
Abstract
The identification of viability-associated long noncoding RNAs (lncRNAs) is a means of uncovering therapeutic approaches for hepatocellular carcinoma (HCC). In addition, aberrant genome-wide hypomethylation has been implicated in HCC initiation and progression. However, the relationship between lncRNA dysregulation and genome-wide hypomethylation in hepatocarcinogenesis has not been fully elucidated. A novel lncRNA named LINC00662 was previously demonstrated to play a role in gastrointestinal cancer. In this study, we demonstrated that this lncRNA was correlated with survival and exhibited oncogenic properties, both in vitro and in vivo. Moreover, we determined that LINC00662 could lead to genome-wide hypomethylation and alter the genomic methylation profile by synchronously reducing the S-adenosylmethionine (SAM) level and enhancing the S-adenosylhomocysteine (SAH) level. Mechanistically, LINC00662 was determined to regulate the key enzymes influencing SAM and SAH levels, namely, methionine adenosyltransferase 1A (MAT1A) and S-adenosylhomocysteine hydrolase (AHCY), by RNA–RNA and RNA–protein interactions. In addition, we demonstrated that some SAM-dependent HCC-promoting genes could be regulated by LINC00662 by altering the methylation status of their promoters via the LINC00662-coupled axes of MAT1A/SAM and AHCY/SAH. Taken together, the results of this this study indicate that LINC00662 could be a potential biomarker for HCC therapy. More importantly, we proposed a new role of lncRNA in regulating genomic methylation to promote oncogene activation.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is a common malignancy and has become the third leading cause of cancer-related death worldwide [1]. However, the options for HCC therapy are limited, and the molecular mechanisms underlying HCC have not been thoroughly elucidated to date [2, 3]. It is widely recognized that both biomolecular dysregulation and aberrant genomic methylation can be detected in HCC initiation and progression [4]. However, the relationships between these phenomena and the role of their correlations in HCC have not been determined. In the last decade, HCC research has garnered attention following the identification of multiple long noncoding RNAs (lncRNAs) using transcriptome profiling [5,6,7]. As a biological molecule, lncRNAs have been demonstrated to be linked to various physiological and pathological functions, including hepatocarcinogenesis [8, 9]. These RNAs may act as an HCC promoter or suppressor by regulating multiple intracellular signaling pathways with different molecular mechanisms. However, the influence of lncRNA on altering genomic methylation has not been heavily investigated. DNA methylation is an enzymatic process involving various chemical substances [10,11,12]. Among these chemical substances, S-adenosylmethionine (SAM), as the major biological methyl donor, has been the focus of HCC research due to its crucial role in multiple biological processes [13,14,15,16]. In mammals, the SAM level is regulated by methionine adenosyltransferase (MAT), which contains two MAT-encoding genes: MAT1A and MAT2A [17, 18]. MAT1A is expressed in quiescent adult hepatocytes, and MAT2A is usually expressed in the proliferating liver, during dedifferentiation and in cancer [19, 20]. Interestingly, MAT1A can upregulate the SAM concentration, while MAT2A has the opposite effect due to the MAT1A/MAT2A switch [13, 17, 18]. Normally, SAM participates in transmethylation reactions to provide methyl, and then SAM is converted to S-adenosylhomocysteine (SAH) [21]. Thus, SAH is a byproduct involved in SAM-dependent transmethylation reactions, while SAH is also a strong feedback inhibitor of most SAM-dependent transmethylation reactions. In addition, S-adenosylhomocysteine hydrolase (AHCY, also known as SAHH) is the only known enzyme that catalyzes the hydrolysis of SAH to homocysteine and adenosine, thereby relieving this inhibition [22]. Thus, altered SAM and SAH levels or aberrant SAM-to-SAH ratios are considered activators that may significantly influence genomic methylation [23, 24]. More importantly, genomic methylation strongly contributes to tumorigenesis [23, 25, 26].
To date, the relationship between lncRNA dysregulation and genomic methylation alteration has not been fully elucidated, and the role of the abnormal genomic methylation caused by a single lncRNA in HCC initiation and progression has rarely been reported. In this study, we identified a functional lncRNA named long intergenic nonprotein coding RNA 662 (LINC00662), which acts as a tumor promoter by regulating SAM and SAH levels to maintain genomic hypomethylation, to provide new insights into HCC research.
Materials and methods
Tissue samples
In this study, we included 70 adult primary HCC tissue samples and paired adjacent normal tissue samples. The samples were obtained from patients who had previously undergone surgical resection in our department. The included patients met the following criteria: (1) clear histopathological diagnosis of primary HCC; (2) no preoperative therapy or other tumors; and (3) no previous pharmacological SAM treatment. All the tissue samples were well-preserved at −80 °C until they were employed in further experiments. The related protocols were approved by the Human Subjects Committee of Zhongnan Hospital according to the Declaration of Helsinki. Written informed consent was obtained from all patients.
In vivo and in vitro studies
Details are described in the Supplementary Data.
Statistical analysis
Dichotomous variables were assessed by χ2 test or Fisher’s exact test. Student’s t test was performed for comparisons of continuous variables between groups. The Kaplan–Meier test was used to estimate the survival rate, and multivariate Cox regression analysis with a backward stepwise approach was used to test for independent prognostic factors. The relationships in the relevant parametric data were evaluated by Spearman’s analysis. Differences with P values <0.05 were considered to be statistically significant. Statistical differences between groups were analyzed using SPSS 22.0 software (IBM, Chicago, IL, USA), and plotting generation was performed using GraphPad Prism 6.0 (GraphPad Software, USA).
Results
LINC00662 was upregulated in HCC and is associated with HCC progression
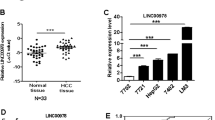
The expression of LINC00662 (accession: NR_027301.1) was detected in HCC tissues and paired adjacent liver tissues from 70 patients by q-PCR. The results showed significantly higher LINC00662 expression in HCC tissues than in adjacent tissues (Fig. 1a). We then analyzed the different clinical characteristics in the groups expressing high and low LINC00662 levels in clinical samples (35 patients in each group). The results from the clinical studies indicated that histological grade and portal vein tumor thrombus exhibited significant differences between the high LINC00662 and low LINC00662 groups (P < 0.05) (Fig. 1b). Moreover, LINC00662 could significantly enhance overall survival (P = 0.0071) and recurrence-free survival rate (P = 0.0064) (Fig. 1c). In addition, multivariate regression analysis revealed that LINC00662 levels were associated with overall survival rate (hazard ratio 2.340; P < 0.05) and recurrence-free survival rate (hazard ratio 3.112; P < 0.05) (Fig. 1d). These findings thus reveal that the expression of LINC00662 has clinical significance and can serve as a potential prognostic biomarker in HCC patients.

a Quantitative-PCR analysis of LINC00662 expression in HCC tissues and paired adjacent liver tissues from 70 patients. b Correlation between the LINC00662 expression levels and HCC clinicopathological features of 70 patients. c Kaplan–Meier analysis of overall survival (left panel) and recurrence-free survival (right panel) based on LINC00662 levels in the high expression group and the low expression group. d Multivariate analysis by Cox proportional hazard regression model for overall survival (upper panel) and recurrence-free survival (lower panel). *P < 0.05, **P < 0.01, ***P < 0.001.
LINC00662 promotes HCC progression in vitro and in vivo
To discover the exact role of LINC0062 in HCC, we performed experiments to investigate its biological functions in vitro and in vivo. We first detected LINC00662 in different hepatoma cell lines by q-PCR. The results showed that LINC00662 was significantly lower in Huh7 cells than in the other cell lines, and HCCLM9 cells had the highest level of LINC00662 (Supplementary Fig. S1A). Thus, we chose HCCLM9 and Huh7 cells for LINC00662 knockdown and overexpression, respectively (Supplementary Fig. S1B, C) according to LINC00662 expression levels. We then tested the proliferation capacity using a Cell Counting Kit-8 assay and discovered that cell proliferation could be upregulated by LINC00662 (Fig. 2a). In addition, flow cytometric analysis indicated that LINC00662 could markedly suppress apoptosis in hepatoma cells (Fig. 2b). Furthermore, cell growth was synchronously determined by clonogenic formation and EdU assays. The number of colonies and EdU incorporation were drastically reduced following LINC00662 downregulation (Fig. 2c, d). Finally, the results of wound healing and transwell assays illustrated that migration and invasion were repressed when LINC00662 was silenced (Fig. 2e, f). Taken together, these results suggest that LINC00662 has oncogenic effects in vitro. On the other hand, in vivo experiments, we conducted tumor growth and metastatic models by employing HCCLM9 cells stably transfected with LINC00662-shRNA (Fig. 3a). After subcutaneously injecting and sacrificing (Fig. 3b), the tumor growth curve showed that LINC00662 may effectively promote tumor size and weight (Fig. 3c). Moreover, comparing with the control group, LINC00662 knockdown dramatically reduced Ki-67 expression, indicated by immunohistochemistry (Fig. 3d). Meanwhile, lung tissues and corresponding lung HE slices revealed that LINC00662 knockdown significantly suppressed extrahepatic lung metastasis (Fig. 3e, f). Considering all this evidence, we may conclude that LINC00662 promotes oncogenic behaviors in vivo and in vivo.

a CCK-8 assay revealed that LINC00662 promoted the proliferation of HCCLM9 and Huh7 cells. Error bars are SD (n = 3). b Flow cytometric analysis determined that LINC00662 could significantly reduce apoptosis in HCCLM9 and Huh7 cells. Error bars are SD (n = 3). c Colony formation was enhanced when LINC00662 expression was increased and decreased when LINC00662 expression was reduced in HCCLM9 and Huh7 cells. Error bars are SD (n = 3). d HCCLM9 cell growth was suppressed 48 h after LINC00662 siRNA transfection, while Huh7 cell growth was promoted 48 h after LINC00662-vector transfection as determined by the EdU assay (bar = 50 μm). Error bars are SD (n = 3). e Representative images of wound healing assays performed after LINC00662 knockdown in HCCLM9 cells and after LINC00662 overexpression in Huh7 cells. Error bars are SD (n = 3). f Transwell assays performed using HCCLM9 cells after LINC00662 knockdown and Huh7 cells after LINC00662 overexpression (bar = 60 μm). Error bars are SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.

a Schematic representation of xenografts based on two different models in vivo. b Representative images of the mice (upper panel) and removed tumors (lower panel) formed in nude mice that were subcutaneously injected with cells stably transfected with LINC00662-shRNA or empty vector. c The effects of LINC00662 on tumor growth are summarized by a tumor growth curve (left panel) and tumor weight graph (right panel). Error bars represent the SD (n = 5). d Microscopic images of HE and immunohistochemical staining patterns for Ki-67 in tumor xenografts from the nude mice in the LINC00662 knockdown group and the control group (scale bars = 24 μm). e Representative images of visible metastatic nodules in the mouse lungs (left panel) and HE staining of metastatic nodules in the lungs (right panel). The black arrows indicate tumors. f Quantitative comparison of metastatic nodules between the scramble group and LINC00662-shRNA group. *P < 0.05, **P < 0.01, ***P < 0.001.
LINC00662 may regulate genomic methylation via aberrant SAM and SAH levels
In recent years, multiple studies have reported that lncRNAs play different roles in regulating HCC progression by, for instance, activating classic signaling pathways. Meanwhile, it has been known for decades that a hallmark of cancer is global hypomethylation [27, 28]. More importantly, genomic hypomethylation could activate oncogenes to promote oncogenic processes. Therefore, since lncRNA dysregulation and global hypomethylation could be synchronously detected, lncRNA may have impacts on genomic methylation in oncogenic processes [29, 30]. As mentioned above, LINC00662 was demonstrated to act as a tumor promoter in vitro and in vivo. Thus, we speculated that LINC00662 may associate with genomic methylation in HCC progression.
To validate our hypotheses, we first compared the 5-methylcytosine (5mC) levels, which represented the degrees of genomic methylation [31], in the included HCC tissues and paired adjacent tissues by enzyme-linked immunosorbent assay (ELISA). The contents of 5mC were verified to be lower in the HCC tissues than in the adjacent tissues (Fig. 4a). In addition, we quantified SAM and SAH levels in HCC tissues and adjacent liver tissues using ultra-performance liquid chromatography (UPLC). The results revealed that the SAM level was lower and the SAH level was significantly higher in the HCC tissues than in the adjacent liver tissues (Fig. 4b, c). It is widely known that genomic methylation is determined by SAM and SAH levels [23, 24], which may be regulated by multiple biomolecules, including lncRNAs [29, 30]. To verify that LINC00662 may regulate genomic methylation via aberrant SAM and SAH levels, we investigated the relationships of LINC00662 with the levels of 5mC, SAM, and SAH. The statistical results illustrated that in both HCC tissues and adjacent liver tissues, LINC00662 was strongly negatively correlated with 5mC contents and SAM levels (Fig. 4d, e) but positively correlated with SAH levels (Fig. 4f).

a ELISA results examining the content of 5mC in 70 HCC tissues and paired adjacent liver tissues. b UPLC results of the SAM concentration in 70 HCC tissues and paired adjacent liver tissues. c UPLC results of the SAH concentration in 70 HCC tissues and paired adjacent liver tissues. d Bivariate correlation analysis of the relationship between 5mC content and LINC00662 expression in 70 HCC tissues (left panel) and paired adjacent liver tissues (right panel). e Bivariate correlation analysis of the relationship between SAM concentration and LINC00662 expression in 70 HCC tissues (left panel) and paired adjacent liver tissues (right panel). f Bivariate correlation analysis of the relationship between SAH concentration and LINC00662 expression in 70 HCC tissues (left panel) and paired adjacent liver tissues (right panel). ***P < 0.001.
To further validate that LINC00662 may alter genomic methylation by regulating SAM and SAH levels in vitro, we analyzed the levels of 5mC, SAM, and SAH in HCCLM9 cells after LINC00662 knockdown and in Huh7 cells after LINC00662 overexpression, respectively. We demonstrated that LINC00662 could downregulate 5mC and SAM levels but could upregulate SAH levels (Fig. 5a, b). In addition, LINC00662 was also verified to reduce the SAM-to-SAH ratios in both cell lines (Fig. 5c). Next, to better elaborate the effects of LINC00662 on genomic methylation patterns, significant differences in aberrant methylation loci were screened and quantitatively analyzed by an Infinium Methylation 850K BeadChip Microarray. We first analyzed the changes in the methylation patterns of HCCLM9 cells after LINC00662 knockdown. The results showed that 97331 loci (not including GAPDH) revealed significant methylation changes after LINC00662 knockdown in HCCLM9 cells. Among these loci, 79.1% of these loci exhibited hypermethylation, and the other 20.9% exhibited hypomethylation (Fig. 5d). We observed the overall distribution of the beta values from the microarray and found that DNA methylation was induced in HCCLM9 cells (Supplementary Fig. S2A). We next compared methylation profiles across different gene regions and found that there was a markedly higher proportion of hypermethylated loci than hypomethylated loci (Supplementary Fig. S2B, C). Among loci with methylation alternations, 30.8% were located at TSS 1500 + 200 regions, and 24.9% were located at CpG island regions (Supplementary Fig. S2D, E) (Fig. 5d). In addition, we investigated methylation alterations in Huh7 cells after LINC00662 overexpression. There were 75733 loci with changes in methylation, 68.5% of which were hypomethylated, and 31.5% of which were hypermethylated (Fig. 5e), as determined by the genomic methylation distributions (Supplementary Fig. S3A). Consistent with this result, hypomethylation was more common in each gene region when LINC00662 was overexpressed (Supplementary Fig. S3B, C). Among all these aberrant methylation loci, 16.1% (Supplementary Fig. S3D) were located in TSS 1500 + 200 regions, and 15.5% (Supplementary Fig. S3E) were located in CpG island regions (Fig. 5e). Moreover, after multiple hypothesis testing, we summarized the methylation alterations in the promoter-related regions of multiple HCC-promoting genes using minimum adjusted P values (Supplementary Fig. S4) in both cell lines. All these findings may indicate that LINC00662 may lead to genomic hypomethylation and alter methylation profile patterns by synchronously enhancing SAM levels and reducing SAH levels. More importantly, these changes may increase the hypomethylation status of the promoter regions of multiple HCC-promoting genes.

a ELISA results examining the content of 5mC after LINC00662 knockdown in HCCLM9 cells or overexpression in Huh7 cells. Error bars are SD (n = 3). b, c UPLC was performed to measure the intracellular levels of SAM and SAH and their ratios after LINC00662 knockdown in HCCLM9 cells or overexpression in Huh7 cells. Error bars are SD (n = 3). d Summary of alterations in the total methylation status and genomic methylation profile patterns in different regulatory regions of genes as determined by the Infinium Methylation 850K BeadChip Microarray after LINC00662 knockdown in HCCLM9 cells or e after LINC00662 overexpression in Huh7 cells. TSS 1500 is 200–1500 bp upstream of the transcription start site (TSS); TSS200 is 1–200 bp upstream of the TSS; 5′UTR is the 5′ untranslated region; the 1st exon is the first translated region; the gene body includes all other exons and all introns; and the 3′UTR is the 3′ untranslated region where translation ends. Regions outside of the TSS 1500 and 3′UTR are considered intergenic, and TSS 1500 + 200 is considered the promoter-related region. CpG islands are defined as having a GC content of 50% or greater and a length greater than 200 bp. The north shore is 0–2000 bp upstream of the CpG island, and the north shelf is 2001–4000 bp upstream of the CGI. The south shore is 0–2000 bp downstream of the CpG island, and the south shelf is 2001–4000 bp downstream of the CpG island. Open sea regions are defined as >4000 bp outside of the CpG islands. **P < 0.01, ***P < 0.001.
LINC00662 alters SAM and SAH levels via regulating MAT1A and AHCY
We have demonstrated that LINC0062 may lead to genome-wide hypomethylation and change methylation profile patterns by synchronously altering SAM and SAH levels. As mentioned above, SAM and SAH could be respectively regulated by their key enzyme MAT1A and AHCY [13, 22]. Therefore, we speculated that LINC00662 may downregulate MAT1A and upregulate AHCY to alter SAM and SAH levels. To support our speculation, we quantified the levels of MAT1A (accession: KR709424.1) and AHCY (accession: KR710248.1) by q-PCR and western blot analysis in HCCLM9 cells after knocking down LINC00662 and in Huh7 cells after LINC00662 overexpression. We demonstrated that LINC00662 could downregulate the mRNA and protein levels of MAT1A and the protein level of AHCY (Fig. 6a, b). Next, we investigated the relationship of LINC00662 and MAT1A and AHCY in HCC tissues. The results revealed that LINC00662 levels were statistically negatively correlated with MAT1A mRNA expression but failed to achieve an association with AHCY mRNA (Fig. 6c). We next conducted western blotting to detect the protein level of the included 70 HCC tissues and determined that the protein level of AHCY was negatively correlated with LINC00662 (Fig. 6d, e). To date, we may conclude that LINC00662 was significantly negatively correlated with MAT1A mRNA and AHCY protein levels.

a q-PCR analyses of the MAT1A and AHCY mRNA levels after LINC00662 knockdown in HCCLM9 cells and overexpression in Huh7 cells. Error bars are SD (n = 3). b Western blot analyses of the MAT1A and AHCY protein levels after LINC00662 knockdown in HCCLM9 cells and overexpression in Huh7 cells. Error bars are SD (n = 3). c Bivariate correlation analysis of the relationship between LINC00662 and MAT1A mRNA (upper panel) and AHCY mRNA (lower panel) in 70 HCC tissues. d Western blot (left panel) and quantitative (right panel) analysis of AHCY protein levels in 70 HCC tissues. Error bars are SD (n = 3). e Bivariate correlation analysis of the relationship between LINC00662 and AHCY protein levels in 70 HCC tissues. ***P < 0.001.
Noncoding RNAs have been shown to target mRNAs via direct or indirect RNA–RNA interactions [32, 33]. Therefore, we proposed some assumptions according to a series of bioinformatics predictions using IntraRNA [34, 35]. Interestingly, the 3′UTR of the MAT1A mRNA was identified as a target of LINC00662 (Fig. 7a). To clarify the interactions among these molecules, RNA fluorescence in situ hybridization (FISH) was conducted after LINC00662 knockdown in HCCLM9 cells and overexpression in Huh7 cells. The results indicated that both MAT1A mRNA and LINC00662 were mainly located in the cytoplasm and that their subcellular localization was not affected by LINC00662 knockdown or overexpression (Fig. 7b). The RNA–RNA Rip (RaPID assay) determined that the 3′ UTR of MAT1A mRNA could significantly enhance the enrichment of LINC00662 with direct combination in both HCCLM9 and Huh7 cells (Fig. 7c). Moreover, actinomycin D (ActD) assay indicated that LINC00662 could markedly promote the decay of MAT1A mRNA (Fig. 7d). In addition, luciferase reporter illustrated that LINC00662 significantly inhibited the luciferase activity of wild-type MAT1A 3′UTR compared with that of MAT1A with mutations in putative targeting sites (Fig. 7e). Based on these findings, we may conclude that LINC00662 could directly interact with the MAT1A 3′UTR and promote the decay of this mRNA to reduce MAT1A levels. On the other hand, as previously described, LINC00662 could downregulate AHCY protein levels but had no impact on its mRNA level. Thus, we deduced that LINC00662 directly interacts with the AHCY protein. To validate our speculation, we performed RNA pulldown and RNA immunoprecipitation (RIP) assays and we observed that LINC00662 directly bound to the AHCY protein (Fig. 8a) and that AHCY could directly enrich LINC00662 in hepatoma cells (Fig. 8b). Next, we conducted immunohistochemistry to identify AHCY subcellular localization, and the results indicated that AHCY was located in the cytoplasm in HCC tissue (Fig. 8c). Then, we performed immunocytochemistry and immunofluorescence assays after LINC00662 knockdown in HCCLM9 cells and overexpression in Huh7 cells. We visually observed that AHCY was still primarily located in the cytoplasm in hepatoma cells (Fig. 8c, d). The expression of AHCY protein seemed to be higher when LINC00662 was expressed at low levels in both HCC tissues and hepatoma cells (Fig. 8c). We then employed cycloheximide (CHX) assay and western blot and illustrated that LINC00662 could promote a reduction in the AHCY protein (Fig. 8e). Finally, to investigate whether the LINC00662-AHCY interaction increased the level of AHCY by affecting the ubiquitination/degradation of AHCY, we examined the ubiquitination and protein level of AHCY in the presence or absence of LINC00662 and/or the proteasome inhibitor MG132. As the results show, LINC00662 induced the ubiquitination of AHCY (anti-AHCY IP product) to decrease the level of AHCY (Fig. 8f). These results suggest that LINC00662 directly interacts with the AHCY protein, enhancing its instabilities by increasing its ubiquitin-mediated degradation.

a Schematic of the predicted RNA–RNA interaction between LINC00662 and the 3′UTR of MAT1A. b RNA FISH depiction of the subcellular localization of LINC00662 and MAT1A mRNA after LINC00662 knockdown in HCCLM9 cells and overexpression in Huh7 cells. (bar = 32 μm). c Schematic representation of the interaction of LINC00662 with the MAT1A 3′UTR in an in vitro RNA–RNA interaction assay (upper panel). LINC00662 was markedly enriched at the MAT1A 3′UTR compared with the control or mutant sequences as determined by q-PCR (lower panel). Error bars are SD (n = 3). d ActD treatment and q-PCR analysis were conducted to verify the posttranscriptional regulation of MAT1A mRNA by LINC00662. Error bars are SD (n = 3). e Luciferase activities were determined by dual luciferase analysis to evaluate the inhibitory effects of LINC00662 on MAT1A. Error bars are SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001. NS not significant.

a Western blot analysis of the specific association of LINC00662 with AHCY from RNA pulldown assays. b RNA immunoprecipitation (RIP) experiments were performed using the AHCY antibody, and specific primers were used to detect LINC00662 with q-PCR. Error bars are SD (n = 3). c, d Representative images of immunohistochemical, immunocytochemical (bar = 30 μm) and immunofluorescence (bar = 32 μm) slices comparing AHCY protein visually between high LINC00662 and low LINC00662 in HCC tissues or hepatoma cells. e Different treatment cell groups were incubated with cycloheximide (CHX) for different time points. The protein levels of AHCY were detected by western blot (left panel) and quantitative (right panel) analysis. Error bars are SD (n = 3). f Ubiquitin levels of AHCY immunoprecipitates measured by the appearance or absence of MG132 and input AHCY protein level tested by western blot analysis. **P < 0.01, ***P < 0.001.
LINC00662 promotes oncogene activation by inducing genomic hypomethylation
Multiple studies have illustrated that both SAM accumulation and SAH reduction contribute to antitumorigenicity by altering the DNA methylation status of multiple oncogenes [26, 36,37,38]. Based on the abovementioned results, we concluded that LINC00662 may induce hypomethylation by coupling the MAT1A/SAM and AHCY/SAH axes, which strongly contribute to carcinogenesis. To validate this hypothesis, we measured the 5mC content after upregulating MAT1A and AHCY in HCCLM9 and Huh7 cells and found that both MAT1A and AHCY could markedly enhance 5mC content. Furthermore, this effect could be enhanced by each other (Fig. 9a). To further clarify the impact of aberrant genomic methylation on tumorigenesis, we investigated the influence of LIN00662 on the expression of oncogenes. According to previous results, in HCCLM9 cells, LINC00662 knockdown led to hypermethylation of the promoters of multiple HCC-promoting genes, including MYC (known as c-myc) (NC_000008.11), HRAS (known as H-ras) (NC_000011.10) and CTNNB1 (known as β-catenin) (NC_000003.12), which were the three genes with the most methylation alternations in our study (Supplementary Fig. S4A). In addition, these three genes also revealed significant hypomethylation in Huh7 cells after LINC00662 overexpression (Supplementary Fig. S4B). Therefore, we selected these three oncogenes to verify the effects of LINC00662 on oncogene expression. We first analyzed the mRNA and protein levels of these three oncogenes after synchronously knocking down LINC00662 and administering SAH treatment in HCCLM9 cells. We discovered that the mRNA and protein levels of MYC, HRAS, and CTNNB1 were affected by LINC00662 and SAH. More importantly, rescue experiments demonstrated that LINC00662 and SAH exhibited synergistic efficacy. However, conflicting effects were observed in Huh7 cells after synchronously overexpressing LINC00662 and administering SAM treatment (Fig. 9b, c). We next performed a methylated DNA immunoprecipitation (MeDIP)-PCR assay to investigate whether these aberrant expression levels correspond to the methylation status of their DNA. The results indicated that both LINC00662 and SAH could influence the relative oncogene methylation status, which determined their subsequent expression in HCCLM9 cells. In addition, the same results were observed in Huh7 cells after LINC00662 overexpression and SAM treatment, and rescue experiments (Fig. 9d). To date, we demonstrated that the expression of these oncogenes was SAM/SAH-dependent and thus could be influenced by the SAM and SAH axes, which play crucial roles in oncogene activation. On the other hand, logically, to construct the abovementioned two axes contributing to tumor promotion, the roles of MAT1A and AHCY in hepatocarcinogenesis need to be identified. It is known that both MAT1A and AHCY participate in 1-carbon metabolism and are downregulated in HCC [39, 40]. Therefore, we overexpressed MAT1A and AHCY in HCCLM9 and Huh7 hepatoma cells to investigate their carcinogenesis in HCC (Supplementary Fig. S5). The results indicated that both MAT1A and AHCY upregulation contribute to the inhibition of oncogenic behaviors (Supplementary Fig. S6, S7). Based on these findings, we speculated that LINC00662 may exhibit tumor-promoting effects by inducing genomic hypomethylation, leading to the activation of multiple oncogenes (Fig. 9e).

a ELISA was performed to detect 5mC levels after MAT1A overexpression and/or AHCY overexpression in HCCLM9 and Huh7 cells. Error bars are SD (n = 3). b, c q-PCR and western blot analyses of the mRNA and protein levels of MYC, HRAS, and CTNNB1 in HCCLM9 cells with siRNA-LINC00662 and SAH treatment or in Huh7 cells with vector-LINC00662 and SAM treatment. Error bars are SD (n = 3). d MeDIP with q-PCR was performed to measure the methylation status of the MYC, HRAS and CTNNB1 genes after LINC00662 knockdown and SAH treatment in HCCLM9 or after LINC00662 overexpression and SAM treatment in Huh7 cells. Error bars are SD (n = 3). e Schematic of the proposed mechanism of the LINC00662-mediated alteration in genomic methylation via regulating SAM and SAH levels to active oncogenes. *P < 0.05, **P < 0.01, ***P < 0.001. NC negative control.
Discussion
Genomic hypomethylation is a hallmark of cancer and is recognized as a prerequisite for oncogene activation [36,37,38, 41]. In addition, lncRNA dysregulation in multiple cancers was also detected, which involves many molecular biological interactions that regulate oncological behaviors; however, the mechanism of lncRNA-mediated tumorigenic behaviors by altered DNA methylation remains largely unknown [28, 41, 42]. The full posttranscriptional length of LINC00662 is 2085 bp, which allows it to participate in many biological processes. Four recent studies reported the cancer-promoting efficacy of LINC00662 in gastric cancer, lung cancer, and oral cancer cells [43,44,45,46]. However, these studies lacked comprehensive and in-depth explanations of oncogene activation and molecular mechanisms. Based on clinical data, the LINC00662 level was demonstrated to be a prognostic factor in HCC for the first time. In addition, LINC00662 revealed tumorigenic behaviors, both in vivo and in vitro. All of these phenomena indicated that LINC00662 could be regarded as a potential biomarker for the diagnosis or treatment of HCC. Moreover, it is known that the dysregulation of cellular metabolism is a hallmark of cancer, and the dysregulation of methionine metabolism is implicated in human liver cancer [15,16,17,18,19,20,21, 47]. Therefore, we considered the relationship between LINC00662 and genomic methylation and attempted to discover a new way to comprehensively interpret the mechanisms of HCC promotion.
Our results indicated that LINC00662 could reduce SAM levels and enhance SAH levels. As the major methyl donor, SAM greatly contributes to maintaining genomic hypermethylation in normal liver [13, 48, 49]. In addition, as a byproduct of SAM-dependent transmethylation, the accumulation of SAH may exert pleiotropic effects on HCC progression and prognosis [25, 50]. Therefore, synchronously reducing SAM and enhancing SAH demonstrated that LINC00662 presented a remarkable means of influencing genomic methylation and revealed its contributions to HCC promotion [25, 26, 36,37,38, 41]. We found that LINC00662 suppresses SAM and enhances SAH by regulating two key enzymes, MAT1A and AHCY, which directly interact with MAT1A mRNA and AHCY protein, respectively. Concerning the upstream regulation, SAM was mainly determined by MAT1A in normal liver [17, 19]. However, MAT1A may be suppressed with liver injury and HCC, resulting in a reduction in SAM levels [18, 51]. Similarly, AHCY is the only enzyme that hydrolyzes SAH to inhibit SAH accumulation and maintain genomic hypermethylation [22]. More importantly, the upregulation of MAT1A and AHCY contributed to antitumorigenesis [25, 51, 52]. We also demonstrated these effects in hepatoma cells in the current study. In addition, previous studies have demonstrated that both SAM accumulation and SAH reduction could lead to tumor suppression because the transcription of multiple oncogenes is dependent on SAM/SAH, which is consistent with our results [25, 26, 36]. Based on these facts, aberrant SAM and SAH levels may play important roles in oncological signaling pathway activation or inhibition. In the current study, the three oncogenes exhibiting the highest levels of aberrant methylation were verified to be associated with the LINC00662-coupled axes. These findings may further support the hypothesis that LINC00662 could activate multiple tumor signaling pathways by altering the DNA methylation profile. This process may represent a new concept to elucidate the initialization and development of HCC. However, although lncRNA was determined to directly interact with mRNA and protein [53, 54], the synchronous effects of lncRNA on posttranscriptional and posttranslational regulation have rarely been reported. These findings may imply that LINC00662 has many unknown potential functions, and we believe that there are other mechanisms underlying the regulation of tumor initialization and promotion.
Notably, this study also illustrated that LINC00662 could alter the patterns of methylation profiles. According to our results, after either knocking down or overexpressing LINC00662, both promoter-related (TSS 1500 + 200) and gene body regions were more susceptible to aberrant methylation than were other gene segments [55]. Moreover, we also found that the proportion of promoter regions with aberrantly methylated loci was consistent with the proportion of CpG islands with aberrantly methylated loci, which might indicate that aberrantly methylated CpG island loci were mainly distributed in the promoter region. It is recognized that the methylation status of CpG islands in promoter regions may directly regulate relative gene expression [56]. Thus, comprehensively, this phenomenon indicates that LINC00662 promotes the expression of oncogenes by altering the methylation of their promoters. In addition, for gene body regions, hypomethylation may lead to a similar influence on gene expression as promoter hypermethylation [57]. Therefore, although the influence of SAM and SAH alterations on genomic methylation appeared to be complex, we can identify some coupled effects on gene expression. However, the underlying mechanism warrants further research.
To date, only two previous papers have elucidated the regulation of different molecular mechanisms by lncRNA and genome-wide methylation [29, 30]. However, these two papers provided a limited description of the molecular mechanisms of aberrant genomic methylation while neglecting to investigate the comprehensive influences of this methylation on tumorigenesis. In the current study, the tumorigenic effect of LINC00662 was determined in HCC for the first time. In addition, LINC00662 is the first lncRNA identified to synchronously regulate SAM and SAH. The remarkable function of LINC00662 could alter genomic methylation profile patterns to achieve tumorigenic activation. Moreover, theoretically, a single lncRNA may have effects on intracellular metabolism or multiple tumor signaling pathways at the same time through different mechanisms. However, we focused on investigating the aberrant intracellular substrate circumstances that affect the expression and function of multiple oncogenes to provide more evidence to predict HCC development. Taken together, the results of our study may provide a new approach to comprehensively investigate tumor initiation and promotion. However, we must admit some shortcomings in our research. For instance, although we theoretically hypothesized that the altered genomic methylation caused by LINC00662 may significantly influence the expression of multiple oncogenes, we only verified this in the three genes with the most aberrant methylation. In addition, due to the complexity and heterogeneity of intracellular dysregulation, our conclusions may not suitably explain the expression of every gene; we only proposed the importance of a single lncRNA in the alteration of the intracellular substrate environment to interpret a macroscopic phenomenon. In addition, we acknowledge that as a tumor promoter, LINC00662 may activate some oncogenes via other mechanisms; elucidating this activity was not the aim of the current study but warrants further research.
In conclusion, the data presented in this study establish LINC00662, which is upregulated in HCC and correlates with patient survival, as a functionally relevant lncRNA in HCC. LINC00662 promotes oncogenic behaviors both in vitro and in vivo. The tumorigenic effects of this lncRNA result from alterations in methylation profiles via synchronously regulating intracellular SAM and SAH levels. Thus, our study addressed a novel functional lncRNA and highlights the novel importance of the role of a single lncRNA in the aberrant DNA methylation that modulates HCC initiation and progression.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–17.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:359–86.
Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424–35.
Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, et al. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE. 2007;2:e845.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27.
Cui H, Zhang Y, Zhang Q, Chen W, Zhao H, Liang J. A comprehensive genome-wide analysis of long noncoding RNA expression profile in hepatocellular carcinoma. Cancer Med. 2017;6:2932–41.
Bao H, Su H. Long noncoding RNAs act as novel biomarkers for hepatocellular carcinoma: progress and prospects. Biomed Res Int. 2017;2017:6049480.
Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma: mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017;67:603–18.
Wu JC, Santi DV. Kinetic and catalytic mechanism of HhaI methyltransferase. J Biol Chem. 1987;262:4778–86.
Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–69.
Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93.
Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32:391–5.
Mato JM, Martínez-Chantar ML, Lu SC. S-Adenosylmethionine metabolism and liver disease. Ann Hepatol. 2013;12:183–9.
Lu SC, Ramani K, Ou X, Lin M, Yu V, Ko K, et al. S-Adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009;50:462–71.
Yan L, Liang X, Huang H, Zhang G, Liu T, Zhang J, et al. S-Adenosylmethionine affects cell cycle pathways and suppresses proliferation in liver cells. J Cancer. 2019;10:4368–79.
Lu SC, Tsukamoto H, Mato JM. Role of abnormal methionine metabolism in alcoholic liver injury. Alcohol. 2002;27:155–62.
Martínez-Chantar ML, García-Trevijano ER, Latasa MU, Martín-Duce A, Fortes P, Caballería J, et al. Methionine adenosyltransferase II beta subunit gene expression provides a proliferative advantage in human hepatoma. Gastroenterology. 2003;124:940–8.
Lu SC, Mato JM. S-Adenosylmethionine in liver health, injury, and cancer. Physiol Rev. 2012;92:1515–42.
Lu SC, Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol. 2005;35:227–34.
Mato JM, Alvarez L, Ortiz P, Pajares MA. S-Adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharm Ther. 1997;73:265–80.
Tehlivets O, Malanovic N, Visram M, Pavkov-Keller T, Keller W. S-Adenosyl-L-homocysteine hydrolase and methylation disorders: yeast as a model system. Biochim Biophys Acta. 2013;1832:204–15.
Calvisi DF, Simile MM, Ladu S, Pellegrino R, De Murtas V, Pinna F, et al. Altered methionine metabolism and global DNA methylation in liver cancer: relationship with genomic instability and prognosis. Int J Cancer. 2007;121:2410–20.
Ponnaluri VKC, Estève PO, Ruse CI, Pradhan S. S-Adenosylhomocysteine hydrolase participates in DNA methylation inheritance. J Mol Biol. 2018;430:2051–65.
Leal JF, Ferrer I, Blanco-Aparicio C, Hernández-Losa J, Ramón Y, Cajal S, et al. S-Adenosylhomocysteine hydrolase downregulation contributes to tumorigenesis. Carcinogenesis. 2008;29:2089–95.
Li TW, Peng H, Yang H, Kurniawidjaja S, Panthaki P, Zheng Y, et al. S-Adenosylmethionine and methylthioadenosine inhibit β-catenin signaling by multiple mechanisms in liver and colon cancer. Mol Pharmacol. 2015;87:77–86.
Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988;48:1159–61.
Dante R, Dante-Paire J, Rigal D, Roizès G. Methylation patterns of long interspersed repeated DNA and alphoid repetitive DNA from human cell lines and tumors. Anticancer Res. 1992;12:559–63.
Zhou J, Yang L, Zhong T, Mueller M, Men Y, Zhang N, et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat Commun. 2015;6:10221.
Guo T, Wang H, Liu P, Xiao Y, Wu P, Wang Y, et al. SNHG6 acts as a genome-wide hypomethylation trigger via coupling of miR-1297-mediated S-adenosylmethionine-dependent positive feedback loops. Cancer Res. 2018;78:3849–64.
Robertson KD. DNA methylation and chromatin—unraveling the tangled web. Oncogene. 2002;21:5361–79.
Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, et al. RNA–RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell. 2014;159:188–99.
Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–5.
Gawronski AR, Uhl M, Zhang Y, Lin YY, Niknafs YS, Ramnarine VR, et al. MechRNA: prediction of lncRNA mechanisms from RNA–RNA and RNA–protein interactions. Bioinformatics. 2018;34:3101–10.
Mann M, Wright PR, Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 2017;45:435–9.
Luo J, Li YN, Wang F, Zhang WM, Geng X. S-Adenosylmethionine inhibits the growth of cancer cells by reversing the hypomethylation status of c-myc and H-ras in human gastric cancer and colon cancer. Int J Biol Sci. 2010;6:784–95.
Zhao Y, Li JS, Guo MZ, Feng BS, Zhang JP. Inhibitory effect of S-adenosylmethionine on the growth of human gastric cancer cells in vivo and in vitro. Chin J Cancer. 2010;29:752–60.
Ou X, Yang H, Ramani K, Ara AI, Chen H, Mato JM, et al. Inhibition of human betaine-homocysteine methyltransferase expression by S-adenosylmethionine and methylthioadenosine. Biochem J. 2007;401:87–96.
Simile MM, Peitta G, Tomasi ML, Brozzetti S, Feo CF, Porcu A, et al. MicroRNA-203 impacts on the growth, aggressiveness and prognosis of hepatocellular carcinoma by targeting MAT2A and MAT2B genes. Oncotarget. 2019;10:2835–54.
Pogribny IP, Dreval K, Kindrat I, Melnyk S, Jimenez L, de Conti A, et al. Epigenetically mediated inhibition of S-adenosylhomocysteine hydrolase and the associated dysregulation of 1-carbon metabolism in nonalcoholic steatohepatitis and hepatocellular carcinoma. FASEB J. 2018;32:1591–601.
Molnár B, Galamb O, Péterfia B, Wichmann B, Csabai I, Bodor A, et al. Gene promoter and exon DNA methylation changes in colon cancer development—mRNA expression and tumor mutation alterations. BMC Cancer. 2018;18:695.
Sadek KM, Lebda MA, Nasr NE, Nasr SM, El-Sayed Y. Role of lncRNAs as prognostic markers of hepatic cancer and potential therapeutic targeting by S-adenosylmethionine via inhibiting PI3K/Akt signaling pathways. Environ Sci Pollut Res Int. 2018;25:20057–70.
Liu Z, Yao Y, Huang S, Li L, Jiang B, Guo H, et al. LINC00662 promotes gastric cancer cell growth by modulating the Hippo-YAP1 pathway. Biochem Biophys Res Commun. 2018;505:843–9.
Gong W, Su Y, Liu Y, Sun P, Wang X. Long non-coding RNA Linc00662 promotes cell invasion and contributes to cancer stem cell-like phenotypes in lung cancer cells. J Biochem. 2018;164:461–9.
Xu D, Chen Y, Yuan C, Zhang S, Peng W. Long non-coding RNA LINC00662 promotes proliferation and migration in oral squamous cell carcinoma. Onco Targets Ther. 2019;12:647–56.
Zhang L, Chen S, Wang B, Su Y, Li S, Liu G, et al. An eight-long noncoding RNA expression signature for colorectal cancer patients’ prognosis. J Cell Biochem. 2019;120:5636–43.
Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76.
Frau M, Tomasi ML, Simile MM, Demartis MI, Salis F, Latte G, et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology. 2012;56:165–75.
Zhang X, Li H, Qiu Q, Qi Y, Huang D, Zhang Y. 2,4-Dichlorophenol induces global DNA hypermethylation through the increase of S-adenosylmethionine and the upregulation of DNMTs mRNA in the liver of goldfish Carassius auratus. Comp Biochem Physiol C Toxicol Pharm. 2014;160:54–9.
Frau M, Feo F, Pascale RM. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J Hepatol. 2013;59:830–41.
Wang R, Jin Y, Yao XH, Fan W, Zhang J, Cao Y, et al. A novel mechanism of the M1-M2 methionine adenosyltransferase switch-mediated hepatocellular carcinoma metastasis. Mol Carcinog. 2018;57:1201–12.
Li J, Ramani K, Sun Z, Zee C, Grant EG, Yang H, et al. Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol. 2010;176:2456–66.
Zhang Y, Pitchiaya S, Cieślik M, Niknafs YS, Tien JC, Hosono Y, et al. Analysis of the androgen receptor-regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat Genet. 2018;50:814–24.
Ma Y, Yang Y, Wang F, Moyer MP, Wei Q, Zhang P, et al. Long non-coding RNA CCAL regulates colorectal cancer progression by activating Wnt/β-catenin signalling pathway via suppression of activator protein 2α. Gut. 2016;65:1494–504.
Eckstein M, Rea M, Fondufe-Mittendorf YN. Transient and permanent changes in DNA methylation patterns in inorganic arsenic-mediated epithelial-to-mesenchymal transition. Toxicol Appl Pharm. 2017;331:6–17.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22.
Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90.
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (No. 2042018kf0079); Hubei Province Natural Science Foundation of China (No. 2018CFB157); National Natural Science Foundation of China (No. 81800522).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics
The related protocols were approved by the Human Subjects Committee of Zhongnan Hospital according to the Declaration of Helsinki. Written informed consent was obtained from all patients.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H.-U. Simon
Rights and permissions
About this article

Cite this article
Guo, T., Gong, C., Wu, P. et al. LINC00662 promotes hepatocellular carcinoma progression via altering genomic methylation profiles. Cell Death Differ 27, 2191–2205 (2020). https://doi.org/10.1038/s41418-020-0494-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0494-3
This article is cited by
-
Emerging role of RNA modification and long noncoding RNA interaction in cancer
Cancer Gene Therapy (2024)
-
Mutual interaction of lncRNAs and epigenetics: focusing on cancer
Egyptian Journal of Medical Human Genetics (2023)
-
LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond
Journal of Experimental & Clinical Cancer Research (2022)
-
Identification of ZBTB9 as a potential therapeutic target against dysregulation of tumor cells proliferation and a novel biomarker in Liver Hepatocellular Carcinoma
Journal of Translational Medicine (2022)
-
METTL3/LINC00662/miR-186-5p feedback loop regulates docetaxel resistance in triple negative breast cancer
Scientific Reports (2022)
