
Abstract
Aberrant activation of cardiac fibroblasts is the main cause and character of cardiac fibrosis, and inhibition of cardiac fibrosis becomes a promising treatment for cardiac diseases. Platelet-activating factor (PAF) and Hippo pathway is recently recognized as key signaling mechanisms in cardiovascular diseases. In this study we explored the potential roles of PAF and Hippo signaling pathway in cardiac fibrosis. Myocardial infarction (MI) was induced in mice by left anterior descending artery ligation. After 28 days, the mice were sacrificed, and the hearts were collected for analyses. We showed that PAF receptor (PAFR) and yes-associated protein 1 (YAP1, a key effector in the Hippo pathway) were significantly increased in the heart of MI mice. Increased expression of PAFR and YAP1 was also observed in angiotensin II (Ang II)-treated mouse cardiac fibroblasts. In mouse cardiac fibroblasts, forced expression of YAP1 increased cell viability, resulted in collagen deposition and promoted fibroblast-myofibroblast transition. We showed that PAF induced fibrogenesis through activation of YAP1 and promoted its nuclear translocation via interacting with PAFR, while YAP1 promoted the expression of PAFR by binding to and activating transcription factor TEAD1. More importantly, silencing PAFR or YAP1 by shRNA, or using transgenic mice to induce the conditional deletion of YAP1 in cardiac fibroblasts, impeded cardiac fibrosis and improved cardiac function in MI mice. Taken together, this study elucidates the role and mechanisms of PAFR/YAP1 positive feedback loop in cardiac fibrosis, suggesting a potential role of this pathway as novel therapeutic targets in cardiac fibrosis.
Similar content being viewed by others

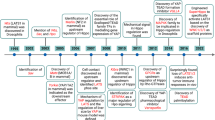

Introduction
Cardiovascular diseases, along with irreversible cardiac fibrosis, are a major cause of death worldwide. Damage to the heart initiates the process of repair; however, sustained impairment exaggerates the proliferation of cardiac fibroblasts (CFs), which leads to the overdeposition of extracellular matrix (ECM), ultimately causing arrhythmia and heart failure [1, 2]. Nevertheless, effective therapy for cardiac fibrosis still needs to be explored due to lack of valid therapeutic methods. Thus, it is urgent to reveal the underlying mechanism of cardiac fibrosis to lay a solid theoretical foundation of theory and provide a possible future treatment.
As one of the most well-known cytokines, PAF participates in the generation and development of multiple cardiovascular diseases, such as arrhythmia, coronary heart disease, and ischemic heart disease [3,4,5]. Upregulation of PAF in patients with heart failure has also been reported [6, 7], but it remains to be determined whether PAF is involved in myocardial fibrosis and, if so, what the molecular and signaling mechanisms are for its actions.
The Hippo pathway plays critical roles in cell proliferation, apoptosis, development and organ size control [8,9,10]. Recently, an increasing amount of evidence suggests that dysregulation of the Hippo pathway contributes to multiple diseases, including heart diseases [11,12,13,14]. Xin et al. found that YAP1, a downstream effector of the Hippo pathway, promoted cardiac regeneration by activating the insulin-like growth factor and Wnt signaling pathways [15]. However, the role of the Hippo pathway in the progression of cardiac fibrosis is not well understood. In addition, the upstream factors that control the Hippo pathway need to be identified. Yu et al. found that the Hippo pathway was regulated by G protein-coupled receptor (GPCR) signaling [16]. We supposed that PAFR, as a classical GPCR, regulates the activities of the Hippo pathway to mediate a profibrotic effect of PAF in the heart.
In this study, we found that PAF promoted fibrogenesis through activation of YAP1 by binding to PAFR. Conversely, YAP1 enhanced the expression of PAFR by binding to the transcription factor TEAD1. Moreover, we found that silencing PAFR or YAP1 attenuated cardiac fibrosis and improved heart function in MI mice. This study elucidated the role and mechanism of the PAFR/YAP1 loop in cardiac fibrosis, indicating the potential for this pathway as a novel therapeutic target for cardiac fibrosis.
Materials and methods
Animals and experimental model of myocardial infarction (MI)
Male C57BL/6 mice (25 g–30 g) were purchased in the standard animal room of the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). YAP1fl/fl and Col1a2CreERT mice were purchased from The Jackson Laboratory (Bar Harbor, USA). Then the YAP1fl/fl mice were crossed into Col1a2CreERT mice, which are referred to as YAP1-cKO mice. Six- to eight-week-old male mice were treated with tamoxifen at a dose of 80 mg/kg intraperitoneally for 5 days, and then, the mice were allowed to rest for 1 week after the last injection to induce the conditional knockout of YAP1 in fibroblasts. Mice were raised under controlled temperature and humidity conditions (45% RH) with a 12-h light/dark cycle. The mice were intraperitoneally injected with Avertin (250 mg/kg), two layers of muscles were separated by hemostatic forceps, and the heart was exposed after opening the chest. The left anterior descending artery was ligated with a 7/0 ligature. All surgical procedures were performed under sterile conditions. Successful occlusion was confirmed by elevation of the S-T segment in lead II. After 28 days, the mice were anesthetized and sacrificed, and then, the hearts were quickly isolated. The portion of the heart bordering the MI was prepared for Masson staining and other experiments. All experiments concerning animals and cells were approved by the Harbin Medical University Experimental Animal Ethics Committee and complied with the Helsinki Declaration of 1975.
Isolation and culture of cardiac fibroblasts
CFs were isolated from 1- to 3-day-old C57BL/6 mice, and hearts were cut into small chunks. The tissue was digested with tryptase/EDTA (Gibco, Grand Island, USA) and PBS at 4 °C for 8–12 h. Type II collagenase digestion was added at 37 °C for 10–15 min. The above steps were repeated 3–4 times. The cells were precipitated by centrifugation and cultured in complete medium (90% DMEM + 10% FBS). After 60 min of incubation in the incubator, the nonadherent cardiomyocytes were discarded, and the primary CFs were cultured in complete medium for 48 h for the next step. Before transfection, the medium was replaced with DMEM, and the cells were transfected with sh-PAFR by Lipofectamine 2000 (Invitrogen, Carlsbad, USA) in Opti-MEM according to the manufacturer’s protocol. Six hours later, the medium was replaced with complete medium.
Masson and HE staining
The heart tissue was fixed with 4% paraformaldehyde for 1 day, embedded with paraffin and then cut into 5 μm slices. The slides were dewaxed with xylene and then rehydrated with graded ethanol for the following staining assay. For Masson staining, the tissue was stained following the protocol of a Masson Trichrome Staining kit purchased from Solarbio (Beijing, China). Tissue slides were stained with hematoxylin iron and then differentiated by acid alcohol. Subsequently, the slides were stained with bluing solution and rinsed with distilled water. Then, the slides were stained with ponceaux and washed with 1% acetic acid solution and phosphomolybdic acid solution. Later, the slides were put into aniline blue solution and washed with 1% acetic acid solution. Finally, the sections were dehydrated and mounted in xylene and then sealed with neutral balsam. For HE staining, the slides were stained with hematoxylin, differentiated with acid alcohol then soaked in tap water. Next, sections were stained by eosin and rinsed with tap water. Finally, the sections were dehydrated and mounted in xylene and then sealed with neutral balsam. The figures were quantified by Image J.
Immunohistochemistry (IHC)
The slides were dewaxed with xylene and rehydrated with graded ethanol. The slides were incubated with 3% H2O2, and the antigen retrieval process was applied with sodium citrate buffer. Next, the slides were blocked with blocking buffer and then incubated with primary antibody overnight at 4 °C. Then, the sections were incubated with the secondary antibody and stained with DAB reagent purchased from ZSGB-BIO (Beijing, China). Finally, the sections were dehydrated and mounted in xylene and then sealed with neutral balsam.
MTT cell viability assay
Cell viability was measured by MTT assay. CFs were cultured in 96-well plates. After treatment, the cells were incubated at 37 °C and 5% CO2 for 48 h, and then 20 μL of MTT (0.5 mg/L) solution was added to the cells. After incubating for another 4 h, the liquid in the well plate was discarded, and 200 μL of dimethyl sulfoxide was added to each well of the 96-well plate. The cells were incubated at room temperature for 15 min. At 490 nm, a microplate reader was used to read the absorbance value of each well. Relative absorbance values (normalized to the control) were used as an indication of cell viability.
Western blotting
Total protein was extracted from cell and heart tissue with a mixture of RIPA lysis buffer (Beyotime, Jiangsu, China) and protease inhibitor. The electrophoresis was performed in an SDS-PAGE gel, and then, the protein was transferred onto nitrocellulose membranes (Pall Life Science). After blocking with 5% skim milk, the protein blots were probed with primary antibody at 4 °C overnight. GAPDH (anti-GAPDH antibody from Kangchen Shanghai, China) was used as an internal control. Antibodies against YAP1 (Cell Signaling Technology, Beverly, MA), PAFR and α-SMA (Abcam Inc., USA), collagen I and Fn1 (Proteintech, Wuhan, China), and CTGF (Santa Cruz, Calif., USA) were used. The next day, the membranes were rinsed with PBST and incubated with the secondary antibody. The results were finally quantified by Odyssey v1.2 Software.
Immunofluorescence staining
The culture medium was discarded and the fibroblasts were fixed in 200 μL 4% paraformaldehyde for 15 min at 37 °C. Then the cells were incubated in 200 μL penetrating solution for 1 h. Cells were washed with PBS and blocked with 200 μL 50% goat serum for 40 min. Primary antibodies with YAP1 (66900-1-Ig, Proteintech, 1:50) and α-SMA (ab7817, Abcam, 1:200) were added to each well at 4 °C for overnight. The cells were incubated in secondary antibody for 2 h in the dark. Cell nuclei were visualized by DAPI for 5 min at room temperature. The cells were observed under fluorescence microscope.
Ultrasound imaging measurements
The mouse was anesthetized by intraperitoneal injection of Avertin (250 mg/kg), and was fixed on the ultrasound operating table. The sensor of the Vevo®2000 high-resolution imaging system was used to obtain the two-dimensional echocardiogram of the animal and the left ventricle (LV). Two-dimensional echocardiographic views of the mid-ventricular short axis and parasternal long Axes M-mode were obtained at baseline and MI. M-mode was recorded to measure LV wall thickness, end-diastolic diameter, and end-systolic diameter. The percentage of fraction shortening (%FS) and ejection fraction (%EF) and other cardiac functions were calculated by the software.
Quantitive real-time RT-PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, USA) and then reverse-transcribed to complementary DNA with a cDNA Synthesis SuperMix for qPCR kit (Transgenebiotech, Beijing, China). qRT-PCR was performed with SYBER GREEN (Roche, Basel, Switzerland) in an ABI 7500 FAST real-time PCR system. The result was evaluated based on the Ct value normalized to GAPDH.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were carried out as previously described [17] using an anti-TEAD1 antibody (Abcam Inc., USA). Ten percent chromatin without immunoprecipitation was used as an input for the positive control, and a nonspecific antibody (rabbit anti-IgG; BD Biosciences) served as a negative control. The precipitated DNAs were subjected to PCR to amplify the TEAD1-binding sites using primers specific for PAFR-1 (forward: 5′-AACCATTTTAGCATTGACAAGG-3′; reverse: 5′-CAGGCTCCATGATAGACGCTG-3′) and PAFR-2 (forward: 5′-CCTCCCCCACCTCTCTCTCCCCTAC-3′; reverse: 5′- CGGAAGTGGAAAGTGCAGTGCCC-3′). The amplified fragments were then resolved electrophoretically on a 2% (w/v) agarose gel and verified by DNA sequencing.
Statistical analysis
Data are presented as mean ± SEM. One way analysis of variance followed by Bonferroni or Dunnett’s post hoc test was used for multiple group comparisons. A two-tailed value of P < 0.05 was considered as statistically significant difference. Data were analyzed using the GraphPad Prism 5.0.
Results
PAF causes myocardial fibrosis by interacting with PAFR
To determine the role of PAF/PAFR in cardiac fibrosis, we developed a cardiac fibrosis model in mice with MI. As shown in Fig. 1a–c, the expression of PAFR was upregulated in the hearts of MI mice. Consistent with the results from the mouse model, the expression of PAFR was also increased in CFs treated with angiotensin II (Ang II) or serum (Fig. 1d–g).

Elevated expression of PAFR at the protein and mRNA levels in the peri-infarct area, measured by immunohistochemistry (IHC) (a), qRT-PCR (b) and Western blot (c). qRT-PCR (d) and Western blot (e) analyses of mRNA and protein levels of PAFR in CFs treated with angiotensin II (Ang II). Increased expression of PAFR at the mRNA (f) and protein levels (g) in response to different concentrations of serum. n = 5. *P < 0.05, **P < 0.01 vs. sham or control.
To investigate whether PAF/PAFR upregulation participates in cardiac fibrosis or is merely a bystander under conditions favoring fibrogenesis, we first examined the effects of PAF on the proliferation of CFs. As shown in Fig. 2a, PAF promoted cell proliferation in a dose-dependent manner at concentrations below 10 nM, but inhibited the proliferation of CFs at concentrations above 1000 nM. In addition, PAF enhanced the expression of PAFR in a dose-dependent manner (Fig. 2b). Moreover, silencing PAFR with a specific shRNA (Fig. 2c, d) or pharmacological inhibition of PAFR with WEB2086 (an antagonist of PAFR) prevented PAF-induced cell proliferation (Fig. 2e). Furthermore, PAF upregulated the mRNA levels of collagen 1α1 and collagen 3α1, and silencing PAFR abrogated this effect of PAF (Fig. 2f, g). Consistent with its action at the mRNA level, PAF increased the protein levels of collagen I, fibronectin 1 (Fn1) and alpha smooth muscle actin (α-SMA), and these effects were abolished by WEB2086 or sh-PAFR (Fig. 2h, i).

a The MTT assay showed that PAF significantly increased CF viability at doses of 3, 10 and 30 nmol/L. b Elevated protein expression of PAFR assessed by Western blot. c, d Western blot and qRT-PCR assays demonstrated the expression of PAFR in CFs were transfected with sh-PAFR. e Cell viability experiments detected a reduced cell proliferation rate when CFs were treated with WEB2086 (an antagonist of PAFR) and sh-PAFR. f, g qRT-PCR analysis indicated altered expression of Col 1α1 and Col 3α1 in CFs. h, i PAF increased the expression of collagen 1, fibronectin 1 (Fn1), and α smooth muscle actin (α-SMA). WEB2086 and sh-PAFR reduced the effects of PAF. n = 4. *P < 0.05, **P < 0.01 vs. control or sh-scramble; #P < 0.05, ##P < 0.01 vs. PAF.
Silencing of PAFR improves heart function and alleviates cardiac fibrosis in MI mice
We next verified these in vitro results by subsequent in vivo experiments. To this end, we injected adenovirus carrying the PAFR shRNA (Adv-sh-PAFR) into mice with MI and then evaluated the effects of PAFR inhibition on cardiac fibrosis (Fig. 3a) compared with the results from sham-operated control mice. As illustrated in Fig. 3a, b, knockdown of PAFR had no obvious effect on collagen deposition and heart function in wild-type mice. However, silencing PAFR significantly improved ejection fraction (EF) and fractional shortening (FS) in MI mice (Fig. 3c, d). Additionally, PAFR silencing attenuated collagen deposition and inhibited the expression of collagen 1α1, collagen 3α1, connective tissue growth factor (CTGF) and fibrotic-related proteins, including collagen I, Fn1, α-SMA and CTGF, as well as PAFR in the hearts of MI mice (Fig. 3e–g). These results suggested that PAF/PAFR upregulation is likely an important contributor to cardiac fibrosis.

a Relative mRNA expression of PAFR, Col 1α1 and Col 3α1 in mice after treatment with Adv-sh-PAFR. n = 5. b Heart function represented by EF (%) and FS (%) showed no change in mice injected with Adv-sh-PAFR. n = 5. c, d Silencing of PAFR improved heart function in MI mice. n = 10. e HE and Masson staining showed decreased collagen deposition and heart structure remodeling in PAFR deletion mice. n = 5. f qRT-PCR analysis demonstrated reduced mRNA expression of fibrosis-related genes and PAFR in mice with knockdown of PAFR. n = 5. g Western blot analysis indicated alleviated expression of fibrosis-associated proteins after injection of Adv-sh-PAFR in MI mice. n = 4. *P < 0.05, **P < 0.01 vs. sham or control; #P < 0.05, ##P < 0.01 vs. MI.
PAF/PAFR promotes cardiac fibrosis through the Hippo/YAP1 pathway
The next question we wanted to answer was how PAF/PAFR could be linked to fibrogenesis. We have mentioned earlier that PAFR belongs to GPCR and Hippo/YAP1 are known to be activated by GPCR [16]. We thus proposed that PAF/PAFR could act through the Hippo/YAP1 pathway. First, we evaluated the role of YAP1 in cardiac fibrosis. As shown in Fig. 4a–c, YAP1 expression was significantly increased at both the mRNA and protein levels in MI mice, as well as in fibrogenic CFs after stimulation with Ang II or serum (Fig. 4d). Transfection of YAP1 markedly increased the proliferation of CFs and the expression of collagen 1α1, collagen 3α1 and CTGF in cultured CFs, as well as some other fibrotic-related genes (Fig. 4e–g). These changes induced the transition of fibroblasts into myofibroblasts (Fig. 4h).

YAP1 was increased at both the mRNA and protein levels in the infarcted heart (a–c) and in CFs treated with Ang II or serum (d). Forced expression of YAP1 with the YAP1 plasmid promoted cell viability (e), increased the expression of collagen 1α1, collagen 3α1, and CTGF (f), and caused fibrogenesis (g) in CFs. h Overexpression of YAP1 promoted fibroblast-to-myofibroblast transition. n = 4–6. *P < 0.05, **P < 0.01 vs. sham, control, or vector.
Moreover, consistent with previous findings, PAF/PAFR upregulated the expression of YAP1 and CTGF, a downstream component of the Hippo/YAP1 signaling pathway, which is known to be a key factor determining cardiac fibrosis (Fig. 5a–c). Moreover, PAF induced the translocation of YAP1 from the cytoplasm into the nucleus, along with an increase in the expression of α-SMA, a marker of myofibroblast formation (Fig. 5d). Moreover, silencing PAFR attenuated these effects of PAF on YAP1 (Fig. 5a, b, d), indicating that PAF regulates the expression and translocation of YAP1 by interacting with PAFR. These effects were further supported by the results obtained from the YAP/TAZ responsive luciferase reporter gene assay (8 × GTIIC-luciferase): PAF robustly enhanced luciferase activity, an effect nearly abolished by PAFR inhibitor or shRNA but not by scrambled control shRNA (Fig. 5e).

a, b Western blot analysis demonstrated reduced protein expression of YAP1 and CTGF when PAFR was blocked by WEB2086 and sh-PAFR. c qRT-PCR showed decreased expression of CTGF mRNA while PAFR was inhibited by WEB2086 and sh-PAFR. d PAF promoted nuclear translocation of YAP1 and fibroblast-to-myofibroblast transition. e PAF increased luciferase reporter activity which contains a synthetic YAP/TAZ responsive element (8 × GTIIC-luciferase). f Western blot analysis demonstrated decreased expression of YAP1 in CFs treated with YAP1 shRNA. g qRT-PCR indicated reduced expression of YAP1 and CTGF after deletion of YAP1 by sh-YAP1. h The MTT assay showed decreased cell viability after transfection with sh-YAP or treatment with verteporfin. i Lower expression of CTGF mRNA was determined by qRT-PCR after silencing YAP1 or treatment with verteporfin in CFs. j, k qRT-PCR and Western blot experiments demonstrated reduced expression of fibrosis-related mRNA and proteins after the disruption of YAP or TEAD1 in CFs. n = 4–6. *P < 0.05, **P < 0.01 vs. control or sh-Scamble; #P < 0.05, ##P < 0.01 vs. PAF.
We then considered whether PAF/PAFR employs YAP1 to participate in the process of cardiac fibrosis. To answer this question, we conducted the following experiments. We first tested three shRNAs for their potency for silencing YAP1. As illustrated in Fig. 5f, while both the #1 and #2 shRNAs were able to repress YAP1 expression at the protein level in the CFs compared with the sh-Scramble group, label #2 gave rise to the most potent effect. Therefore, it was used for all subsequent experiments and was designated sh-YAP1. Along with the silencing of YAP1, CTGF mRNA was also diminished by sh-YAP1 (Fig. 5g). Sh-YAP1 mitigated PAF-induced cell proliferation in CFs (Fig. 5h), as did verteporfin (an inhibitor of YAP1-mediated gene expression). Moreover, inhibition of YAP1 retarded the fibroblast-to-myofibroblast transition (Fig. 5d) and finally alleviated fibrosis evoked by PAF (Fig. 5i–k).
PAFR is transcriptionally regulated by YAP1 via a positive feedback loop
The above data suggested that YAP1 mediated the profibrotic action of PAF/PAFR. YAP1 exerts its biological function by binding to the transcription factor TEAD as its transcriptional coactivator to transcriptionally regulate gene expression [18]. Intriguingly, our computational analysis revealed that there are three putative TEAD1-binding sites within the promoter region of the PAFR gene (Fig. 6a). This finding indicated that YAP1/TEAD1 has the potential to regulate PAFR expression at the transcriptional level. Our chromatin immunoprecipitation (ChIP) and qRT-PCR experiments indeed demonstrated the physical interaction between TEAD1 and the promoter region of PAFR encompassing the TEAD1-binding sites and the transcriptional activation of PAFR expression, respectively (Fig. 6b, c). To determine which of the three binding sites plays a key role in the interaction, we constructed luciferase reporter plasmids containing a wild-type or truncated TEAD1-binding site upstream of the PAFR coding sequences. We found that deletion of TEAD1-binding site 1 almost eliminated the activation effect of TEAD1 on PAFR (Fig. 6d), suggesting that site 1 has a crucial role in initiating transcription. We then constructed a luciferase reporter containing the 2110-bp region (positions −2000 to +110) (p-PAFR-luc) and a 12-bp TEAD1-binding site 1 mutant (p-mPAFR-luc) and cotransfected the reporter constructs with vector and TEAD1 into CFs. As shown in Fig. 6e, forced expression of TEAD1 increased p-PAFR-luc activity, whereas this effect was eliminated when we used the 12-bp TEAD1-binding site 1 mutant. Furthermore, our qRT-PCR and Western blot analyses further confirmed that forced expression of either YAP1 or TEAD1 remarkably boosted up the expression of PAFR at both the mRNA and protein levels (Fig. 6f, g). On the other hand, interrupting the binding between YAP1 and TEAD1 by overexpressing YAP1-S94A (a loss-of-function mutant of YAP1) or applying verteporfin dampened the transactivation of PAFR induced by YAP1 in CFs (Fig. 6f, g). Taken together, our results indicate that YAP1 enhances PAFR expression through TEAD1-binding to the PAFR promoter.

a The putative binding sites for TEAD1 in the promoter region of the PAFR gene. ChIP (b) and qRT-PCR (c) analyses revealed that TEAD1 was able to bind to the PAFR promoter region and promoted PAFR expression in CFs. d Luciferase reporter assays were performed on CFs cotransfected with reporter plasmids containing truncated versions of the promoter region of the PAFR gene, as indicated, and pRL-TK. e Luciferase reporter assays were performed on CFs transfected with a recombinant vector containing the base pair (bp) −2000 to +110 PAFR region to the luciferase gene, referred to as p-PAFR-luc. A similar vector with a 12-bp mutation of the predicted TEAD1-binding site is referred to as p-mPAFR-luc. qRT-PCR (f) and Western blot (g) analyses showing that forced expression of either YAP1 or TEAD1 increased the expression of PAFR at the mRNA and protein levels. n = 5. *P < 0.05, **P < 0.01 vs. vector; #P < 0.05 vs. YAP1.
Inhibition of YAP1 ameliorates cardiac fibrosis in mice after MI
To investigate whether suppression of YAP1 in mice could abate myocardial fibrosis, adenovirus carrying the sh-YAP1 fragment (Adv-sh-YAP1) was administered to mice 3 days after coronary artery ligation and sham-operated mice (Fig. 7a). qRT-PCR analysis showed that silencing of YAP1 by Adv-sh-YAP1 had no effect on the expression of fibrosis-associated genes such as Col 1α1 and Col 3α1 in wild-type mice (Fig. 7b), and cardiac function also showed no change (Fig. 7c). However, knockdown of YAP1 significantly improved EF and FS and thus recovered cardiac function in MI mice (Fig. 7d, e). Simultaneously, inhibition of YAP1 markedly alleviated the deposition of collagen and reduced fibrosis-related proteins, including collagen I, α-SMA and CTGF (Fig. 7f–h). In conclusion, our results revealed that the suppression of YAP1 in CFs alleviated cardiac fibrosis.

a, b Relative mRNA level of YAP1, Col 1α1 and Col 3α1 after Adv-sh-YAP1 administration in mice. c Cardiac function manifested by EF (%) and FS (%) in mice injected with Adv-sh-YAP1. d, e Ultrasound imaging showed improved EF (%) and FS (%) in MI mice with knockdown of YAP1. f HE and Masson’s staining indicated reduced collagen deposition in YAP1 silenced mice subjected to MI. g, h qRT-PCR and Western blot demonstrated decreased expression of fibrosis-related genes and proteins in mice with Adv-sh-YAP1. n = 6–13. *P < 0.05, **P < 0.01 vs. Control of Sham; #P < 0.05, ##P < 0.01 vs. MI.
YAP1 promotes cardiomyocyte proliferation during the process of myocardial infarction and hence facilitates heart regeneration [19,20,21]. Hereby, we intended to investigate the expression of YAP1 in the CFs of MI mice. As shown in Fig. 8a, b, elevated expression of YAP1 was observed in CFs isolated from MI mice. Furthermore, we constructed an exogenous estrogen-induced fibroblast-specific deletion of YAP1 transgenic mice, YAP1fl/fl; Col1a2CreERT transgenic mice (Fig. 8c), hereinafter referred to as YAP1-cKO mice to elucidate the role of YAP1 in CFs during cardiac fibrosis. To investigate the knockout of YAP1 in CFs, we performed agarose gel electrophoresis, and the results showed positive homozygous bands for YAP1fl/fl at 270 bp and positive bands for Col1a2CreERT at 186 bp (Fig. 8d). Furthermore, qRT-PCR and Western blot analysis demonstrated markedly decreased expression of YAP1 at the mRNA and protein levels in CFs isolated from YAP1-cKO mice (Fig. 8e, f). As shown in Fig. 8g, h, knockout of YAP1 in CFs dramatically improved cardiac function in MI mice. In addition, conditional deletion of YAP1 significantly reduced collagen deposition (Fig. 8i). Moreover, immunohistochemistry analysis indicated decreased expression of α-SMA in the hearts of YAP1-cKO mice compared to those of wild-type mice subjected to MI (Fig. 8i). YAP1 silencing strikingly alleviated the expression of fibrosis-related proteins, such as Fn1 and Collagen I, as well as fibrotic genes including Col 1α1, Col 3α1 and Fn1 (Fig. 8j, k). In conclusion, our results shed light on the loss of YAP1 in CFs alleviating cardiac fibrosis and improving heart function in MI mice.

a, b Increased mRNA and protein expression of YAP1 in the CFs of MI mice. c Strategy for the construction of YAP1-cKO mice. d The agarose gel electrophoresis indicated the mutant-type bands of YAP1fl/fl at 270 bp and Col1a2CreERT at 186 bp. e, f qRT-PCR and western blot assays demonstrated reduced protein and gene expression of YAP1 in cardiac fibroblasts separated from YAP1-cKO mice. g, h Improved EF (%) and FS (%) in YAP1-cKO mice subjected to MI detected by ultrasound imaging. i Masson’s staining and IHC showed decreased collagen deposition and α-SMA expression in hearts of MI mice with YAP1 knockout. j, k qRT-PCR and Western blotting demonstrated reduced expression of fibrosis-associated proteins and genes in YAP1-cKO mice subjected to MI. l Proposed model for the role of the PAF/YAP1 positive feedback loop in cardiac fibrosis. n = 3–6. *P < 0.05.
Discussion
In the present study, we characterized the profibrotic property of PAF/PAFR in the setting of MI, which is known to be associated with enhanced adverse cardiac fibrosis, and we elucidated the molecular events for the fine regulation of this signaling mechanism. We found that PAF and its receptor PAFR were both remarkably upregulated during MI and that this upregulation promoted cardiac fibrogenesis in CFs. This profibrotic action of PAF/PAFR was mediated by upregulation of YAP1 expression and functional activation of YAP1. In contrast, knockdown of either PAFR or YAP1 alleviated cardiac fibrosis and improved heart function in MI mice. On the other hand, YAP1 was found to transactivate the expression of PAFR by interacting with TEAD as its coactivator. Hence, these new findings allowed us to reach the following conclusions. First, the upregulated expression of YAP1 promoted cardiac fibrosis as a result of PAFR activation via PAF. Second, PAFR and YAP1 constituted a regenerative feedback loop to elevate their expression to amplify the cardiac fibrosis properties of this axis. Finally, our data revealed a novel signaling axis leading to cardiac fibrosis: PAF → PAFR↑ \(\rightleftarrows\) YAP1↑* → CTGF↑* → fibrosis↑ (Fig. 8l).
As a phospholipid, PAF possesses powerful biological functions, such as platelet secretion and aggregation [22]. It has been reported that PAF initiates Hedgehog signaling and promotes kidney fibrosis by activating PAFR, a G protein-coupled receptor [23]. Moreover, Song et al. found that the PAF/PAFR axis was involved in the process of hepatic fibrosis [24]. Although there is evidence of PAF upregulation in heart failure [6, 7], its role and mechanism in cardiac fibrosis are not completely known. In this study, we observed an increase in PAFR in MI mice and in CFs treated with PAF, Ang II or serum. Furthermore, our data showed that PAF led to fibrosis in vitro and in vivo by binding to its receptor PAFR. More importantly, interrupting the PAF/PAFR axis using PAFR-specific shRNA significantly alleviated fibrosis and improved cardiac function, indicating that targeting the PAF/PAFR axis may be a novel therapeutic strategy for the treatment of cardiac diseases associated with fibrosis.
Increasing evidence has shown that dysregulation of the Hippo pathway contributes to various diseases [15, 25, 26]. However, there is controversy and misunderstanding about the role of the Hippo pathway in the progression of cardiovascular disease, especially in cardiac fibrosis. Aharonov et al. revealed that the overexpression of ERBB2 in cardiomyocytes activated YAP and then led to heart regeneration by inducing an EMT-like regeneration response which improved cytoskeleton rebuilding and inhibited cell migration and ECM deposition [27]. In addition, it has been reported that the deletion of Lats1/2, which is upstream of YAP, in CFs results in the transition to myofibroblasts, which ultimately gives rise to spontaneous and self-sustaining fibrosis [28]. These findings confirmed the essential role of YAP1 in CFs during the course of cardiac fibrosis. Then, the question aries: does YAP1 exert the accordant function in all cell types of the heart? Del et al. found that ectopic expression of Rassf1A in cardiomyocytes promoted apoptosis and cardiac fibrosis by activating of Mst1 (an upstream regulator of YAP1), and cardiomyocyte-specific deletion of Rassf1A attenuated hypertrophy and fibrosis caused by TAC. Surprisingly, overexpression of Rassf1A in CFs repressed CF proliferation, whereas systemic ablation of Rassf1A aggravated TAC-induced hypertrophy and fibrosis [29]. These results implied that the effect of the Rassf1A/Mst1 pathway in cardiac hypertrophy and fibrosis is cell type dependent in the heart. Thus, as a downstream effector, we assumed that the action of YAP1 in the heart is also cell type dependent. Although several studies showed the protective role of YAP1 in the heart when YAP1 was overexpressed in cardiomyocytes [15, 26, 30], our results revealed that enhanced expression of YAP1 in CFs caused proliferation and transformation of fibroblasts, and then resulted in collagen deposition and cardiac fibrosis. Moreover, inhibition of YAP1 attenuated cardiac fibrosis and improved cardiac function in the hearts of mice after MI. Our results indicated that YAP1 might be a key factor in cardiac regeneration and fibrogenesis. At the beginning of ischemic injury, activation of YAP1 stimulates cardiomyocyte regeneration in cardiomyocytes and promotes wound repair and scar formation in CFs. However, continuous activation of YAP1 results in interstitial fibrosis because of excessive activation of fibroblasts and delayed termination of the scar repair. Overall, our study suggests that suppressing YAP1 in CFs could be a novel strategy for the treatment of myocardial infarction or heart failure, yet the precise effect of YAP1 in cardiac fibrosis should be done in future studies by constructing mice with fibroblast-specific gain- and loss-of-function of YAP1.
Although the Hippo pathway is involved in the progression of development, organ size regulation and diseases, upstream regulators that control the Hippo pathway remain elusive. Recently, Yu et al. found that GPCRs regulate the activation of the Hippo pathway. They proposed that Gs-coupled receptors stimulate Lats1/2 kinase, and then increase YAP/TAZ phosphorylation. However, G12/13-, Gq/11-, and Gi/o-coupled receptors inhibited Lats1/2 kinase and promoted the dephosphorylation and nuclear localization of YAP1 [16]. In this study, we unraveled, for the first time, the regulation of PAFR, which belongs to the GPCR family, on the Hippo pathway. We found that PAF stimulated the nuclear shuttling of unphosphorylated YAP1 by binding to PAFR, whereas YAP1 increased the expression of PAFR by binding to TEAD1 simultaneously. Our study elucidated the involvement of the positive feedback loop of PAFR/YAP1 in cardiac remodeling after MI. In response to stimuli, this loop was activated and promoted fibrogenesis in cultured CFs, which finally contributed to cardiac fibrosis and heart failure.
In summary, our study highlights that PAFR mediates PAF-induced cardiac fibrosis by promoting the nuclear translocation of YAP1. We revealed the involvement of the PAFR/YAP1 positive feedback loop in cardiac fibrosis. In a series of in vitro and in vivo studies, we revealed the role of the PAF/PAFR-YAP1 axis in cardiac fibrosis and illuminated the regulatory relationship between them. These findings indicate that interfering with this axis may be considered as a novel strategy for the prevention and treatment of cardiac fibrosis and the associated pathological processes.
References
Hinderer S, Schenke-Layland K. Cardiac fibrosis—a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. 2019;146:77–82.
Passaro F, Tocchetti CG, Spinetti G, Paudice F, Ambrosone L, Costagliola C, et al. Targeting fibrosis in the failing heart with nanoparticles. Adv Drug Deliv Rev. 2021;174:461–81.
Tao YK, Zhao SP, Yu PL, Shi J, Gu CD, Sun HT, et al. Elevated platelet activating factor level in ischemia-related arrhythmia and its electrophysiological effect on myocardium. Biomed Environ Sci. 2013;26:365–70.
Zheng GH, Xiong SQ, Mei LJ, Chen HY, Wang T, Chu JF. Elevated plasma platelet activating factor, platelet activating factor acetylhydrolase levels and risk of coronary heart disease or blood stasis syndrome of coronary heart disease in Chinese: a case control study: a case-control study. Inflammation. 2012;35:1419–28.
Penna C, Bassino E, Alloatti G. Platelet activating factor: the good and the bad in the ischemic/reperfused heart. Exp Biol Med. 2011;236:390–401.
Detopoulou P, Nomikos T, Fragopoulou E, Chrysohoou C, Antonopoulou S. Platelet activating factor in heart failure: potential role in disease progression and novel target for therapy. Curr Heart Fail Rep. 2013;10:122–9.
Detopoulou P, Nomikos T, Fragopoulou E, Antonopoulou S, Kotroyiannis I, Vassiliadou C, et al. Platelet activating factor (PAF) and activity of its biosynthetic and catabolic enzymes in blood and leukocytes of male patients with newly diagnosed heart failure. Clin Biochem. 2009;42:44–9.
Wang Y, Yu A, Yu FX. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell. 2017;8:349–59.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28.
Hong L, Li X, Zhou D, Geng J, Chen L. Role of Hippo signaling in regulating immunity. Cell Mol Immunol. 2018;15:1003–9.
Driskill JH, Pan D. The Hippo pathway in liver homeostasis and pathophysiology. Annu Rev Pathol. 2021;16:299–322.
Tamura T, Kodama T, Sato K, Murai K, Yoshioka T, Shigekawa M, et al. Dysregulation of PI3K and Hippo signaling pathways synergistically induces chronic pancreatitis via CTGF upregulation. J Clin Invest. 2021;131;e143414.
Li FL, Guan KL. The two sides of Hippo pathway in cancer. Semin Cancer Biol. 2021;S1044-579X(21)00200-5.
Wang J, Liu S, Heallen T, Martin JF. The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nat Rev Cardiol. 2018;15:672–84.
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA. 2013;110:13839–44.
Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–91.
Zhao X, He L, Li T, Lu Y, Miao Y, Liang S, et al. SRF expedites metastasis and modulates the epithelial to mesenchymal transition by regulating miR-199a-5p expression in human gastric cancer. Cell Death Differ. 2014;21:1900–13.
Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17.
Flinn MA, Link BA, O’Meara CC. Upstream regulation of the Hippo-Yap pathway in cardiomyocyte regeneration. Semin Cell Dev Biol. 2020;100:11–9.
Li Y, Feng J, Song S, Li H, Yang H, Zhou B, et al. gp130 controls cardiomyocyte proliferation and heart regeneration. Circulation. 2020;142:967–82.
Ma WY, Song RJ, Xu BB, Xu Y, Wang XX, Sun HY, et al. Melatonin promotes cardiomyocyte proliferation and heart repair in mice with myocardial infarction via miR-143-3p/Yap/Ctnnd1 signaling pathway. Acta Pharmacol Sin. 2021;42:921–31.
Yang Y, Nemoto EM, Harvey SA, Subbotin VM, Gandhi CR. Increased hepatic platelet activating factor (PAF) and PAF receptors in carbon tetrachloride induced liver cirrhosis. Gut. 2004;53:877–83.
Latchoumycandane C, Hanouneh M, Nagy LE, McIntyre TM. Inflammatory PAF receptor signaling initiates Hedgehog signaling and kidney fibrogenesis during ethanol consumption. PLoS ONE. 2015;10:e0145691.
Song M, Zhang H, Chen Z, Yang J, Li J, Shao S, et al. Shikonin reduces hepatic fibrosis by inducing apoptosis and inhibiting autophagy via the platelet-activating factor-mitogen-activated protein kinase axis. Exp Ther Med. 2021;21:28.
Yang S, Zhang L, Purohit V, Shukla SK, Chen X, Yu F, et al. Active YAP promotes pancreatic cancer cell motility, invasion and tumorigenesis in a mitotic phosphorylation-dependent manner through LPAR3. Oncotarget. 2015;6:36019–31.
Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ Res. 2014;115:354–63.
Aharonov A, Shakked A, Umansky KB, Savidor A, Genzelinakh A, Kain D, et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat Cell Biol. 2020;22:1346–56.
Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33:1491–505.
Del Re DP, Matsuda T, Zhai P, Gao S, Clark GJ, Van Der Weyden L, et al. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J Clin Invest. 2010;120:3555–67.
Del Re DP, Yang Y, Nakano N, Cho J, Zhai P, Yamamoto T, et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J Biol Chem. 2013;288:3977–88.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81900225, 82170299, 81870211, 81872863); the HMU Marshal Initiative Funding (HMUMIF-21023); the Major Scientific Fund Project of Heilongjiang Province (ZD2019H001); and the CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-078).
Author information
Authors and Affiliations
Contributions
HHL and HLS conceived the project, designed the experimental scheme and edited the manuscript. TYL, WS and LLL planned the experiments, composed the manuscript and managed experimental data. TYL, WS, XGZ, MQH, QZ, WHJ and XYS carried out cytological and molecular biological experiments, analyzed data and made charts. TYL, LLL, NY, JXG, XL, JZ were responsible for animals breeding, performed animal researches and executed data analysis. XGZ, JXG, JZ and NY assisted data analysis with constructive recommendation. YHZ and XLL gave suggestions on writing and revising the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Li, Ty., Su, W., Li, Ll. et al. Critical role of PAFR/YAP1 positive feedback loop in cardiac fibrosis. Acta Pharmacol Sin 43, 2862–2872 (2022). https://doi.org/10.1038/s41401-022-00903-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00903-9
Keywords
This article is cited by
-
Preventive effects of Ramelteon on bleomycin-induced pulmonary fibrosis in mice
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
-
The Hippo signalling pathway and its implications in human health and diseases
Signal Transduction and Targeted Therapy (2022)
