
Abstract
Understanding etiology of human neurological and psychiatric diseases is challenging. Genomic changes, protracted development, and histological features unique to human brain development limit the disease aspects that can be investigated using model organisms. Hence, in order to study phenotypes associated with human brain development, function, and disease, it is necessary to use alternative experimental systems that are accessible, ethically justified, and replicate human context. Human pluripotent stem cell (hPSC)-derived brain organoids offer such a system, which recapitulates features of early human neurodevelopment in vitro, including the generation, proliferation, and differentiation of neural progenitors into neurons and glial cells and the complex interactions among the diverse, emergent cell types of the developing brain in three-dimensions (3-D). In recent years, numerous brain organoid protocols and related techniques have been developed to recapitulate aspects of embryonic and fetal brain development in a reproducible and predictable manner. Altogether, these different organoid technologies provide distinct bioassays to unravel novel, disease-associated phenotypes and mechanisms. In this review, we summarize how the diverse brain organoid methods can be utilized to enhance our understanding of brain disorders.
Facts
-
Brain organoids offer an in vitro approach to study aspects of human brain development and disease.
-
Diverse brain organoid techniques offer bioassays to investigate new phenotypes associated with human brain disorders that are difficult to study in monolayer cultures.
-
Brain organoids have been particularly useful to study phenomena and diseases associated with neural progenitor morphology, survival, proliferation, and differentiation.
Open question
-
Future brain organoid research needs to aim at later stages of neurodevelopment, linked with neuronal activity and connections, to unravel further disease-associated phenotypes.
-
Continued improvement of existing organoid protocols is required to generate standardized methods that recapitulate in vivo-like spatial diversity and complexity.
Similar content being viewed by others

Introduction
The complex architecture and function of the human brain enables humans to perform higher cognitive functions. Abnormalities in the structure or function of the brain can lead to severe neurological and psychiatric disorders. It is becoming increasingly clear that many neurological and psychiatric disorders have their roots in neurodevelopment [1, 2]. However, understanding the neurodevelopmental cause and mechanisms of these brain disorders is challenging, due to the limited access to the human brain tissues and the complexity of the human brain. While primary material derived from patient biopsies provides crucial insights into the disease pathology, it often represents the end point of the disease and does not reveal the causative mechanisms. On the other hand, access to fetal tissue from terminated pregnancies is limited. Studies using model organisms, especially rodents, have majorly advanced our understanding of how certain genetic mutations and environmental conditions impact neurodevelopment, neuronal circuit formation and behavior [3]. However, given the large evolutionary distance between mouse and human and the immense elaboration of the primate brain in size and complexity, there are many features unique to human brain development and diseases that cannot be recapitulated in rodent systems [4]. Primate models also suffer from major limitations as experimental systems due to the high ethical considerations, requirement of immense resources, and rare availability of the samples. Therefore, to study human brain development, function, and disease, it is necessary to use alternative experimental systems that closely mimic the human context and are also accessible and ethically just.
Protocols for culture and differentiation of human pluripotent stem cell (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), have enabled in vitro generation of diverse human cell types including neurons and glia, thereby allowing the investigation of biological processes directly in the human context [5]. hiPSC technologies have further made it possible to study the impact of healthy and disease-linked genotypes on the development and physiology of human cells. In addition, genetic manipulations using zinc finger nucleases, TALENs, and CRISPR-Cas9 have facilitated recapitulation of disease-linked mutations in cultured cells and repair of genetic mutations in patient-derived hiPSCs [6]. These approaches have been a boon for studying human neurobiology, revealing cellular phenotypes linked with brain disorders [7]. Nevertheless, the lack of tissue architecture and tissue environment limits the disease phenotypes that can be recapitulated and studied using monolayered culture systems. To overcome these issues, efforts in suspension culture of hPSCs led to the culture protocols to generate 3-D embryoid bodies (EB) [8, 9] and self-organizing organoid systems of different regions of the human central nervous system, including the cortex [10] and the retina [11].
Human brain organoids are hPSC-derived self-organizing structures that recapitulate the neurodevelopmental scheme to generate 3-D tissue architectures that mimic various features of the developing fetal brain pertaining to cellular composition and tissue structure [12]. hPSCs cultured in appropriate media conditions form an EB [9] or a spheroid [13] and undergo neural induction to adopt the neuroectodermal fate [10, 13, 14]. The neuroectodermal progenitors self-organize into multiple 3-D structures featuring apical lumens called neural rosettes or neural buds reminiscent of the neural tube. Exposure of these neuroectodermal cells to Matrigel, an extracellular matrix rich gel, can boost formation of larger neural rosettes with continuous neuroepithelium [14]. Subsequently, the neuroectodermal cells give rise to the neural stem and progenitor cells that proliferate and differentiate to generate neurons and glia of the human brain (Fig. 1). Importantly, organoid development mimics the temporal neurodevelopmental trajectory similar to the fetal development and recapitulates the corresponding cell types and gene expression profiles in a similar timeframe. This has been verified in multiple studies by immunohistochemical analysis as well as by comparing bulk and single-cell transcriptomic and epigenomic profile of fetal and organoid samples [13,14,15,16,17,18,19,20,21,22,23,24,25,26]. Corroborating their functional maturity, neurons generated in the organoids have been reported to form synaptic structures as revealed by electron microscopy [20, 27]. In addition, action potentials and calcium transients that are sensitive to synaptic blockers are suggestive of functional synapses [10, 13,14,15, 19, 20, 27]. Furthermore, brain organoids can also recapitulate development of glial cells— astrocytes and oligodendrocytes [13, 28]. Thus, brain organoids offer an experimentally amenable system to visualize and investigate aspects of human neurodevelopment in a dish. Some latest reviews have covered the history of the organoid technology [12, 29, 30] and the use of organoids to study brain evolution and neurodevelopment [31, 32]. In this review, we summarize how the diverse brain organoid methods can be utilized to screen for novel neurological phenotypes and to enhance our understanding of brain diseases.

Two-dimensional cultured hPSCs are dissociated and reaggregated to form an embryoid body (EB) or a spheroid in hPSC culture medium supplemented with Rho kinase inhibitor. Subsequent culture in a neural induction medium boosts the neuroectodermal fate. The neuroectodermal cells self-organize into multiple 3-D structures featuring apical lumens called “neural rosettes” or “neural buds,” reminiscent of the neural tube. This process is further boosted upon exposure to Matrigel, an extracellular matrix rich gel. Subsequently, the neuroectodermal cells give rise to the neural stem and progenitor cells that proliferate and differentiate to give rise to neurons and glia. The insets show zoomed in pictures with nuclei represented in shades of blue. For region-specific organoid protocols, patterning factors are added to the culture media at specific stages.
Recapitulating human context using brain organoids
hPSC-based organoid systems facilitate investigation of phenotypes specific to the human context by combining two important determinants of the human features—the human genome and the associated human developmental timeline.
Human genomic context
Genetic mutations are a major driving force of neurological and psychiatric disorders [1, 33]. Large-scale genetic mapping studies using genome-wide associations and whole genome or exome sequencing have identified many SNPs, InDels, and structural variants associated with diverse brain diseases [34, 35]. These disease-linked variants extend across a broad spectrum ranging from rare, highly penetrant mutations to common variants with low effect size. While it is possible to recapitulate specific rare mutations for monogenic mendelian disorders in animal model systems, it poses a big challenge for polygenic/complex diseases, in which a large number of loci contribute to the disease phenotype. Furthermore, during human evolution the genome has undergone many changes, including gene duplications, losses, and special divergence in the regulatory noncoding regions [36]. These evolutionary genomic changes have possibly led to novel gene regulatory mechanisms and subsequent innovations in neurodevelopment, resulting in dramatic structural and compositional changes in the human brain, especially the cerebral cortex [37,38,39,40,41,42]. These evolutionary sequence polymorphisms are also implicated in various psychiatric disorders [43]. Hence, to understand the impact of disease-associated variants and the etiology of human diseases, it is crucial to consider the human genomic context and the evolutionarily diversified neurological features.
hPSC-derived organoids maintain the human genomic context and enable studying the impact of disease-associated monogenic as well as polygenic genomic changes. For instance, Down syndrome (DS)/trisomy 21 is a human aneuploidy with complex neurological consequences [44]. The unique combination of genes on the human chromosome 21 makes it difficult to mimic all the aspects of the disorder using animal models, where the same genes are scattered across different chromosomes. Although there have been insightful studies that mimic the trisomy of these genes in mice, hiPSC-based approach has further boosted DS research [44, 45]. Recently, using brain organoids grown from hiPSCs of DS patients, Xu et al. showed that misregulated expression of OLIG2 gene in the ventral forebrain progenitors leads to overproduction of inhibitory interneurons in the DS brain organoids, possibly linking it to the imbalance of excitatory and inhibitory neurotransmission implicated in DS [45, 46]. Importantly, replication of the human genomic context is not restricted to hiPSC-based methods but can also be coupled with genome editing methods and reporter assays to screen impact of disease-relevant genetic variants.
Human developmental time
A prominent difference in human and rodent developmental scheme is developmental time. Human gestation period is highly protracted and requires 40 weeks compared with around 20 days for mouse. Similar protraction is reflected in human neurogenesis, which spans beyond 20 weeks in contrast to a week in mouse [47], and the subsequent neuronal migration and maturation. Hence, recapitulating the impact on temporal phenotypes such as cell cycle dynamics, duration of neurogenic period, and cell migration rate is challenging using model organisms. Interestingly, brain organoid culture recapitulates species-related developmental timing in vitro, giving an opportunity to assay phenotypes unique to the human neurodevelopmental timeline. For instance, upon comparing the cell cycle dynamics of neural progenitors in organoids grown from hiPSCs from control and autism spectrum disorder (ASD) probands, Mariani et al. found that the cell cycle in the ASD proband organoids was significantly shorter, resulting in increased neuronal differentiation and overproduction of neurons [48]. Furthermore, protraction of human neurodevelopment makes it more susceptible to environmental insults. Indeed, maternal starvation and chronic exposure to toxic compounds can lead to severe neurological disorders and increased risk of psychiatric diseases [49]. Impact of chronic exposure to chemical agents can be studied using human brain organoids [15, 50, 51], which is otherwise difficult to study in animal models with short gestational periods.
Diverse palette of human brain organoid culture methods and their applications
The initial brain organoid culture protocols pioneered by the Sasai [10, 52] and Knoblich [14] groups paved the way to a new field of research. Within the last few years, multiple brain organoid protocols and related techniques, such as the spheroid protocols [13], have been developed to mimic aspects of embryonic and fetal brain development. Current protocols range from generation of simpler brain organoids focused on specific brain regions, to complex brain organoids that recapitulate cell interactions and interconnectivity between multiple brain regions (Fig. 2 and Box 1). We highlight the major organoid culture methodologies available currently, and their suitability and limitations for studying neurodevelopmental phenomena and analyzing novel neurological disease-associated phenotypes.

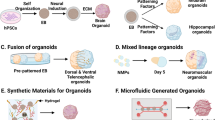
Schematic representation of currently available organoid methodologies. The schematics show a section of an organoid and distinct colors represent different brain regions. a Cerebral organoid protocol with minimalistic culture media generates self-patterned organoids containing diverse regions of the brain. b Region-specific organoids are generated by addition of patterning factors to the culture media to direct the regional identities. c Organoid fusions are generated by culturing young organoids with distinct patterning together in a microwell or by embedding in a single Matrigel droplet. The fusion approach allows investigation of interaction between the cells of differently patterned organoids. d Axially patterned organoids can be generated using a two-step process of EB formation. A small EB generated from transgenic hPSCs, capable of inducible expression of a morphogen (green cells) is seeded with additional control hPSCs (grey cells) to form a larger EB harboring an inducible signaling center. Upon induction of morphogen expression, the secreted morphogen diffuses in the tissue and patterns the organoid tissue in a gradient manner. e To incorporate cells of nonneuroectodermal origin, coculture protocols have been developed. (i) Cells added to the organoid culture medium, e.g., hiPSC-derived microglia (magenta), infiltrate the organoid tissue. (ii) Mixing of genetically altered cells during EB/spheroid formation is used to incorporate cells designed to follow a different developmental trajectory in the organoid. For example, upon mixing control hESCs with hESCs expressing inducible version of hETV2, a reprogramming factor for endothelial fate, cells expressing hETV2 (purple) adopted endothelial fates in the brain organoids [90].
Self-patterned cerebral organoids
According to the “neural default pathway”, hPSCs cultured in absence of external patterning factors adopt the neuroectodermal fate to generate neural tissue [53]. Based on this principle, Lancaster et al. developed the cerebral organoid protocol with minimalistic media [14, 54]. In this protocol, the self-organization and self-patterning of hPSCs, coupled with matrix-embedding, generate primarily neuroectodermal tissue arranged in the form of neural rosettes and recapitulate the tissue architecture of the germinal zones of neural stem and progenitor cells with the differentiated neurons migrating outwards. Hence, phenotypes related to tissue architectures can be studied using this protocol. For instance, recently hiPSCs from patients carrying mutations in DCSH1 and FAT4 genes were used to model periventricular heterotopia using the cerebral organoid protocol [55]. While analyzing the impact of the DCSH1 and FAT4 mutations, the authors found a defect in the morphology of the radial glia cells that provide a scaffold for the radial migration of the cortical neurons. In addition, scRNA-seq analysis with lineage reconstruction revealed that an altered neuronal state, with differentially expressed neuronal migration and navigation-related genes, was enriched in the mutant organoids. This observation was corroborated by occurrence of a subset of neurons showing migratory defects in the mutant organoids [55].
Due to the self-patterning-based development, the neuroectodermal regions originating in cerebral organoids have diverse spatial identities across forebrain, midbrain, and hindbrain, including organizer regions like the hem and antihem that secrete morphogens to pattern the organoid tissue [16]. As a result, the organoid morphology and brain region composition varies across individual cerebral organoids and especially across organoid batches and hiPSC lines (Fig. 2a) [16, 20, 23, 24]. However, regardless of these factors, scRNA-seq analysis has revealed that the transcriptomic identity of the same cell type generated across different cerebral organoids is highly similar at the level of individual cells [20, 24, 56]. Therefore, in combination with scRNA-seq-based interrogation, cerebral organoids can reveal potential disease-related phenotypes and affected cell types to postulate novel hypothesis about the disease mechanisms. Thus, cerebral organoids can be a good starting point to study human neurological disorders or function of disease-associated genetic elements.
Region-specific organoids
During development, the identity of different brain regions is established by patterning of the neural tube through the action of developmental signaling pathways activated or repressed by signaling molecules/morphogens secreted in vivo by cells of the organizer regions [53]. Applying the same principles in vitro, self-patterning in the organoids can be restricted and the cells in the developing organoid can be guided to adopt fates of specific brain regions by supplementing the culture media with patterning molecules that stimulate or block the reciprocal developmental signaling at appropriate developmental stages (Figs. 2b and 3).

During development, the identity of different brain regions is established by patterning of the neural tube through the action of developmental signaling pathways. Applying the same principles in vitro, the cells in the developing organoid can be guided to adopt fates of specific brain regions by supplementing the culture media with patterning molecules. The schematic drawing represents the different regions of the human neural tube (right) and a transverse section of the telencephalon (left) with the corresponding crucial patterning factors. The boxes include patterning strategies from key studies that pioneered region-specific patterning protocols.
Using previous knowledge from developmental biology and 2-D hPSC differentiation protocols [57], region-specific patterning media are designed based on the anterior–posterior (AP) and dorso–ventral (DV) location of the desired region in the neural tube (Fig. 3). The “primary molecular cocktail” is supplemented to guide efficient neural induction and the identity along the AP axis of the neural tube. Inhibition of BMP and TGF-β signaling by “the dual-SMAD strategy” efficiently guides the hPSCs to neuroectodermal identity [13, 15, 58]. Alternatively, TGF-β inhibition with simultaneous WNT inhibition boosts neuroectodermal and mainly telencephalic identity (Fig. 3) [10, 52]. High-throughput scRNA-seq profiling has revealed that both strategies reproducibly generate telencephalic organoids [21, 23]. To induce more caudal fates, early treatment with insulin and early WNT activation have been used in a dose-dependent manner to generate thalamic [59], hypothalamic [15, 60], midbrain [15, 60, 61] and cerebellar fates (Fig. 3) [62]. The “second molecular cocktail” is supplemented to guide the identity along the DV axis of the neural tube. For telencephalic organoids, a pulse of WNT activity at later stages enriches cerebral cortex tissue [15, 60, 63] and combination of WNT and BMP treatment enriches medial pallium fates to derive hippocampal tissue (Fig. 3) [64]. On the other hand, ventral fates crucially depend on sonic hedgehog (SHH), secreted by the notochord and the floorplate [65]. Early SHH activation is used to drive ventral fates of the telencephalon [19, 66, 67], hypothalamus, and midbrain [15, 61]. In addition to this broad scheme, additional region-specific morphogens are included in some patterning media (Fig. 3).
While these molecular cocktails establish the broad identities of brain regions, generating sub-region-specific organoids is challenging. For instance, the mammalian cortex is subdivided into multiple functional areas like the prefrontal cortex in the frontal lobe and visual cortex in the occipital lobe that differ in their gene expression profiles, cell diversity, and function [68, 69]. In addition, specific areas of the cortex such as the prefrontal cortex are exceptionally expanded in humans [70] and implicated in psychiatric disorders [71, 72]. However, the current cortical organoid protocols generate cortical tissues with mixed identities [10, 52, 73, 74]. Novel protocols that generate organoids of specific areas of the cortex will be useful to further boost the investigation of human brain diseases. On these lines, treatment of cortical organoids with anterior patterning factor FGF8 was shown to rostralise the cortical tissue [10, 52]. However, given the involvement of the same signaling molecules in patterning of different brain regions, the current challenge is to identify chemically-defined molecular cocktails and respective timeframes of treatment, which specify brain regions of interest in a cost-effective and reproducible manner.
Region-specific organoids are useful to study the impact of genetic defects or chemical treatments on a specific disease-associated brain region. For example, Parkinson’s disease (PD) is a neurodegenerative disorder characterized by loss of dopaminergic neurons in the midbrain [75]. Protocols to generate midbrain-floorplate organoids successfully generate TH positive dopaminergic neurons implicated in the disease [61] and help to further investigate the disease mechanisms by growing midbrain-like organoids from PD patient-derived hiPSCs [76, 77]. However, it should be noted that region-specific organoids require prior knowledge or hypothesis about the disease mechanism and restrict investigation of interregion interactions, a crucial feature of brain function.
Organoid fusions
The complex circuitry of the brain is generated by interaction between cells of different, often very distant, brain regions. This challenge of finding the right functional partner is overcome by two kinds of cell behaviors: (a) active migration of cells to their target sites (e.g., the cortical interneurons born in the ventral telencephalon migrate dorsally to the cerebral cortex [78]) and (b) long-range projections via axonal tracts (e.g., neurons of the thalamic nuclei project to the cortex via the “thalamocortical tract” to relay the sensory information [79]). Defects in neuronal migration or axon pathfinding can result in cortical malformations and severe congenital disorders [80, 81]. However, interregion cell interactions cannot be recapitulated with region-specific organoids, and the variability of the cerebral organoid protocol prevents consistent formation of the interacting regions of interest. To overcome this problem, several groups developed organoid fusion methods, wherein prepatterned EB/young organoids are co-cultured either in a microwell or a tube [19, 59, 67] or embedded together in a single Matrigel droplet [66]. Eventually, the two regions fuse together and exhibit complex cell interactions (Fig. 2c). Fusion of organoids with different genetic backgrounds allows studying cell autonomous and nonautonomous effects of the genetic background on neuronal behavior. Furthermore, live imaging of fused organoids or their organotypic slices enables visualization of these complex cell behaviors including neuronal migration, axon pathfinding, and long-range projections.
The organoid fusion approach has been particularly insightful to study migration of cortical interneurons [19, 66, 67]. Fusion of dorsal and ventral telencephalic organoids (DV fusions) made it possible to visualize the characteristic saltatory migration of cortical interneurons and to study the impact of genetic and chemical perturbation on their migratory properties. Birey et al. used the DV fusion approach to study the impact of gain-of-function mutation in a L-type calcium channel that causes Timothy syndrome (TS) characterized by ASD and epilepsy [19]. When ventral telencephalic organoids grown from TS patient-derived hiPSCs were fused with control dorsal telencephalic organoids, the interneurons carrying the TS mutation showed cell autonomous defects in saltatory migration. Applying a chemical agent that reduced the activity of the L-Type calcium channel lowered the impact of the mutation and rescued the migratory defects [19]. Another study used the fusion approach to recapitulate long-range corticothalamic and thalamocortical projections by fusing region-specific cortical and thalamic organoids [59]. The thalamocortical projections further showed colocalization with synaptic markers suggestive of synaptic connection between cortical and thalamic neurons. Thus, organoid fusion methods are powerful techniques to visualize and study complex interregion interactions that are not possible to study using 2-D monolayered culture. However, it should be noted that the organoids lack the exact spatial map of attractive and repulsive cues present in the fetal brain and thus the neurons are unlikely to replicate the stereotypic paths observed in vivo. Nevertheless, organoid fusions can be used to build assays to screen for cell behaviors such as neuronal migration and formation of neuronal connections. In the future, we anticipate that an extension of these fusion approaches will help to build more complex brain circuitry and neuronal connections with nonneural organoids or primary tissues.
Axially patterned organoids
The enormous diversity of the neurons in the brain is generated by gradients of morphogen activity in AP and DV direction [57]. However, any morphogen added to the medium is delivered in a uniform concentration, possibly restricting the diversity of fates. To establish a gradient of morphogen activity and downstream patterning, Cederquist et al. established a novel way to generate axially patterned organoids using a two-step process of EB formation [82]. Initially, a small EB was generated from transgenic hPSCs, capable of inducible expression of a morphogen. Subsequent seeding of additional hPSCs led to formation of a larger EB harboring an inducible signaling center or organizer (Fig. 2d). Upon induction of morphogen expression, the secreted morphogen diffused in the tissue to create a gradient. Cederquist et al. used this method to create a gradient of the ventral-inducing morphogen SHH and successfully generated telencephalic organoids with a spatially arranged DV axis [82]. In the future, this new technology will pave the way to pattern longer developmental axes such as the AP axis and to combine different axes to mimic in vivo patterning. Axial patterning combined with organoid fusion will help to recreate the complexity of brain in a regulated manner.
Coculture with other cell types and tissues
The brain also contains cells with nonneuroectodermal origin, namely microglia and the cells of the vasculature. Microglia are brain resident macrophages that play a crucial role in neuronal maturation and functioning. Microglial dysfunction is implicated in neurodegenerative disorders and psychiatric disorders such as schizophrenia and autism [83]. However, since microglia originate in the yolk sac, it is difficult to generate them simultaneously with the neuroectoderm in the current organoid systems. A recent study reported presence of microglial-like cells in cerebral organoids, originating from a few mesodermal cells arising in the cerebral organoid protocol [84]. However, for the broad applicability to diverse brain organoids methods, other groups have co-cultured organoids with hiPSC-derived microglia that infiltrate the organoid (Fig. 2e) [85, 86]. Recently, such coculture paradigm was used to study clearance of the amyloid β protein aggregates in organoids carrying familial Alzheimer’s disease mutations [87]. Further improvements and tests with these methods will boost in vitro recapitulation of microglial functions in the organoids.
Brain vasculature develops from endothelial cells to build the blood–brain barrier and provides oxygen and nutrients to the developing brain tissue [88]. Due to the lack of vasculature, the current organoids protocols often exhibit a necrotic core in the center of the organoid, posing a limit on the organoid growth capacity. Implantation of human brain organoids in the adult mouse brain, resulted in the vascularization of the organoid tissue and replacement of necrotic core with healthy neurons [89]. However, the generation of brain vasculature in vitro has been challenging. In a recent study, Cakir et al. generated organoids by mixing control hESCs with hESCs expressing inducible version of hETV2, a reprogramming factor for endothelial fate. Upon induction, the cells expressing hETV2 in the brain organoids adopted endothelial fates and gene expression profiles (Fig. 2e) [90]. These “endothelium-containing brain organoids” implanted in the mouse formed functional blood vessels in vivo. Alternatively, in a new preprint, coculture of brain organoids with human umbilical vein endothelial cells has been proposed as a strategy to vascularize organoids [91]. It will be interesting to test to what extent these methods can recapitulate the blood–brain barrier in the organoids. Recently, co-culturing techniques have also been used to recapitulate glioblastoma pathology by coculture of patient-derived glioblastoma stem cells with cortical organoids [92, 93]. Thus, organoid coculture methods expand the usability of brain organoid systems.
Given this great diversity of brain organoid culture methods, it can be challenging for the investigators to choose the right method. Indeed, there is no one brain organoid protocol that fits all purposes. Instead, these diverse methods offer a collection of bioassays, each having their own benefits and limitations. It is advisable to choose the organoid assay based on the balance between the complexity vs minimalism of the organoid system required for the biological question under investigation and to interpret the results keeping in mind the limitations of each assay. A combination of different organoid assays will help to unravel diverse phenotypes associated with the biological question at hand.
Modeling human neurodevelopmental and neurological disorders using brain organoids
The abovementioned organoid technologies equip us with tools to investigate mechanisms of brain diseases and novel disease phenotypes, which can eventually lead to better diagnostic methods and therapies. For an overview of diverse brain disorders modeled using organoids refer to Table 1 and also some of the latest excellent reviews [94, 95]. Here we focus on a few prominent neurological disorders related to cortical progenitor biology for which organoids have been instrumental to unravel biological mechanisms related to cell proliferation, cell death, and neuronal differentiation.
Abnormalities in cortical progenitor function can have detrimental effects on brain development [96]. Both proliferative capacity and morphology of the cortical progenitors are crucial to ensure normal neurogenesis and neuronal migration, to ultimately determine the cortical cytoarchitecture. Briefly, the stem and progenitor cells of the developing mammalian cortex are located in the neuroepithelium that lines the developing ventricles, forming the ventricular zone. Initially, these progenitors, referred to as apical/ventricular radial glia undergo symmetric divisions to expand their pool. Later during development, they switch to asymmetric divisions to generate either neurons via direct neurogenesis or intermediate progenitors with limited self-renewal capacity. Intermediate progenitors delaminate from the ventricular zone and localize basally to form the subventricular zone, where they undergo neurogenic divisions to give rise to the cortical neurons [97]. Compared with rodents, the number, proliferative capacity and diversity of the neural stem and progenitor cells are greater in the developing human cerebral cortex [98]. The subventricular zone is specially expanded in humans due to a higher number of intermediate progenitors and enrichment of a special class of transit amplifying intermediate progenitors called basal/outer radial glia, which show increased self-renewal capacity and neuronal output. Owing to these differences in the genetic landscape, progenitor make-up and tissue architecture, often the mouse models carrying similar disease-like mutations only partially recapitulate different aspects of cortical defects. Notably, brain organoids recapitulate the diverse classes of human cortical stem and progenitor cells as well as their temporal birth order [14, 17]. Organoids also recapitulate the ventricular zone and subventricular zone cytoarchitecture and newborn neurons migrate outwards to more basal locations to form the cortical plate-like structure [52, 63]. In the following sections, we illustrate how brain organoids have been useful to study diseases associated with progenitor dysfunction caused by genetic mutations as well as viral pathogens.
Primary microcephaly
Primary microcephaly is a congenital genetic disorder characterized by highly reduced head and brain size with a prominent phenotype in the cerebral cortex [99]. Studies using model organisms, especially rodents, have implied perturbed proliferation of cortical progenitors as the underlying disease mechanism [100]. However, for many microcephaly-linked genes, the extent of the microcephaly phenotype differs across species. For instance, patients carrying a mutation in the NDE1 gene have a severely reduced brain size, while the Nde1 deletion mutant mice show only a mild size reduction [101, 102]. Hence, organoids offer a possibility to understand the human-specific aspects of various microcephaly associated-mutations. Lancaster et al. generated cerebral organoids using hiPSCs derived from a primary microcephaly patient carrying truncating mutations in the microcephaly gene CDK5RAP2 [14]. Cerebral organoids grown from these cells were significantly smaller than the isogenic controls and contained higher number of neurons at a younger developmental stage compared with the controls, indicative of premature neurogenesis. It was found that the loss of CDK5RAP2 function leads to premature shift of progenitor division mode from symmetric to asymmetric divisions, leading to premature neurogenesis, exhaustion of the stem cell pool, and ultimately the microcephalic phenotype. This study for the first time showcased the disease modeling ability of the brain organoids [14]. Recently, Zhang et al. recapitulated the microcephalic phenotype using hPSCs gene-edited to cause loss-of-function mutations in a crucial microcephaly gene WDR62 [103]. In the WDR62−/− organoids, the proliferation of the neural stem cells was severely perturbed, with reduced symmetric proliferative divisions and occurrence of premature differentiation. Reduction in proliferation and increased apoptosis was not only observed in ventricular progenitors but also for human-enriched outer radial glia, which probably adds to the severity of the human phenotype over the mouse.
Viral infection associated-microcephaly
Viral infections during pregnancy can have detrimental effects on the development of the fetus and result in congenital defects and higher risk of developing psychiatric disorders later in life [104]. Organoids played an important role in establishing the link between maternal Zika virus infection and congenital microcephaly [105, 106]. Studies from multiple groups established that human neural progenitors are susceptible to Zika virus infection. Infection of neural progenitors in the organoids affected their proliferative capacity, led to premature neurogenesis and increased cell death resulting into reduced organoid size, reminiscent of the microcephalic phenotype [15, 105, 106]. Subsequent studies used Zika infection in organoids to screen compounds for their ability to neutralize Zika virus and ameliorate the neurological defects [107,108,109,110]. Thus, organoids can also be used to dissect impact of pathogens that affect brain development and to identify potential therapies.
Macrocephaly
On the opposite spectrum of progenitor-related disorders is macrocephaly, which originates from higher proliferation of neural progenitors through deregulated growth factor signaling [96]. Heterozygous loss-of-function mutations in PTEN were found in patients with macrocephaly and associated ASD [111, 112]. Cerebral organoids grown from hPSCs carrying PTEN loss-of-function mutation displayed increased downstream AKT signaling, transiently delayed neuronal differentiation and expanded progenitor pool of both ventricular progenitors and intermediate progenitors [113]. While similar phenotypes were also seen in mouse organoids, the PTEN−/− human cerebral organoids exhibited an additional phenotype of folds on the organoid surface, which was dependent on AKT signaling strength, indicative of human-specific severity of the disease [113].
Outlook of brain organoid research
The field of brain organoid research is still young and continues to progress with the emergence of improved methodologies. Combined with new experimental approaches, the brain organoid technology has the potential to develop functional assays to analyze even more complex phenotypes. At the same time, to enhance the usability of brain organoid data, the field needs to agree on standardized procedures on protocols, quality controls, and data management. Here, we discuss a few prominent directions that can enhance the scope and applicability of brain organoid research.
Bioengineering
While the current organoid methodologies are well suited for generation of various progenitor and neuronal cell types, the final tissue architecture of the human brain is highly complex and not recapitulated in the organoids. For instance, despite the generation of all major cell types, the six-layered architecture of the cerebral cortex is not recapitulated in the organoids. The use of Matrigel has been shown to support formation of cortical plate-like structure in the brain organoids [52, 63]. However, whether and how the poorly defined and variable composition of Matrigel impacts organoid development is unknown. Well-defined synthetic matrices used in other organoid systems [114, 115] may offer an interesting avenue to overcome the short comings of Matrigel. Use of bioengineered fiber scaffolds has been useful to facilitate formation of larger and continuous cortical areas [63]. In addition, long term culture of organotypic slices of brain organoids is reported to enhance neuronal survival and segregation of upper and deep layer cortical neurons [22, 116]. However, additional improvements are needed to generate cortical organoids that recapitulate cortical layering. Further efforts are also needed to generate organoids with standardized shapes and sizes to bring uniformity in phenotypic characterizations and to suit high-throughput genetic and chemical screens. Combination of bioengineering, biomaterials, microfluidics, and automation will be key in this direction [117].
Neuronal connectivity in organoids
Understanding the neuronal microcircuits and network function of the human brain is crucial to understand the etiology of abnormal neuronal function and connectivity in conditions such as epilepsy. Multiple studies have reported that brain organoids contain neurons exhibiting intrinsic excitability and spontaneous activity sensitive to synaptic blockers, suggestive of functional synapses [10, 13,14,15, 19, 20, 27]. To this end, visualization of calcium transients, intracellular whole cell patch-clamp recordings and high density large extracellular probes, e.g. silicon probes, have been used. These methods are promising to study how the electrophysiological properties of neurons in the organoids change across the developmental timeline [27] or in disease conditions [19, 118, 119]. Recent studies using high density large extracellular probes, planar multielectrode array recordings and calcium transients have reported occurrence of spontaneous synchronous neuronal events, suggestive of neuronal networks [20, 27, 119]. However, the prevalence, composition, properties, and relevance of these neuronal connections remain to be tested and occurrence of in vivo-like complex neuronal activity and functional circuitry remains elusive. While decoration of synaptic markers is used to indicate presence of structural synapses, ultramicroscopy, and ultimately, neuronal tracing methods will be key to unravel the connectivity in organoids and to understand the impact of genetic and chemical perturbations on human synaptogenesis, neuronal activity and network function.
Genomic and single-cell technologies in organoids
Current organoid studies are focused on studying human gene function using patient-derived hiPSCs and genome editing of limited genetic loci. Pooled CRISPR screens widely used in 2-D culture systems [120] will help to screen and prioritize genetic elements and their variants in a massively parallel manner for complex phenotypes that cannot be studied in 2-D. Furthermore, single-cell technologies have enhanced our ability to analyze molecular phenotypes at cellular resolution and to detect emergent phenotypes that are difficult to discern with traditional investigation methods [121]. Currently, scRNA-seq is routinely used in the brain organoid field as regular scheme of analysis and to compare variability of generated cell types across protocols. Further adaptation of diverse single-cell cell technologies to brain organoids will expand the set of discernable phenotypes otherwise hidden in bulk analysis such as novel cell types/states altered in a disease.
In vivo validation
Although improvements in organoid technologies generate more sophisticated in vivo-like tissue, it still remains an in vitro experimental system with certain limitations. Recent observations using scRNA-seq analysis have highlighted that many cells in brain organoids display broad transcriptomic signatures with poor sub-type specific features and co-expression of markers of progenitors and differentiated cells [74]. In addition, compared with primary tissue, organoid cells were found to exhibit higher expression of glycolytic and ER stress-related genes, owing to in vitro culture [73, 74]. It is yet to be determined if these cells show other well-defined features of ER stress, such as unfolded protein response [122] and whether higher expression of glycolytic genes is related to stress or instead an adaptation to the in vitro culture medium. Nevertheless, it is important to consider these properties of organoids while interpreting the organoid phenotypes. It is clear that organoids are an in vitro system to facilitate biomedical research but in no way lessen the importance of animal models and primary tissues. The new details on human biology revealed by organoid systems require in vivo validation. To this end, whenever possible, validation using fetal samples will be crucial.
To conclude, the combination of different brain organoid technologies together with clinical data and experiments in model organisms will reveal novel disease mechanisms and contribute to potential diagnostic methods and therapies.
References
Hu WF, Chahrour MH, Walsh CA. The diverse genetic landscape of neurodevelopmental disorders. Annu Rev Genom Hum Genet. 2014;15:195–213.
Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 2016;89:248–68.
Leung C, Jia Z. Mouse genetic models of human brain disorders. Front Genet. 2016;7:40.
Zhao X, Bhattacharyya A. Human models are needed for studying human neurodevelopmental disorders. Am J Hum Genet. 2018;103:829–57.
Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–82.
Hotta A, Yamanaka S. From genomics to gene therapy: induced pluripotent stem cells meet genome editing. Annu Rev Genet. 2015;49:47–70.
Sterneckert JL, Reinhardt P, Schöler HR. Investigating human disease using stem cell models. Nat Rev Genet. 2014;15:625–39.
Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000;6:88–95.
Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–6.
Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Stem Cell. 2008;3:519–32.
Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Stem Cell. 2012;10:771–85.
Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345:1247125.
Pașca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat methods. 2015;12:671–8.
Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–9.
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell. 2016;165:1238–54.
Renner M, Lancaster MA, Bian S, Choi H, Ku T, Peer A, et al. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017;36:1316–29.
Camp JG, Badsha F, Florio M, Kanton S, Gerber T, Wilsch-Bräuninger M, et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA. 2015;112:15672–7.
Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, Ecker JR. Cerebral organoids recapitulate epigenomic signatures of the human fetal. Brain Cell Rep. 2016;17:3369–84.
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature. 2017;545:54–9.
Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Yang SM, Berger DR, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545:48–53.
Yoon S-J, Elahi LS, Pașca AM, Marton RM, Gordon A, Revah O, et al. Reliability of human cortical organoid generation. Nat methods. 2018;16:75–78.
Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat Neurosci. 2019;22:669–79.
Velasco S, Kedaigle AJ, Simmons SK, Nash A, Rocha M, Quadrato G, et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature. 2019;570:523–7.
Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature. 2019;574:418–22.
Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F et al. Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science. 2018;362. https://doi.org/10.1126/science.aat6720.
Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon S-J, Huber N, Pritchard JK, et al. Chromatin accessibility dynamics in a model of human forebrain development. Science. 2020;367:eaay1645.
Trujillo CA, Gao R, Negraes PD, Gu J, Buchanan J, Preissl S, et al. Complex oscillatory waves emerging from cortical organoids model early human brain network development. Stem Cell. 2019;25:558–69.e7.
Madhavan M, Nevin ZS, Shick HE, Garrison E, Clarkson-Paredes C, Karl M, et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat Methods. 2018;110:20284.
Benito-Kwiecinski S, Lancaster MA. Brain organoids: human neurodevelopment in a dish. Cold Spring Harb Perspect Biol. 2019. https://doi.org/10.1101/cshperspect.a035709.
Simian M, Bissell MJ. Organoids: a historical perspective of thinking in three dimensions. J Cell Biol. 2016;216:31–40.
Qian X, Song H, Ming G-L. Brain organoids: advances, applications and challenges. Development. 2019;146:dev166074–12.
Heide M, Huttner WB, Mora-Bermúdez F. Brain organoids as models to study human neocortex development and evolution. Curr Opin Cell Biol. 2018;55:8–16.
Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13:537–51.
Sullivan PF, Geschwind DH. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell. 2019;177:162–83.
Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362. https://doi.org/10.1126/science.aat7615.
Reilly SK, Noonan JP. Evolution of gene regulation in humans. Annu Rev Genom Hum Genet. 2016;17:45–67.
Doan RN, Shin T, Walsh CA. Evolutionary changes in transcriptional regulation: insights into human behavior and neurological conditions. Annu Rev Neurosci. 2018;41:185–206.
Heide M, Long KR, Huttner WB. Novel gene function and regulation in neocortex expansion. Curr Opin Cell Biol. 2017;49:22–30.
Florio M, Borrell V, Huttner WB. Human-specific genomic signatures of neocortical expansion. Curr Opin Neurobiol. 2017;42:33–44.
Franchini LF, Pollard KS. Human evolution: the non-coding revolution. BMC Biol. 2017;15:89.
Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, et al. Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science. 2015;347:1155–9.
Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond Ser B. 2013;368:20130025.
Doan RN, Bae BI, Cubelos B, Chang C, Hossain AA, Al-Saad S, et al. Mutations in human accelerated regions disrupt cognition and social behavior. Cell. 2016;167:341–54 e12.
Antonarakis SE. Down syndrome and the complexity of genome dosage imbalance. Nat Rev Genet. 2017;18:147–63.
Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91.
Xu R, Brawner AT, Li S, Liu J-J, Kim H, Xue H, et al. OLIG2 drives abnormal neurodevelopmental phenotypes in human iPSC-based organoid and chimeric mouse models of Down syndrome. Stem Cell. 2019;24:908–26.e8.
Toma K, Wang T-C, Hanashima C. Encoding and decoding time in neural development. Dev Growth Differ. 2016;58:59–72.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–90.
Schmitt A, Malchow B, Hasan A, Falkai P. The impact of environmental factors in severe psychiatric disorders. Front Neurosci. 2014;8. https://doi.org/10.3389/fnins.2014.00019.
Lee C-T, Chen J, Kindberg AA, Bendriem RM, Spivak CE, Williams MP, et al. CYP3A5 mediates effects of cocaine on human neocorticogenesis: studies using an in vitro 3D self-organized hPSC model with a single cortex-like unit. Neuropsychopharmacol. 2017;42:774–84.
Wang Y, Wang L, Zhu Y, Qin J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip. 2018;18:851–60.
Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci USA. 2013;110:20284–9.
Muguruma K, Sasai Y. In vitro recapitulation of neural development using embryonic stem cells: From neurogenesis to histogenesis. Dev Growth Differ. 2012;54:349–57.
Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9:2329–40.
Klaus J, Kanton S, Kyrousi C, Ayo-Martin AC, Di Giaimo R, Riesenberg S, et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat Med. 2019;25:561–8.
Tanaka Y, Cakir B, Xiang Y, Sullivan GJ, Park I-H. Synthetic analyses of single-cell transcriptomes from multiple brain organoids and fetal brain. Cell Rep. 2020;30:1682–9.e3.
Suzuki IK, Vanderhaeghen P. Is this a brain which I see before me? Modeling human neural development with pluripotent stem cells. Development. 2015;142:3138–50.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–80.
Xiang Y, Tanaka Y, Cakir B, Patterson B, Kim K-Y, Sun P, et al. hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Stem Cell. 2019;24:487–97.e7.
Qian X, Jacob F, Song MM, Nguyen HN, Song H, Ming G-L. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat Protoc. 2018;13:565–80.
Jo J, Xiao Y, Sun AX, Cukuroglu E, Tran H-D, Göke J, et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell. 2016;19:248–57.
Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10:537–50.
Lancaster MA, Corsini NS, Wolfinger S, Gustafson EH, Phillips AW, Burkard TR, et al. Guided self-organization and cortical plate formation in human brain organoids. Nat Biotechnol. 2017;35:659–66.
Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, et al. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat Commun. 2015;6:8896–11.
Lupo G, Harris WA, Lewis KE. Mechanisms of ventral patterning in the vertebrate nervous system. Nat Rev Neurosci. 2006;7:103–14.
Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods. 2017;14:743–51.
Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell. 2017;21:383–98 e7.
Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, et al. New insights into the development of the human cerebral cortex. J Anat. 2018;235:432–51.
Clowry GJ, Alzu’bi A, Harkin LF, Sarma S, Kerwin J, Lindsay SJ. Charting the protomap of the human telencephalon. Semin Cell Dev Biol. 2018;76:3–14.
Smaers JB, Gómez-Robles A, Parks AN, Sherwood CC. Exceptional evolutionary expansion of prefrontal cortex in great apes and humans. Curr Biol. 2017;27:714–20.
Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017;18:727–40.
Gamo NJ, Arnsten AFT. Molecular modulation of prefrontal cortex: rational development of treatments for psychiatric disorders. Behav Neurosci. 2011;125:282–96.
Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell. 2019;176:743–56.e17.
Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 2020;110:20284–33.
Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87.
Kim H, Park HJ, Choi H, Chang Y, Park H, Shin J, et al. Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids. Stem Cell Rep. 2019;12:518–31.
Smits LM, Reinhardt L, Reinhardt P, Glatza M, Monzel AS, Stanslowsky N, et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 2019;5:5–8.
Silva CG, Peyre E, Nguyen L. Cell migration promotes dynamic cellular interactions to control cerebral cortex morphogenesis. Nat Rev Neurosci. 2019;20:318–29.
López-Bendito G, Molnár Z. Thalamocortical development: how are we going to get there? Nat Rev Neurosci. 2003;4:276–89.
Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu Rev Neurosci. 2001;24:1041–70.
Engle EC. Human genetic disorders of axon guidance. Cold Spring Harb Perspect Biol. 2010;2:a001784.
Cederquist GY, Asciolla JJ, Tchieu J, Walsh RM, Cornacchia D, Resh MD, et al. Specification of positional identity in forebrain organoids. Nat Biotechnol. 2019;37:436–44.
Beggs S, Salter MW. SnapShot: microglia in disease. Cell. 2016;165:1294–1294.e1.
Ormel PR, Vieira de Sá R, van Bodegraven EJ, Karst H, Harschnitz O, Sneeboer MAM, et al. Microglia innately develop within cerebral organoids. Nat Commun. 2018;9:4167–14.
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. 2017;94:278–93.e9.
Song L, Yuan X, Jones Z, Vied C, Miao Y, Marzano M, et al. Functionalization of brain region-specific spheroids with isogenic microglia-like cells. Sci Rep. 2019;9:11055–18.
Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–54.e7.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96.
Mansour AA, Gonçalves JT, Bloyd CW, Li H, Fernandes S, Quang D, et al. An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol. 2018;36:432–41.
Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang Y-J, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods. 2019;16:1169–75.
Shi Y, Sun L, Liu J, Zhong S, Wang M, Li R, et al. Vascularized human cortical organoids model cortical development in vivo. 2019. https://doi.org/10.1101/682104.
Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 2019;26:3203–11.e5.
Bhaduri A, Di Lullo E, Jung D, Müller S, Crouch EE, Espinosa CS, et al. Outer radial glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Stem Cell. 2020;26:48–63.e6.
Amin ND, Paşca SP. Building models of brain disorders with three-dimensional organoids. Neuron. 2018;100:389–405.
Di Lullo E, Kriegstein AR. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 2017;18:573–84.
Sun T, Hevner RF. Growth and folding of the mammalian cerebral cortex: from molecules to malformations. Nat Rev Neurosci. 2014;15:217–32.
Taverna E, Götz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502.
Florio M, Huttner WB. Neural progenitors, neurogenesis and the evolution of the neocortex. Development. 2014;141:2182–94.
Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. AJHG. 2005;76:717–28.
Jayaraman D, Bae B-I, Walsh CA. The genetics of primary microcephaly. Annu Rev Genom Hum Genet. 2018;19:177–200.
Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, et al. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet. 2011;88:536–47.
Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–93.
Zhang W, Yang S-L, Yang M, Herrlinger S, Shao Q, Collar JL, et al. Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat Commun. 2019;10:2612–14.
Silasi M, Cardenas I, Kwon J-Y, Racicot K, Aldo P, Mor G. Viral infections during pregnancy. Am J Reprod Immunol. 2015;73:199–213.
Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016;352:816–8.
Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature. 2016;534:267–71.
Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Stem Cell. 2016;19:258–65.
Xu Y-P, Qiu Y, Zhang B, Chen G, Chen Q, Wang M, et al. Zika virus infection induces RNAi-mediated antiviral immunity in human neural progenitors and brain organoids. Cell Res. 2019;29:265–73.
Watanabe M, Buth JE, Vishlaghi N, la Torre-Ubieta de L, Taxidis J, Khakh BS, et al. Self-organized cerebral organoids with human- specific features predict effective drugs to combat Zika virus. Infect Cell Rep. 2017;21:517–32.
Zhou T, Tan L, Cederquist GY, Fan Y, Hartley BJ, Mukherjee S, et al. High-content screening in hPSC-neural progenitors identifies drug candidates that inhibit Zika virus infection in fetal-like organoids and adult. Brain Stem Cell. 2017;21:274–83.e5.
Butler MG. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–21.
Marchese M, Conti V, Valvo G, Moro F, Muratori F, Tancredi R, et al. Autism-epilepsy phenotype with macrocephaly suggests PTEN, but not GLIALCAM, genetic screening. BMC Med Genet. 2014;15:26–7.
Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, et al. Induction of expansion and folding in human cerebral organoids. Stem Cell. 2017;20:385–96.e3.
Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordóñez-Morán P, et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 2016;539:560–4.
Cruz-Acuña R, Quirós M, Farkas AE, Dedhia PH, Huang S, Siuda D, et al. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat Cell Biol. 2017;19:1326–35.
Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell. 2020. https://doi.org/10.1016/j.stem.2020.02.002.
Brassard JA, Lutolf MP. Engineering stem cell self-organization to build better organoids. Cell Stem Cell. 2019;24:860–76.
Ghatak S, Dolatabadi N, Trudler D, Zhang X, Wu Y, Mohata M, et al. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. eLife. 2019;8:1567–22.
Sun AX, Yuan Q, Fukuda M, Yu W, Yan H, Lim GGY, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019;366:1486–92.
Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311.
Camp JG, Platt R, Treutlein B. Mapping human cell phenotypes to genotypes with single-cell genomics. Science. 2019;365:1401–5.
Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2018;286:241–78.
Li R, Sun L, Fang A, Li P, Wu Q, Wang X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell. 2017;8:823–33.
Gabriel E, Wason A, Ramani A, Gooi LM, Keller P, Pozniakovsky A, et al. CPAPpromotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016;35:803–19.
Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, et al. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Stem Cell. 2017;21:319–31.e8.
Allende ML, Cook EK, Larman BC, Nugent A, Brady JM, Golebiowski D, et al. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J Lipid Res. 2018;59:550–63.
Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial. Glia Stem Cell. 2017;20:435–49.e4.
Iefremova V, Manikakis G, Krefft O, Jabali A, Weynans K, Wilkens R, et al. An organoid-based model of cortical development identifies non-cell-autonomous defects in wnt signaling contributing to Miller-Dieker Syndrome. Cell Rep. 2017;19:50–59.
Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA, et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods. 2018;15:631–9.
Ogawa J, Pao GM, Shokhirev MN, Verma IM. Glioblastoma model using human cerebral organoids. Cell Rep. 2018;23:1220–9.
Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci. 2019;22:243–55.
Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2017;23:1051–65.
Raja WK, Mungenast AE, Lin Y-T, Ko T, Abdurrob F, Seo J, et al. Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS ONE. 2016;11:e0161969–18.
Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry. 2018;23:2363–74.
Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, et al. Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J Neurosci. 2017;37:9917–24.
Srikanth P, Lagomarsino VN, Muratore CR, Ryu SC, He A, Taylor WM, et al. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl Psychiatry. 2018;8:77–14.
Ye F, Kang E, Yu C, Qian X, Jacob F, Yu C, et al. DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis. Neuron. 2017;96:1041–54 e5.
Blair JD, Hockemeyer D, Bateup HS. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med. 2018;24:1568–78.
Pașca AM, Park J-Y, Shin H-W, Qi Q, Revah O, Krasnoff R, et al. Human 3D cellular model of hypoxic brain injury of prematurity. Nat Med. 2019;25:784–91.
Boisvert EM, Means RE, Michaud M, Madri JA, Katz SG. Minocycline mitigates the effect of neonatal hypoxic insult on human brain organoids. Cell Death Dis. 2019;10:325–11.
Paşca SP. Building three-dimensional human brain organoids. Nat Neurosci. 2018. https://doi.org/10.1038/s41593-018-0107-3.
Acknowledgements
We would like to thank the Knoblich lab members for their feedback on the manuscript. We apologize to the authors, whose work could not be cited due to the space limitations. Work in the Knoblich laboratory is supported by the Österreichischen Akademie der Wissenschaften, the Austrian Science Fund (Z_153_B09), and an advanced grant from the European Research Council under the European Union’s Horizon 2020 research and innovation program (GA 695642). JS was supported by EMBO long term fellowship (EMBO ALTF 794-2018).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
About this article

Cite this article
Sidhaye, J., Knoblich, J.A. Brain organoids: an ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ 28, 52–67 (2021). https://doi.org/10.1038/s41418-020-0566-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0566-4
This article is cited by
-
Humanized brain organoids-on-chip integrated with sensors for screening neuronal activity and neurotoxicity
Microchimica Acta (2024)
-
Human 3D brain organoids: steering the demolecularization of brain and neurological diseases
Cell Death Discovery (2023)
-
Interrogation of the mammalian gut–brain axis using LC–MS/MS-based targeted metabolomics with in vitro bacterial and organoid cultures and in vivo gnotobiotic mouse models
Nature Protocols (2023)
-
Establishment and characterization of human pluripotent stem cells-derived brain organoids to model cerebellar diseases
Scientific Reports (2022)
-
Human microglial models to study HIV infection and neuropathogenesis: a literature overview and comparative analyses
Journal of NeuroVirology (2022)
