
Abstract
X-linked myotubular myopathy (XLMTM) is a severe congenital myopathy characterised by generalised weakness and respiratory insufficiency. XLMTM is associated with pathogenic variants in MTM1; a gene encoding the lipid phosphatase myotubularin. Whole genome sequencing (WGS) of an exome-negative male proband with severe hypotonia, respiratory insufficiency and centralised nuclei on muscle biopsy identified a deep intronic MTM1 variant NG_008199.1(NM_000252.2):c.1468-577A>G, which strengthened a cryptic 5′ splice site (A>G substitution at the +5 position). Muscle RNA sequencing was non-diagnostic due to low read depth. Reverse transcription PCR (RT-PCR) of muscle RNA confirmed the c.1468-577A>G variant activates inclusion of a pseudo-exon encoding a premature stop codon into all detected MTM1 transcripts. Western blot analysis establishes deficiency of myotubularin protein, consistent with the severe XLMTM phenotype. We expand the genotypic spectrum of XLMTM and highlight benefits of screening non-coding regions of MTM1 in male probands with phenotypically concordant XLMTM who remain undiagnosed following exome sequencing.
Similar content being viewed by others


Introduction
X-linked myotubular myopathy (XLMTM) is a severe congenital centronuclear myopathy with an estimated incidence of 1/50,000 male births [1]. Affected males present neonatally with generalised hypotonia and profound respiratory difficulties, often leading to death within the first year of life [1]. XLMTM is caused by mutations in the MTM1 gene which encodes myotubularin, a phosphoinositide phosphatase thought to be involved in sarcoplasmic reticulum remodelling [1]. Characteristic features of XLMTM in muscle histopathology include small rounded muscle fibres with centrally located nuclei which may be surrounded by a region lacking in myofibrils. Oxidative enzyme staining can show mitochondria grouped towards the centre of the fibre with an almost colourless peripheral halo devoid of mitochondria [2].
Causative MTM1 variants identified in XLMTM cases are typically detected by exomic sequencing approaches; next-generation sequencing of the exome or candidate panel of genes, or Sanger sequencing of MTM1 exons [1, 3, 4]. While these approaches cover intronic sequences directly flanking each exon, they fail to detect deep intronic variants. Pathogenic deep intronic variants are becoming increasingly recognised as a molecular basis for disease [5], including for MTM1 [6]. Most of these intronic variants result in the inclusion of pseudo-exons into mRNA transcripts by activating cryptic splice sites or splice enhancers, further than 100 base pairs (bp) into the intron [5].
Here, we report a novel deep intronic MTM1 variant NG_008199.1(NM_000252.2):c.1468-577A>G identified by whole genome sequencing (WGS) in an individual with severe hypotonia, no respiratory effort at birth and centralised nuclei on muscle biopsy.
Materials and methods
This study was approved by the Children’s Hospital at Westmead Human Research Ethics Committee (Biospecimen Bank_10/CHW/45) with informed, written consent. Whole exome sequencing (WES), PCR-free WGS and muscle RNA-seq was performed and analysed at the Broad Institute of Harvard and MIT as previously described [7]. In silico prediction algorithms available in Alamut® Visual v2.9.0 (Interactive Biosoftware, Rouen, France) were used for splicing predictions. RNA was isolated from muscle cryosections and RT-PCR performed as previously described [8]. RT-PCR primers used; MTM1-Ex11F 5′-CATCCTTGGCCATGCTGATG-3′ with MTM1-Ex13/14R 5′-AGTCCTTTCTGTAACCTTCTGTC-3′, MTM1-Ex14R 5′-AAGTGACGCATACTGGCAAC-3′ with MTM1-Ex12/13F 5′-TGTCAAAACAGTTCCCTACAGC-3′ or MTM1-Ex13/In13F 5′-TGCTCGAGAAAGACAGTTTATGG-3′, and GAPDH-Ex3F 5′-TCACCAGGGCTGCTTTTAAC-3′ with GAPDH-Ex6R 5′-GGCAGAGATGATGACCCTTT-3′. Cycling conditions were 95 °C for 120 s, 25, 30 or 35 cycles 95 °C 30 s, 59 °C 30 s, 72 °C 60 s, then 72 °C 8 min. Western blotting was performed as previously described [9]. Antibodies used were against myotubularin (PA5-17972, ThermoFisher Scientific) and GAPDH (MAB374, Merck Millipore). The c.1468-577A>G MTM1 variant was submitted to the Leiden Open Variation Database (individual: 00303956, DB-ID: MTM1_000334, https://databases.lovd.nl/shared/individuals/00303956). Exons are numbered as per NG_008199.1.
Results
Clinical history
The proband (II:3) was the third male child born to non-consanguineous parents of European descent with no relevant family history of a neuromuscular disorder (Fig. 1a). The pregnancy was complicated by polyhydramnios at 32 weeks and vaginal delivery was induced at 38 weeks gestation. II:3 presented at birth with severe central and peripheral hypotonia, scaphocephaly, myopathic facies, congenital ptosis, severe limitation of lateral eye movements, severe facial weakness (unable to close eyelids during sleep), undescended testes, a small chest and thin ribs, mild hip dysplasia, and no respiratory effort. He required immediate resuscitation and ventilatory support. Brain magnetic resonance imaging on day 5 of life was normal.

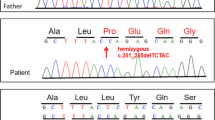
a Family pedigree for II:3. b Histopathology of muscle biopsies from II:3 (i) Haemotoxylin and Eosin (H&E) staining at 8½ weeks of age showed multiple fibres with central nuclei (×400 actual magnification), (ii) succinate dehydrogenase (SDH) staining at 8½ weeks of age showed some fibres with reduced activity at the fibre centres and others with pale sub-sarcolemmal halos (×400 actual magnification) and (iii) H&E staining at 11 years of age showed extensive fat replacement of spinal muscles (×100 actual magnification). c Schematic of MTM1 exons 11–15 showing the c.1468-577A>G variant strengthens a naturally occurring cryptic 5′ splice site to activate spliceosomal inclusion of an intron 13 pseudo-exon encoding a TAA stop codon. The boxed region denotes the pseudo-exon, flanked by cryptic 3′ and 5′ splice sites. Splicing prediction scores from the splicing module available in Alamut® Visual v2.9.0: SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder are annotated showing strengthening of the cryptic 5′ splice site by the de novo c.1468-577A>G variant, which substitutes a less favoured A (WT) at the +5 position for a highly favoured G (in II:3).
The proband had severe to profound muscle weakness from birth, with only very limited antigravity movements in upper and lower limbs, and subsequently developed multiple limb contractures. There was a slow but definite improvement in limb muscle strength during the first 2 years of life but no improvement in respiratory muscle strength, and he has minimal distal antigravity movement, with no ability to sit or roll. A tracheostomy was inserted at 4 months of age to facilitate long term ventilation. II.3 suffered recurrent left lower lobe lung collapse during the first 18 months of life. He had severe restrictive lung disease, with a forced vital capacity of 20% normal at 2½ years of age. Severe bulbar weakness necessitated insertion of a percutaneous endoscopic gastrostomy feeding tube at 10 weeks of age. An upper thoracic kyphosis was first noted on X-ray at 10 days of age, in the absence of vertebral segmentation anomalies. This progressed to a severe kyphoscoliosis which required surgical fixation at 12 years of age. The proband also developed mild to moderate conductive hearing loss requiring insertion of myringotomy tubes.
Muscle biopsy (site not stated) at 8½ weeks revealed an increase in centralised nuclei in small rounded fibres (average fibre diameter 9.8 µm, Fig. 1bi). The central aspect of the fibres showed reduced ATPase and oxidative enzyme studies revealed some fibres with increased activity centrally and others with a peripheral halo of increased activity (Fig. 1bii). Type 1 and 2 fibres were present with a slight predominance of type 1 fibres. Spinal muscle taken at the time of orthopaedic surgery at 11 years old, showed extensive fat replacement with only scant residual severely atrophic fibres only 7–30 µm in diameter, with one showing a central nucleus and others appearing as peripheral nuclei (Fig. 1biii).
Genetic investigations and findings
Sanger sequencing of the coding regions of MTM1 and WES analysis were both negative. Subsequent WGS and RNA-seq were initially negative (patient N27 in [7]). Careful reanalysis of the trio WGS data identified a de novo hemizygous single nucleotide variant in intron 13 of MTM1, ChrX(GRCh37):g.149831329A>G, NG_008199.1(NM_000252.2):c.1468-577A>G. In silico prediction algorithms predicted the c.1468-577A>G variant strengthens a potential cryptic 5′ splice site 48 nt downstream of a high-scoring potential cryptic 3′ splice site (Fig. 1c). The c.1468-577A>G variant was absent in the Genome Aggregation Database (gnomAD) [10].
RNA-seq, RT-PCR and western blot studies
Suspicion of a possible variant activated pseudo-exon (encoding a premature stop codon) induced by the c.1468-577A>G MTM1 variant led us to interrogate the available RNA-seq data. Despite acceptable quality scores for the RNA library [7], RNA-seq data for II:3 had a low mapping rate and slightly elevated duplicate read and ribosomal RNA rate. Manual examination of the MTM1 gene showed poor read depth, providing insufficient evidence for either normal splicing or mis-splicing (Fig. 2a). RT-PCR was therefore performed to further investigate potential mis-splicing.

a Sashimi plots of MTM1 in RNA sequencing data show II:3 (red) had a much lower read count for MTM1 region compared to a GTEx muscle RNA-seq control (blue). b RT-PCR of muscle derived RNA from II:3 (8½ weeks, muscle origin not documented on biopsy report) and three controls; C1 (4 months, muscle origin not documented), C2 (6½ years, quadriceps), C3 (20½ years, muscle origin not documented) using: (i) primers flanking MTM1 intron 13, which amplifies the pseudo-exon (298 bp band) in II:3 and normal splicing in the controls (250 bp band); (ii) primers specifically targeting normal MTM1 exon 13 to exon 14 splicing by bridging the exon 13/14 splice junction (329 bp band), only detectable in control samples; (iii) primers specifically targeting the MTM1 pseudo-exon in II:3 (189 bp band), absent in control samples; (iv) amplification of GAPDH demonstrate cDNA loading. Replicate samples were subject to PCR for 25 or 30 cycles in order to confirm the PCR cycling conditions were sub-saturating and able to detect lower levels or quality of a specimen. RT-PCRs for i–iii used 35 cycles. c Sanger sequencing of the excised bands for II:3 and C1 in b(i) shows the 48 bp inclusion of the variant activated pseudo-exon in II:3 and normal MTM1 exon 13 to exon 14 splicing in C1. d Western blotting of muscle lysates from II:3 and two male age-matched controls (C1, age 4 months, muscle origin not documented; and C2, age 4 months, muscle origin not documented) using an antibody directed against myotubularin and GAPDH (loading control). Western blot analysis revealed undetectable levels of myotubularin (65 kDa) in II:3 compared to a standard curve of the controls.
RT-PCR of muscle derived RNA from II:3 and three age and gender-matched controls using primer pairs flanking intron 13 showed an abnormal band of higher molecular weight in II:3 that was not detected in controls (Fig. 2bi). Sanger sequencing of the excised band revealed inclusion of a 48 nt pseudo-exon from intron 13 into MTM1 mRNA transcripts (r.1467_1468ins1468-629_1468-582, Fig. 2c). There was no evidence for normal exon 13–14 splicing when using a reverse primer bridging the exon 13–14 junction (Fig. 2bii), implicating the pseudo-exon is incorporated into all, or the vast majority, of MTM1 transcripts in II:3. A forward junctional primer spanning exon 13 and the pseudo-exon amplified a positive band only in II:3 (absent from controls), indicating that inclusion of the pseudo-exon is uniquely activated by the c.1468-577A>G variant and does not occur at detectable levels in controls (Fig. 2biii). There was no evidence in RNA sequencing in control GTEx muscle RNA [11] of pseudo-exon junctional reads (data not shown). A western blot performed on a skeletal muscle lysate from II:3 and two age-matched controls with an anti-myotubularin antibody revealed undetectable levels of myotubularin in II:3, and no evidence of a truncated product (Fig. 2d).
Discussion
The MTM1 c.1468-577A>G variant affects the +5 position of a natural cryptic 5′ splice site within intron 13, strengthening splice site strength through substitution of a less favoured A for preferred G [12] (Fig. 1c). Spliceosomal use of this strengthened cryptic 5′ splice site, in combination with a naturally occurring 3′ splice site, results in incorporation of a 48 nt pseudo-exon (r.1467_1468ins1468-629_1468-582) into all detectable MTM1 mRNA transcripts. The pseudo-exon encodes a premature stop codon (TAA) located >50 nt upstream of the last exon junction complex and is predicted to result in nonsense-mediated decay [13]. Western blot confirms absence of myotubularin in II:3 skeletal muscle lysates, consistent with nonsense-mediated decay of MTM1 transcripts and the severe XLMTM phenotype presented. Collective evidence from functional genomics investigations resulted in classification of c.1468-577A>G as a pathogenic variant.
A deep intronic MTM1 variant (c.529-909A>G) is previously described to activate inclusion of an intron 7 pseudo-exon containing a stop codon, into MTM1 transcripts [6]. The previous c.529-909A>G variant and the c.1468-577A>G variant described herein were both missed by initial sequencing of MTM1 exons, highlighting the utility of clinical phenotyping to target screening of non-coding regions within the most common and/or phenotypically concordant disease genes. Approximately 30% of centronuclear myopathy (CNM) cases remain undiagnosed after exomic screening of CNM-associated genes [2, 6, 14] and it is plausible that the underlying genetic cause of CNM in these cases are deep intronic variants of known CNM genes. Importantly, deep intronic pathogenic variants are ideal candidates for splice-switching antisense oligonucleotide therapies which have started entering clinical use [15].
RNA-seq is emerging as a powerful adjunct test in Mendelian disorders, providing a diagnosis for 7.5–36% of cases undiagnosed with previous genetic testing [7, 16,17,18]. Theoretically, RNA-seq should be capable of detecting the MTM1 pseudo-exon inclusion in II:3. However, low read depth across the entire transcriptome for II:3 compounded by low mRNA expression of MTM1 in muscle prevented the detection of this mis-splicing event. This reflects the diagnostic challenges of RNA-seq when analysing genes with low mRNA expression in the biologically relevant tissue, and mRNA samples with poor read depth.
Our work expands the genotypic spectrum of XLMTM and highlights the benefits of RNA and protein adjunct tests for hard-to-diagnose cases. For this case, and others among our neuromuscular cohort [7,8,9, 19,20,21], an available muscle biopsy was the cornerstone for a research-driven clinical diagnosis. Importantly, conventional RT-PCR provides increased sensitivity over RNA-seq and the ability to amplify and sequence cDNA products spanning multiple exons provides strong diagnostic certainty of the mis-splicing event. In combination, these interdisciplinary techniques lead to the genetic diagnosis for II:3.
References
Longo G, Russo S, Novelli G, Sangiuolo F, D’Apice MR. Mutation spectrum of the MTM1 gene in XLMTM patients: 10 years of experience in prenatal and postnatal diagnosis. Clin Genet. 2016;89:93–8.
Romero NB. Centronuclear myopathies: a widening concept. Neuromuscul Disord. 2010;20:223–8.
Al-Hashim A, Gonorazky HD, Amburgey K, Das S, Dowling JJ. Clinical/Scientific notes: novel intronic mutation in MTM1 detected by RNA analysis in a case of xlinked myotubular myopathy. Neurol Genet. 2017;3:e182.
Johannsen J, Hempel M, Diehl T, Haack TB, Denecke J. Exome sequencing is a valuable approach in critically ill patients with suspected monogenic disease: Diagnosis of X-linked centronuclear myopathy in preterm twins. Pediatr Neonatol. 2017;58:458–9.
Vaz-Drago R, Custódio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet. 2017;136:1093–1111.
Tosch V, Vasli N, Kretz C, Nicot AS, Gasnier C, Dondaine N, et al. Novel molecular diagnostic approaches for X-linked centronuclear (myotubular) myopathy reveal intronic mutations. Neuromuscul Disord. 2010;20:375–81.
Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9:eaal5209.
Bryen SJ, Ewans L, Pinner J, MacLennan SC, Donkervoort S, Castro D, et al. Recurrent TTN metatranscript‐only c.39974‐11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. 2019. https://doi.org/10.1002/humu.23938.
Bryen SJ, Joshi H, Evesson FJ, Girard C, Ghaoui R, Waddell LB, et al. Pathogenic abnormal splicing due to intronic deletions that induce biophysical space constraint for spliceosome assembly. Am J Hum Genet. 2019;105:573–87.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019. Accessed 19 Feb 2020. https://www.biorxiv.org/content/10.1101/531210v3.
GTEx Consortium TGte. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–5.
Zhang S, Samocha KE, Rivas MA, Karczewski KJ, Daly E, Schmandt B, et al. Base-specific mutational intolerance near splice sites clarifies the role of nonessential splice nucleotides. Genome Res. 2018;28:968–74.
Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–9.
Fattori F, Maggi L, Bruno C, Cassandrini D, Codemo V, Catteruccia M, et al. Centronuclear myopathies: genotype–phenotype correlation and frequency of defined genetic forms in an Italian cohort. J Neurol. 2015;262:1728–40.
Wai H, Douglas AGL, Baralle D. RNA splicing analysis in genomic medicine. Int J Biochem Cell Biol. 2019;108:61–71.
Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824.
Gonorazky HD, Naumenko S, Ramani AK, Nelakuditi V, Mashouri P, Wang P, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am J Hum Genet. 2019;104:466–83.
Frésard L, Smail C, Ferraro NM, Teran NA, Li X, Smith KS, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med Nat Publ Group. 2019;25:911–9.
Sandaradura SA, Bournazos A, Mallawaarachchi A, Cummings BB, Waddell LB, Jones KJ, et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat. 2018;39:383–8.
Jones HF, Bryen SJ, Waddell LB, Bournazos A, Davis M, Farrar MA, et al. Importance of muscle biopsy to establish pathogenicity of DMD missense and splice variants. Neuromuscul Disord. 2019;29:913–9.
Riley LG, Waddell LB, Ghaoui R, Evesson FJ, Cummings BB, Bryen SJ, et al. Recessive DES cardio/myopathy without myofibrillar aggregates: intronic splice variant silences one allele leaving only missense L190P-desmin. Eur J Hum Genet. 2019;27:1267–73.
Acknowledgements
We thank the families for their invaluable contributions to this research, and the clinicians and health care workers involved in their assessment and management.
Funding
This study was supported by the National Health and Medical Research Council of Australia (APP1048816 and APP1136197 STC, APP1080587 STC, DGM, GNT1090428 ECO). SJB is supported by a Muscular Dystrophy New South Wales PhD scholarship. WES, WGS and RNA-seq was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 to DGM and Heidi Rehm. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health (https://commonfund.nih.gov/GTEx), and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS.
Author information
Authors and Affiliations
Contributions
SJB: performed RNA studies, prepared manuscript and figures. ECO: clinical data and management of family, manuscript editing. FJE: western blot analysis, preparation of Fig. 2. JKL: analysis of genomic data, preparation of figures. LBW: ethics, analysis of genomic data, manuscript editing. HJ: analysis of RNA sequencing data. MMR: clinical data and management of family, manuscript editing. BBC: analysis of RNA sequencing data, manuscript editing. CAM: performed histological analysis, manuscript editing. DGM: broad collaboration, WES, WGS, RNA-seq. AJK: clinical data and management of family. STC: oversight of analysis and experimentation, supervision of research personnel, analysis, manuscript editing.
Corresponding author
Ethics declarations
Conflict of interest
STC is director of Frontier Genomics Pty Ltd (Australia). Frontier Genomics has not traded (as of March 2020). Frontier Genomics Pty Ltd (Australia) has no existing financial relationships that will benefit from publication of these data. The remaining co-authors do not have any relationships, financial or otherwise, that may result in a perceived conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bryen, S.J., Oates, E.C., Evesson, F.J. et al. Pathogenic deep intronic MTM1 variant activates a pseudo-exon encoding a nonsense codon resulting in severe X-linked myotubular myopathy. Eur J Hum Genet 29, 61–66 (2021). https://doi.org/10.1038/s41431-020-00715-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00715-7
