
Abstract
Skeletal muscle mitochondria readily accumulate Ca2+ in response to SR store-releasing stimuli thanks to the activity of the mitochondrial calcium uniporter (MCU), the highly selective channel responsible for mitochondrial Ca2+ uptake. MCU positively regulates myofiber size in physiological conditions and counteracts pathological loss of muscle mass. Here we show that skeletal muscle-specific MCU deletion inhibits myofiber mitochondrial Ca2+ uptake, impairs muscle force and exercise performance, and determines a slow to fast switch in MHC expression. Mitochondrial Ca2+ uptake is required for effective glucose oxidation, as demonstrated by the fact that in muscle-specific MCU−/− myofibers oxidative metabolism is impaired and glycolysis rate is increased. Although defective, mitochondrial activity is partially sustained by increased fatty acid (FA) oxidation. In MCU−/− myofibers, PDP2 overexpression drastically reduces FA dependency, demonstrating that decreased PDH activity is the main trigger of the metabolic rewiring of MCU−/− muscles. Accordingly, PDK4 overexpression in MCUfl/fl myofibers is sufficient to increase FA-dependent respiration. Finally, as a result of the muscle-specific MCU deletion, a systemic catabolic response impinging on both liver and adipose tissue metabolism occurs.
Similar content being viewed by others
Introduction
Mitochondrial Ca2+ uptake has pleiotropic roles. By buffering [Ca2+] increases, it contributes to the regulation of Ca2+-dependent functions that take place in the cytosol. In addition, within the organelle, Ca2+ positively regulates key dehydrogenases of the TCA cycle thus promoting ATP synthesis. On the other hand, excessive mitochondrial [Ca2+] has a pro-apoptotic function [1]. Mitochondria are placed in close contact with the endoplasmic reticulum/sarcoplasmic reticulum (ER/SR) Ca2+ stores, thus sensing microdomains of high [Ca2+] that enable prompt Ca2+ uptake [2]. Studies on this mechanism have been greatly facilitated by the identification of the long sought mitochondrial calcium uniporter (MCU), the highly selective channel responsible for Ca2+ accumulation by the organelle [3, 4]. MCU is composed of pore-forming subunits (MCU, MCUb) associated with essential MCU regulator, and of regulatory proteins (MICU1–3) that, together, contribute to the fine-tuned regulation of the channel activity [5,6,7,8].
We previously highlighted the positive role of mitochondrial Ca2+ uptake in controlling muscle trophism. Overexpression of MCU in the murine hindlimb triggers muscle hypertrophy and exerts a protective effect against denervation-induced atrophy, while MCU silencing causes muscle atrophy [9]. Importantly, in trained elderly subjects the improvement of muscle function is associated with increased MCU expression [10]. Moreover, the MCU complex has a peculiar composition in the skeletal muscle. Indeed, a muscle-specific MICU1 splicing variant (i.e., MICU1.1), by binding Ca2+ with higher affinity, confers myofiber mitochondria sustained Ca2+ uptake and ATP production required for contraction [11]. Finally, skeletal muscle force is decreased in total MCU−/− mice, underlying the crucial role of mitochondrial Ca2+ uptake for energy production required for muscle work [12].
To further clarify the role of skeletal muscle mitochondrial Ca2+ uptake in organ physiology and metabolism and to unveil potential systemic effects of skeletal muscle MCU deletion, we have generated both a constitutive and an inducible skeletal muscle-specific MCU knockout mouse model. Locally, loss of MCU in skeletal muscle triggered decreased muscle performance, a fiber-type switch toward fast myosin heavy chains (MHCs), and a metabolic rewiring toward preferential fatty acid (FA) oxidation. Systemically, a catabolic response impinging on liver and adipose tissue metabolism occurred.
Materials and methods
DNA plasmids
The following plasmids have been previously described: mtGCaMP6f [13] and R-GECO1 [14].
Flag-PDK4 and Flag-PDP2 were amplified by PCR from mouse liver cDNA with the following primers and cloned in pcDNA3.1:
Flag-PDK4 primers: Fw: 5’-TAATGGATCCGCCACCATGAAGGCAGCCCGCTTCGTGATG-3’;
Rev: 5’-TAATGAATTCTCACTTATCGTCGTCATCCTTGTAATCCACTGCCAGCTTCTCCTTCGC-3’;
Flag-PDP2 primers: Fw: 5’-TAATGGATCCGCCACCATGTCAAGTACTGTGTCCTACTGGATCTTC-3’; and
Rev: 5’-TAATGAATTCTCACTTATCGTCGTCATCCTTGTAATCACCCTCTTTACAGTAGGTATCAATTGA-3’.
Animals
All animal experiments were approved and performed in accordance with the Italian law D. L.vo n_26/2014. C57BL/6N-Mcutm1a mice were either produced by blastocyst microinjection of mutant embryonic cell lines or purchased from International Mouse Phenotyping Consortium (Mcu HEPD0762_7_B01). The former were derived by microinjection into C57BL/6NCrl blastocysts of mutagenized embryonic stem (ES) cell line from the EUCOMM consortium (C57BL/6N-Mcutm1a(EUCOMM)Hmgu) in our laboratory, the second was produced by the consortium using similar ES cells, but different clones, obtained in the same recombination experiment. The mutant allele presents a cassette composed of a FRT site followed by lacZ sequence and a loxP site. This first loxP site is followed by neomycin under the control of the human beta-actin promoter, SV40 polyA, followed by a FRT site and a second loxP site. The third loxP site is located downstream exon 5 of MCU gene (Ccdc109a). To generate the C57BL/6N-Mcutm1c line (here referred to as MCUfl/fl line), C57BL/6N-Mcutm1a mice were crossed with an FLP-expressing line. FLP-recombinase excited both the LacZ and neomycin-resistance cassettes restoring MCU expression. Homozygous MCUfl/fl mice were crossed with transgenic mice expressing the Cre recombinase under the control of the Mlc1f promoter (Mlc1f-Cre) [15] or the Cre recombinase fused to the mutated ligand-binding domain of the human estrogen receptor under the HSA promoter (HSA-Cre-ERT2) [16]. MCUfl/fl mice from both procedures displayed similar phenotypes after Cre-recombination. For the present work, MCUfl/fl mice derived from purchased C57BL/6N-Mcutm1a mice were used. In all, 1.5 mg/30 g body weight (BW) of tamoxifen (Sigma-Aldrich) dissolved in 10% EtOH and 90% sunflower oil (Sigma-Aldrich) was intraperitoneally (i.p.) injected every 3 days for 6 weeks. In addition, the same animals were fed with a tamoxifen-containing diet (Envigo TAM400/CreER) that allowed administration of tamoxifen of 40 mg/kg BW/die. In some key experiments, the Cre control groups were used (mlc1f-Cre and HSA-Cre-ERT2 mice). All the mice were fed ad libitum and always sacrificed before 9:30 a.m.
In vivo DNA transfection of mouse skeletal muscle
Adult male mice were used in all experiments. First, the animal was anesthetized. Hyaluronidase solution (2 mg/ml) (Sigma-Aldrich) was injected under the hindlimb footpad. After 30 min, 20 μg of plasmid DNA in 20 µl of physiological solution was injected with the same procedure of the hyaluronidase. Then one gold-plated acupuncture needle was placed under the skin at heel, and a second one at the base of the toes, oriented parallel to each other and perpendicular to the longitudinal axis of the foot and connected to the BTX porator (Harvard apparatus). The muscles were electroporated by applying 20 pulses, 20 ms each, 1 s of interval to yield an electric field of 100 V. Single fibers cultures were carried out 7–10 days later.
Mouse exercise studies
For acute concentric exercise studies, 3- or 6-month-old mice were acclimated to and trained on a 10° uphill LE8700 treadmill (Harvard apparatus) for 2 days. On day 1, mice ran for 5 min at 8 m/min and on day 2 mice ran for 5 min at 8 m/min followed by another 5 min at 10 m/min. On day 3, mice were subjected to a single bout of running starting at the speed of 10 m/min. Forty minutes later, the treadmill speed was increased at a rate of 1 m/min every 10 min for a total of 30 min and then increased at the rate of 1 m/min every 5 min until mice were exhausted. Exhaustion was defined as the point at which mice spent >5 s on the electric shocker without attempting to resume running. Total running time and total running distance were recorded for each mouse.
For eccentric exercise training, 3-month-old mice were acclimated to and trained on a 10° downhill for 2 days. On day 1, mice ran for 5 min at 8 m/min and on day 2 mice ran for 5 min at 8 m/min followed by another 5 min at 10 m/min. On days 3–5, mice were subjected to a single bout of running starting at the speed of 10 m/min. Forty minutes later, the treadmill speed was increased at a rate of 1 m/min every 10 min for a total of 30 min and then increased at the rate of 1 m/min every 5 min until mice were exhausted. Exhaustion was defined as above. Total running time and total running distance were recorded for each mouse.
Muscle force and fatigue measurements
To measure muscle force in living animals, the contractile performance of gastrocnemius muscle in vivo was measured as described previously [17]. Briefly, anesthetized mice were placed on a thermostatically controlled table, keeping the knee stationary, and the foot firmly fixed to a footplate, which was connected to the shaft of the motor of a muscle-lever system (305B, Aurora Scientific). Contraction was elicited by electrical stimulation of sciatic nerve. Teflon-coated seven-stranded steel wires (AS 632, Cooner Sales) were implanted with sutures on either side of the sciatic nerve proximal to the knee before its branching. At the distal ends of the two wires, the insulation was removed, and the proximal ends were connected to a stimulator (S88, Grass). To avoid recruitment of the dorsal flexor muscles, the common peroneal nerve was cut.
The fatigue protocol consists of a tetanic stimulation (frequency 100 Hz, 4–6 V) lasting 300 ms each second for a total time of 120 s.
Histology and fluorescence microscopy
For fiber size measurements, 20 μm-thick cryosections were fixed in 4% formaldehyde for 20 min, quenched with 50 mM NH4Cl in phosphate-buffered saline (PBS) and blocked in PBS containing 0.5% bovine serum albumin (BSA) for 20 min. Sections were then incubated with primary antibody anti-Laminin (Sigma-Aldrich) to label the sarcolemma for 1 h at 37 °C and washed 3 times in PBS. Alexa Fluor 488-conjugated secondary antibody (Thermo Fisher Scientific) was used. Fiber size measurements were performed with the Fiji distribution of ImageJ [18].
For fiber-type measurements, 20 μm-thick cryosections were blocked in M.O.M. working solution (Vector Laboratories). Sections were then incubated with primary antibody sc-71 (Hybridoma bank) 1:100 in 0.5% BSA in PBS to label the type IIA myosins for 1 h at 37 °C and washed 3 times in PBS. Alexa Fluor 488-conjugated secondary antibody (Life Technologies) was used. Then sections were incubated with primary antibody Bf-f3 (Hybridoma bank) 1:100 in 0.5% BSA in PBS to label the type IIB myosins for 1 h at 37 °C and washed 3 times in PBS. Alexa Fluor 555-conjugated secondary antibody (Life Technologies) was used. Finally, sections were then incubated with primary antibody Bad5 (Hybridoma bank) 1:100 in 0.5% BSA in PBS to label the type I myosins for 1 h at 37 °C and washed 3 times in PBS. Alexa Fluor 647-conjugated secondary antibody (Thermo Fisher Scientific) was used. Fiber-type analyses were performed with the Fiji distribution of ImageJ [18].
For hematoxylin and eosin (H&E) staining, 20 μm-thick cryosections were stained using Rapid Frozen Sections H&E staining Kit (Bio-Optica) according to manufacturer’s instructions. Briefly, cryosections were incubated in hematoxylin solution for 60 s. Then, after 3 washes, they were incubated in eosin solution for 30 s. Finally, cryosections were dehydrated.
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was extracted from tibialis anterior (TA) muscles, livers, and visceral adipose tissue (VAT) using the SV Total RNA Isolation Kit (Promega) following the manufacturer’s instructions. The RNA was quantified with Nanodrop (Thermo Fisher Scientific). Complementary DNA was generated from 500 nmol of total RNA with a cDNA Synthesis Kit SuperScript II (Thermo Fisher Scientific). Oligo(dT)12–18 primers (Thermo Fisher Scientific) were used as primer for first-strand cDNA synthesis with reverse transcriptase. The obtained cDNA was analyzed by real-time PCR using the IQ5 thermocycler and the SYBR green chemistry (Bio-Rad). The primers were designed and analyzed with Primer3 [19]. The housekeeping gene Gapdh was used as an internal control for cDNA normalization. For quantification, expression levels were calculated by the ΔCt method. Real-time PCR primer sequences were as follows:
Mcu: Fw 5’-AAAGGAGCCAAAAAGTCACG-3’; Rv 5’-AACGGCGTGAGTTACAAACA-3’.
Gapdh: Fw 5’-CACCATCTTCCAGGAGCGAG-3’; Rv 5’-CCTTCTCCATGGTGGTGAAGAC-3’.
Pgc1α-tot: Fw 5’-CGCTGCTCTTGAGAATGGAT-3’; Rv 5’-CGCAAGCTTCTCTGAGCTTC-3’.
Pgc1α-1: Fw 5’-GGACATGTGCAGCCAAGACTCT-3’; Rv 5’-CACTTCAATCCACCCAGAAAGCT-3’.
Pgc1α-4: Fw 5’-TCACACCAAACCCACAGAAA-3’; Rv 5’-CTGGAAGATATGGCACAT-3’.
G6pase: Fw 5’-ACGCCCGTATTGGTGGGTCCT-3’; Rv 5’-GCCAGAGGGACTTCCTGGTCCG-3’.
Pck1: Fw 5’-CTGCATAACGGTCTGGACTTC-3’; Rv 5’-CAGCAACTGCCCGTACTCC-3’.
Pnpla2: Fw: 5’-CTGAGAATCACCATTCCCACATC-3’; Rv: 5’-CACAGCATGTAAGGGGGAGA-3’.
Lipe: Fw: 5’-AGGATCGAAGAACCGCAGTC-3’; Rv: 5’-GTCTTCTGCGAGTGTCACCA-3’.
Abhd5: Fw: 5’-TGGTGTCCCACATCTACATCA-3’; Rv: 5’-CAGCGTCCATATTCTGTTTCCA-3’.
Mgll: Fw: 5’-CACGTGGACACCATCCAGAA-3’; Rv: 5’-GCCACTAGGATGGAGATGGC-3’.
Hmgcs2: Fw: 5’-CCGGTGTCCCGTCTAATGG-3’; Rv: 5’-GCAGATGCTGTTTGGGTAGC-3’.
Bdh1: Fw: 5’-ATAGGGCCTGAGAGGGAAGG-3’; Rv: 5’-GCAGTACAAATGCATCCCGC-3’.
IL-6: Fw: 5’-TAGTCCTTCCTACCCCAATTTCC-3’; Rv: 5’-TTGGTCCTTAGCCACTCCTTC-3’.
Western blotting and antibodies
To monitor protein levels, frozen muscles and frozen livers were pulverized by means of Qiagen Tissue Lyser and protein extracts were prepared in an appropriate buffer containing: muscle lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 10% glycerol, 2% sodium dodecyl sulfate (SDS), 1% Triton X-100, Complete EDTA-free protease inhibitor mixture (Roche), 1 mM PMSF, 1 mM NaVO3, 5 mM NaF, and 3 mM β-glycerophosphate) or RIPA buffer for liver extracts (125 mM NaCl, 25 mM Tris-Cl pH7.4, 1 mM EGTA-Tris pH 7.4, 1% Triton-X100, 0.5% sodium deoxycholate, 0.1% SDS, and Complete EDTA-free protease inhibitor mixture (Roche)). In all, 40 μg of total proteins were loaded, according to BCA quantification. Proteins were separated by SDS-polyacrylamide gel electrophoresis, in commercial 4–12% acrylamide gels (Thermo Fisher Scientific) and transferred onto nitrocellulose membranes (Thermo Fisher Scientific) by wet electrophoretic transfer. Blots were blocked 1 h at room temperature (RT) with 5% non-fat dry milk (Bio-Rad) in TBS-tween (0.5 M Tris, 1.5 M NaCl, 0.01% Tween) solution and incubated at 4 °C with primary antibodies. Secondary antibodies were incubated 1 h at RT. The following antibodies were used: anti-phosphoAKT (1:1000, Cell Signaling), anti-AKT (1:1000, Cell Signaling) anti-ACTIN (1:20,000, Santa Cruz), anti-FLAG (1:1000, Cell Signaling) anti-phosphoGSK3α/β (1:1000, Cell Signaling), anti-GSK3α/β (1:1000, Cell Signaling), anti-MCU (1:1000, Sigma-Aldrich), anti-phosphoPDH (1:5000, Abcam), anti-PDH (1:1000, Cell Signaling) anti-GRP75 (1:5000, Santa Cruz), and anti-TOM20 (1:20,000, Santa Cruz). Secondary horseradish peroxidase-conjugated antibodies were purchased from Bio-Rad and used at 1:5000 dilution.
Real-time imaging of mitochondrial and cytosolic Ca2+ in flexor digitorum brevis (FDB) fibers
Real-time imaging
FDB fibers were isolated 7–10 days after in vivo transfection. Muscles were digested in collagenase A (4 mg/ml) (Roche) dissolved in Tyrode’s salt solution (pH 7.4) (Sigma-Aldrich) containing 10% fetal bovine serum (Thermo Fisher Scientific). Single fibers were isolated, plated on laminin-coated glass coverslips, and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with HEPES (42430 Thermo Fisher Scientific), supplemented with 10% fetal bovine serum, containing penicillin (100 U/ml), streptomycin (100 μg/ml). Fibres were maintained in culture at 37 °C with 5% CO2.
Mitochondrial and cytosolic Ca2+ measurements
FDB muscles were electroporated with a plasmid encoding 4mtGCaMP6f together with R-GECO1. After single fiber isolation, real-time imaging was performed. During the experiments, myofibers were maintained in Krebs-Ringer modified buffer (135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 20 mM HEPES, 1 mM MgSO4, 0.4 mM KH2PO4, 1 mM CaCl2, 5.5 mM glucose, pH 7.4) at RT, in the presence of 75 μM N-benzyl-P-toluenesulfonamide (BTS, Sigma-Aldrich) to avoid the fiber contraction. In all, 20 mM caffeine (Sigma-Aldrich) was added when indicated to elicit Ca2+ release from intracellular stores. Experiments were performed on a Zeiss Axiovert 200 microscope equipped with a ×40/1.3 N.A. PlanFluor objective. Excitation was performed with a DeltaRAM V high-speed monochromator (Photon Technology International) equipped with a 75 W xenon arc lamp. Images were captured with a high-sensitivity Evolve 512 Delta EMCCD (Photometrics). The system is controlled by MetaMorph 7.5 (Molecular Devices) and was assembled by Crisel Instruments. 4mtGCaMP6f and R-GECO1 were alternatively excited every second at 490 and 560 nm, respectively, and images were acquired through a dual band emission filter (520/40 and 630/60) (Chroma). Exposure time was set to 50 ms (4mtGCaMP6f) and 150 ms (R-GECO1). Acquisition was performed at binning 1 with 200 of EM gain. Image analysis was performed with Fiji distribution of the ImageJ software [18]. Images were background subtracted and linear unmixing was performed to get rid of bleed through of the two fluorochromes. Data are expressed as F/F0 where F0 is the mean intensity at the beginning of the experiment.
Cytosolic Ca2+ measurements
Fibers were dissected and loaded with 2 μM fura-2/AM (Thermo Fisher Scientific) diluted in Krebs-Ringer modified buffer (described above) containing 0.02% pluronic acid for 20 min at 37 °C and then washed with Krebs-Ringer modified buffer in presence of 75 μM BTS (Sigma-Aldrich) to avoid the fiber contraction. 20 mM caffeine (Sigma-Aldrich) was added when indicated to elicit Ca2+ release from intracellular stores. Experiments were performed on a Zeiss Axiovert 200 microscope equipped with a ×40/1.3 N.A. PlanFluor objective. Excitation was performed with a DeltaRAM V high-speed monochromator (Photon Technology International) equipped with a 75 W xenon arc lamp. Images were captured with a high-sensitivity Evolve 512 Delta EMCCD (Photometrics). The system is controlled by MetaMorph 7.5 (Molecular Devices) and was assembled by Crisel Instruments. Images were collected by alternatively exciting the fluorophore at 340 and 380 nm and fluorescence emission recorded through a 515/30 nm band-pass filter (Semrock). Exposure time was set to 50 ms. Acquisition was performed at binning 1 with 200 of electron-multiplying gain. Image analysis was performed with Fiji distribution of the ImageJ software. Images were background subtracted. Changes in fluorescence (340/380 nm ratio) was expressed as R/R0, where R is the ratio at time t and R0 is the ratio at the beginning of the experiment.
OCR (oxygen consumption rate) measurements
For OCR experiments, FDB fibers were isolated. Muscles were digested in collagenase A (4 mg/ml) (Roche) dissolved in Tyrode’s salt solution (pH 7.4) (Sigma-Aldrich) containing 10% fetal bovine serum (Thermo Fisher Scientific). Single fibers were isolated, plated on laminin-coated XF24 microplate wells and cultured in DMEM (D5030 Sigma-Aldrich), supplemented with 1 mM NaPyr, 5 mM glucose, 33 mM NaCl, 15 mg phenol red, 25 mM HEPES, and 1 mM of L-Glu. Fibres were maintained for 2 h in culture at 37 °C in 5% CO2.
To measure exogenous FA utilization, the fibers were cultured in DMEM (D5030 Sigma-Aldrich), supplemented with 0.1 mM NaPyr, 5 mM glucose, 33 mM NaCl, 15 mg phenol red, 25 mM HEPES, 1 mM of L-Glu, 0.5 mM carnitine, and 100 μM palmitate:BSA. Fibres were maintained for 2 h in culture at 37 °C in 5% CO2.
The rate of oxygen consumption was assessed in real-time with the XF24 Extracellular Flux Analyzer (Agilent), which allows to measure OCR changes after up to four sequential additions of compounds. Fibers were plated as reported above. A titration with the uncoupler FCCP was performed in order to utilize the FCCP concentration (0.6 μM) that maximally increases OCR.
The results were normalized for the fluorescence of Calcein (Sigma-Aldrich). Fibers were loaded with 2 μM Calcein for 30 min. Fluorescence was measured using a Perkin Elmer EnVision plate reader in well scan mode using 480/20 nm filter for excitation and 535/20 nm filter for emission.
Glycolysis rate measurements
Glycolysis was measured by monitoring the conversion of 5-3H-glucose to 3H2O, as described previously [20]. Briefly, TA muscle fibers were incubated in a medium that contained [5-3H]-glucose (PerkinElmer) at a specific activity of 10 μCi. Labeled glucose metabolized by glycolysis produces 3H2O that is released into the media. Following incubation for 3 h at 37 °C, media was transferred to uncapped tubes containing 0.5 ml of 0.2 M HCl. The tube was transferred to a scintillation vial containing 1 ml of H2O such that the water in the vial and the contents of the tube were not allowed to mix. The vials were sealed, and diffusion was allowed for 24 h. The amount of diffused 3H2O was determined by scintillation counting.
Lactate measurements
Freshly isolated TA muscle fibers were cultured in DMEM (D5030 Sigma-Aldrich), supplemented with 1 mM NaPyr, 10 mM glucose, 33 mM NaCl, 15 mg phenol red, 5 mM HEPES, and 1 mM of L-Glu. Once an hour, an aliquot of medium was collected and lactate concentration was measured by means of the colorimetric L-Lactate Assay Kit (Abcam) according to the manufacturer’s instructions.
Muscle glycogen and FA content
Glycogen amount was measured by means of the colorimetric Glycogen Assay Kit II (Abcam) according to the manufacturer’s instructions. FA amount was measured by means of the Free FA Quantification Kit (Abcam) according to the manufacturer’s instructions.
Blood metabolite quantification
Blood was collected from the orbital sinus in heparin-coated Pasteur pipettes and centrifuged immediately after collection. FAs and β-hydroxy-butyrate were dosed using an automated spectrophotometer Cobas Fara II (Roche) according to the manufacturer’s instruction. Blood glucose levels were measured with an YSI 2300 STAT PlusTM glucose and lactate analyzer (YSI Life Sciences, Yellow Springs, OH) according to the manufacturer’s instruction.
Lactate levels were measured using Stat Strip Xpress measuring system (Nova Biomedical).
Glucose tolerance test
For the glucose tolerance test, mice were fasted for 5 h. Glucose (2 mg/g BW) was administered i.p. and the blood glucose levels were followed during 2:30 h using ContourXT (Bayer).
Glucose uptake measurements
Glucose uptake in tissues was measured using a non-radioactive colorimetric method [21] by means of the 2-Deoxy-d-Glucose Uptake Measurement Kit (Cosmo Bio Co., LTD, Tokyo, Japan). Mice were fasted 5 h before the beginning of the experiment. Mice were then injected i.p. with a solution containing d-glucose (1 g/kg) and 2-deoxy-d-glucose (0.027 g/kg). One hour after injection, mice were killed by cervical dislocation, and tissues (soleus, TA, and liver) were rapidly removed and frozen in liquid nitrogen. Soleus and TA muscles and 150 mg of liver were used for the assay. Tissues were subsequently weighed and homogenized in 1 ml of Tris-HCl (10 mM, pH 8), with TissueLyser II (Qiagen), by shaking at maximum speed for 3 min, and then heated at 95 °C for 15 min. Samples were then centrifuged at 16,000 × g for 15 min at 4 °C to remove tissue debris. Supernatants were finally collected, and a portion was diluted in 20 µl (final volume; 4 µl for soleus, 5 µl for TA, and 2 µl for liver) with the sample dilution buffer provided with the kit. The assay was then performed following the manufacturer’s instructions.
Metabolomics analysis
Gastrocnemius muscles of adult mice were harvested and underwent untargeted metabolomics analysis performed by Metabolon, Inc. Briefly, samples were prepared using the automated MicroLab STAR® system from Hamilton Company. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC-MS/MS with negative ion mode ESI, one for analysis by hydrophiliic interaction liquid chromatography/UPLC-MS/MS with negative ion mode ESI, and one sample was reserved for backup.
Interleukin (IL)-6 enzyme-linked immunosorbent assay
IL-6 serum levels were measured by means of the mouse IL-6 Kit (Abcam) according to the manufacturer’s instructions.
Statistical analysis of the data
Statistical data are presented as mean ± SD, unless otherwise specified. Significance was calculated by Student’s t test or Mann–Whitney rank-sum test.
Results
Constitutive muscle-specific MCU deletion causes decreased muscle performance and fiber-type switching
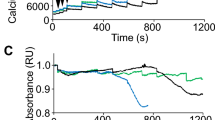
We have previously demonstrated that MCU silencing in skeletal muscle causes decreased myofiber size and inhibition of trophic signaling routes [9]. To discern the contribution of skeletal muscle mitochondrial Ca2+ uptake to muscle physiology and whole-body metabolism, we generated a skeletal muscle-specific MCU−/− mouse model. We crossed mice in which the exon 5 of the MCU gene is flanked by loxP sequences with mice expressing the Cre recombinase under the control of the myosin-light chain 1f promoter (mlc1f-Cre) [15], which is expressed since embryogenesis (Fig. 1a). Expression of MCU in hindlimb muscles of adult mlc1f-Cre::MCUfl/fl (skMCU−/−) mice was essentially absent indicating that efficient recombination occurred (Fig. 1b and S1A-B). MCU deletion is expected to blunt mitochondrial Ca2+ transients triggered by Ca2+ release from SR stores. To confirm this notion, FDB muscles were simultaneously transfected in vivo with 4mtGCaMP6f, a Ca2+-sensitive probe targeted to the mitochondria matrix [13], together with R-GECO, a cytosolic Ca2+-probe [14]. Upon caffeine-induced Ca2+ release from the SR, MCUfl/fl myofibers showed robust cytosolic Ca2+ transients followed by effective mitochondrial Ca2+ uptake (Fig. 1c). In contrast, skMCU−/− mitochondria were unable to take up Ca2+ despite normal cytosolic Ca2+ increases (Fig. 1c). Quantitative analysis of cytosolic [Ca2+] highlighted a slight reduction of Ca2+ transients in skMCU−/− myofibers, suggesting a negative effect of chronic MCU deletion on global Ca2+ homeostasis (Fig. 1d). However, basal cytosolic [Ca2+] was unaffected by MCU deletion (Fig. 1e).

Constitutive muscle-specific MCU deletion decreases muscle performance and causes fiber-type switching. a Mlc1f-Cre mice were crossed with mice in which exon 5 of the MCU gene is flanked by loxP sequences. Expression of the Cre recombinase in embryogenesis determines the deletion of the MCU gene specifically in skeletal muscle. For the sake of simplicity, throughout the manuscript the Mlc1f-Cre::MCUfl/fl mice are named skMCU−/−. b Western blot analyses demonstrated efficient MCU deletion in hindlimb muscles. c Cytosolic and mitochondria [Ca2+] were simultaneously measured in adult FDB myofibers. While in MCUfl/fl myofibers caffeine-induced cytosolic Ca2+ increase was followed by mitochondrial Ca2+ uptake (left representative traces), in skMCU−/− mice mitochondrial Ca2+ uptake was abolished (right representative traces). Histogram on the right reports mean mitochondrial Ca2+ peaks upon caffeine treatment. *p < 0.05, t test (two-tailed, unpaired) of >20 fibers per condition. Data are presented as mean ± SD. d Ratiometric measurements of cytosolic Ca2+ transients upon caffeine treatment highlighted a reduction in peak cytosolic [Ca2+] of skMCU−/− FDB myofibers compared to MCUfl/fl controls. *p < 0.05, t test (two-tailed, unpaired) of >20 fibers per condition. Data are presented as mean ± SD. Representative traces are reported on the right side. e Resting cytosolic [Ca2+] was unaltered in skMCU−/− FDB myofibers compared to MCUfl/fl controls. Data are presented as mean ± SD (>20 fibers per condition). f Fiber size was reduced in skMCU−/− hindlimb muscles compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. g Mean maximal running distance in a single bout of run on a treadmill of MCUfl/fl and skMCU−/− mice indicated that MCU deletion negatively affects exercise performance. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. h Immunofluorescence analyses of soleus myofibers with antibodies specific for different myosin heavy chains (anti-MyHC1 and anti-MyHC2A) demonstrated a reduction in slow-twitch type 1 myosin-positive fibers and an increase in fast-twitch type 2A myosin-positive fibers in skMCU−/− samples. *p < 0.05, Mann–Whitney rank-sum test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. Representative images are reported on the right side. Scale bar 100 μm. i In vivo fatigue of gastrocnemius muscles was measured in skMCU−/− mice compared to MCUfl/fl mice. Six animals per condition. Data are presented as mean ± SD (five animals per condition). Mean maximal running distances after each of 3 days of downhill run on a treadmill of MCUfl/fl and skMCU−/− mice. *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD
skMCU−/− TA muscles did not show any sign of damage (Figure S1C) but fiber size was reduced (Fig. 1f), although at a milder degree than AAV-shMCU-treated muscles [9]. Moreover, phosphorylation levels of Akt and mRNA expression levels of PGC1α4 were decreased (Figure S1D-E). Overall, these results indicate that skMCU−/− mice mirror the effects of MCU silencing [9], thus representing an ideal model for the analysis of both local and systemic physiologic and metabolic outcomes of skeletal muscle mitochondrial Ca2+ uptake inhibition.
Next, we evaluated muscle performance. In vivo analysis showed a decline in tetanic force of skMCU−/− compared to MCUfl/fl mice (Figure S1F). We then measured the distance run on a treadmill during a single bout of uphill strenuous exercise. In line with the constitutive MCU−/− mice [12], muscle-specific MCU deletion caused a deficit in running capacity, both at 3 and 6 months of age (Fig. 1g and S1G-H), thus demonstrating that impaired exercise performance is caused by an intrinsic skeletal muscle defect. In addition, a significant fiber-type switch was apparent, as opposed to the constitutive MCU ablation [12]. Indeed, quantitative analysis demonstrated reduced expression of the slow-twitch type 1 MHC and an increase of the fast-twitch type 2A MHC in the soleus muscle of skMCU−/− mice (Fig. 1h). To assess the response to repeated isometric tetanic contractions, we performed 120 isometric contractions of a duration of 300 ms once every second. However, the relative decrease in force generation was not different between MCUfl/fl and skMCU−/− animals (Fig. 1i), indicating that the differences in exercise performance is not due to a different resistance to fatigue. Finally, repeated bouts of exhausting downhill running in 3 consecutive days revealed a progressive decrease in performance that was similar between MCUfl/fl and skMCU−/− mice, indicating that lengthening contraction-induced muscle damage occurs independently of MCU (Figure S1I).
Muscle-specific MCU deletion triggers a metabolic shift toward FA utilization
Mitochondrial Ca2+ uptake is expected to control oxidative metabolism [1]. To test whether decreased mitochondrial Ca2+ accumulation influences skeletal muscle respiration, we measured OCR in freshly isolated FDB muscle myofibers. Myofibers were treated consecutively with oligomycin, to measure ATP-linked respiration; FCCP, to measure maximal oxygen consumption; and finally with rotenone and antimycin, to verify the extent of non-mitochondrial respiration. skMCU−/− fibers showed a decrease in the basal, maximal, and ATP-linked respiration compared to MCU-expressing fibers (Fig. 2a and S2A), in line with the role of mitochondrial Ca2+ uptake in regulating oxidative metabolism. Both MCUfl/fl and skMCU−/− myofibers showed little spare respiratory capacity (Fig. 2a). We hypothesized that this effect might be due to the use of freshly isolated myofibers that are still actively contracting. To verify this hypothesis, we measured OCR in myofibers treated with BTS to avoid fiber contraction [9]. In these conditions, myofibers display large spare respiratory capacity, as expected (Figure S2B). Next, in skMCU−/− mice blood lactate levels were increased (Fig. 2b and S2C). These data suggest that the absence of mitochondrial Ca2+ uptake drastically reduces pyruvate utilization by mitochondria, which is rather converted into lactate. To directly determine the proportion of the glycolytic rate, we incubated MCUfl/fl and skMCU−/− myofibers with 5-3H-glucose, and we measured the β-emission of 3H2O produced by glycolysis in the cell medium, as reported [20]. skMCU−/− samples released higher [3H2O] compared to MCUfl/fl fibers, demonstrating increased glycolysis (Fig. 2c). Accordingly, skMCU−/− TA muscle myofibers showed increased lactate production compared to MCUfl/fl over time (Figure S2D).

Muscle-specific MCU deletion triggers a metabolic shift toward fatty acid utilization. a OCR measurements indicated reduced respiratory capacity in skMCU−/− FDB myofibers compared to controls. Left: representative traces. Right: quantification. To calculate basal and maximal respiration, non-mitochondrial O2 consumption was subtracted from absolute values. ATP linked respiration was calculated as the difference between basal and oligomycin-insensitive O2 consumption. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of ten samples per condition. Data are presented as mean ± SD. b Blood lactate concentration was increased in skMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. c The rate of glycolysis was determined by measuring the conversion of 5-3H-glucose to 3H2O. skMCU−/− TA myofibers showed increased glycolysis rate relative to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. d Phosphorylation levels of PDH were increased in skMCU−/− TA muscles compared to controls. e Glucose dependent-respiration was decreased in skMCU−/− FDB myofibers compared to controls. Left: OCR measurements were performed as in a. UK5099 was added to inhibit the mitochondrial pyruvate carrier (MPC). Right: Glucose dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. f Fatty acid (FA) content was increased in skMCU−/− TA muscles relative to controls. *p < 0.05, t test (two-tailed, unpaired) of six animals per condition. Data are presented as mean ± SD. g FA-dependent respiration was increased in skMCU−/− FDB myofibers compared to controls. Left: OCR measurements were performed as in a. Etomoxir was added to inhibit FA utilization. Right: FA dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. h OCR measurement demonstrated differential substrates utilization in skMCU−/− myofibers. Left: during OCR measurements UK5099 was added to freshly isolated myofiber followed by etomoxir. Right: Glucose dependency and FA dependency were calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. i Phosphorylation levels of ACC were increased in skMCU−/− TA muscles compared to controls. j Untargeted metabolomics analysis of skMCUfl/fl and skMCU−/− muscles. Metabolites set enrichment analysis revealed significant upregulation of acylcarnitine and diacylglycerol lipids in skMCU−/− muscles (pathway p values are reported). Metabolites that are significantly increased are labeled. The size of the circles is proportional to the fold change (fold change values are reported in Table 1). p Value is shown in the figure (Welch’s two-sample t test of nine animals per condition)
Mitochondrial Ca2+ uptake positively regulates the activity of three critical mitochondrial dehydrogenases. Isocitrate dehydrogenase and α-ketoglutarate dehydrogenase are directly modulated by mitochondrial Ca2+, instead pyruvate dehydrogenase (PDH) is indirectly activated by Ca2+, which stimulates the activity of PDH phosphatase isoform 1 (PDP1) [22]. In line with the total MCU−/− mouse muscles [12] and the AAV-shMCU-infected muscles [9], in skMCU−/− muscles phosphorylation levels of PDH were increased, suggesting the inhibition of its enzymatic activity (Fig. 2d and S2E). If pyruvate utilization is impaired in skMCU−/− muscles, in MCUfl/fl mitochondria inhibition of pyruvate uptake should mimic the effects of MCU deletion, while it should have little effect in skMCU−/− myofibers. To test this hypothesis, we performed OCR measurements in myofibers treated with UK5099, an inhibitor of the mitochondrial pyruvate carrier (MPC) [23,24,25,26]. MPC inhibition had small effect on basal OCR of skMCU−/− myofibers, while it reduced O2 consumption of MCUfl/fl fibers to the levels of untreated skMCU−/− myofibers (Fig. 2e). These data demonstrating the aberrant glucose oxidation in skMCU−/− muscles suggest that other energy sources may be utilized by MCU-depleted mitochondria. FA metabolism could represent such alternative [27]. FA concentration was indeed elevated in skMCU−/− muscles compared to skMCUfl/fl controls (Fig. 2f). To determine the genuine contribution of FA oxidation to mitochondrial respiration, we measured OCR upon treatment with etomoxir, an inhibitor of carnitine palmitoyltransferase-1 and thus of FA oxidation. While in MCUfl/fl myofibers, FA accounted for 40% of the basal respiration, in skMCU−/− fibers 80% of basal OCR was dependent on FA (Fig. 2g). Treatment with UK5099 followed by etomoxir further confirmed the differential substrates utilization (Fig. 2h). In addition, acetyl-CoA carboxylase (ACC) phosphorylation was increased in skMCU−/− muscles, suggesting decreased ACC activity and thus FA synthesis (Fig. 2i). These data demonstrate that MCU deletion impairs glucose oxidation and triggers a metabolic rewiring that favors FA utilization. Nonetheless, OCR measurements in the presence of exogenous palmitate demonstrated that the overall capacity to oxidize FA is actually reduced in skMCU−/− myofibers compared to controls, because of the Ca2+-dependent regulation of the above-mentioned tricarboxylic acid (TCA) cycle enzymes (Figure S2F). Accordingly, untargeted metabolomics analysis of MCUfl/fl and skMCU−/− muscles revealed significant changes in lipid-related metabolites, while the other metabolic pathways were mostly unaffected. In particular, we detected increased levels of diacylglycerols, suggestive of elevated triacylglycerol lipolysis, and acylcarnitines (Fig. 2j and Table 1). Altogether these data demonstrate that, despite lower absolute OCR values, β-oxidation in MCU−/− muscles accounts for more than half of total OCR, in contrast with MCUfl/fl muscles, which mainly rely on glucose oxidation.
PDH inhibition underlies the metabolic switch of skMCU− /− myofibers
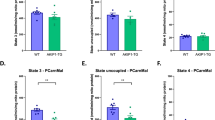
We wished to know whether the increased PDH phosphorylation (Fig. 2d) is sufficient to trigger the metabolic switch of skMCU−/− muscles. To this aim, we performed two complementary experiments. First, we overexpressed PDH kinase 4 (PDK4) in MCUfl/fl FDB muscles to increase PDH phosphorylation (Fig. 3a) and thus inhibit PDH activity. Similarly to MCU deletion, PDK4 overexpression caused a drastic increase in FA dependency, as demonstrated by the great reduction in OCR upon etomoxir treatment (Fig. 3b), that was barely observed in control-transfected muscles. This result indicates that, in the absence of pyruvate decarboxylation, FA metabolism contributes to replenish the mitochondrial acetyl-CoA pool. As complementary approach, we transiently overexpressed the Ca2+-independent PDH phosphatase (PDP2) [22] in skMCU−/− FDB muscles to rescue PDH activity. PDP2 overexpression triggered PDH dephosphorylation (Fig. 3c) in line with increased PDH activation. This was sufficient to rescue pyruvate oxidation in skMCU−/− myofibers, as demonstrated by the barely detectable effect of etomoxir treatment on basal OCR in PDP2-overexpressing skMCU−/− myofiber compared to control-transfected fibers (Fig. 3d). These results conclusively demonstrate that loss of MCU causes a metabolic switch toward increased FA oxidation by decreasing PDH activity.

Pyruvate dehydrogenase inhibition underlies the increase in FA oxidation of skMCU−/− muscles. a Western blot analysis showed that Flag-PDK4 overexpression in MCUfl/fl FDB muscles increased the phosphorylation levels of PDH (pcDNA3.1 was used as control). b FA-dependent respiration was increased in MCUfl/fl PDK4-overexpressing FDB myofibers. Left: OCR measurements were performed as in Fig. 2a. Etomoxir was added to inhibit FA utilization. Right: FA dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. c Western blot analysis showed that Flag-PDP2 overexpression in skMCU−/− FDB muscles decreased the phosphorylation levels of PDH (pcDNA3.1 was used as control). d FA-dependent respiration was decreased in skMCU−/− PDP2-overexpressing FDB myofibers. Left: OCR measurements were performed as in Fig. 2a. Etomoxir was added to inhibit FA utilization. Right: FA dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD
Muscle-specific MCU deletion impinges on systemic metabolism
To verify whether altered energy substrate metabolism due to the loss of skeletal muscle MCU triggers systemic adaptations, we measured blood concentration of key metabolites. Blood glucose levels were lower in skMCU−/− mice compared to MCUfl/fl (Fig. 4a and S3A). These data suggest that, despite defective glucose oxidation, skMCU−/− muscles are perfectly capable to take up glucose. Accordingly, skMCU−/− TA and soleus muscles accumulated 2-deoxy-d-glucose even more efficiently than MCUfl/fl muscles (Fig. 4b). In line with defective carbohydrates utilization, in skMCU−/− muscles glycogen levels were increased (Fig. 4c). In light of the role of skeletal muscle activity in controlling systemic metabolism, we hypothesized that muscle-specific deletion of MCU would impinge on hepatic glucose metabolism. Indeed, the liver of skMCU−/− mice accumulated less glucose than MCUfl/fl mice (Fig. 4d), pointing to a systemic metabolic adaptation in which the liver might contribute to euglycemia by counteracting the excessive muscle glucose uptake. The mRNA levels of the gluconeogenesis enzymes glucose-6-phosphatase (G6pase) and phosphoenolpyruvate carboxykinase 1 (Pck1) were upregulated in skMCU−/− liver compared to MCUfl/fl controls (Fig. 4e). At the same time, liver glycogen content was decreased (Fig. 4f). Glycogen synthase activity was presumably inhibited, in line with decreased phosphorylation, and thus increased activation of glycogen synthase kinase 3β (GSK3β). GSK3 is a target of Akt that, coherently, was also dephosphorylated in skMCU−/− livers (Fig. 4g and S3B). To verify whether a general catabolic response occurs, we analyzed liver lipases' expression. Hormone-sensitive lipase (Lipe) and monoglyceride lipase (Mgll) mRNA levels were increased in skMCU−/− liver compared to controls, and 1-acylglycerol-3-phosphate O-acyltransferase (ABHD5) and patatin-like phospholipase domain-containing 2 (Pnpla2) had an increasing tendency (Fig. 4h). Liver catabolism triggered by muscle-restricted MCU deletion was finally assessed by increased ketogenesis, as demonstrated by increased expression of ketogenic enzymes, i.e., 3-hydroxybutyrate dehydrogenase 1 (Bdh1) and hydroxymethylglutaryl-CoA synthase (Hmgcs2) (Fig. 4i), and of blood β-hydroxybutyric acid levels (Fig. 4j). Finally, blood FA levels were higher in skMCU−/− compared to MCUfl/fl samples (Fig. 4k). The latter result may be suggestive not only of a hepatic response but could also implicate adipose tissue remodeling. While BW was unchanged (Figure S3C), VAT weight was decreased in skMCU−/− relative to control mice (Fig. 4l) and the expression of Lipe and Pnpla2 lipases was increased (Fig. 4m), indicating increased adipose tissue catabolism.

Muscle-specific MCU deletion impinges on systemic metabolism. a Glucose tolerance test of MCUfl/fl and skMCU−/− mice. skMCU−/− mice had decreased glycemia at all time points compared to MCUfl/fl *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD. b In vivo measurements of 2-deoxy-d-glucose uptake revealed that skMCU−/− TA and SOL muscles accumulated more glucose than MCUfl/fl muscles. *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD. c Glycogen content was increased in skMCU−/− muscles relative to controls. *p < 0.05, Mann–Whitney rank-sum test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. d In vivo measurements of 2-deoxy-d-glucose uptake revealed that skMCU−/− liver took up less glucose than controls. *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD. e Relative mRNA expression levels of the gluconeogenesis enzymes G6pase and Pck1 in skMCU−/− liver compared to MCUfl/fl liver. *p < 0.05, t test (two-tailed, unpaired) of nine animals per condition. Data are presented as mean ± SD. f Glycogen content was decreased in skMCU−/− mice liver compared to controls. *p < 0.05, Mann–Whitney rank-sum test (two-tailed, unpaired) of six animals per condition. Data are presented as mean ± SD. g Phosphorylation levels of GSK3 α/β and AKT were decreased in skMCU−/− livers compared to controls. Total AKT and GSK3 α/β were used as control. h Relative mRNA expression levels of the lipases Lipe, Abhd5, Mgll, and Pnpla2 in skMCU−/− liver compared to MCUfl/fl liver. *p < 0.05, t test (two-tailed, unpaired) of nine animals per condition. Data are presented as mean ± SD. i Relative mRNA expression levels of the ketogenesis enzymes Bdh1 and Hmgcs2 in skMCU−/− liver compared to MCUfl/fl liver. *p < 0.05, t test (two-tailed, unpaired) of nine animals per condition. Data are presented as mean ± SD. j Plasma beta-hydroxybutyric acid concentration was increased in skMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. k Plasma FA concentration was increased in skMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. l The amount of visceral adipose tissue (VAT) was decreased in skMCU−/− mice compared to MCUfl/fl animals. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. m Relative mRNA expression levels of the lipases Mgll, Lipe, and Pnpla2 in skMCU−/− VAT compared to MCUfl/fl liver. *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD
We wondered whether these systemic responses are solely due to increased skeletal muscle glucose demand or whether muscle-specific cytokines (myokines) are secreted in response to MCU deficiency. In particular, IL-6 has been shown to be produced by skeletal muscle during exercise and to enhance FA oxidation, insulin-stimulated glucose uptake, and to contribute to hepatic glucose production during exercise [28]. IL-6 levels were increased in skMCU−/− serum and muscle samples compared to controls (Figure S3D-E). These data indicate that combined effects might be responsible for the systemic phenotype of skMCU−/− mice. However, a comprehensive analysis of the myokinome will shed further light.
Overall, these data reveal that the skeletal muscle-restricted defect in mitochondrial metabolism is partially compensated through both (i) the exploitation of metabolic flexibility within the defective tissue (shift toward FA oxidation) and (ii) the triggering of a whole-body rewiring of metabolic pathways.
MCU deletion in adult skeletal muscle causes atrophy, impairs exercise performance, and triggers a fiber-type switch
Constitutive gene deletion could trigger adaptive developmental responses that may hinder a peculiar phenotype. To overcome this issue, we developed an inducible muscle-specific MCU knockout mouse (HSA-Cre-ERT2::MCUfl/fl), in which the MCUfl/fl mouse is mated with the HSA-Cre-ERT2 mouse, i.e., a mouse carrying the Cre recombinase fused to a mutated ligand-binding domain of the human estrogen receptor α and controlled by the human skeletal actin promoter [16]. Upon tamoxifen treatment, the Cre enzyme translocates into the nucleus . The HSA-Cre-ERT2::MCUfl/fl mouse, treated with tamoxifen during adulthood, was named iskMCU−/− (Fig. 5a). However, even long-term tamoxifen administration was only partially effective in deleting MCU. While in some muscles MCU synthesis was inhibited, either totally or partially, in others (e.g., the diaphragm) it was unaffected (Fig. 5b and S4A). Coherently, fiber size reduction was proportional to the degree of MCU downregulation (Fig. 5c). Nevertheless, performance of iskMCU−/− mice on an uphill exhaustion exercise protocol was significantly reduced (Fig. 5d). Finally, in the soleus muscle, the expression of slow-twitch type 1 MHC was decreased, whereas that of type 2A MHC expression was increased (Fig. 5e).

Inducible muscle-specific MCU deletion impairs skeletal muscle performance and triggers fiber-type switch. a HSA-Cre-ERT2 mice were crossed with mice in which exon 5 of the MCU gene is flanked by loxP sequences (MCUfl/fl). At 8 weeks of age, all mice received tamoxifen treatment. Upon tamoxifen binding, Cre recombinase determines the deletion of the MCU gene specifically in the skeletal muscle. For the sake of simplicity, the tamoxifen-treated HSA-Cre-ERT2::MCUfl/fl mice are named iskMCU−/−. b Western blot analyses of different iskMCU−/− muscles revealed variable downregulation of MCU expression. c Fiber size of iskMCU−/− hindlimb muscles was reduced proportionally to the degree of MCU ablation detected in b. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. d Mean maximal running distance in a single bout of run on a treadmill of MCUfl/fl and iskMCU−/− mice indicated that MCU deletion negatively affects exercise performance. *p < 0.05, t test (two-tailed, unpaired) of five animals per condition. Data are presented as mean ± SD. e Immunofluorescence analysis of soleus myofibers with antibodies specific for different myosin heavy chains (anti-MyHC1 and anti-MyHC2A) demonstrated a reduction in slow-twitch type 1 myosin-positive fibers and an increase in fast-twitch type 2A myosin-positive fibers in iskMCU−/− samples relative to controls. *p < 0.05, Mann–Whitney rank-sum test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. Representative images are reported on the right side. Scale bar 100 μm
MCU deletion in adult skeletal muscle triggers local and systemic metabolic adaptations
Similarly to skMCU−/− mice, basal, maximal, and ATP-linked OCR of iskMCU−/− FDB myofibers were reduced compared to control fibers (Fig. 6a and S5A). p-PDH in FDB muscle was increased (Fig. 6b). In agreement with impaired pyruvate utilization, the drastic reduction of basal OCR triggered by UK5099-dependent MPC inhibition in MCUfl/fl myofibers (Figs 2e and 6c) was not observed in iskMCU−/− fibers (Fig. 6c). In addition, FA dependency of basal OCR was double in iskMCU−/− fibers compared to controls (Fig. 6d). Although fasting glycemia was unaltered (Fig. 6e and S5B), blood lactate levels were increased in iskMCU−/− mice relative to controls (Fig. 6f and S5C), coherently with impaired glucose oxidation. In addition, similarly to skMCU−/− mice, loss of MCU in adult skeletal muscle caused systemic metabolic effects, as demonstrated by the increased blood FA (Fig. 6g) and ketone bodies (Fig. 6h). The incomplete MCU deletion prevented any further biochemical and physiological analysis, although the metabolic adaptations were anyway partially evident.

MCU deletion in adult skeletal muscle impairs whole-body metabolism. a OCR measurements indicated reduced respiratory capacity in iskMCU−/− FDB myofibers compared to controls. Left: representative traces. Right: quantification. To calculate basal and maximal respiration, non-mitochondrial O2 consumption was subtracted from absolute values. ATP linked respiration was calculated as the difference between basal and oligomycin-insensitive O2 consumption. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of ten samples per condition. Data are presented as mean ± SD. b Phosphorylation levels of PDH were increased in iskMCU−/− soleus muscles compared to controls. c Glucose dependent-respiration was decreased in iskMCU−/− FDB myofibers compared to controls. Left: OCR measurements were performed as in a. UK5099 was added to inhibit MPC. Right: Glucose dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. d FA-dependent respiration was increased in iskMCU−/−. Left: OCR measurements were performed as in a. Etomoxir was added to inhibit FA utilization. Right: FA dependency was calculated and expressed as the percentage of basal OCR. Data are normalized on mean Calcein fluorescence. *p < 0.05, t test (two-tailed, unpaired) of five samples per condition. Data are presented as mean ± SD. e iskMCU−/− mice had unaltered fasting glycemia compared to MCUfl/fl controls. Data are presented as mean ± SD (five animals per condition). f Blood lactate concentration was increased in iskMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. g Plasma FA concentration was increased in iskMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD. h Plasma beta-hydroxybutyric acid concentration was increased in iskMCU−/− mice compared to controls. *p < 0.05, t test (two-tailed, unpaired) of four animals per condition. Data are presented as mean ± SD
Discussion
The role of Ca2+ uptake by the skeletal muscle mitochondria was recently investigated [9, 10, 12], but outstanding questions still remain.
First, the causes underlying the reduced exercise performance consequent to MCU deletion are unclear. In our study, deletion of MCU specifically in the skeletal muscle impaired exercise performance, demonstrating that the decreased running capacity is solely due to a skeletal muscle defect. In vivo measurements demonstrated reduced tetanic force of skMCU−/− muscles. However, the strength developed at frequencies recruited during treadmill exercise is unaffected by loss of MCU. In addition, our results exclude the contribution of impaired fatigue resistance to the skMCU−/− phenotype. Coherently with the decrease in exercise performance, a slow-to-fast MHC shift characterizes both skMCU−/− and iskMCU−/− soleus muscles, thus excluding a compensatory effect occurring during development. Since muscle remodeling toward faster MHCs occurs in disuse conditions [29], one possible explanation is that the inefficient substrate oxidation of MCU−/− muscles impairs muscle activity, which in turn determines MHC expression remodeling.
The shift in MHC expression is coherent with a metabolic switch toward decreased oxidative metabolism (Fig. 7a). In line with this, we measured a major OCR decrease in MCU-deleted fibers, indicating that MCU loss can limit the supply of reducing equivalents to the electron transport chain. This effect is partially mitigated by upregulation in lipid catabolism. Similarly, inhibition of MPC has been shown to trigger a profound metabolic rewiring and to boost FA oxidation in muscle cells [27], thus suggesting that the impairment in pyruvate catabolism is the main trigger of the ongoing metabolic rewiring. To confirm this notion, we here show that reduced PDH activity is per se sufficient to shift substrate preference from carbohydrates toward lipids. Nonetheless, the reduced maximal OCR in PDK4-overexpressing fibers suggests that optimal glucose oxidation is required to sustain high-energy demand, which is not otherwise met by the alternative FA metabolism. Accordingly, overexpression of the Ca2+-independent PDP2 isoform in skMCU−/− fibers was sufficient to inhibit the preferred lipid oxidation. Altogether, these data indicate that MCU controls muscle substrate preference and warrants a high level of plasticity, by impinging on the activity level of PDH. In the absence of MCU, pyruvate to acetyl-CoA conversion slows down, thus limiting the TCA supply, and rewiring of cell metabolism toward preferential lipid catabolism partially compensates this defect.

Model. a Schematic representation of the metabolic consequences of MCU deletion in the skeletal muscle. Left: Ca2+ enters mitochondria through the MCU complex and stimulates the PDH activity. PDH converts pyruvate into acetyl-CoA, which in turn feeds the TCA cycle. Right: in MCU−/− muscles mitochondrial Ca2+ uptake is inhibited, PDH is inactive, and pyruvate oxidation is impaired. In turn, TCA cycle activity decreases. Increased FA metabolism partially compensates the impairment in glucose oxidation. In addition, FA oxidation produces reducing equivalents that fuel the respiratory chain. b The primary skeletal muscle mitochondria defect triggered by MCU deletion has an impact on whole-body metabolism. The decrease in muscle glucose oxidation translates into increased glycolysis, glycogen storage, and lactate production. The aberrant glucose utilization signals to the liver and the adipose tissue, which in turn mobilize nutrient depots. skMCU−/− mice have decreased muscle force, impaired exercise performance, and display MyHC1 → MyHC2A expression switch
However, MCU loss causes an additional bioenergetics defect, because mitochondrial Ca2+ level is a positive allosteric regulator of two additional TCA cycle enzymes (i.e., isocitrate and α-ketoglutarate dehydrogenases). OCR measurements in the presence of palmitate, directly demonstrated that, although MCU−/− muscles mostly rely on β-oxidation to sustain respiration, their overall capacity to oxidize FA is actually reduced. Thus, in MCU−/− myofibers, the flux of FA-derived acetyl-CoA through the TCA cycle is slowed down because of isocitrate and α-ketoglutarate dehydrogenase inhibition, decreasing the availability of reducing equivalents fuelling basal respiration. Overall, the consequences of MCU deletion on mitochondria function in the skeletal muscle are apparent already in basal conditions, although metabolic rewiring mitigates potential more severe phenotype.
In light of the function of skeletal muscle activity on whole-body metabolism, the metabolic defect of MCU-deleted muscles translates into a systemic catabolic response (Fig. 7b). Whether this is a direct effect of altered serum metabolites concentrations, or rather circulating factors (i.e., myokines) play a role, is not clear yet.
By means of the iskMCU−/− mouse, we wished to uncover potential phenotypic features otherwise lost by developmental adaptations. However, complete MCU deletion was achieved only in some muscles. Nonetheless, iskMCU−/− mouse phenotype was similar to skMCU−/−, suggesting that either embryonic compensations are negligible or that complete MCU ablation in the adult would be required to uncover further defects.
Finally, our models are in accordance with observations made by cardiomyocyte-specific conditional MCU deletion [30, 31] in terms of lack of overt basal phenotype and absolute requirement of effective mitochondrial Ca2+ uptake to match the metabolic output with increased energy demand. However, contrary to skMCU and iskMCU muscles, baseline p-PDH was unaltered in MCUfl/fl-MCM hearts [30, 31], maybe reflecting either the lower MCU currents or the high dependency on FA oxidation of cardiac mitochondria.
Overall, our data indicate that a complex metabolic rewiring occurs in skMCU−/− muscles. Mitochondrial Ca2+ uptake is required for efficient glucose oxidation and sustains rapid flux through the TCA cycle. The impairment in muscle performance of skMCU−/− mice is mainly due to reduced glucose oxidation consequent to inhibition of PDH and thus TCA cycle activity, which is accompanied by fiber-type remodeling toward faster MHCs. Systemic adaptations may also contribute to the decreased exercise capacity. Metabolic flexibility in terms of increased FA oxidation at least partially sustains muscle activity and compensates for the impairment in carbohydrates' utilization. On the one hand, this metabolic rewiring contributes to explain the mild phenotype of MCU-deficient mouse models. On the other, despite these profound metabolic adaptations, the selective loss of MCU in skeletal muscle still shows a negative impact on muscle strength and performance, thus further underlining the physio-pathological relevance of mitochondrial Ca2+ homeostasis.
References
Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–78.
Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–7.
De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–40.
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5.
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–76.
Sancak Y, Markhard ALL, Kitami T, Kovacs-Bogdan E, Kamer KJJ, Udeshi NDD, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–82.
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca2+uptake. Nature. 2010;467:291–6.
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE. 2013;8:e55785.
Mammucari C, Gherardi G, Zamparo I, Raffaello A, Boncompagni S, Chemello F, et al. The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep. 2015;10:1269–79.
Zampieri S, Mammucari C, Romanello V, Barberi L, Pietrangelo L, Fusella A, et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol Rep. 2016;4:e13005.
Vecellio Reane D, Vallese F, Checchetto V, Acquasaliente L, Butera G, De Filippis V, et al. A MICU1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol Cell. 2016;64:760–73.
Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72.
Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol Med. 2016;8:569–85.
Zhao Y, Araki S, Wu J, Teramoto T, Chang Y-F, Nakano M, et al. An expanded palette of genetically encoded Ca2+ indicators. Science (80-). 2011;333:1888–91.
Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–6.
Schuler M, Ali F, Metzger E, Chambon P, Metzger D. Temporally controlled targeted somatic mutagenesis in skeletal muscles of the mouse. Genesis. 2005;41:165–70.
Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, et al. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–905.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86.
TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. 2014;542:91–114.
Saito K, Lee S, Shiuchi T, Toda C, Kamijo M, Inagaki-Ohara K, et al. An enzymatic photometric assay for 2-deoxyglucose uptake in insulin-responsive tissues and 3T3-L1 adipocytes. Anal Biochem. 2011;412:9–17.
Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta Bioenerg. 2009;1787:1309–16.
Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, et al. Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab. 2015;22:669–81.
McCommis KS, Chen Z, Fu X, McDonald WG, Colca JR, Kletzien RF, et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab. 2015;22:682–94.
Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen Y-C, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science. 2012;337:96–100.
Herzig S, Raemy E, Montessuit S, Veuthey J-L, Zamboni N, Westermann B, et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science. 2012;337:93–96.
Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, et al. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol Cell. 2014;56:425–35.
Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 2013;280:4131–48.
Ciciliot S, Rossi AC, Dyar KA, Blaauw B, Schiaffino S. Muscle type and fiber type specificity in muscle wasting. Int J Biochem Cell Biol. 2013;45:2191–9.
Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22.
Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34.
Acknowledgements
Research was supported by fundings from the European Union (ERC mitoCalcium, no. 294777 to RR), the Italian Ministry of Education, University, and Research (PRIN 2015W2N883_003 to CM), the French Muscular Dystrophy Association AFM-Téléthon (18857 to CM), the Italian Telethon Foundation (GGP16029 to RR), and the Italian Association for Cancer Research (IG 18633 to RR). We thank Margherita Franceschini for assistance with Ca2+ measurements, Elisabetta Iori for assistance in metabolites measurements, and the laboratory of Jorge Ruas for sharing primer sequences.
Author contributions
GG performed most of the experiments, LN and BB performed force measurements, SC performed in vivo 2-deoxy-d-glucose uptake experiments, PBr and PBo performed microinjections for the production of the MCUfl/fl mouse, GPF supervised metabolites measurements, DDS supervised metabolomics experiments, RR and CM conceived the research, and CM directed the research and wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Edited by N Chandel
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Gherardi, G., Nogara, L., Ciciliot, S. et al. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ 26, 362–381 (2019). https://doi.org/10.1038/s41418-018-0191-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0191-7
This article is cited by
-
MCU-independent Ca2+ uptake mediates mitochondrial Ca2+ overload and necrotic cell death in a mouse model of Duchenne muscular dystrophy
Scientific Reports (2024)
-
Reduced mitochondrial calcium uptake in macrophages is a major driver of inflammaging
Nature Aging (2023)
-
The mitochondrial calcium uniporter of pulmonary type 2 cells determines severity of acute lung injury
Nature Communications (2022)
-
Mitochondrial calcium uptake regulates tumour progression in embryonal rhabdomyosarcoma
Cell Death & Disease (2022)
-
Mitochondrial Calcium Uniporter (MCU) deficiency reveals an alternate path for Ca2+ uptake in photoreceptor mitochondria
Scientific Reports (2020)
