
Abstract
Ferroptosis induced by detrimental accumulation of lipid peroxides has been recently linked to a variety of pathological conditions ranging from acute tissue injuries to chronic degenerative diseases and suppression of ferroptosis by small chemical inhibitors is beneficial to the prevention and treatment of these diseases. However, in vivo applicable small chemical ferroptosis inhibitors are limited currently. In this study, we screened an alkaloid natural compound library for compounds that can inhibit RSL3-induced ferroptosis in HT1080 cells and identified a group of bisbenzylisoquinoline (BBIQ) compounds as novel ferroptosis-specific inhibitors. These BBIQ compounds are structurally different from known ferroptosis inhibitors and they do not appear to regulate iron homeostasis or lipid ROS generation pathways, while they are able to scavenge 1,1-diphenyl-2-picryl-hydrazyl (DPPH) in cell-free reactions and prevent accumulation of lipid peroxides in living cells. These BBIQ compounds demonstrate good in vivo activities as they effectively protect mice from folic acid-induced renal tubular ferroptosis and acute kidney injury. Several BBIQ compounds are approved drugs in Japan and China for traditional uses and cepharanthine is currently in clinical trials against SARS-CoV-2, our discovery of BBIQs as in vivo applicable ferroptosis inhibitors will expand their usage to prevent ferroptotic tissue damages under various pathological conditions.
Similar content being viewed by others
Introduction
Ferroptosis is a non-apoptotic cell death that is triggered by lethal accumulation of lipid peroxides generated from membrane polyunsaturated fatty acid (PUFA) or polyunsaturated ether phospholipids (PUFA-ePLs) by lipoxygenases or non-enzymatic Fenton reaction in an iron-dependent manner [1,2,3]. To defense against ferroptotic cell death, cells have evolved to use multiple antioxidant pathways to remove detrimental lipid peroxides. Among them, the GPX4/GSH pathway is the best characterized pathway that plays a central role in anti-ferroptosis defense. Disruption of this pathway by inhibition of GPX4 (by RSL3) or GSH metabolism (by erastin) is able to induce ferroptosis in a variety of cell types [1, 4]. Additional pathways such as the FSP1/CoQ10 pathway [5, 6], the GCH1/BH4 pathway [7, 8], and the DHODH pathway [9] have been recently identified to play defensive roles against ferroptosis under certain conditions. In addition, small chemical radical trapping antioxidants (RTAs) such as ferrostatin-1 and liproxstatin-1 have been developed as ferroptosis-specific inhibitors and are widely used in in vitro studies [1, 10].
Investigation of Gpx4-deficiency mice reveals multiple in vivo roles of ferroptosis in development and diseases. Gpx4 is highly expressed in the proximal tubules of kidney and inducible disruption of Gpx4 leads to ferroptotic cell death of proximal tubular epithelia and acute kidney failure in mice [10]. Ferroptosis is also observed in renal tubular cells under ischemia-reperfusion injury [11], or acute kidney injury (AKI) induced by oxalate crystal [11], folic acid [12], or cisplatin [13]. Chemotherapeutic compound such as doxorubicin or ischemia/reperfusion-induced ferroptosis is involved in cardiomyopathy as well [14]. Tissue-specific functions of Gpx4 have been investigated in additional knockout mouse lines. For example, neuron-specific knockout of Gpx4 in mice results in motor neuron degeneration and paralysis [15]. Hepatocyte-specific knockout of Gpx4 induces extensive liver degeneration [16]. Heterozygous disruption of Gpx4 in intestinal epithelial cells sensitizes mice to PUFA-rich diet-induced mucosal inflammation and enteritis [17] and ferroptosis is also involved in dextran sodium sulfate (DSS) induced ulcerative colitis [18]. In most of these abovementioned studies, inhibition of ferroptosis is generally able to ameliorate tissue damages, indicating that ferroptosis promotes the pathogenesis of these diseases.
The involvement of ferroptosis in multiple diseases promotes the development of small chemical ferroptosis inhibitors. Several RTAs have been developed as ferroptosis inhibitor thus far. While being very effective in cell culture, their activities in vivo are limited. In this study, we aimed to identify ferroptosis inhibitors with novel chemical structures and improved in vivo activities. We performed high throughput screen in an alkaloid natural compound library for compounds that are capable of inhibiting RSL3-induced ferroptosis in HT1080 cells and identified a group of BBIQ compounds as novel ferroptosis inhibitors. Several of them demonstrates promising in vivo activities and might be used as lead compounds for further anti-ferroptosis drug development.
Results
Identification of BBIQ compounds as ferroptosis inhibitors
To identify potential small chemical ferroptosis inhibitors from natural products, we first screened an alkaloid natural compound library for compounds that can inhibit RSL3-induced ferroptosis in HT1080 cells. Cells cultured in 96-well plates were co-treated with RSL3 (2 µM) and a testing compound (5 µM) for 24 h and cell viability was then determined using the CCK-8 Kit. Assays were repeated three times (Tested compounds and their rescuing activities are listed in Supplementary Table 1) and the top 20 hits from the screen were further tested for their abilities to inhibit cell death induced by a variety of reagents (ferroptosis induced by RSL3 and erastin, necrosis induced by H2O2 and rotenone, apoptosis induced by staurosporine and autophagic cell death induced by rapamycin) (Fig. 1A). We found these compounds effectively inhibit RSL3 or erastin-induced ferroptosis but are less effective to cell death induced by other cell death stimuli tested, indicating they are ferroptosis-specific inhibitors. We analyzed the chemical structures of these compounds and found that, interestingly, eight out of the top 20 hits (berbamine, fangchinoline, cepharanthine, neferine, liensinine, isoliensinine, daurisoline, and dauricine) share similar scaffold structure (two benzylisoquionline units linked via oxygen bridge(s)) and are members of the BBIQ alkaloids (Fig. 1B). These BBIQ compounds are structurally different from known ferroptosis inhibitors such as ferrostatin-1 or liproxstatin-1 and might constitute a novel type of ferroptosis inhibitor, so we further characterized them in this study. We evaluated cellular toxicity of these compound in HT1080 cells and found they do not induce obvious toxicity at concentrations up to 10 µM while they show variable degrees of cytotoxicity at 15 µM or higher concentrations), which is generally in between ferrostatin-1 and liproxstatin-1, two widely used ferroptosis inhibitors (Supplementary Fig. 1A). We then analyzed dose response of these BBIQs to block ferroptosis and found that cepharanthine or fanchinoline at 0.5 µM is sufficient to inhibit RSL3-induced ferroptosis, while other compounds need higher dose (5–10 µM) to effectively inhibit ferroptosis (Fig. 1C and Supplementary Fig. 1B). We then tested their anti-ferroptosis activities in additional cell lines and found they also effectively inhibit RSL3-induced ferroptosis in cell lines such as lung carcinoma cell line H1975, glioma cell line U251, breast adenocarcinoma cell line MDA-MB-231 and hepatocellular carcinoma cell line HepG2 (Supplementary Fig. 2). Together, these results suggest BBIQs as a novel class of ferroptosis inhibitors.

A An overview of the top 20 hit compounds from our anti-ferroptosis screen to block cell-death induced by various stimuli in HT1080 cells. Cells were treated with RSL3 (2 µM), erastin (5 μM), staurospourine (1 μM), rotenone (3 μM), H2O2 (50 μM), or rapamycin (10 μM) in the presence or absence of a testing compound (5 µM) and cell viability analyzed at 24 h after treatment. Compounds are listed in descending order based on their rescuing activity to RSL3-induced ferroptosis. BBIQ compounds are labeled in red. B Structure of the eight BBIQ compounds investigated in this study. C Comparison of the anti-ferroptosis activities of listed compounds at 0.5 and 5 µM. Data represent mean ± s.d. from three independent experiments and p value (vs. RSL3 alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ns, no significance; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
BBIQs block RSL3-induced accumulation of lipid ROS
Lipid peroxidation is a hallmark of ferroptosis and known ferroptosis inhibitors generally function to reduce the accumulation of detrimental lipid peroxides by either blocking lipid peroxidation (such as iron chelators) or functioning as radical-trapping antioxidants (such as ferrostatin-1 and liproxstatin-1). In previous studies, many BBIQ family compounds have been reported to modulate cellular ROS levels (either decrease or increase ROS level depending on samples analyzed). We speculated that BBIQs might regulate the level of various ROS species during RSL3-induced ferroptosis. To test this hypothesis, we first analyzed cytosolic ROS level upon RSL3 treatment. HT1080 cells were treated with RSL3 in the presence or absence of BBIQ compound for 4 h then cellular ROS levels were determined by DCFH-DA immunofluorescence staining and FACS analysis. We found that RSL3 treatment elevates cytosolic ROS level while co-treatment with ferrostatin-1 (positive control), cepharanthine, or other BBIQs all block the induction of cytosolic ROS (Fig. 2A and Supplementary Fig. 3A). FACS analysis confirmed the suppression of RSL3-induced cytosolic ROS by BBIQs (Fig. 2B, C). We next analyzed lipid ROS levels in those treated cells by C11-BODIPY staining and found that these BBIQs effectively block RSL3-induced accumulation of lipid ROS (Fig. 3 and Supplementary Fig. 3B). On the other hand, we found that BBIQs are not able to block rotenone-induced mitochondrial superoxide (detected by MitoSOX staining) (Fig. 4 and Supplementary Fig. 3C). Together, these results indicate that BBIQs inhibit ferroptosis through their downregulation of lipid ROS.

A Representative images of cytosolic ROS immunofluorescence staining in HT1080 cells. Cells cultured on coverslips were treated with RSL3 (2 μM) plus a testing compound (5 μM) or ferrostatin-1 (5 μM) for 4 h, labeled with the DCFH-DA probe (10 μM for 30 min) then cytosolic ROS levels were analyzed by confocal imaging. Scale bar: 50 μm. B Cytosolic ROS assessed by flow cytometry. Cells were treated as in (A) then analyzed using the BD Accuri C6 Plus Flow Cytometer. C Statistical results for (B). Data represent mean ± s.d. from three biological repeats and p value (vs. RSL3 alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ***, p < 0.001.

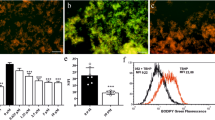
A Representative images of immunofluorescence staining of lipid ROS. Cells were treated as described in Fig. 2A then stained with the C11 BODIPY 581/591 probe (2 μM for 20 min) and analyzed by confocal imaging. Scale bar: 50 μm. B Flow cytometry analysis of RSL3-induced oxidation of C11-BODIPY. C Statistical results for (B). Data represent mean ± s.d. from three independent repeats. p value (vs. RSL3 alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ***, p < 0.001.

A Representative immunofluorescence staining of mitochondrial ROS. Cells were treated with rotenone (2 μM) in the presence of a testing BBIQ compound (5 μM) or NAC (5 mM) for 4 h, stained with the MitoSOX Red Mitochondrial Superoxide Indicator (5 μM for 10 min) then analyzed by confocal imaging. Scale bar: 50 μm. B Flow cytometry analysis of rotenone-induced mitochondrial ROS. C Statistical results for (B). Data represent mean ± s.d. from three independent repeats and p value (vs. rotenone alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ns, no significance; **, p < 0.01; ***, p < 0.001.
BBIQs have intrinsic ROS scavenging activities
Previous studies have identified multiple pathways involved in the homeostasis of lipid ROS and regulation of ferroptosis. We analyzed whether or not BBIQs modulate ferroptosis through these known pathways. We first determined protein levels of key ferroptosis-related factors by western blot and found that, either in the unstimulated or RSL3-stimulated cells, protein levels of key factors involved in iron metabolism (TFRC, FTH1), lipid synthesis and peroxidation (ACSL4, ALOX15), antioxidant defense (SLC7A11, GPX4, FSP1, NFE2L2) and additional ferroptosis regulator such as TP53 are not affected by BBIQ treatment (Fig. 5A). These results indicate that BBIQs do not block ferroptosis through their regulation of these pathways. We then investigated whether BBIQs have intrinsic antioxidant activities and can act as RTAs to remove ROS in cell-free assays. We found that these BBIQs (at 5, 10, or 50 µM) all have radical scavenging activities to the stable free radical DPPH in cell-free reactions (Fig. 5B). On the other hand, in a hydroxyl free radical scavenging capacity assay, we found that BBIQs (50 µM) are not effective in reducing hydroxyl free radical generated from H2O2 through Fenton reaction (Fig. 5C). Together with the observation that BBIQs do not protect cells from H2O2-induced cell death (Fig. 1A), these data indicate that BBIQs function preferentially as RTAs for lipid ROS which underlies their anti-ferroptotic activities.

A Representative western blots for key ferroptosis regulators. BBIQ compounds do not affect the homeostasis of all proteins examined in either unstimulated or RSL3-stimulated HT1080 cells. Assays were repeated twice with similar results. B Cell-free DPPH free radical scavenging assay. BBIQs were tested at 1, 5, 10, and 50 μM. Z-VAD-FMK and necrostatin-1 were tested at 50 μM and NAC at 5 mM. Data represent mean ± s.d. from three independent repeats and p value (vs. DMSO) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ns, no significance; **, p < 0.01; ***, p < 0.001. C Cell-free hydroxyl radical scavenging capacity assay. The concentration for NAC is 5 mM. Data represent mean ± s.d. from three independent repeats and p value (vs. DMSO) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ns, no significance; ***, p < 0.001.
BBIQs protect folic acid-induced acute kidney injury (FA-AKI) in mice
A variety of in vivo activities have been documented in literatures for BBIQ compounds, ranging from antiarrhythmic, antioxidants, antitumor, anti-inflammation, to protective activities against tissue damages induced by ischemia/reperfusion injury, chemotherapeutic reagents, etc. Our revelation of BBIQs as ferroptosis inhibitors indicate that some of their abovementioned functions might be attributed to their anti-ferroptotic activities in vivo. To evaluate whether BBIQs can be effectively used as ferroptosis inhibitors in vivo, we evaluated their cyto-protective activities in the nephrotoxic folic acid-induced acute kidney injury model where ferroptosis is the primary cause of tissue damage [12]. Cepharanthine, fangchinoline, daurisoline, and isoliensinine were chosen for this in vivo experiment because of their high in vitro anti-ferroptosis activity (cepharanthine and fangchinoline, Fig. 1C) or relative low cytotoxicity (daurisoline and isoliensinine, Supplementary Fig. 1A). Mice were pretreated intragastrically with cepharanthine, fangchinoline, daurisoline, or isoliensinine (all at 20 mg/kg) then injected intraperitoneally with a single dose of folic acid (250 mg/kg) and mice were analyzed 48 h after folic acid treatment. We first compared the levels of blood urea nitrogen (BUN) and plasma creatinine between control and treatment groups and found that the four BBIQs tested all reduce the BUN and plasma creatinine levels in folic acid-treated animals (Fig. 6A). Furthermore, the folic acid-induced expression of acute kidney injury marker ngal is also blocked by these BBIQs as determined by qRT-PCR (Supplementary Fig. 4A), indicating reduced renal injury and improved renal function in BBIQ-treated animals. We then performed histological analysis of kidney tissue in these animals and found the folic acid-induced renal damages is also reduced in BBIQ-rescued groups (Fig. 6B). Immunohistochemistry staining of kidney injury molecule-1 (KIM-1), an early biomarker of nephrotoxicity, further confirmed that BBIQs ameliorate the folic acid-induced AKI (Fig. 6C). Furthermore, we observed folic acid-induced renal infiltration of CD68 or F4-80 positive monocytes/macrophages is also inhibited by BBIQ treatment (Fig. 6D), indicating reduced inflammation in BBIQ-treated animals. We then analyzed the expression levels of several pro-inflammatory cytokines/chemokines by qRT-PCR and found that BBIQs effectively blocked the folic acid-induced renal expression of mcp-1, il-6, and tnf-α (Supplementary Fig. 4B), which further supports that BBIQ treatment ameliorates folic acid-induced renal inflammation and tissue damage.

C57BL/6J mice were randomly grouped (7 animals per group) and pretreated with the indicated testing compounds (po, 20 mg/kg) then treated with folic acid (ip, 250 mg/kg) for 48 h. Mice were then sacrificed and samples prepared for further analysis. Assays were repeated once. A Assessment of renal function in all treated mice by levels of blood urea nitrogen (BUN) and plasma creatinine. Data represent mean ± s.d. of 14 animals from two independent repeats and p value (vs. folic acid alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ns, no significance; *, p < 0.05; **, p < 0.01; ***, p < 0.001. B Representative H&E staining images showing kidney tubular injuries in folic acid-treated mice which is ameliorated by administration of BBIQs. C Representative immunohistochemistry staining images for kidney injury marker KIM-1 in samples analyzed in (B). D Immunohistochemistry analysis for the infiltration of CD68 or F4-80 positive monocytes/macrophages in samples as in (B). Scale bars in (B–D): 50 µm.
Our in vitro discovery that BBIQs inhibit ferroptosis by preventing the accumulation of detrimental lipid ROS indicates that similar mechanism could underlie renal protective function of BBIQs in FA-AKI. To test this possibility, we performed immunohistochemistry staining of 4-hydroxynonenal (4-HNE), a byproduct of lipid peroxidation, and revealed that all BBIQs reduce the formation of folic acid-induced 4-HNE in renal sections (Fig. 7A). We then analyzed cell death levels in the same samples by TUNEL assay and found the folic acid-induced cell death is also blocked by BBIQ treatment (Fig. 7B, C). Together, these date indicate that BBIQs are able to suppress accumulation of lipid ROS, reduce renal cell death and damage-induced inflammation and improve renal function in folic acid-treated mice.

A Representative immunohistochemistry staining results for 4-HNE, which is a byproduct of lipid peroxidation and a marker for oxidative stress. Scale bar: 50 µm. B Representative images of TUNEL assay to assess the level of cell death in renal sections. Scale bar: 50 µm. C Quantitative results for (B). Data represent mean ± s.d. of (TUNEL-positive count per 1000 cells) in sections from 7 mice for each treatment and p value (vs. folic acid alone) is determined by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. ***, p < 0.001.
Discussion
Several BBIQ alkaloids identified as novel ferroptosis inhibitors in this study have been extensively investigated in previous studies. A variety of functions (sometimes contradictory) from cyto-protective activities (such as antioxidant activity, anti-inflammation, and immune-modulation, etc.) to anticancer activities (anti-proliferation and metastasis, antidrug resistance, induction of cell death, etc) have been attributed to them [19,20,21,22,23]. As long as ferroptosis is concerned, cepharanthine, dauricine, and neferine are recently reported to be involved in the regulation of ferroptosis in certain cells. Cepharanthine inhibits RSL3-induced ferroptosis in endothelial cells and attenuate brain injury after subarachnoid hemorrhage (SAH), presumably through downregulation of ALOX15 which is a positive regulator of ferroptosis [24]. However, ALOX15 has been shown to be dispensable for ferroptosis induced by GPX4-depletion in fibroblasts, T-cells, or renal tissues [10, 25] and a prevailing role of ALOX15 in ferroptosis is debatable [26]. Thus, it remains to be investigated whether cepharanthine can protect cells from ferroptosis in an ALOX15-independent manner. Meanwhile, dauricine protects ferroptotic brain damage after intracerebral hemorrhage through upregulating GPX4 and glutathione reductase (GSR) [27]. On the other hand, neferine which is another BBIQ compound we investigated in this study, is reported to promote apoptosis and ferroptosis in thyroid cancer cell lines through downregulation of NRF2 pathway [28]. We reported here that, in the well-documented RSL3-induced ferroptosis assay in HT1080 cells, BBIQs including cepharanthine, dauricine, and neferine do not affect protein levels of ferroptosis regulators including ALOX15, GPX4, or NRF2 (also known as NFE2L2). Instead, they can directly function as RTAs to scavenge lipid ROS. This observation suggests BBIQs to be protective against all ferroptotic stimuli. Thus, we propose that the lipid ROS scavenging activities of cepharanthine and dauricine contribute to their reported protective roles in hemorrhage-induced brain injury and other BBIQs we identified here are expected to have similar protective activity as well.
BBIQs have been reported to modulate ROS species in various cell types or cell-free systems either positively or negatively. For example, cell-free studies indicate cepharanthine can directly scavenge hydroxyl radicals or DPPH and inhibit lipid peroxidation in mitochondria initiated by Fe2+/ADP [29, 30]. Meanwhile, studies in certain cancer cell lines reported that cepharanthine can stimulate ROS generation and promote cell death in doxorubicin-resistant murine leukemia cells [31], human hepatocellular carcinoma cells [32], non-small-cell lung cancer cells [33], or glioma and neuronal cells [34]. Thus, the ROS scavenging activity in cell-free systems does not always predict a ROS-reducing effect in living cells. In this study, we found that cepharanthine scavenges DPPH (but not hydroxyl radical) and inhibits RSL3 or erastin-induced lipid peroxidation in HT1080 cells. Can ROS scavenging activity in DPPH assay be used to predict anti-ferroptosis capacity? A recent study revealed that many compounds with high radical scavenging activity in cell-free DPPH assay do not inhibit ferroptosis in cells [35], possible due to factors such as the relatively high compound dose used in cell-free assays which is likely to induce artificial effect, the radical species and their distribution in cell-free system are very different from those critical for ferroptosis, cell-free assay cannot evaluate the ability of a compound to partition into lipid environments in living cells, etc. Thus, the potential anti-ferroptosis capacity of a ROS scavenger need to be directly evaluated in well-defined and well-controlled experimental systems.
Although the reported functions of BBIQ alkaloids in cancer cells or other in vitro cultured cells are often contradictory, a general theme from in vivo studies is that BBIQs protect tissue damages under a variety of pathogenic conditions. For example, cepharanthine have been reported to protect renal injuries induced by ischemia/reperfusion [36, 37], chemotherapeutic reagent cisplatin [38], streptozotocin-induced diabetic nephropathy [39], transient middle cerebral artery occlusion (tMCAO) or subarachnoid hemorrhage induced brain injury [24, 40] or dextran sulfate sodium (DSS)-induced ulcerative colitis [41]. Protective functions of fangchinoline have been reported in models of oxidative glutamate toxicity [42], streptozotocin-induced diabetic nephropathy [43] as well as or LPS-induced cardiac dysfunction [44]. In addition to its well-studied anti-hypertension and anti-arrhythmia function [45], berbamine is reported to protect ischemia/reperfusion-induced heart injury [46, 47] and ethanol-induced liver damage [48]. Neferine is reported to attenuate ischemia/reperfusion or LPS-induced AKI [49], cerebral ischemia-induced brain damage [50], and DSS-induced ulcerative colitis [51]. Liensinine is reported to ameliorate doxorubicin-induced cardiomyopathy [52] and isoliensinine inhibits bleomycin-induced pulmonary fibrosis [53]. Although a variety of mechanisms such as anti-oxidation, immune-modulation, or modulation of gut microbiota have been proposed, direct targets and prevailing pathways responsible for the cyto-protective functions of BBIQs in these disease models remain to be established. In light of recent discoveries about the involvement of ferroptosis in an expanding list of pathological conditions [26, 54, 55], our discovery of BBIQs as ferroptosis inhibitors provide novel opportunities to investigate tissue protective mechanisms of BBIQs in these disease models.
The coronavirus disease 2019 (COVID-19) pandemic caused by the infection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) promotes the identification and development of small chemical compounds with antiviral activities. In vitro cell culture-based SARS-CoV-2 infection assays have been used by many research laboratories to screen a variety of compound libraries and several BBIQs were repeatedly identified as antiviral candidate compounds. For example, cepharanthine and berbamine were identified as potential anti-SARS-CoV-2 agents by several groups [56,57,58]. Further studies reported that cepharanthine blocks SARS-CoV-2 entry into target cells [59, 60] while berbamine inhibits SAR-CoV-2 infection by interfering endolysosomal trafficking of ACE2 [61] or blocking the S protein-mediated membrane fusion [62]. Neferine suppresses SARS-CoV-2 infection by inhibiting Ca2+-dependent membrane fusion and virus entry [63]. Daurisoline inhibits replication of SARS-CoV-2 in host cells possibly through an autophagy-related mechanism [64]. These studies revealed a conserved antiviral activity of these BBIQs while the proposed antiviral mechanism for each compound is largely based on previously reported activity of the compound. Our finding that these BBIQs have a common mechanism as RTAs to restrict lipid peroxidation and block ferroptosis indicates that BBIQs might have additional function and mechanism during virus infection, especially in light of possible ferroptotic tissue damages induced by SARS-CoV-2 infection as reported in recent studies [65,66,67].
In summary, our study identified a group of in vivo applicable ferroptosis inhibitors which may facilitate in vivo ferroptosis studies and provide novel opportunity to protect ferroptotic tissue damages under a variety of pathological conditions.
Materials and methods
Compounds
Alkaloid natural product library was obtained from TargetMol (TargetMol, Shanghai, China, L6110). RSL3 (Selleckchem, Houston, TX, S8155) and erastin (Selleckchem, S7242) were used to induce ferroptosis. Staurosporine (Selleckchem, S1421), rapamycin (Selleckchem, S1039), rotenone (Selleckchem, S2348)/H2O2 (Sigma-Aldrich, MO, 31642) were used to induce apoptosis, autophagy, and necrosis, respectively. Ferrostatin-1 (Selleckchem, S7243)/liproxstatin-1 (Selleckchem, S7699), Z-VAD-FMK (Selleckchem, S8102), 3-methyladenine (Selleckchem, S2767), necrostatin-1 (Selleckchem, S8037) were used to inhibit ferroptosis, apoptosis, autophagic cell death and necrosis, respectively. N-acetylcysteine (NAC) (Selleckchem, S1623) is an antioxidant that removes ROS. BBIQ compounds used in the study were all obtained from Selleckchem which include fangchinoline (S3611), cepharanthine (S4238), berbamine (S9141), dauricine (S9295), daurisoline (S9150), neferine (S5144), isoliensinine (S9247), liensinine (S9411).
Cell culture
HT1080 (ATCC, CCL-121), MDA-MB-231 (ATCC, CRM-HTB-26), and H1975 (ATCC, MD, CRL-5908) cells were purchased from ATCC. HepG2 and U251 cells were from stocks maintained in our lab. Cells were routinely tested for mycoplasma contamination. HT1080, U251, and HepG2 cells were cultured in DMEM High Glucose cell culture medium (HyClone, UT, SH3022.01) supplemented with 10% fetal bovine serum (NTC, Argentina, SFBE) and 1% Penicillin-Streptomycin Solution (HyClone, SV30010). MDA-MB-231 and H1975 cells were cultured in RPMI 1640 cell culture medium (Gibico, CA, C11875500BT) supplemented with 10% fetal bovine serum (NTC, SFBE) and 1% Penicillin-Streptomycin Solution (HyClone, SV30010). Cells were all cultured at 37 °C with 5% CO2 (Thermo, MA, HERACELL150i).
Compound treatment
For alkaloid natural product library screen, HT1080 cells in 80% confluency were trysinized to single cell suspension, seeded on 96-well plates at the density of 8000 cells per well for 24 h, then treated with RSL3 (2 μM) plus a testing compound (5 μM) diluted in culture media. Cell viability was determined 24 h after treatment by CCK-8 assay as described below. Assays were performed in triplicates and repeated three times. The top 20 hits from the screen were further tested for their abilities to protect HT1080 cells from cell death induced additional stimuli which included erastin (5 μM), staurosporine (1 μM), rapamycin (10 μM), rotenone (3 μM) or H2O2 (50 μM). Ferrostatin-1/liproxstatin-1 (5 μM), NAC (1 mM), Z-VAD-FMK (50 μM), 3-methyladenine (5 mM), and necrostatin-1 (50 μM) were used as control. For the eight BBIQ compounds in the top 20 hits, dose responses in range of 0.5 to 30 μM were assayed in HT1080 cells and their anti-ferroptotic activity were further tested in additional cell lines (H1975, U251, MDA-MB-231, and HepG2). Cells cultured in their own growth media were treated and assayed using the same protocol described above.
Cell viability assay
Cell viability was assayed using the Cell Counting Kit-8 (CCK-8) (Biosharp, Beijing, China, BS350B) with manufacturer’s protocol. Briefly, the CCK-8 stock solution was diluted (1:10) in complete cell culture medium to make the working solution. At the end of compound treatment, cell culture medium was removed and replaced with 110 μl of CCK-8 working solution and cells were cultured at 37 °C in dark for 1 h then put into an automatic microplate reader (Thermo, Epoch 2) to measure OD450. Cell viability was then calculated as following: Cell Viability = [(Test Group OD450 − Blank group OD450)/(Control Group OD450 − Blank Group OD450)] × 100%. All assays were performed in triplicates and repeated at least three times.
Flow cytometry analysis of ROS
HT1080 cells were seeded on 12-well plates at the density of 15,000 cells per well overnight then treated with RSL3 (2 μM) or rotenone (2 μM) in the presence or absence of ferrostatin-1 (5 μM), NAC (5 mM), or a testing BBIQ compound (5 μM). Cells were treated for 4 h at 37 °C, then washed 3 times with FBS-free DMEM cell and ready for fluorescent dye labeling. For detection of cytosolic ROS, DCFH-DA probe (Beyotime, Shanghai, China, S0033S) was diluted in serum-free medium to final concentration of 10 μM. Cells at the end of drug treatment were incubated in DCFH-DA working solution for 30 min at 37 ˚C in dark. For detection of lipid ROS, cells were incubated in the C11 BODIPY 581/591 lipid ROS probe (Invitrogen, NY, D3861) diluted in serum-free media at the final concentration of 2 μM for 20 min in dark. For detection of mitochondrial ROS, cells were stained in 5 μM of MitoSOX Red Mitochondrial Superoxide Indicator (Invitrogen, M36008) for 10 min in dark. After dye incubation, cells were washed 3 times with serum-free cell culture medium, trypsinized with 100 μl 0.25% Trypsin-EDTA (Gibico, 25200056). Trypsinization was terminated by adding 1 ml of complete DMEM culture medium and cells were centrifuged at 1300 rpm for 3 min and then resuspended in sterile PBS containing 5% FBS. Cell suspension was filtrated by a 70-μm filter, and then analyzed using the BD Accuri C6 Plus Flow Cytometer (BD, NJ) and the BD FlowJo V10 software.
Immunofluorescence staining of ROS
HT1080 cells were seeded on coverslip (WPI, FL, FD35–100) at the density of 150,000 cells per 30-mm dish or 24 h. Cells were then treated and stained with probes for various ROS species as described above. After ROS labeling, cells were washed then incubated in Hoechst 33258 (1:1000 diluted in serum-free DMEM medium) for 5 min. Cells were then washed with DMEM medium and imaged using the LSM800 fluorescence confocal microscope (Zeiss, Germany).
Western blotting
Cells cultured in 30-mm dish were treated with the indicated compound as described above. After compound treatment, 300 µl RIPA (Thermo, 89901) containing 1% PMSF (Yeasen, Shanghai, China, 20104ES03) and 1% protease inhibitor cocktail (Yeasen, 20123ES50) was added to cell culture and samples were put on ice for 10 min. The mixture was then centrifuged at 12,000 pm for 10 min at 4 °C. The supernatant was transferred to a new EP tube, mixed thoroughly with 1/4 volume of the loading dye (Beyotine, P0015), and incubated in boiling water for 15 min. Samples were centrifuged at 13,000 rpm for 3 min and supernatants were separated by 12% SDS-PAGE, transferred to PVDF membranes (EMD Millipore, MA, IPVH00010). Membranes were blocked in solution (5% skim milk in TBST) for 1 h, washed with TBST for 3 times, then incubated with a primary antibody diluted in QuickBlock™ Primary Antibody Dilution Buffer for Western Blot (Beyotime, P0256) overnight at 4 °C. Blots were then washed with TBST and incubated with a HRP-conjugated secondary antibodies (Goat Anti-Mouse IgG (H + L) (Proteitech, Wuhan, China, SA00001-1) or Goat Anti-Rabbit IgG (H + L) (Proteitech, SA00001-2)) for at RT for 1 h. Blots were then washed with TBST extensively and detected using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, WBKLS0500). The primary antibodies used in this study were: anti-GPX4 (1:1000, Abcam, Cambridge, UK, ab125066), anti-SLC7A11 (1:1000, Proteintech, 26864-1), anti-ACSL4 (1:1000, Abcam, ab205199), anti-FTH1 (1:1000, Abcam, ab75973), anti-TFRC (1:1000, Abcam, ab269513), anti-ALOX15 (1:1000, GeneTex, CA, GTX33001), anti-FSP1 (1:1000, Affinity, OH, DF8636), anti-TP53 (1:1000, Affinity, BF8013), anti-NFE2L2 (1:1000, Abcam, ab62352), anti-GAPDH (1:1000, Proteintech, HRP-60004).
DPPH assay
The free radical scavenging capacity of BBIQ compounds was assayed using the DPPH Assay Kit (Solarbio, Beijing, China, BC4755). Briefly, testing compounds were diluted in the provided solution to the concentration of 1, 5, 10, or 50 µM. An aliquot of testing compound solution (10 µl) was mixed thoroughly with 190 µl of DPPH working solution in 96-well plate and incubated at RT in dark for 30 min. After reaction, the absorbance (A) of the reaction mixture at the wave length of 515 nm was assayed using an automatic microplate reader (Thermo, Epoch 2). The DPPH scavenging rate is calculated by the formula: positive control: DVC% = [(A blank − A positive control)/A blank] × 100%; sample: D% = [[A blank − (A sample − A control)]/A blank] × 100%.
Hydroxyl radical assay
The hydroxyl radical scavenging capacity of a compound was assayed with the hydroxyl radical scavenging ability detection Kit (Solarbio, BC1320). Briefly, a testing compound was diluted in the provided solution to the final concentration of 50 µM. NAC (5 mM) was used as positive control. Compound solution was mixed with the hydroxyl radical working solution and incubated in 37 °C water bath for 60 min. Samples were then centrifuged at 10,000 rpm at RT for 10 min, supernatant was taken to measure absorbance at 536 nm. The hydroxyl radical scavenging rate is calculated by the formula: D% = (A sample − A control)/(A blank − A control) × 100%.
Folic acid-induced acute kidney injury (FA-AKI) model
Animal procedures were approved by the Institutional Animal Care and Use Committee of Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Science (Issue No. 2021034). Ferroptosis is the primary cause of folic acid-induced acute kidney injury so we used this protocol to evaluate in vivo anti-ferroptosis activity of BBIQs. C57BL/6J mice (8–10 weeks old) were purchased from Vital River Laboratory Animal Technology Company (Zhejiang, China) and were adapted to the housing conditions for one week before compound treatment. To induce AKI, mice (7 per experimental group) received a single intraperitoneal injection of FA (Sigma-Aldrich, F7876) dissolved in 0.3 M sodium bicarbonate at the dosage of 250 mg/kg. The control group was injected with vehicle (0.3 M sodium bicarbonate, 200 µl). BBIQs generally have limited solubility and bioavailability in vivo, so mice were pretreated with BBIQs before FA treatment. Specifically, cepharanthine, fangchinoline, daurisoline, or isoliensinine were firstly dissolved in DMSO (100 mg/ml) then diluted to 1.25 mg/ml in 0.5% CMC-Na (Sigma, 9004-32-4). Mice were dosed intragastrically with cepharanthine, fangchinoline, daurisoline, or isoliensinine (all at 20 mg/kg, based on results from our preliminary experiments) daily from 2-day prior to FA injection to 1-day after that (48, 24, 2 h before FA treatment plus 24 h after FA treatment). Mice were then euthanized 48 h after FA injection. Plasma samples were collected at the time of euthanasia. Kidneys were perfused in situ with cold saline before removal and one kidney was frozen in liquid nitrogen for RNA preparation and the other was perfused with cold 4% PFA and then fixed for paraffin or frozen section.
Renal function assessment
Renal function was assessed by measuring the plasma creatinine and blood urea nitrogen (BUN) levels, two routinely used parameters for detecting acute kidney injury. The blood samples collected in heparin sodium-coated blood collection tube were centrifuged at 1000 rpm for 10 min then the plasma was transferred to new 1.5 ml EP tube. Plasma creatinine levels were assayed using the Creatinine Assay Kit (Nanjing Jiancheng Bioengineering Institute, C011-2-1). Plasma BUN levels were assayed with the Urea Assay Kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China, C013-1-1) according to the manufacturer’s instructions.
H&E and immunohistochemistry staining
For H&E staining, kidneys fixed in 4% PFA were embedded in paraffin then sectioned at the thickness of 5 µm. Haematoxylin and eosin (H&E) staining of sections was performed using the protocol provided by the manufacturer (Beyotime, C0105M). For immunohistochemistry, paraffin sections were fixed in pre-cooled acetone at 4 °C for 10 min then washed with PBS. After three washes, sections were incubated in 3% H2O2/methanol for 20 min at RT, followed by PBS wash (twice, 5 min each). Sections were blocked in 5% BSA/PBS for 10 min at RT then incubated with a primary antibody (anti-KIM-1 (1:100, Immunoway, TX, YT5888), anti-F4/80 (1:100, CST, MA, 70076S), anti-CD68 (1:100, CST, 97778S), anti-4-HNE (1:100, Abcam, ab48506)) overnight at 4 °C, and then followed by secondary antibody incubation (30 min at 37 °C). Three times of PBS wash were performed before and after incubation of secondary antibody. Finally, sections were incubated with the DAB solution (Beyotime, P0202) in dark for 10 min at RT, washed with tap water, and then counterstained with haematoxylin (Beyotime, C0107). Sections were then examined and photographed with the LSM 800 fluorescence confocal microscope (Zeiss).
RNA extraction and qRT-PCR
The frozen kidney tissues were firstly homogenized in 500 µl cold saline and the FastPure Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, RC112) was used for RNA extraction according to manufacturer’s instructions. Reverse transcription of RNA was performed using the HiScript III RT SuperMix for qPCR (+gDNA wiper) (Vazyme, R323). qPCR was performed using the CFX96 Touch Real-Time PCR Detection System (BioRad, CA). Primers used in this study were: GAPDH, 5′-ATCATCCCTGCATCCACT-3′ and 5′-ATCCACGACGGACACATT-3′; NGAL, 5′-AATGTCACCTCCATCCTGGT-3′ and 5′-ATTTCCCAGAGTGAACTGGC-3′; TNF-α, 5′-CAGGCGGTGCCTATGTCTC-3′ and 5′-CGATCACCCCGAAGTTCAGTAG-3′; MCP-1, 5′-CCTGCTGTTCACAGTTGCC-3′ and 5′-ATTGGGATCATCTTGCTGGT-3′; IL-6, 5′-AGTCCGGAGAGGAGACTTCA-3′ and 5′-ATTTCCACGATTTCCCAGAG-3′.
TUNEL assay
Frozen sections were prepared from kidneys fixed in 4% PFA using the Leica CM3050S Microtome. Sections were fixed with 4% PFA again for 30 min followed by 2 PBS wash. Sections were incubated with 0.5% Triton X-100/PBS for 5 min then incubated in TUNEL solution (Beyotime, C1090) in dark at 37 °C for 60 min. Sections were counterstained with DAPI (1:5000 in PBS, Beyotime, C1005) for 5 min, washed then imaged using the LSM800 fluorescence confocal microscope (Zeiss). At least three sections from three different mice were analyzed for each treatment.
Statistical analyses
Statistical analyses were performed using the GraphPad Prism 8.0.2 software (GraphPad Software, CA). Data were collected from 3 independent biological repeats and expressed as means ± s.d. Statistical significance was determined using one-way or two-way ANOVA, and p values <0.05 were considered statistically significant. Sample sizes subjected to statistical analysis were at least 7 animals per group.
Data availability
The data in the article and its supplementary information files are available from the authors on reasonable request.
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–75.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020;585:603–8.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–31.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688–92.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693–8.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021;593:586–90.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111:16836–41.
Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrology. 2017;28:218–29.
Hu Z, Zhang H, Yi B, Yang S, Liu J, Hu J, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020;11:73.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. 2019;116:2672–80.
Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290:28097–106.
Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22–31.
Mayr L, Grabherr F, Schwarzler J, Reitmeier I, Sommer F, Gehmacher T, et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun. 2020;11:1775.
Chen Y, Zhang P, Chen W, Chen G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol Lett. 2020;225:9–15.
Rogosnitzky M, Danks R. Therapeutic potential of the biscoclaurine alkaloid, cepharanthine, for a range of clinical conditions. Pharmacol Rep. 2011;63:337–47.
Bailly C. Cepharanthine: an update of its mode of action, pharmacological properties and medical applications. Phytomedicine: Int J Phytother Phytopharmacol. 2019;62:152956.
Merarchi M, Sethi G, Fan L, Mishra S, Arfuso F, Ahn KS. Molecular targets modulated by fangchinoline in tumor cells and preclinical models. Molecules 2018;23:10.
Cheng Y, Li HL, Zhou ZW, Long HZ, Luo HY, Wen DD, et al. Isoliensinine: a natural compound with “drug-like” potential. Front Pharmacol. 2021;12:630385.
Marthandam Asokan S, Mariappan R, Muthusamy S, Velmurugan BK. Pharmacological benefits of neferine—a comprehensive review. Life Sci. 2018;199:60–70.
Gao S, Zhou L, Lu J, Fang Y, Wu H, Xu W, et al. Cepharanthine attenuates early brain injury after subarachnoid hemorrhage in mice via inhibiting 15-lipoxygenase-1-mediated microglia and endothelial cell ferroptosis. Oxid Med Cell Longev. 2022;2022:4295208.
Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–68.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–37.
Peng C, Fu X, Wang K, Chen L, Luo B, Huang N, et al. Dauricine alleviated secondary brain injury after intracerebral hemorrhage by upregulating GPX4 expression and inhibiting ferroptosis of nerve cells. Eur J Pharmacol. 2022;914:174461.
Li S, Zhang Y, Zhang J, Yu B, Wang W, Jia B, et al. Neferine exerts ferroptosis-inducing effect and antitumor effect on thyroid cancer through Nrf2/HO-1/NQO1 inhibition. J Oncol. 2022;2022:7933775.
Kogure K, Goto S, Abe K, Ohiwa C, Akasu M, Terada H. Potent antiperoxidation activity of the bisbenzylisoquinoline alkaloid cepharanthine: the amine moiety is responsible for its pH-dependent radical scavenge activity. Biochim Biophys Acta. 1999;1426:133–42.
Kogure K, Tsuchiya K, Abe K, Akasu M, Tamaki T, Fukuzawa K, et al. Direct radical scavenging by the bisbenzylisoquinoline alkaloid cepharanthine. Biochim Biophys Acta. 2003;1622:1–5.
Furusawa S, Wu J, Fujimura T, Nakano S, Nemoto S, Takayanagi M, et al. Cepharanthine inhibits proliferation of cancer cells by inducing apoptosis. Methods Find Exp Clin Pharmacol. 1998;20:87–97.
Biswas KK, Tancharoen S, Sarker KP, Kawahara K, Hashiguchi T, Maruyama I. Cepharanthine triggers apoptosis in a human hepatocellular carcinoma cell line (HuH-7) through the activation of JNK1/2 and the downregulation of Akt. FEBS Lett. 2006;580:703–10.
Hua P, Sun M, Zhang G, Zhang Y, Tian X, Li X, et al. Cepharanthine induces apoptosis through reactive oxygen species and mitochondrial dysfunction in human non-small-cell lung cancer cells. Biochem Biophys Res Commun. 2015;460:136–42.
Cierluk K, Szlasa W, Rossowska J, Tarek M, Szewczyk A, Saczko J, et al. Cepharanthine induces ROS stress in glioma and neuronal cells via modulation of VDAC permeability. Saudi Pharm J. 2020;28:1364–73.
Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, et al. A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. 2021;17:665–74.
Kusaka J, Hagiwara S, Hasegawa A, Kudo K, Koga H, Noguchi T. Cepharanthine improves renal ischemia-reperfusion injury in rats. J Surgical Res. 2011;171:212–7.
Kao MC, Chung CY, Chang YY, Lin CK, Sheu JR, Huang CJ. Salutary effects of cepharanthine against skeletal muscle and kidney injuries following limb ischemia/reperfusion. Evid Based Complement Alternat Med. 2015;2015:504061.
Sogawa N, Hirai K, Sogawa C, Ohyama K, Miyazaki I, Tsukamoto G, et al. Protective effect of cepharanthin on cisplatin-induced renal toxicity through metallothionein expression. Life Sci. 2013;92:727–32.
Samra YA, Said HS, Elsherbiny NM, Liou GI, El-Shishtawy MM, Eissa LA. Cepharanthine and Piperine ameliorate diabetic nephropathy in rats: role of NF-kappaB and NLRP3 inflammasome. Life Sci. 2016;157:187–99.
Zhao J, Piao X, Wu Y, Liang S, Han F, Liang Q, et al. Cepharanthine attenuates cerebral ischemia/reperfusion injury by reducing NLRP3 inflammasome-induced inflammation and oxidative stress via inhibiting 12/15-LOX signaling. Biomed Pharmacother. 2020;127:110151.
Wang HG, Zhang MN, Wen X, He L, Zhang MH, Zhang JL, et al. Cepharanthine ameliorates dextran sulphate sodium-induced colitis through modulating gut microbiota. Microb Biotechnol. 2022;15:2208–22.
Bao F, Tao L, Zhang H. Neuroprotective effect of natural alkaloid fangchinoline against oxidative glutamate toxicity: involvement of keap1-Nrf2 axis regulation. Cell Mol Neurobiol. 2019;39:1177–86.
Jiang Y, Liu J, Zhou Z, Liu K, Liu C. Fangchinoline protects against renal injury in diabetic nephropathy by modulating the MAPK signaling pathway. Exp Clin Endocrinol Diabetes. 2020;128:499–505.
Chen H, Shi Z, Xing Y, Li X, Fu F. Fangchinoline attenuates cardiac dysfunction in rats with endotoxemia via the inhibition of ERK1/2 and NF-kappaB p65 phosphorylation. Ann Transl Med. 2020;8:1167.
Guo ZB, Fu JG. Progress of cardiovascular pharmacologic study on berbamine. Zhongguo Zhong xi yi jie he za zhi Zhongguo Zhongxiyi jiehe zazhi = Chin J Integr Traditional West Med. 2005;25:765–8.
Zhang CM, Gao L, Zheng YJ, Yang HT. Berbamine protects the heart from ischemia/reperfusion injury by maintaining cytosolic Ca(2+) homeostasis and preventing calpain activation. Circulation J. 2012;76:1993–2002.
Zheng Y, Gu S, Li X, Tan J, Liu S, Jiang Y, et al. Berbamine postconditioning protects the heart from ischemia/reperfusion injury through modulation of autophagy. Cell Death Dis. 2017;8:e2577.
Liu XY, Chen GN, Du GM, Pan Y, Song WQ, Jiang TW, et al. Berbamine ameliorates ethanol-induced liver injury by inhibition of hepatic inflammation in mice. Chin J Nat Med. 2020;18:186–95.
Li H, Chen W, Chen Y, Zhou Q, Xiao P, Tang R, et al. Neferine attenuates acute kidney injury by inhibiting NF-kappaB signaling and upregulating klotho expression. Front Pharmacol. 2019;10:1197.
Wu C, Chen J, Yang R, Duan F, Li S, Chen X. Mitochondrial protective effect of neferine through the modulation of nuclear factor erythroid 2-related factor 2 signalling in ischaemic stroke. Br J Pharmacol. 2019;176:400–15.
Min X, Guo Y, Zhou Y, Chen X. Protection against dextran sulfate sodium-induced ulcerative colitis in mice by neferine, a natural product from nelumbo nucifera gaertn. Cell J. 2021;22:523–31.
Liang X, Wang S, Wang L, Ceylan AF, Ren J, Zhang Y. Mitophagy inhibitor liensinine suppresses doxorubicin-induced cardiotoxicity through inhibition of Drp1-mediated maladaptive mitochondrial fission. Pharmacol Res. 2020;157:104846.
Xiao JH, Zhang JH, Chen HL, Feng XL, Wang JL. Inhibitory effects of isoliensinine on bleomycin-induced pulmonary fibrosis in mice. Planta Med. 2005;71:225–30.
Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30:478–90.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Fan HH, Wang LQ, Liu WL, An XP, Liu ZD, He XQ, et al. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin Med J. 2020;133:1051–6.
Jeon S, Ko M, Lee J, Choi I, Byun SY, Park S, et al. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother. 2020;64:e00819–20.
Zhang ZR, Zhang YN, Li XD, Zhang HQ, Xiao SQ, Deng F, et al. A cell-based large-scale screening of natural compounds for inhibitors of SARS-CoV-2. Signal Transduct Target Ther. 2020;5:218.
Chen CZ, Xu M, Pradhan M, Gorshkov K, Petersen JD, Straus MR, et al. Identifying SARS-CoV-2 entry inhibitors through drug repurposing screens of SARS-S and MERS-S pseudotyped particles. ACS Pharmacol Transl Sci. 2020;3:1165–75.
Ohashi H, Watashi K, Saso W, Shionoya K, Iwanami S, Hirokawa T, et al. Potential anti-COVID-19 agents, cepharanthine and nelfinavir, and their usage for combination treatment. iScience. 2021;24:102367.
Huang L, Yuen TT, Ye Z, Liu S, Zhang G, Chu H, et al. Berbamine inhibits SARS-CoV-2 infection by compromising TRPMLs-mediated endolysosomal trafficking of ACE2. Signal Transduct Target Ther. 2021;6:168.
Zhang ZR, Zhang YN, Zhang HQ, Zhang QY, Li N, Li Q, et al. Berbamine hydrochloride potently inhibits SARS-CoV-2 infection by blocking S protein-mediated membrane fusion. PLoS Neglected Tropical Dis. 2022;16:e0010363.
Yang Y, Yang P, Huang C, Wu Y, Zhou Z, Wang X, et al. Inhibitory effect on SARS-CoV-2 infection of neferine by blocking Ca(2+)-dependent membrane fusion. J Med Virol. 2021;93:5825–32.
Wang P, Luo R, Zhang M, Wang Y, Song T, Tao T, et al. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis. 2020;11:1042.
Fratta Pasini AM, Stranieri C, Girelli D, Busti F, Cominacini L. Is ferroptosis a key component of the process leading to multiorgan damage in COVID-19? Antioxidants. 2021;10:1677.
Han Y, Zhu J, Yang L, Nilsson-Payant BE, Hurtado R, Lacko LA, et al. SARS-CoV-2 infection induces ferroptosis of sinoatrial node pacemaker cells. Circulation Res. 2022;130:963–77.
Zhang R, Sun C, Chen X, Han Y, Zang W, Jiang C, et al. COVID-19-related brain injury: the potential role of ferroptosis. J Inflamm Res. 2022;15:2181–98.
Acknowledgements
We thank members of our lab for technical support. This work was supported by Science and Technology Planning Project of Guangdong Province (2020B1212060052) and Guangzhou Regenerative Medicine and Health Guangdong Laboratory Grant (2018GZR110104008).
Author information
Authors and Affiliations
Contributions
YF and YZ: designed and performed most of the experiments, analyzed the results, and wrote the paper; KS, SC, and DP provided technical and material support; XS: conceptualized and supervised the project, analyzed the results, wrote and revised the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fan, Y., Zhang, Y., Shi, K. et al. Identification of a group of bisbenzylisoquinoline (BBIQ) compounds as ferroptosis inhibitors. Cell Death Dis 13, 1000 (2022). https://doi.org/10.1038/s41419-022-05447-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05447-8
