
Abstract
Addressing the devastating threat of drug-resistant pathogens requires the discovery of new antibiotics with advanced action mechanisms and/or novel strategies for drug design. Herein, from a biophysical perspective, we design a class of synthetic antibacterial complexes with specialized architectures based on melittin (Mel), a natural antimicrobial peptide, and poly(ethylene glycol) (PEG), a clinically available agent, as building blocks that show potent and architecture-modulated antibacterial activity. Among the complexes, the flexibly linear complex consisting of one Mel terminally connected with a long-chained PEG (e.g., PEG12k–1*Mel) shows the most pronounced improvement in performance compared with pristine Mel, with up to 500% improvement in antimicrobial efficiency, excellent in vitro activity against multidrug-resistant pathogens (over a range of minimal inhibitory concentrations of 2–32 µg mL−1), a 68% decrease in in vitro cytotoxicity, and a 57% decrease in in vivo acute toxicity. A lipid-specific mode of action in membrane recognition and an accelerated “channel” effect in perforating the bacterial membrane of the complex are described. Our results introduce a new way to design highly efficient and low-toxicity antimicrobial drugs based on architectural modulations with clinically available agents.
Similar content being viewed by others


Introduction
Drug design is an extensively researched field in medicinal science that is essential for human health1. In its early stages, drug design relied on the structural transformation of natural active ingredients2, and then it was further developed so that the drug’s molecular structure was modified based on the principles of organic chemistry3. Currently, drug design involves the comprehensive application of medicinal chemistry4, molecular biology5, and quantum chemistry6. Recent developments in biophysics have helped to gain a deep understanding of the chemical mechanisms of the biological activities of a drug, particularly the modes of action of different functional units. This knowledge provides an alternative means for the design of new lines of agents with higher requirements, which is essential since antimicrobial resistance has become a substantial problem. In a recent study, β-hairpin, a macrocycle that targets the β-barrel outer membrane protein LptD of gram-negative bacteria, was successfully integrated with the peptide macrocycle of colistin (a compound regarded as the last resort for fighting increasingly resistant gram-negative bacteria) that binds to the lipid A portion of lipopolysaccharides (LPSs) in the outer leaflet. The integrated drug can be used against colistin-resistant pathogens, and it has been tested in preclinical toxicology studies for potential medicinal use7. Moreover, self-assembled protein- (or peptide-) based hybrid nanostructures, especially hydrogels or nanoparticles such as collagen-gold hydrogels8, Fmoc amino acid-Ag biometallohydrogels9, chlorine e6-doxorubicin (Ce6-DOX) nanoparticles10, and glutaraldehyde-assisted dipeptide-chlorine e6-heparin particles11, promise a combination of antimicrobial, antitumor, and tissue culture applications12. Although the integration of existing functional units can combine and even improve the performance of the original components13, stronger biofunctions can be achieved by designing units with specialized architectures inspired by natural-protein-derived scaffolds. Herein, we use melittin (Mel; Fig. 1a)14, a typical natural antimicrobial peptide (AMP), and poly(ethylene glycol) (PEG), a clinically available agent15, as building blocks for the preparation of composite molecules with designed architectures. These molecules exhibit potent architecture-modulated antibacterial activity.

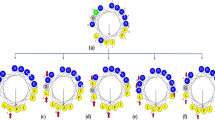
a Amino acid sequences, conformational images, and helical wheel diagrams of Mel. The hydrophobic (in gray), hydrophilic (in orange), and charged (in blue) residues are shown differently. A kinked region exists between the two helixes in the amphiphilic α–helical conformation, and the helical wheel diagrams show the distribution of a polar face and a nonpolar face. b Conjugation route of PEGw–m*peptide complexes with different architectures, including linear (left), crossed (middle), and multibranched (right) complexes.
Materials and methods
Synthesis of complexes
All PEGs were modified with Mel via covalent conjugation between the active site of PEG and the amino group of the peptide (molecular ratio of active site:peptide = 2.5:1; Supplementary Information). Subsequently, the complexes were dialyzed (MW 3500) and concentrated, and then they were analyzed by dynamic light scattering (DLS, DynaPro, Malvern), sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (JY300, JunYi, Beijing, China), UV–Vis absorption spectroscopy (UV3600, Shimadzu), and Fourier transform infrared spectroscopy (FT-IR; Tensor II, Bruker).
Dynamic giant unilamellar vesicle leakage assay
Giant unilamellar vesicles (GUVs) composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and/or 1,2-dioleoyl-sn-glycero-3-phospho-(1ʹ-rac-glycerol) (DOPG) and labeled with 1 mol% fluorescent 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh-PE) were prepared via the conventional electroformation method (Supplementary Information) and were observed using a confocal microscope (LSM 710, Zeiss). After drug addition, the dynamic process of calcein entry into a GUV was monitored by recording the fluorescence intensity of the GUV interior at each time point. The recorded fluorescence intensities were normalized to that of the surrounding environment and plotted as a function of time.
Antimicrobial minimal inhibitory concentration and time-dynamic bactericidal tests
The minimal inhibitory concentrations (MICs) of the agents were determined according to the standardized broth microdilution method16,17. At least three independent MIC measurements were performed for each agent. The time-dynamic bactericidal test was used to assess the antimicrobial effectiveness of different agents over the duration of the treatment process based on the standardized protocol.
Acute toxicity test in mice
Pristine Mel (5 mg kg−1), PEG12k–1*Mel (5 mg kg−1), or an equal volume of phosphate-buffered saline (PBS, control) were intraperitoneally injected into C57BL/6 mice (20 ± 2 g, female). After 24 h, the levels of alanine aminotransferase (ALT) and creatinine in eyeball blood serum were tested. Tissue sections of liver and kidneys were collected, stained with hematoxylin and eosin (H&E), and imaged by light microscopy (Nikon Eclipse TE2000-S, Japan).
Molecular dynamic simulations
Molecular dynamic (MD) simulations were performed using the Martini coarse grained (CG) force field (version 2.2)18 with the Gromacs 5.1.4 package19. The 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) lipid was CG based on a 4-to-1 mapping scheme, and PEG was built using Martini SP0 beads with CG parametrization. The periodic boundary conditions were applied in all three directions.
Statistical analysis
The simulations and experimental measurements were repeated three to five times for each condition/data point. Analysis of variance was performed using OriginPro 9.0 (OriginLab software, Northampton, MA), and the values are expressed as the mean ± standard deviation. For all experimental datasets, the samples were compared to controls unless otherwise stated. Further details regarding the experiments and simulations are available in the Supplementary Information.
Results and discussion
Fabrication and antimicrobial performance of the PEG12k–1*Mel complex
As one of the most representative natural AMPs, Mel has an amphiphilic and α-helical structure when it is bound to a cell membrane (Fig. 1a). The chemical conjugation of Mel peptides with the predesigned active sites (e.g., the nonnatural organic group maleimide) of different PEG molecules (varying structures and molecular weights) results in the formation of PEGw–m*Mel complexes that are characterized by various architectures. In the PEGw–m*Mel complexes, w refers to the molecular weight of PEG, and m represents the peptide number in each complex (more details in Fig. 1b and S1). For example, in the PEG12k–1*Mel complex, each Mel peptide is terminally conjugated to one single-chained PEG molecule with a mean molecular weight of 12,000 (Fig. 2a). DLS, UV-Vis absorption, FT-IR, and SDS-polyacrylamide gel electrophoresis analyses of the complex confirmed the successful modification of Mel with PEG molecules (Fig. S2). Moreover, the concentration-dependent size distribution of PEG12k–1*Mel in the DLS test, e.g., from 500 nm at 0.1 µg mL−1 to 800 nm at 5.0 µg mL−1, indicates the formation of self-assembled aggregates of the complex in comparison to pure Mel, which might exist as dispersed random coils in aqueous solution.

a Conjugation route of the PEG12k–1*Mel complex. b Representative SEM images of E. coli and S. aureus bacteria after 16 h of exposure to native Mel, PEG12k–1*Mel (peptide concentration of 64 µg mL−1 (with respect to the peptide)), or no agent exposure. Arrows indicate lesions and collapses of the bacterial membrane. c MIC distributions of pure Mel and PEG12k–1*Mel against E. coli and S. aureus. P-values were calculated by ANOVA (**P < 0.01, ***P < 0.001). The insets depict the time-dynamic antimicrobial assays showing the effects of native Mel and PEG12k–1*Mel treatments (0, 3, 6, or 16 h) on bacterial viability before determining the colony-forming unit (CFU). d Standard Alamar blue cell proliferation assay of MGC-803 cells cultured with pure Mel or PEG12k–1*Mel for 24 h. The concentrations of the agents varied between 1.6 and 200.0 µg mL−1. The data show the normalized mean and standard error of at least three independent experiments. e Antimicrobial activities (MICs of other commercial antibiotics are shown in Table S1 of Supplementary Information).
To test the bactericidal and antibacterial activity of PEG12k–1*Mel, the complex was applied to living Escherichia coli CGMCC 1.12883 and Staphylococcus aureus CGMCC 1.10755 cells, which represent gram-negative and gram-positive bacteria, respectively. After exposure to the complex (peptide concentration of 64.0 µg mL−1 with respect to the peptide) or to pure Mel for 16 h, scanning electron microscopy (SEM) images of the bacteria were recorded. As shown in Fig. 2b, the membranes of the treated bacteria exhibit obvious morphological changes and disruptions compared to the natural bacterial membranes. In particular, the bacteria exposed to PEG12k–1*Mel have seriously damaged membranes with leakage of the bacterial content. Based on MIC tests, the activities of pure Mel and PEG12k–1*Mel against E. coli and S. aureus are much greater than those of the widely used polymyxin B (against E. coli) and fusidic acid (against S. aureus) antibiotics16,17 (Figs. 2c and S3a). The mean MIC values of PEG12k–1*Mel against E. coli and S. aureus are 6 and 17 µg mL−1, respectively, compared to the values of 32 and 45 µg mL−1, respectively, for pure Mel. These values clearly show that PEG–Mel complexation enhances the antibacterial efficiency of Mel. To assess the antimicrobial effectiveness of different agents, time-dynamic bactericidal tests20,21 were also performed using a drug concentration of 64.0 µg mL−1. The obtained results demonstrate that the viability of bacteria incubated with PEG12k–1*Mel for 0, 3, 6, or 16 h is less than that of bacteria exposed to pure Mel for the same duration (Fig. 2c-inset and Fig. S3b). This indicates that the bactericidal and antibacterial effects of PEG12k–1*Mel are better than those of Mel. Furthermore, repeated treatment with PEG12k–1*Mel (up to five cycles) does not lead to an increased MIC, which suggests that the bacteria do not readily develop resistance to this drug. In addition to E. coli and S. aureus, the PEG12k–1*Mel complex shows potent activity against other pathogens, including Acinetobacter baumannii ATCC 19606 and Pseudomonas aeruginosa ATCC 27853, as well as many other drug-resistant isolates (three strains of clinically isolated A. baumannii and methicillin-resistant S. aureus). The MICs of the complex against these pathogens vary in the range of 2–32 µg mL−1, and they are much lower than the MICs of pristine Mel and most commercial antibiotics (Fig. 2e and Table S1).
Toxicity tests demonstrate that the cytotoxicity of PEG12k–1*Mel is much less than that of pristine Mel. Specifically, the standard Alamar blue assays16 performed on mouse gastric cancer (MGC)-803 cells show that PEG12k–1*Mel treatment decreases cellular mortality by approximately 68% compared to pure Mel treatment (200 µg mL−1) (Figs. 2d and S3c). Moreover, higher concentrations of PEG12k–1*Mel do not increase cellular mortality, unlike pure Mel. The acute toxicity test in C57BL/6 mice shows similar results (Fig. S4). Specifically, the PEG12k–1*Mel-treated mice show significantly decreased ALT (decreased by ~57%) and creatinine (with values similar to the control) levels in comparison with the pristine Mel-treated mice, although at the same peptide concentration (e.g., 5 mg kg−1). Histopathological studies show little kidney and liver injury in mice. Overall, the experiments conducted on living cells indicate that PEG12k–1*Mel exhibits a significantly enhanced and rapid antimicrobial effect compared to pristine Mel, and it is less cytotoxic (especially at high concentrations).
Dynamic interactions between the PEG12k–1*Mel complex and cellular membranes
Having established that the PEG12k–1*Mel drug induces serious damage to bacterial membranes, we next assessed the dynamic interactions between the drug and a cellular membrane. The assessment was conducted in real time using a novel setup that was specifically designed based on the interfacial photoelectric response effect (Fig. S5)22. In this setup, a silicon wafer photoelectrode coated with a DOPC bilayer, which serves as a model cell membrane23, via the traditional vesicle fusion method24,25 was taken as the working electrode. Moreover, a Ag/AgCl electrode was used as the counter electrode. Using this double-electrode system, drug-induced changes in the output photogenerated voltage (U) can be monitored in real time based on changes in the electrical double layer at the solid–liquid interface corresponding to the working electrode. The change in voltage values directly reflects modifications to the structure and properties of the lipid membrane (a simplified model of the test system and a detailed analysis are available in the Supplementary Information). Therefore, the dynamic interaction process at the drug–membrane interface, i.e., the structural deformation and poration of the lipid membrane due to drug exposure, can be demonstrated in real time based on the U–t profiles. As shown in Fig. 3a, U remains constant with time in the absence of a drug, which indicates that the membrane–solution interface is in a steady state. When Mel is added, U decreases with time, and the rate of decrease is enhanced by using PEG12k–1*Mel instead of Mel. The profiles depicted in Fig. 3a clearly show three stages of changes in U upon drug addition. The first stage is an exponential decrease (t = 0–10 min for Mel), followed by a sudden drop and a slower exponential decrease (t = 10 to 30 min for Mel), finally reaching equilibrium (t > 30 min for Mel). As mentioned earlier, the changes in U are indicative of a dynamic interaction between the drug and the membrane. This interaction disrupts the membrane structure (e.g., thinning and extending) via the adsorption and accumulation of the agent on the bilayer surface and via the formation of transmembrane pores due to drug insertion. Thus, the final structure of the stabilized membrane contains inserted and rearranged molecules. The sudden drop in voltage at t = 10 min for pure Mel and at t = 2 min for PEG12k–1*Mel signifies the commencement of transmembrane pore formation.

a Typical U–t profiles of DOPC membranes exposed to DI water, pure Mel (5.0 µg mL−1) or PEG12k–1*Mel (0.5 µg mL−1). The yellow arrows indicate the beginning of poration. The profile is fitted in stages to the exponential function U = A1 × e−t/τ + U0. b Confocal fluorescence images of representative GUVs showing the peptide-induced transmembrane leakage of calcein from the GUVs due to the addition of pristine Mel (3.0 µg mL−1) or PEG12k–1*Mel (1.0 µg mL−1). An image of the original calcein-loaded GUVs (PC with 1 mol% RhB-PE for fluorescence labeling) is also shown for reference. Green and red colors represent calcein and lipids, respectively. c Representative profiles showing variations in the normalized GUV fluorescence intensity as a function of time after treatment with Mel or PEG12k–1*Mel. The variation profiles are fitted to the equation y = a/(1 + exp(−k × (x−xc)). The bilayer is composed of pure PC or PC/PG (85/15). d Relative distribution of the molar entry rate of calcein into GUVs composed of pure PC or PC/PG after the addition of pristine Mel or PEG12k–1*Mel at various concentrations. The inset shows the histogram with an amplified y-axis. The concentrations of the agents are marked correspondingly (in µg mL−1). Data points in d are the average of at least five replicates.
Permeabilization (e.g., poration) of bacterial membranes is essential for the antimicrobial effectiveness of AMPs such as Mel26. Herein, we show that PEG12k–1*Mel exhibits faster perforation and enhanced poration ability than Mel. Based on photovoltage tests carried out using varying concentrations of the complex, the most efficient working concentration of PEG12k–1*Mel was determined to be 0.1 µg mL−1. Surprisingly, the poration speed and efficiency of the complex at concentrations higher or lower than this value were reduced. The observation of a clear stage and a sharp turning point between the stages of the U–t profile confirms that photovoltage tests are a sensitive and powerful technique for real-time in situ monitoring of interfacial interactions, particularly membrane poration. These assessments cannot be readily conducted using other techniques.
To further evaluate the effects of Mel and PEG12k–1*Mel on cellular membranes, a dynamic GUV leakage assay27 was performed. The first image in Fig. 3b shows a representative GUV composed of DOPC labeled with 1 mol% of fluorescent Rh-PE. This GUV contains calcein, a water-soluble and membrane-impermeable fluorophore that is normally used to test the permeability of membranes16. In the absence of an agent, the GUV-encapsulated calcein shows no leakage, even after 4 h. Similarly, in the presence of a low concentration of pristine Mel, variations in the calcein content of vesicles are not detected by confocal microscopy observation. Calcein release is first observed in ~40% of the GUVs at the threshold Mel concentration of 3.0 µg mL−1 (peptide-to-lipid ratio P/L ≈ 1/24, with the dynamic transmembrane leakage process of calcein being quantitatively analyzed later). Considering that at this concentration, the vesicles and membranes remain undamaged, the leakage of calcein may be attributed to the formation of transmembrane pores. At higher peptide concentrations, the membrane perforation phenomenon is amplified, and more leakage occurs. Compared to pure Mel, the threshold working concentration of PEG12k–1*Mel is significantly lower (1.0 µg mL−1; P/L ≈ 1/72; P/L ≈ 1/128 based on MD simulations, as detailed in the next section), and calcein leakage is observed for ~65% of the GUVs at this concentration. Note that peptides such as Mel normally show concentration-dependent antimicrobial efficiency; this decrease in the threshold working concentration demonstrates a significantly enhanced activity of the complex compared with pristine Mel. Beyond the threshold, calcein release is intensified; however, unlike Mel treatment, PEG12k–1*Mel treatment also induces membrane remodeling behavior28. At a peptide concentration of 2.5 µg mL−1, pure Mel and PEG12k–1*Mel exhibit different effects on supported multilamellar membranes (SMMs). Specifically, Mel induces lipid fibril formation, while PEG12k–1*Mel promotes the formation of large vesicles (Fig. S6a). These results confirm that the membrane poration and disruption ability of the complex is significantly greater than that of pristine Mel.
The effect of drug exposure on the transmembrane diffusion of calcein provides additional proof of the drug–membrane interactions, which can be quantitatively reflected. Figures 3c and S6 show the dynamic transmembrane diffusion process of calcein due to drug exposure. This effect is characterized in stages, wherein the first stage corresponds to membrane binding of agents and pore formation, and the second linear increase stage refers to the steady state of calcein transfer through pores with a constant number and size. When PEG12k–1*Mel is used as a drug agent instead of Mel, the stages become more pronounced. The stable platform observed in the initial stage reflects the dominating liposome repellency effect of the complex due to the highly hydrated structure of the PEG moiety. Moreover, the sharp turn after the platform (before a linear increase) represents a highly accelerated membrane poration process, which is consistent with the interfacial photovoltage experiments. Moreover, the membrane activity of complexes demonstrates a lipid-specific dependence. Compared to the electrically neutral membrane composed of pure DOPC lipids, the negatively charged membrane consisting of DOPC and DOPG (PC/PG = 85/15 by mol) is affected more by the PEG12k–1*Mel complex, with a stable molar entry rate (0.5 µg mL−1, P/L ≈ 1/144) that is ~15.7 times higher than that of the pure DOPC bilayer (3.0 µg mL−1; Fig. 3d). Considering that the bacterial membrane is more negatively charged than the membranes of mammalian cells, it is expected that the complex may effectively target bacteria with minimal effects on mammalian cells. These results are in good agreement with the antibacterial and cytotoxicity assays.
Molecular mechanism of the improved antibacterial performance
The molecular mechanism underlying the improved antibacterial performance of the PEG12k–1*Mel complex was analyzed based on MD simulations. The simulation results (Fig. S7) are similar to those determined experimentally, and they show that the membrane poration ability of PEG12k–1*Mel is greater than that of Mel. Specifically, the complex is characterized by a lower threshold P/L ratio (1/128) and results in larger pores (~16 nm2 at P/L = 1/43) than the pristine peptide (P/L = 1/43 and pore size ≈2 nm2). Moreover, similar to the experiments, the simulations indicate that the PEG12k–1*Mel complex exhibits a lipid-specific action mode. It is expected that this mode of action is strongly associated with the enhanced membrane poration and binding capacity of the complex. The highly hydrated PEG corona formed at high agent concentrations (e.g., the case with P/L = 1/26, Fig. S7a) and the compact structure of the aggregated complex hinder the interaction of PEG12k–1*Mel with an electroneutral bilayer. However, an anionic membrane would expose the functional unit of the complex, thereby promoting its activity (Fig. S7c). Free energy calculations confirm that the membrane-binding process of the complex has an energy barrier whose value is significantly decreased upon replacing the pure PC membrane with a PC/PG membrane (Fig. S8).
In addition to the hydrophilicity of the PEG moiety, the membrane poration ability of PEG12k–1*Mel is enhanced by the “channel” effect observed during the membrane insertion stage due to the cooperation between the flexible PEG chain and the Mel peptide. As shown in Fig. 4, when the complex adsorbs on the membrane surface, the Mel part of the complex changes its conformation (becomes “U”-shaped) so that it is slightly embedded in the bilayer. Meanwhile, the PEG part of the complex remains in the hydrophilic headgroup layer of the outer leaflet, as evidenced by the high interaction energy between PEG and the lipid head of the outer leaflet (t = 0–800 ns in Fig. 4). Subsequently, the aggregated Mel helices form a central “channel” that protects PEG from the hydrophobic core region of the bilayer and facilitates its passage across the membrane (t = 800–3200 ns, which is the time during which the interaction energy between PEG and the lipids approaches zero). Suddenly, PEG binds to the lipid headgroups of the inner leaflet along the N-terminus of the peptide (t > 3200 ns), which further drives the full transmembrane insertion of the Mel part of the complex and the formation of a stable transmembrane pore. Therefore, the formation of a “peptide channel” with deformed and aggregated Mel helices and the binding of PEGs to the lipid heads in both leaflets of the membrane are essential for efficient complex insertion. Without PEG, pristine Mel cannot readily penetrate through the membrane, even though it forms the required “U”-shaped conformation upon adsorption29. Consequently, pristine Mel can form only transient pores on membranes.

a Snapshots of the process of PEG12k–1*Mel penetration into a membrane. The Mel and PEG parts of the complex are shown as magenta lines and blue beads, respectively. The lipid heads are shown as gray beads, but the lipid tails are not shown for clarity. Yellow cylinders are used to highlight the “channel” formed by aggregated Mel in the membrane, while yellow ellipses represent the surface binding state of the PEG part. b Temporal evolution of the locations of Mel and PEG (“terminal” and “linker” ends) relative to the bilayer and the corresponding interaction energies between PEG and the lipids (head or tail) of the outer or inner leaflet. A background color (pink) is used to highlight the “channel”-mode poration process. C1–C4 refer to the states shown in a. Dashed lines in b represent the two headgroup layers of the membrane. P/L=1/128.
Molecular architecture-modulated complex biofunctions
From a biophysical perspective, the functions of biomacromolecules are determined by their structures. Therefore, a series of PEG–Mel complexes with varying architectures were constructed by decorating Mel with PEG. The designed complexes can be divided into three groups (as shown in Figs. 1 and S1): linear architecture complexes (i.e., each Mel is terminally conjugated with one single-chained PEG), crossed architecture complexes (i.e., two or four Mel helices are conjugated with a two-armed or four-armed PEG), and multibranched architecture complexes (i.e., many modified peptides are on the PEG main chain). The molecular weights (MWs) of the PEGs were also varied by including flexible long chains (MW ≥ 5000) or rigid short chains (MW ≤ 200). PEGw-m*Mel complexes were applied to living cells to test their bactericidal and cytotoxic activities and applied to GUVs or SMMs to assess their membrane poration activity. As shown in Table 1 and Fig. S3, the complexes have architecture-regulated performance (further details are available in the Supplementary Information).
Conclusion
In summary, the emergence of drug-resistant bacteria has created an urgent need for the development of a new line of antibacterial agents. As part of the natural defense system of many organisms, AMPs such as Mel kill pathogens via a physical attack that is almost impossible for bacteria or even superbugs to adapt to. The nonprotein-specific membrane destabilization activity of AMPs facilitates the creation of a new class of antimicrobial agents30. However, at present, the clinical use of these agents is hindered by their low antimicrobial efficiency, proteolytic degradation behavior, and toxicity to mammalian cells31. Due to their masking effect15, PEG chains have been used to improve the dispersibility and stability of drugs in aqueous solution and reduce the in vivo clearance or the possible adsorption of plasma proteins32,33. However, despite its advantages, PEG modification tends to weaken the biological activity of the decorated molecule34,35,36. In this study, we show that the conjugation of Mel with different PEG structures results in the formation of complexes that exhibit enhanced antibacterial efficiency against gram-negative and gram-positive bacteria. Additionally, these complexes, particularly those having a flexible linear architecture (e.g., PEG12k–1*Mel), are effective against multidrug-resistant pathogens, and their toxicity and side effects are less than those of pristine Mel. Covalent bonding occurs between PEG and Mel to form complexes with specialized architectures, which further assemble into aggregates via noncovalent interactions for a cell-(lipid-) specific action over bacterial or mammalian cells37. The experimental and simulation results reported herein indicate that the complexes interact with cellular membranes via a phased process that involves sharp turning points between stages. Based on the collected data, the improved antibacterial performance of PEG12k–1*Mel and similar complexes can be attributed to the lipid-specific mode of action and to the “channel” effect that allows for stable pore formation by facilitating the passage of the PEG part of the complex across the cellular membrane. Considering that clinically available agents (PEG, polylactic acid, etc.) and AMPs (magainin, LL-37, etc.) are cost-effective and that their structures and properties can be tailored, the drug design strategy proposed herein can be used to design novel antimicrobial agents and to develop new drugs with improved performance based on the known functional units (e.g., clinical drugs and natural molecules).
References
Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013).
Kuntz, I. D. Structure-based strategies for drug design and discovery. Science 257, 1078 (1992).
Rodrigues, T., Reker, D., Schneider, P. & Schneider, G. Counting on natural products for drug design. Nat. Chem. 8, 531–541 (2016).
Sliwoski, G., Kothiwale, S., Meiler, J. & Lowe, E. W. Computational methods in drug discovery. Pharmacol. Rev. 66, 334 (2014).
López-Igual, R., Bernal-Bayard, J., Rodríguez-Patón, A., Ghigo, J.-M. & Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. 37, 755–760 (2019).
Schnaare, R. S. & Martin, A. N. Quantum chemistry in drug design. J. Pharm. Sci. 54, 1707–1713 (1965).
Luther, A. et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 576, 452–458 (2019).
Xing, R. et al. An injectable self‐assembling collagen–gold hybrid hydrogel for combinatorial antitumor photothermal/photodynamic therapy. Adv. Mater. 28, 3669–3676 (2016).
Song, J. et al. Multifunctional antimicrobial biometallohydrogels based on amino acid coordinated self-assembly. Small 16, 1907309 (2020).
Zhang, R. et al. Carrier-free, chemophotodynamic dual nanodrugs via self-assembly for synergistic antitumor therapy. ACS Appl. Mater. Interfaces 8, 13262–13269 (2016).
Ma, K. et al. Injectable self-assembled dipeptide-based nanocarriers for tumor delivery and effective in vivo photodynamic therapy. ACS Appl. Mater. Interfaces 8, 30759–30767 (2016).
Gelain, F., Luo, Z. & Zhang, S. Self-assembling peptide EAK16 and RADA16 nanofiber scaffold hydrogel. Chem. Rev. 120, 13434–13460 (2020).
Yuan, B. et al. Self-assembly of highly oriented lamellar nanoparticle-phospholipid nanocomposites on solid surfaces. J. Am. Chem. Soc. 129, 11332–11333 (2007).
Raghuraman, H. & Chattopadhyay, A. Melittin: a membrane-active peptide with diverse functions. Biosci. Rep. 27, 189–223 (2007).
Hong, W. et al. PEGylated self-assembled nano-Bacitracin A: probing the antibacterial mechanism and real-time tracing of target delivery in vivo. ACS Appl. Mater. Interfaces 10, 10688–10705 (2018).
Lu, X. et al. Designing melittin-graphene hybrid complexes for enhanced antibacterial activity. Adv. Healthc. Mater. 8, 1801521 (2019).
Xiao, S. et al. Graphene oxide as antibacterial sensitizer: Mechanically disturbed cell membrane for enhanced poration efficiency of melittin. Carbon 149, 248–256 (2019).
Marrink, S. J., Risselada, H. J., Yefimov, S., Tieleman, D. P. & de Vries, A. H. The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824 (2007).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Balouiri, M., Sadiki, M. & Ibnsouda, S. K. Methods for in vitro evaluating antimicrobial activity: a review. J. Pharm. Anal. 6, 71–79 (2016).
Zhang, X. et al. Quaternized silicon nanoparticles with polarity-sensitive fluorescence for selectively imaging and killing gram-positive bacteria. Adv. Funct. Mater. 26, 5958–5970 (2016).
Dou, Y. et al. Photo-voltage transients for real-time analysis of the interactions between molecules and membranes. ACS Appl. Bio Mater. 4, 620–629 (2021).
Gudheti, M. V., Mlodzianoski, M. & Hess, S. T. Imaging and shape analysis of GUVs as model plasma membranes: Effect of trans DOPC on membrane properties. Biophys. J. 93, 2011–2023 (2007).
L’Heureux, A. et al. Aminodifluorosulfinium salts: selective fluorination reagents with enhanced thermal stability and ease of handling. J. Org. Chem. 75, 3401–3411 (2010).
Lu, N., Yang, K., Yuan, B. & Ma, Y. Molecular response and cooperative behavior during the interactions of melittin with a membrane: dissipative quartz crystal microbalance experiments and simulations. J. Phys. Chem. B 116, 9432–9438 (2012).
Lee, M.-T., Sun, T.-L., Hung, W.-C. & Huang, H. W. Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. USA 110, 14243 (2013).
Krauson, A. J., He, J. & Wimley, W. C. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 134, 12732–12741 (2012).
Liu, J. et al. Molecular details on the intermediate states of melittin action on a cell membrane. Biochim. Biophys. Acta Biomembr. 1860, 2234–2241 (2018).
Deng, Z., Li, J., Yuan, B. & Yang, K. Residue-specialized membrane poration kinetics of melittin and its variants: Insight from mechanistic landscapes. Commun. Theor. Phys. 71, 887–902 (2019).
Hong, J. et al. How melittin inserts into cell membrane: conformational changes, inter-peptide cooperation, and disturbance on the membrane. Molecules 24, 1775 (2019).
Sancho-Vaello, E. & Zeth, K. Antimicrobial peptides: has their time arrived? Future Microbiol. 10, 1103–1106 (2015).
Suk, J. S., Xu, Q., Kim, N., Hanes, J. & Ensign, L. M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 99, 28–51 (2016).
Guiotto, A. et al. PEGylation of the antimicrobial peptide nisin A: problems and perspectives. Il Farmaco 58, 45–50 (2003).
Veronese, F. M. Peptide and protein PEGylation: a review of problems and solutions. Biomaterials 22, 405–417 (2001).
Gullapalli, R. P. & Mazzitelli, C. L. Polyethylene glycols in oral and parenteral formulations—A critical review. Int. J. Pharm. 496, 219–239 (2015).
Imura, Y., Nishida, M., Ogawa, Y., Takakura, Y. & Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta Biomembr. 1768, 1160–1169 (2007).
Zhao, L., Liu, Y., Chang, R., Xing, R. & Yan, X. Supramolecular photothermal nanomaterials as an emerging paradigm toward precision cancer therapy. Adv. Funct. Mater. 29, 1806877 (2019).
Acknowledgements
This work is supported by the National MCF Energy R&D Program (2018YFE0306105) and the National Natural Science Foundation of China (Nos. 21422404, 21774092, U1532108, U1932121, 51725204, 21771132, 21471106, 51972216, and 21728502). K.Y. and B.Y. acknowledge the support of the Natural Science Foundation of Jiangsu Province of China (Nos. BK20171207 and BK20171210). Z.K. acknowledges the Natural Science Foundation of Jiangsu Province (BK20190041, BK20190828), the Key-Area Research and Development Program of Guangdong Province (2019B010933001), the Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the 111 Project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yuan, B., Liu, J., Deng, Z. et al. A molecular architectural design that promises potent antimicrobial activity against multidrug-resistant pathogens. NPG Asia Mater 13, 18 (2021). https://doi.org/10.1038/s41427-021-00287-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-021-00287-y
This article is cited by
-
Insight into Biophysicochemical Principles of Biopolymers through Simulation and Theory
Chinese Journal of Polymer Science (2023)
