts compared to P2 and P3 fibroblasts, normalized to HDAC as a loading control (one-way ANOVA, P2 vs. control, **p = 0.0040; P3 vs. control, **p = 0.0028, mean ± SD, n = 2 independent experiments).
The MCM 2–7 subunits assemble as a complex to form the MCM helicase, and structural modeling indicated the MCM7 missense variants could impact either subunit interactions or protein structure, which could destabilize the complex. We therefore undertook a co-immunoprecipitation assay to quantify effects of these missense variants (Fig. 3A, B). Canonical MCM7 or variant-MCM7,tagged by 2×Strep-V5, together with the remaining MCM subunits (each tagged with 3×FLAG), co-transfected into HEK293FT cells. Following 24 h transfection, the target protein and complexes were immunoprecipitated using magnetic streptavidin beads, and levels of interacting complex members quantified through immunoblotting.

A A representative immunoblot resulting from a pull-down of the MCM2-7 helicase by MCM7-2×Strep-V5 (encoding either the canonical protein sequence or with patient substitutions) highlighting the effect of MCM7 patient variants. *MCM7 degradation. B Quantification of the MCM3-6 helicase complex pulled-down, and C quantification of a single subunit, MCM5, for each of the pull-downs from A. MCM5 was used as a proxy for the MCM3-MCM7 interaction. Pull-downs involved co-transfection of HEK293 cells with 3×FLAG-tagged MCM subunits for MCM2-6, and MCM7-2XStrep-V5 (encoding either the canonical protein sequence or with patient substitutions). Samples were immunoprecipitated using MCM7-2×Strep-V5. Band intensities were measured using Image Studio software. MCM bands were normalized to HDAC and then relative intensities to wildtype calculated. Differences were statistically analysed using a one way ANOVA with Dunnett’s multiple comparisons test (mean ± SEM). MCM3-6, *p = 0.0158 (n = 3 independent experiments); MCM5, *p = 0.0191 (n = 2 independent experiments). D Representative flow cytometry plots in control, P2 and P-1 over a 6 h time course. E Quantification of control vs. P1 (left) and P2, P3 (right) cells in early S-phase, relative to t = 0 time point. (mean ± SEM, n = 6 independent experiments, **p = 0.0047, *p = 0.0122, two-way ANOVA).
For MCM7, we observed that the p.Gly259Ala substitution caused a significantly reduced interaction with the MCM3-6 subunits (p = 0.034) (Fig. 3B). The structural modeling suggested that this substitution could disrupt MCM7 protein structure; this manifested as altered interaction efficiency with the other subunits. For the MCM7 p.Tyr539Cys variant, we observed an average decrease in the interaction strength of the MCM complex, however this was not statistically significant. From our structural modeling, this MCM7 variant is predicted to alter the interaction surface with MCM3. The size of MCM3 made it difficult to visualize amongst the other MCM subunits with similar molecular weights, however MCM5 and MCM3 form a subcomplex within the larger MCM complex [31,32,33], and therefore we measured MCM5 levels as a proxy for MCM3. The p.Tyr539Cys variant caused a significant reduction in immunoprecipitation of MCM5 (p = 0.014) (Fig. 3C). This result likely contributed to the reduced levels observed when testing the larger MCM3-6 complex immunoprecipitation with this variant MCM7. Together, these experiments demonstrate that the identified variants in MCM7 are deleterious at either transcript or protein levels and would reduce functionality of the MCM complex in the cell.
MCM7 variants perturb S-phase progression in patient-derived cells
We next examined whether variants in MCM3 or MCM7 affected the ability to efficiently progress through S phase in patient-derived fibroblasts. A decrease in S-phase progression has been observed in cells from other individuals with variants in MGORS-associated genes ORC1 and MCM5 [6, 10]. To capture active DNA replication, we pulsed control and patient-derived fibroblasts with the nucleotide analog EdU for 30 min and we then harvested cells between 0 and 6 h later, using flow cytometry to quantify cells in early S-phase. While there was no difference between control and fibroblasts from P1, fibroblasts from both P2 and P3 displayed delays in S phase progression (P2: p = 0.0047, P3: p = 0.0122) (Fig. 3D, E).
Discussion
Here, we report the identification of variants in both MCM3 and MCM7 in individuals with either MGORS or an overlapping congenital disorder. Through molecular genetics and functional assays, we confirm the MCM7 variants are deleterious to functioning and impact the cell’s ability to efficiently form MCM complexes, which consequently impairs S-phase progression. While the S phase progression was significantly different in MCM7 patient cells compared to the control, we observed that the impact appeared more subtle than that measured in cells derived from other MGORS patients. S phase progression rates are likely to be influenced by variant severity and cell origin specific proliferative pressure—for example, previous MGORS studies have often used immortalized lymphoblastoid cells rather than primary fibroblasts for similar experiments [6, 10].
A biallelic MCM3 variant was identified in P1 through genome sequencing. P1 has clinically typical MGORS, with proportionately reduced growth and head size, small ears and the characteristic facial features normally associated with MGORS. No other candidate variants were identified in the established MGORS-associated genes, nor in other genes encoding replication-associated components. All proteins linked to MGORS thus far function in the initiation of DNA replication, supporting MCM3 as a candidate. Absence in control populations together with the conservation of p.Gln806 across diverse species and a non-conservative substitution from a polar to a hydrophobic amino acid, supports this variant being deleterious. The winged-helix domain of MCM3 (within which Gln806 lies) is particularly important; through binding to ORC2/CDC6, this region of MCM3 leads the loading of the MCM2-7 hexamer on to chromatin and plays an important role in modulating ATPase activity of the ORC1-6/CDC6 complex [34]. Despite our investigation of both P1-derived fibroblasts and in vitro assays studying the missense variant, we observed no deleterious effect on protein stability. Similarly, we observed no consequences to S-phase progression in P1 fibroblasts. Unlike other forms of microcephalic primordial dwarfism, individuals with MGORS do not show a progressive growth deficit [12], suggesting there is a specific developmental timespan in utero within which cells may be most sensitive to a reduction in DNA replication initiation. Our cellular assays may not recapitulate such developmental pressure, preventing definitive evidence confirming pathogenicity for the MCM3 variant. Further investigation testing the preRC interactions as well as studying this variant using an animal model such as zebrafish [6, 35, 36], could well provide this level of evidence.
Most interestingly, we confirmed biallelic pathogenic variants in MCM7 in two patients who presented with strikingly different phenotypes. P2 has clinically typical MGORS, aligning with phenotypes of patients with pathogenic variants in other members of the preRC. In contrast, P3 has progeria, lipodystrophy and adrenal insufficiency—all features confirmed absent in P2, but observed in other patients with pathogenic variants in genes encoding pre-initiation and replisome components [37]. Progeria and lipodystrophy are associated with de novo POLD1 variants (encoding the catalytic subunit of DNA polymerase-δ) in patients with either atypical Werner syndrome [38] or Mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) syndrome [39], and in fact, Werner syndrome was originally considered as a differential diagnosis in this child. In P3, while the progeria was apparent at birth, it has become less obvious over time. Adrenal insufficiency is associated with variants in MCM4, GINS1, or POLE, with hypogonadism (which can be linked to abnormal adrenal functioning) observed in patients with POLA1 or POLD1 variants [14,15,16, 19, 21, 24, 39]. While many of the preIC or replisome components are also associated with immunodeficiency, especially in NK cells, neither P2 nor P3 had any clinical indicators of immunodeficiency, except for the adrenal insufficiency in P3.
Patients P2 and P3 each inherited a null allele in trans with different missense variants, which affects amino acids in different domains of the MCM7 protein (Fig. 1A). Gly259 (P2: MGORS) lies in the N-terminal domain of MCM7. A double hexamer of MCM7 is loaded in a step-wise fashion to chromatin, with the single hexamers binding in a head-to-head fashion through their N-terminal domains[40]. This N-terminal region of Mcm7 mediates a head-to-head interaction with Mcm5 [40], using the zinc finger subdomain which Gly259 is in close proximity to. The reduced affinity of the MCM3-6 complex observed in our co-immunoprecipitation could reflect a reduced ability to form double heterohexamers through these N-terminal interactions. In contrast, Tyr539 (P3) lies near the surface of the AAA+ domain, which through interaction with Mcm3, is required for ATP binding [34]. Our co-immunoprecipitation assay demonstrated a reduced affinity for MCM3, via measuring MCM5 as a proxy for this interaction. While Mcm2-7 ATPase activity is required for complex loading to chromatin [41, 42], Mcm7 AAA+ domain mutants do not show significant loading defect but instead cause significant effects on helicase activation [41], suggesting the impact of this patient variant might be at activation/unwinding stages of DNA replication, rather than preRC loading. This hypothesis, while speculative, aligns with the phenotype observed in P3 and linked to other members of the pre-initiation complex and the replisome [37]. In contrast to these proposed links between variant and phenotype, the missense variant identified in MCM5 in a clinically typical case of MGORS [10] is not so straightforward to correlate. This variant lies within the highly conserved pre-sensor I hairpin region, which is required for MCM2-7 complex activity and translocation of DNA [10, 40], in contrast to other MGORS-associated proteins having licensing roles. This example illustrates the complexity of the action of the MCM2-7 complex within the cell, and that further biochemical studies of these specific substitutions are required to further delineate the specific disruption to the different steps of DNA replication initiation, ranging from chromatin loading to CMG helicase activation and replication fork progression. Additional patients would also help strengthen such putative genotype–phenotype insights.
While reduced growth is an almost universally observed association, additional features in MGORS are related to anomalies in cartilagenesis or skeletogenesis, which could be linked to recently described cellular interactions between SHH or HOX genes and DNA replication initiation factors [43, 44]. Conversely, multiple members of the preIC and the replisome are involved in various aspects of immune system development and adrenal functioning [37], for which the developmental pathophysiology remains unclear. Together, human genetics studies have revealed different developmental requirements for specific proteins or aspects of this fundamental pathway; future studies focusing on teasing apart these finer details will add further knowledge to the role of DNA replication initiation in organism development. Our novel variants also serve to illustrate the broadening phenotype spectrum that is becoming apparent across Mendelian disorders, as exome and genome sequencing becomes routinely utilized and more clinically atypical variant-positive individuals are identified.
References
Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, et al. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci USA. 2009;106:20240–5.
Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719–30.
Duzdevich D, Warner MD, Ticau S, Ivica NA, Bell SP, Greene EC. The dynamics of eukaryotic replication initiation: origin specificity, licensing, and firing at the single-molecule level. Mol Cell. 2015;58:483–94.
Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, et al. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146:931–41.
Bicknell LS, Bongers EM, Leitch A, Brown S, Schoots J, Harley ME, et al. Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43:356–9.
Bicknell LS, Walker S, Klingseisen A, Stiff T, Leitch A, Kerzendorfer C, et al. Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet. 2011;43:350–5.
Burrage LC, Charng WL, Eldomery MK, Willer JR, Davis EE, Lugtenberg D, et al. De novo GMNN mutations cause autosomal-dominant Primordial Dwarfism associated with Meier-Gorlin syndrome. Am J Hum Genet. 2015;97:904–13.
Fenwick AL, Kliszczak M, Cooper F, Murray J, Sanchez-Pulido L, Twigg SR, et al. Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause Meier-Gorlin syndrome and craniosynostosis. Am J Hum Genet. 2016;99:125–38.
Knapp KM, Sullivan R, Murray J, Gimenez G, Arn P, D’Souza P, et al. Linked-read genome sequencing identifies biallelic pathogenic variants in DONSON as a novel cause of Meier-Gorlin syndrome. J Med Genet. 2020;57:195–202.
Vetro A, Savasta S, Russo Raucci A, Cerqua C, Sartori G, Limongelli I, et al. MCM5: a new actor in the link between DNA replication and Meier-Gorlin syndrome. Eur J Hum Genet. 2017;25:646–50.
Reynolds JJ, Bicknell LS, Carroll P, Higgs MR, Shaheen R, Murray JE, et al. Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat Genet. 2017;49:537–49.
de Munnik SA, Otten BJ, Schoots J, Bicknell LS, Aftimos S, Al-Aama JY, et al. Meier-Gorlin syndrome: growth and secondary sexual development of a microcephalic primordial dwarfism disorder. Am J Med Genet A. 2012;158A:2733–42.
de Munnik SA, Hoefsloot EH, Roukema J, Schoots J, Knoers NV, Brunner HG, et al. Meier-Gorlin syndrome. Orphanet J Rare Dis. 2015;10:114.
Casey JP, Nobbs M, McGettigan P, Lynch S, Ennis S. Recessive mutations in MCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J Med Genet. 2012;49:242–5.
Hughes CR, Guasti L, Meimaridou E, Chuang CH, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest. 2012;122:814–20.
Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. 2012;122:821–32.
Wang X, Ishimi Y. Function of the amino-terminal region of human MCM4 in helicase activity. J Biochem. 2018;164:449–60.
Sheu YJ, Stillman B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol Cell. 2006;24:101–13.
Cottineau J, Kottemann MC, Lach FP, Kang YH, Vely F, Deenick EK, et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J Clin Invest. 2017;127:1991–2006.
Frugoni F, Dobbs K, Felgentreff K, Aldhekri H, Al Saud BK, Arnaout R, et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J Allergy Clin Immunol. 2016;137:635–8 e1.
Logan CV, Murray JE, Parry DA, Robertson A, Bellelli R, Tarnauskaite Z, et al. DNA polymerase Epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet. 2018;103:1038–44.
Mace EM, Paust S, Conte MI, Baxley RM, Schmit MM, Patil SL, et al. Human NK cell deficiency as a result of biallelic mutations in MCM10. J Clin Invest. 2020;130:5272–86.
Pachlopnik Schmid J, Lemoine R, Nehme N, Cormier-Daire V, Revy P, Debeurme F, et al. Polymerase epsilon1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J Exp Med. 2012;209:2323–30.
Starokadomskyy P, Wilton KM, Krzewski K, Lopez A, Sifuentes-Dominguez L, Overlee B, et al. NK cell defects in X-linked pigmentary reticulate disorder. JCI Insight. 2019;4:21.
Thiffault I, Saunders C, Jenkins J, Raje N, Canty K, Sharma M, et al. A patient with polymerase E1 deficiency (POLE1): clinical features and overlap with DNA breakage/instability syndromes. BMC Med Genet. 2015;16:31.
Kortum F, Caputo V, Bauer CK, Stella L, Ciolfi A, Alawi M, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet. 2015;47:661–7.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303.
Yuan Z, Riera A, Bai L, Sun J, Nandi S, Spanos C, et al. Structural basis of Mcm2-7 replicative helicase loading by ORC-Cdc6 and Cdt1. Nat Struct Mol Biol. 2017;24:316–24.
Tocilj A, On KF, Yuan Z, Sun J, Elkayam E, Li H, et al. Structure of the active form of human origin recognition complex and its ATPase motor module. Elife. 2017;6:e20818.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Schwacha A, Bell SP. Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol Cell. 2001;8:1093–104.
Lee JK, Hurwitz J. Isolation and characterization of various complexes of the minichromosome maintenance proteins of Schizosaccharomyces pombe. J Biol Chem. 2000;275:18871–8.
Sherman DA, Forsburg SL. Schizosaccharomyces pombe Mcm3p, an essential nuclear protein, associates tightly with Nda4p (Mcm5p). Nucleic Acids Res. 1998;26:3955–60.
Frigola J, Remus D, Mehanna A, Diffley JF. ATPase-dependent quality control of DNA replication origin licensing. Nature. 2013;495:339–43.
Ryu S, Holzschuh J, Erhardt S, Ettl AK, Driever W. Depletion of minichromosome maintenance protein 5 in the zebrafish retina causes cell-cycle defect and apoptosis. Proc Natl Acad Sci USA. 2005;102:18467–72.
Yao L, Chen J, Wu X, Jia S, Meng A. Zebrafish cdc6 hypomorphic mutation causes Meier-Gorlin syndrome-like phenotype. Hum Mol Genet. 2017;26:4168–80.
Bellelli R, Boulton SJ. Spotlight on the replisome: aetiology of DNA replication-associated genetic diseases. Trends Genet. 2020. https://doi.org/10.1016/j.tig.2020.09.008.
Lessel D, Hisama FM, Szakszon K, Saha B, Sanjuanelo AB, Salbert BA, et al. POLD1 germline mutations in patients initially diagnosed with Werner syndrome. Hum Mutat. 2015;36:1070–9.
Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45:947–50.
Li N, Zhai Y, Zhang Y, Li W, Yang M, Lei J, et al. Structure of the eukaryotic MCM complex at 3.8 A. Nature. 2015;524:186–91.
Kang S, Warner MD, Bell SP. Multiple functions for Mcm2-7 ATPase motifs during replication initiation. Mol Cell. 2014;55:655–65.
Coster G, Frigola J, Beuron F, Morris EP, Diffley JF. Origin licensing requires ATP binding and hydrolysis by the MCM replicative helicase. Mol Cell. 2014;55:666–77.
Lewis EMA, Sankar S, Tong C, Patterson ES, Waller LE, Gontarz P, et al. Geminin is required for Hox gene regulation to pattern the developing limb. Dev Biol. 2020;464:11–23.
Tamayo-Orrego L, Gallo D, Racicot F, Bemmo A, Mohan S, Ho B, et al. Sonic hedgehog accelerates DNA replication to cause replication stress promoting cancer initiation in medulloblastoma. Nat Cancer. 2020;1:840–54.
Cole TJ. The LMS method for constructing normalized growth standards. Eur J Clin Nutr. 1990;44:45–60.
Acknowledgements
The authors thank the families for their involvement in this study. Thank you to Katie Young for assistance with flow cytometry and Anna Pawluchin for assistance with Sanger sequencing.
Funding
KK is supported by the Deutsche Forschungsgemeinschaft (KU 1240/6-2 and KU 1240/10-1). This research, KMK and LSB are supported by the Marsden Fund; LSB is supported by a Rutherford Discovery Fellowship, both administered by the Royal Society of New Zealand. DEJ is supported by the Neurological Foundation of New Zealand and RS is supported by the Health Research Council of New Zealand; both awarded to LSB.
Sources of support
Deutsche Forschungsgemeinschaft (KU 1240/6-2 and KU 1240/10-1). Marsden Fund; Rutherford Discovery Fellowship, both administered by the Royal Society of New Zealand; Neurological Foundation of New Zealand; Health Research Council of New Zealand.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Knapp, K.M., Jenkins, D.E., Sullivan, R. et al. MCM complex members MCM3 and MCM7 are associated with a phenotypic spectrum from Meier-Gorlin syndrome to lipodystrophy and adrenal insufficiency. Eur J Hum Genet 29, 1110–1120 (2021). https://doi.org/10.1038/s41431-021-00839-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00839-4
This article is cited by
-
The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome
European Journal of Human Genetics (2023)
-
Comment on: “The expanding genetic and clinical landscape associated with Meier-Gorlin syndrome” by Nielsen-Dandoroff et al.
European Journal of Human Genetics (2023)
-
Unwinding the Role of the CMG Helicase in Inborn Errors of Immunity
Journal of Clinical Immunology (2023)
-
De novo MCM6 variants in neurodevelopmental disorders: a recognizable phenotype related to zinc binding residues
Human Genetics (2023)
-
Clinical genetics—it’s polygenic
European Journal of Human Genetics (2021)
