
Abstract
Electrochemical reduction of CO2 into value-added chemicals has been envisioned as a promising strategy to alleviate the issue of increasing CO2 emissions. However, the sluggish oxygen evolution reaction (OER), as the anodic reaction, typically consumes approximately 90% of the electricity input, necessitating the development of an efficient OER for energy-saving purposes. Herein, we developed a unique heterostructure of multi-double (bi)-shelled Co-based spheres via a facile template-free method, in which each bi-shelled structure is composed of Co9Se8/Co9S8/CoO (Co-S-Se) with a symmetric configuration. These heterogeneous nanospheres possess both sufficient heterointerfaces and a high density of active sites and exhibit excellent OER activity in alkaline media with a low overpotential of 226 mV at 10 mA cm−2, a small Tafel slope of 46.5 mV dec−1, and long-term durability over 15 h. As a proof and concept, when coupled with a cathodic CO2 reduction reaction, the electrochemical performance of Pd nanosheets (NSs) for CO2 reduction can be significantly enhanced in terms of product selectivity and energy input. Our study might provide insight into the development of efficient OER electrocatalysts for practical CO2 reduction reactions.
Similar content being viewed by others

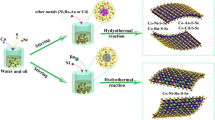

Introduction
The rising atmospheric CO2 concentration and subsequent global warming are widespread concerns that have motivated numerous studies focusing on the capture, utilization, and storage of CO2 (CCUS)1,2. Considering the potentially accidental release of CO2 from storage, the efficient conversion of CO2 into valuable chemicals has recently been considered a reliable and cost-effective method3. Electrochemical CO2 reduction (ECR) uses water as the hydrogen source instead of costly H2 gas and thus has attracted much attention4,5. Two reactions are generally involved in the ECR system: the cathodic CO2 reduction and anodic oxygen evolution reaction (OER). Cathodic CO2 reduction is an efficient process in which electrons are successively transferred to CO2 or CO2-based ions, resulting in the production of diverse products (e.g., CO, CH4, C2H4, C2H5OH)6,7,8. In contrast, the anodic OER involving four sequential proton-coupled electron steps and oxygen–oxygen bond formation is kinetically sluggish, leading to a large overpotential and high energy input (~90% of the overall electricity input for OERs during the ECR process)9,10,11. Therefore, considering the total energy consumption, enhancing OER activity is highly desirable for the development of an energy-saving ECR process. Although some alternative reactions with lower potentials (e.g., the ethanol oxidation reaction, glycerol oxidation) have been reported12, they have not been well studied relative to the OER. In addition, the hydrogen evolution reaction (HER) at the cathodic electrode is a highly competitive reaction that must be inhibited to improve the faradaic efficiency (FE) of ECR. Compared to the generally high performance in acidic media, the kinetics of the HER become rather sluggish in alkaline media. Therefore, it is reasonable to develop advanced OER catalysts that work efficiently in alkaline media, which can be beneficial for promoting the ECR process as well as reducing the overall energy input13.
Currently, nanostructured iridium oxides (IrO2) and ruthenium oxides (RuO2) represent state-of-the-art OER catalysts. Nevertheless, they are limited in scalable utilization due to their scarcity and high cost14. To expand the scope of nonprecious OER catalysts, intensive efforts have been devoted to the development of low-cost 3d transition metals (Ni, Co, Fe, and Mn) or their compounds15,16,17,18,19,20. Among these catalysts, cobalt-based composites have shown potential as low-cost active catalysts for the OER with general overpotentials of 350–450 mV to deliver a current density of 10 mA cm−2 21,22,23,24. To further improve their OER activity, the chemisorption and dissociation of OH− and the intermediates (OH*, OOH*, and H+) on the catalyst surface need to be optimized by tailoring the surface electronic structures25,26. Recently, the essential role of the interfacial effect has been recognized in several catalytic processes27,28. For example, the catalytic activity of the Co9S8/MoS2 composite was demonstrated to be directly proportional to the contact surface area between the metal sulfides Co9S8 and MoS229,30. Oxygen-incorporated cobalt sulfides (CoS4.6O0.6) also showed enhanced O* adsorption and thus superior OER performance relative to the pure cobalt sulfides, benefiting from the reconfigured electron interactions between those components31. Nevertheless, challenges remain regarding the elegant design of cobalt-based heterostructures, of which the surface electronic structure can be regulated to modulate the electrocatalytic properties.
Herein, we propose a facile template-free strategy to develop multi-bishelled spheres with sufficient heterointerfaces and large surface areas. Each bi-shelled structure was generated by the slow split of the Co3O4 shell through the well-controlled Kirkendall effect during the sulfurization process, followed by further deposition of ultrafine Co9Se8 nanoparticles. Therefore, abundant heterointerfaces were formed between those ternary components (CoO, Co9S8, and Co9Se8) in the hollow hybrid structures. As supported by the density functional theory (DFT) results, the strong interactions between the Co-based components synergistically lower the chemisorption energy barriers of the oxygen-containing intermediates and thus effectively promote the catalytic reactions. Their abundant exposed active sites can also maximize the interaction between the catalyst and electrolyte. Benefiting from these favorable features, Co-S-Se spheres served as an efficient OER catalyst for promoting the ECR process catalyzed by Pd NSs.
Materials and methods
Synthesis of the multi-shelled Co3O4 spheres
Typically, 5.0 mmol of Co(NO3)2·6H2O was dissolved into 100 ml of deionized (DI) water, and the mixture was stirred at 80 °C until complete dissolution. Then, 5.0 mL of acac was added, followed by the slow addition of 5.0 mL of N2H4·H2O. After aging for 1 h, a purple product was formed and washed with DI water three times. Then, it was dried in air at 70 °C for 12 h. Subsequently, the as-obtained purple product was annealed at 450 °C for 1 h in air at a heating rate of 1 °C min−1.
Synthesis of the multi-bishelled CoO/Co9S8 spheres
First, 0.144 g of the as-prepared Co3O4 and 0.23 g of Na2S were added into 40 mL of DI water, and the mixture was stirred for 30 min before being transferred to a 50 mL Teflon-lined stainless steel autoclave. Then, the autoclave was put into an electric oven at 90 °C for 30 h. After cooling to room temperature, the product was washed with DI water and dried at 50 °C in a vacuum oven for 12 h. Finally, the as-obtained product was annealed at 400 °C for 1 h under an argon atmosphere.
Synthesis of the multi-bishelled Co-S-Se spheres
The as-obtained multi-bishelled CoO/Co9S8 spheres and Se powder were loaded in two separate quartz boats with a mass ratio of 1:1. Selenium powder was placed upstream of the furnace, and the CoO/Co9S8 spheres were placed downstream at a distance of 5.0 cm to the selenium powder. Then, the furnace was programmed with a heating rate of 2 °C and kept at 400 °C for 1 h under an argon atmosphere. Then, the product was collected after naturally cooling to room temperature.
Synthesis of Pd nanosheets
Pd NSs were prepared based on a previously reported protocol with modification32. In a typical synthesis, 10 mL of N,N-dimethylformamide (DMF) containing 16 mg of Pd (acac)2, 170 mg of citric acid (CA), 60 mg of cetrimonium bromide (CTAB), and 30 mg of poly(vinyl pyrrolidone) (PVP) were put into a 100-mL three-neck flask with magnetic stirring at room temperature for 1 h. Then, 100 mg of W(CO)6 was carefully added into the solution, followed by purging with argon for 15 min. After that, the well-sealed flask was placed into a preheated oil bath at a temperature of 64 °C for 1 h without stirring. The product was collected by centrifugation, washed twice with a mixture of ethanol and acetone (v/v: 1/4), and then dispersed in ethanol for further use.
Characterization methods
SEM images were obtained using field-emission scanning electron microscopy (FE-SEM, JEOL JSM-7600F). X-ray diffraction (XRD) measurements were performed on a Shimadzu XRD-600 with Cu Kα radiation (λ = 0.15404 nm) operated at 40 kV and 30 mA. TEM images were obtained using a JEOL-2100F at an acceleration voltage of 200 kV. A surface area and porosimetry distribution system (ASAP 3020) was used to survey the nitrogen adsorption/desorption isotherms at −196 °C, and all samples were degassed at 120 °C for 8 h under vacuum before testing.
Electrochemical measurements
Electrochemical measurements were performed in a standard three-electrode system using a CHI 760E potentiostat (CH Instruments). Ink was prepared with 5 mg of active materials dispersed in 1 mL of ethanol and 20 μL of Nafion (5%). A mass loading of 1.0 mg cm−2 on a glassy carbon electrode (5 mm in diameter) was used for each catalyst, above which the overpotential at a current density of 10 mA cm−2 (η10) no longer changed. The electrocatalytic OER properties were investigated in a typical three-electrode configuration in O2-saturated 1 M KOH solution with Hg/HgO and a graphite rod as the reference electrodes (REs) and the counter electrodes, respectively. Linear sweep voltammetry (LSV) was conducted at a scan rate of 2 mV s−1. The Hg/HgO electrode was calibrated in a three-electrode system, in which Pt wires worked as both the working electrode and counter electrode, and Hg/HgO served as the reference electrode. O2-saturated 1.0 M KOH was employed as the electrolyte. LSV was then conducted in the range of −0.8 to −1.0 V at a scan rate of 0.5 mV s−1. The potential at which the current crossed zero was taken as the thermodynamic potential (vs. Hg/HgO) for the hydrogen electrode. Therefore, all the potentials were calibrated to a reversible hydrogen electrode (RHE) by using the equation ERHE = EHg/HgO + 0.917 with overpotential η = ERHE − 1.23 V.
Full-cell electrolysis was conducted in an H-type cell separated by a Nafion 117 membrane. A glassy carbon plate with a size of 1.0 cm × 1.0 cm was used for loading Co-S-Se and IrO2/C as electrocatalysts for anodic OER, and an L-shaped glassy carbon electrode with a diameter of 1.0 cm was used for loading the cathodic catalysts. The Pd NSs were loaded on carbon black (Vulcan XC-72) through ultrasonication in an ice bath with a weight ratio of 4:1 (Pd:C), which were then collected by centrifugation and redispersed in ethanol for further testing. The cathodic electrolyte was CO2-saturated KHCO3 (0.1 M, 40 mL), and the anodic electrolyte was an O2-saturated KOH solution (1.0 M, 40 mL). The cathode was used as the working electrode, and the anode was used as the counter and reference electrode to measure the cell voltage. The gaseous products from the cathodic compartment were monitored by online gas chromatography (GC).
Results and discussion
The rational synthesis of the multi-bishelled Co-S-Se spheres proceeded through three steps, as schematically illustrated in Fig. 1. First, uniform multi-shelled Co3O4 hollow spheres were prepared via calcination of the cobalt complex spheres at an optimized heating rate according to our previous report33. Subsequently, the as-obtained Co3O4 spheres were chemically transformed into multi-bishelled CoO/Co9S8 spheres by a mild sulfurization process. During this process, each shell of the Co3O4 hollow spheres was divided into a bi-shelled structure with a symmetric configuration. Finally, ultrafine Co9Se8 nanoparticles were further deposited on the outside surface of each bi-shell structure using Se powder as the selenization source. Before the sulfurization and selenization processes, the X-ray diffraction (XRD) peaks (Fig. S1a) were indexed to Co3O4 (JCPDS card no. 74–1657) with negligible detectable impurities. The scanning electron microscopy (SEM) image (Fig. 2a) revealed their uniform spherical morphology with a porous surface (marked by the arrows), indicating their hollow structure. The transmission electron microscopy (TEM) image (Fig. 2b) further presented a multi-shelled structure with a shell thickness of ~20 nm. The lattice fringes with a spacing of 0.466 nm (Fig. S1b) are consistent with the (111) planes of cubic Co3O4, revealing its good crystallinity.

(1) Multi-shelled Co3O4 spheres were transformed into multi-bishelled CoO/Co9S8 spheres via a mild solution-based sulfurization process. (2) The multi-bishelled Co-S-Se spheres were further prepared by depositing ultrafine Co9Se8 nanoparticles on the CoO/Co9S8 spheres via a selenization process.

a SEM image of the multi-shelled Co3O4 spheres. The yellow arrows represent the porous surface. b TEM image of the multi-shelled Co3O4 spheres. The dashed circles represent the multi-shelled structure. c Dark field scanning and d, e bright field TEM images of the multi-bishelled CoO/Co9S8 spheres. f High-resolution TEM image of the multi-bishelled CoO/Co9S8 spheres.
After the sulfurization process, the XRD pattern (Figure S2) of the as-obtained product revealed the formation of CoO (JCPDS card no. 48–1719) and Co9S8 (JCPDS card no. 09–0233). The as-prepared sample is labeled CoO/Co9S8. The representative dark-field scanning TEM image (Fig. 2c) demonstrated that the uniform hollow sphere was well maintained with a diameter of ~400 nm. Furthermore, this image shows the presence of a bi-shelled structure, where each shell of the original Co3O4 was split into two. The typical bright-field TEM image (Fig. 2d) further shows the bi-shelled structure with clear contrast between the shells and gaps. The gap width of the bi-shelled structure was 10~15 nm (Fig. 2e). The d spacings of both d1 (0.286 nm) near the surface and d2 (0.213 nm) from the inner region are consistent with the (222) plane of Co9S8 and the (200) plane of CoO, respectively, indicating the partial transformation of cobalt oxide to cobalt sulfides. The transformation mainly relies on the controlled Kirkendall effect originating from the different diffusion rates of the reaction species, i.e., the faster outwardly diffused Co2+ cations and the slower inwardly diffused S2− ions. In general, the Kirkendall effect is used as a unique self-templated strategy to prepare hollow structures from solid nanocrystals33. It remains challenging to be further applied to hollow structures considering the ready collapse or aggregation of hollow structures. By controlling the reaction temperature (e.g., 90 °C in the aqueous system) and preparing porous hollow precursors that ensure the complete permeation of reaction species, we demonstrated the capability to engineer the volume-void ratio and build a unique multi-bishelled hollow structure with hybrid compositions in the confined geometry.
To further enrich the heterointerfaces and promote charge transfer, ultrafine metallic cobalt selenide nanoparticles were deposited on the surface of each bi-shell structure. The average diameter of ~400 nm was revealed by the typical dark-field TEM image (Fig. 3a). The bi-shelled structure was still observed, as marked by the double red dashed lines. The deposition of cobalt selenides was confirmed by the additional minor peaks located at 16.7°, 29.6°, and 34.3° in the XRD patterns (Fig. S2), corresponding to the standard spectrum of Co9Se8 (JCPDS card no. 74–1657). The average size (D) of Co9Se8 was calculated from the (200) plane at 2θ = 16.7° using Scherer’s equation as shown below:

a Dark field scanning TEM image. b Bright field TEM image. c High-resolution TEM image. d The corresponding scanned-transmission electron microscopy energy-dispersive X-ray spectrometry (STEM/EDS) elemental mapping images.
D = kλ/βcosθ, where constant k = 0.89, λ = 1.5406 Å, and β is the full width at half maximum (FWHM) of the (200) diffraction peak. The size was determined to be 4.3 ± 1.9 nm. The as-obtained product is labeled CoO/Co9S8/Co9Se8 (Co-S-Se). A gap width of ~10 nm was determined, as shown in the high-magnification TEM image (Fig. 3b). The (422) and (400) planes of Co9Se8 were observed at the surface, and the size of the deposited Co9Se8 nanoparticles was ~3 nm, which is in agreement with the calculated results from Scherer’s equation. The scanned-transmission electron microscopy energy-dispersive X-ray spectrometry (STEM/EDS) elemental mapping in Fig. 3d revealed the uniform distribution of Co, S, Se, and O elements, which is in good agreement with the observed multi-bishelled Co-S-Se architecture. Considering that Se vapor was used for the selenization process, X-ray photoelectron spectroscopy (XPS) measurements were conducted to explore whether Se remained in Co-S-Se. The peaks of Se located at 53.8 eV (Se 3d5/2) and 54.5 eV (Se 3d3/2) (Fig. S3a) were lower in energy than those of elemental Se34, revealing the dominant presence of Co-Se units. In addition, the weight ratios of CoO, Co9S8 and Co9Se8 in the Co-S-Se composite were determined by inductively coupled plasma (ICP) and elemental analysis to be 24.3%, 63.2%, and 12.5%, respectively.
To probe the favorable features of the multi-bishelled Co-S-Se spheres as OER catalysts, their electrocatalytic properties were first investigated in a typical three-electrode configuration in an O2-saturated 1.0 M KOH solution. Figure 4a shows the iR-corrected linear sweep voltammetry (LSV) curves for Co3O4, CoO/Co9S8, Co-S-Se, and commercial IrO2/C. The anode current increased as the potential became positive, corresponding to the electrocatalytic O2 evolution process. Co-S-Se showed a much smaller onset potential of 0.12 V (at a current density of 0.1 mA cm−2) compared to the other catalysts. Furthermore, it reached a current density of 10 mA cm−2 at an overpotential of 226 mV (η10, 1.456 V vs. RHE), which was lower than those of CoO/Co9S8 (300 mV; 1.53 V vs. RHE), Co3O4 (397 mV, 1.627 V vs. RHE), and IrO2/C (326 mV; 1.556 V vs. RHE). The derived plots of their overpotentials (η10 values) reaching a current density of 10 mA cm−2 are shown in Fig. 4b.

a Polarization curves. b Derived plots of their overpotentials (η10). c Tafel plots. d Chronopotentiometry measurements at a current density of 20 mA cm−2 over 20 h.
The OER electrocatalytic kinetics were further examined via Tafel plots by plotting the overpotential against log(J) (Fig. 4c). The Co-S-Se spheres show the lower Tafel slope of ~46.5 mV dec−1 with respect to Co3O4 (72.2 mV dec−1) and CoO/Co9S8 (63.4 mV dec−1), suggesting their favorable OER kinetics benefiting from both their compositional and structural features. Impressively, the outstanding OER activity of Co-S-Se is among the best Co-based or other transition-metal-based OER catalysts reported recently, as listed in Table S1. The stable operating potential (only 0.3% change) of Co-S-Se (Fig. 4d) was also observed during 20 h of testing at a constant current density of 20 mA cm−2, suggesting excellent long-term stability in alkaline solution. The structural stability of the Co-S-Se composite was also demonstrated, as shown in Fig. S4. Broken shells, as marked by arrows, further confirmed the remaining hollow structures.
In light of the important role of the active site number, the electrochemically effective surface area (ECSA) was evaluated via cyclic voltammetry (CV) at different scan rates (Fig. S5). By plotting the capacitive currents (janodic−jcathodic) at 0.35 V (vs. Hg/HgO) against various scan rates, the ECSA was represented by the linear slope (twice the electrochemical double-layer capacitance, Cdl). As shown in Fig. 5a, the Co-S-Se spheres exhibited a higher Cdl of 13.23 mF cm−2 compared to those of the multi-shelled Co3O4 (6.78 mF cm−2) and the multi-bishelled CoO/Co9S8 (10.21 mF cm−2). The high ECSA of the Co-S-Se spheres could be mainly attributed to the step-by-step nanostructure engineering on the multi-shelled Co3O4 and thus increased exposure of the active sites. Furthermore, as shown in Fig. 5b, even normalized with the increased ECSA of Co-S-Se spheres (1.29 times that of CoO/Co9S8 and 1.95 times that of Co3O4), the Co-S-Se spheres still show a much higher current density (47.8 mA cm−2) at an overpotential of 300 mV, which was 4.78 and 39.07 times that of CoO/Co9S8 (10.0 mA cm−2) and Co3O4 (1.29 mA cm−2), respectively. This result suggests that the electrochemical enhancement benefits from not only the increased active surface area but also the intrinsically favorable surface properties of the Co-S-Se spheres. In addition, their mass and specific activities (Fig. 5c) were normalized by mass loading and Brunauer–Emmett–Teller (BET) surface area (Fig. S6), respectively. The results showed that the Co-S-Se spheres have both the highest mass activity (400.04 mA mg−1) and specific activity (1.74 mA cm−2) among those catalysts. To gain further insight into the kinetics of these catalysts, electrochemical impedance spectroscopy (EIS) was carried out in the frequency range of 100 kHz~0.1 Hz in 1.0 M KOH solution, and the Nyquist plots of the different catalysts are shown in Fig. S7. Co-S-Se exhibited a smaller charge transfer resistance (Rct, 9.89 Ω) than Co3O4 (93.7 Ω) and CoO/Co9S8 (27.3 Ω), indicating enhanced charge transfer due to the metallic compositions (cobalt sulfide and selenides) as well as the synergistic effect from their abundant heterointerfaces.

a Capacitive current density at a potential of 0.35 V (vs. Hg/HgO) at different scan rates. b Current density normalized to ECSA at η = 300 mV. c Mass activity and specific activity at 1.5 V (vs. RHE).
To establish a fundamental understanding of the enhanced OER activities of the Co-S-Se architectures, density functional theory (DFT) calculations were further employed to study the synergistic effect in this heterostructure. Co9S8 and Co-S-Se slab models (Fig. S8) were built using Co9S8 (311) (as observed by HRTEM) with optimized surface termination as a representative plane and replacement of the S atom with a Se atom in Co9S8 (311). According to the calculated Gibbs free energy diagrams of the OER process (Fig. 6a, b), the final elementary reaction step (OOH* + OH− → O2 + * + H2O + e−) was determined to be the rate-determining step (RDS) for both models due to its highest energy barrier among the four steps. It was found that Co-S-Se shows much weaker binding abilities toward the different intermediates (OH*, O*, and OOH*, Figs. S9 and 10) compared to the Co9S8 surfaces (Table S2). Therefore, the energy barrier of Co-S-Se for the RDS (1.84 eV and 0.61 eV at applied potentials of 0 V and 1.23 V, respectively) was significantly reduced with respect to that of pure Co9S8 (2.23 eV and 1.00 eV), demonstrating its favorable electronic structure for the overall OER activity. Density of states (DOS) was further analyzed to probe the intrinsic features of Co-S-Se for the OER. As shown in Fig. 6c, the d-band center of Co-S-Se shifted to the low-energy level, which can account for the weaker binding energies to reaction intermediates during the OER process. The charge density difference distribution diagram (Fig. 6d) shows that surface Se is located in the charge accumulation area, while the nearby Co displays charge depletion, indicating charge redistribution at the heterointerface due to electron transfer from Co9S8 to Se. The charge transfer can also be verified by the positive shift of Co, especially Co2+, in the XPS spectrum of Co-S-Se relative to that of CoO/Co9S8 (Fig. S3b). Therefore, it was concluded that the presence of the effective heterointerfaces in Co-S-Se can significantly modulate the surface electronic structure by shifting the d-band center of Co, resulting in the weakened adsorption of OER intermediates and lowering the reduced energy barrier in the rate-determining step, thus enhancing the overall OER activity accordingly.

Gibbs free energy diagrams of the OER process for Co9S8 and Co-S-Se at (a) 0 V and (b) 1.23 V. c Density of states (DOS) of Co9S8 and Co-S-Se (the red and blue dashed lines represent the d-band centers of Co9S8 and Co-S-Se, respectively). d The charge density difference at the interface in Co-S-Se. Light pink and light green represent charge accumulation and depletion, respectively.
Combining the electrochemical measurements for the OER activities and theoretical calculations, it was inferred that the multi-bishelled Co-S-Se architecture features the following main advantages: (i) the unique multi-bishelled hollow structure provides abundant active sites; (ii) the configuration of exposed metallic cobalt sulfides and selenides on the surface assures fast charge transfer, and, importantly, (iii) the unique heterostructure creates sufficient heterointerfaces, and their strong interaction and charge redistribution synergistically lowers the chemisorption energy barriers of oxygen-containing intermediates.
As a proof of concept, we further evaluated the role of this catalyst to promote CO2 reduction motivated by the excellent OER performance of Co-S-Se in alkaline solution. Nanostructured Pd with high surface areas has been extensively studied for ECR due to its good activity and high selectivity for the C1 product35. Therefore, we conducted full-cell electrolysis by coupling Co-S-Se as the anode and Pd NSs (Fig. 7a) as the cathode in a two-compartment cell (H-cell electrolyzer, Fig. 7b) as a representative to explore the role of anodic electrocatalysts. The cathodic electrolyte was CO2-saturated KHCO3 (0.1 M), and the anodic electrolyte was O2-saturated KOH (1.0 M). The anode was initially used as the working electrode, and the cathode was used as the counter and reference electrode to measure the cell voltage. As shown in Fig. 7c, the electrolyzer using Co-S-Se as the anode electrode needs a lower potential of 1.97 V than that of IrO2 (2.02 V) and Pt mesh (2.07 V) to achieve a low current of 0.5 mA. It should be noted that the cell-voltage difference increases with increasing current, demonstrating the excellent performance of Co-S-Se in reducing the energy input of the ECR system. To further investigate the effect of anodic catalysts on the ECR, the gaseous products from Pd NS/C coupled with different anodes at various potentials were monitored by online gas chromatography (GC). As shown in Fig. S11, CO and H2 were the main products, as the sum of their Faradaic efficiency (FE) was close to 100%. The highest FE(CO) of 93.1% (Fig. 7d) was achieved when Pd NS/C was paired with Co-S-Se at a potential of −0.7 VRHE, which is higher than that of the IrO2/C (85.2%) and Pt mesh (76.3%). In addition, when the Pd NS/C worked at −0.7 V, the whole voltage of the cell using Co-S-Se (2.19 V) was much lower than that of IrO2/C (2.27 V) and Pt mesh (2.53 V), demonstrating the improved energy efficiency (Table S3). The improved Faradaic Efficiency of CO could result from the enhanced OER activities from Co-S-Se, which leads to slight pH changes in the catholyte36,37. Therefore, it can be concluded that this efficient OER can not only save the energy input of the ECR system by reducing the cell voltage but also play a key role in improving product selectivity.

a TEM image of Pd NSs as the cathodic electrocatalyst for ECR. b Setup of the H-cell electrolyzer. c LSV of the current against cell voltage from different anodes coupled with Pd NSs. d CO Faradaic efficiency of Pd NSs coupled with different anodes.
Conclusions
In summary, unique multi-bishelled Co-S-Se hollow spheres were developed based on a facile template-free method. In this multi-bishelled architecture, each bi-shelled structure was composed of ternary Co-based components with a symmetric configuration, featuring abundant heterointerfaces and large surface areas. Owing to these merits, the Co-S-Se hollow spheres exhibited outstanding OER properties. In particular, when coupled with ECR, the electrochemical performance of Pd NS/C toward CO2 reduction can be significantly improved in terms of FE(CO) and energy consumption. This work might provide a new strategy for the development of highly efficient OER electrocatalysts for CO2 reduction reactions.
References
Ran, J., Jaroniec, M. & Qiao, S. Cocatalysts in semiconductor‐based photocatalytic CO2 reduction: achievements, challenges, and opportunities. Adv. Mater. 30, 1704649 (2018).
Zhang, S., Fan, Q., Xia, R. & Meyer, T. CO2 reduction: from homogeneous to heterogeneous electrocatalysis. Acc. Chem. Res. 53, 255–264 (2020).
Wu, J., Huang, Y., Ye, W. & Li, Y. CO2 reduction: from the electrochemical to photochemical approach. Adv. Sci. 4, 1700194 (2017).
Fan, J., Han, N. & Li, Y. Electrochemical carbon dioxide reduction in flow cells. J. Electrochem. 24, 510–520 (2020).
Gu, Z. et al. Efficient electrocatalytic CO2 reduction to C2+ alcohols at defect-site-rich Cu surface. Joule 5, 429–440 (2021).
Zhang, W. et al. Electrochemical reduction of carbon dioxide to methanol on hierarchical Pd/SnO2 nanosheets with abundant Pd–O–Sn interfaces. Angew. Chem. Int. Ed. 57, 9475–9479 (2018).
Li, J. & Cheng, F. Electrolyte tailoring for electrocatalytic reduction of stable molecules. J. Electrochem. 26, 474–485 (2020).
Fan, L. et al. Strategies in catalysts and electrolyzer design for electrochemical CO2 reduction toward C2+ products. Sci. Adv. 6, eaay3111 (2020).
Zhou, G. et al. Spin-sate reconfiguration induced by alternating magnetic field for efficient oxygen evolution reaction. Nat. Commun. 12, 1–9 (2021).
Liu, C. et al. Oxygen evolution reaction over catalytic single-site Co in a well-defined brookite TiO2 nanorod surface. Nat. Catal. 4, 36–45 (2021).
Cui, M. et al. High‐entropy metal sulfide nanoparticles promise high‐performance oxygen evolution reaction. Adv. Energy Mater. 11, 2002887 (2021).
Verma, S., Lu, S. & Kenis, P. J. A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 4, 466–474 (2019).
Jiang N., Zhu Z., Xue W., Xia B. Y., You B. Emerging electrocatalysts for water oxidation under near‐neutral CO2 reduction conditions. Adv. Mater. 34, 2105852 (2021).
Chen, S., Duan, J., Jaroniec, M. & Qiao, S. Z. Nitrogen and oxygen dual‐doped carbon hydrogel film as a substrate‐free electrode for highly efficient oxygen evolution reaction. Adv. Mater. 26, 2925–2930 (2014).
Du, P. & Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: recent progress and future challenges. Energy Environ. Sci. 5, 6012–6021 (2012).
Gong, M. et al. An advanced Ni–Fe layered double hydroxide electrocatalyst for water oxidation. J. Am. Chem. Soc. 135, 8452–8455 (2013).
Song, F. & Hu, X. Exfoliation of layered double hydroxides for enhanced oxygen evolution catalysis. Nat. Commun. 5, 4477 (2014).
Liang, Y. et al. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780 (2011).
Xu, S., Li, P. & Lu, Y. In situ atomic-scale analysis of Rayleigh instability in ultrathin gold nanowires. Nano Res 11, 625–632 (2018).
Li, X. et al. Nitrogen‐doped cobalt oxide nanostructures derived from cobalt–alanine complexes for high‐performance oxygen evolution reactions. Adv. Funct. Mater. 28, 1800886 (2018).
Hamdani, M. & Singh, R. Chartier P. Co3O4 and Co-based spinel oxides bifunctional oxygen electrodes. Int. J. Electrochem. Sci. 5, 556–577 (2010).
Lee, S. R. et al. Palladium@Platinum concave nanocubeswith enhanced catalytic activity toward oxygen reduction. ChemCatChem 8, 3082–3088 (2016).
Zou, X. et al. Efficient oxygen evolution reaction catalyzed by low-density Ni-doped Co3O4 nanomaterials derived from metal-embedded graphitic C3N4. Chem. Commun. 49, 7522–7524 (2013).
Xiao, X. et al. A general approach to cobalt-based homobimetallic phosphide ultrathin nanosheets for highly efficient oxygen evolution in alkaline media. Energy Environ. Sci. 10, 893–899 (2017).
Wang, J. et al. Recent progress in cobalt‐based heterogeneous catalysts for electrochemical water splitting. Adv. Mater. 28, 215–230 (2016).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Xue, Z. et al. Interfacial electronic structure modulation of nite nanoarrays with nis nanodots facilitates electrocatalytic oxygen evolution. Adv. Mater. 31, e1900430 (2019).
Niu, S. et al. Se-doping activates FeOOH for cost-effective and efficient electrochemical water oxidation. J. Am. Chem. Soc. 141, 7005–7013 (2019).
Ramos, M., Berhault, G., Ferrer, D. A., Torresa, B. & Chianellia, R. R. HRTEM and molecular modeling of the MoS2–Co9S8 interface: understanding the promotion effect in bulk HDS catalystsw. Catal. Sci. Technol. 2, 164–178 (2012).
Zhu, H. et al. When cubic cobalt sulfide meets layered molybdenum disulfide: a core–shell system toward synergetic electrocatalytic water splitting. Adv. Mater. 27, 4752–4759 (2015).
Cai, P., Huang, J., Chen, J. & Wen, Z. Oxygen-containing amorphous cobalt sulfide porous nanocubes as high-activity electrocatalysts for the oxygen evolution reaction in an alkaline/neutral medium. Angew. Chem. Int. Ed. 56, 4858–4861 (2017).
Li, Y. et al. Size-controlled synthesis of Pd nanosheets for tunable plasmonic properties. CrystEngComm 17, 1833–1838 (2015).
Tianou, H. et al. Inflating hollow nanocrystals through a repeated Kirkendall cavitation process. Nat. Commun. 8, 1261 (2017).
Lei, R., Zhang, H., Tan, P., Chen, Y. & Zhang, Z. Hexagonal selenium nanowires synthesized via vapor-phase growth. J. Phys. Chem. B 108, 4627–4630 (2004).
Gao, D. et al. Pd-containing nanostructures for electrochemical CO2 reduction reaction. ACS Catal. 8, 1510–1519 (2018).
Clark, R., Moore, A., Maclnnis & Bertin, E. Investigation of urea oxidation as a potential anode reaction during CO2 electrolysis. J. Appl. Electrochem. 51, 1583–1590 (2021).
Lee, W. et al. Carbon-Supported IrCoOx nanoparticles as an efficient and stable OER electrocatalyst for practicable CO2 electrolysis. Appl. Catal. B 269, 118820 (2020).
Acknowledgements
This work was supported by the Singapore Ministry of Education AcRF Tier 1: 2020-T1-001-031, Shenzhen Science and Technology Innovation Committee (JCYJ20180507183818040), the National Natural Science Foundation of China (22109044), the Natural Science Foundation of Shanghai, China (22ZR1418500), and start-up funds from East China University of Science and Technology.
Author information
Authors and Affiliations
Contributions
Y. Zhang designed the study and wrote the manuscript. Y. Zhang and P.Z. performed the experimental synthesis and materials characterizations. P.Z. and K.N.D. performed the electrochemical measurements. X.Q. performed the calculations and interpreted the related data. J.Y. and Y. Zheng performed the SEM observation. M.S. and Q.Y. supervised the project. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Y., Zheng, P., Qin, X. et al. Constructing a multi-bishelled cobalt-based electrocatalyst for the oxygen evolution reaction in CO2 electrolysis. NPG Asia Mater 14, 55 (2022). https://doi.org/10.1038/s41427-022-00398-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-022-00398-0
