
Abstract
Heat shock protein 90 (Hsp90) is the most common molecular chaperone that controls the maturation of many oncoproteins critical in tumor development. Hsp90 has been considered as a promising target for cancer treatment, but the clinical significance of Hsp90 and the mechanisms of Hsp90 regulating the tumor-promoting effects in hepatocellular carcinoma (HCC) remain obscure. Previous studies have shown that curcumin, a polyphenol derived from the plant turmeric (Curcuma longa), inhibits tumor growth, which may provide an effective alternative therapy for HCC. Compared to curcumin, a novel derivative of curcumin, 3,5-(E)-Bis(3-methoxy-4-hydroxybenzal)-4-piperidinone hydrochloride (C0818) that is more potent in Hsp90 inhibition and antitumor activity. In this study, we investigated the effect of C0818 on HCC cells in vitro and its relation to Hsp90 inhibition. We showed that C0818 concentration-dependently inhibited the proliferation, the colony formation and induced apoptosis in HepG2 and Sk-Hep-1 cells. C0818 concentration-dependently inhibited DNA synthesis and induced G2/M phase arrest in HepG2 and Sk-Hep-1 cells. We further demonstrated that C0818 induced ROS- and caspase-dependent apoptosis in HCC cells through the mitochondrial-mediated pathway. C0818 induced the degradation of Hsp90 client proteins as RAS, C-Raf, P-C-Raf, Erk, P-ERK, MEK, P-MEK, Akt and P-Akt, which led to subsequent inhibition of the RAS/RAF/MEK/ERK and PI3K/AKT pathways. We revealed that C0818 could inhibit the binding of Hsp90 with its clients without affecting their transcription, which subsequently induced the degradation of Hsp90 clients by the proteasome rather than the lysosome. These results are of potential importance for elucidating a novel Hsp90 inhibitor targeting HCC.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related deaths worldwide and one of the most common malignancies in China [1, 2]. The most common options for treatment are liver transplantation and surgical resection; however, the incidence of HCC has expanded recently [3, 4]. Therefore, it is critical to identify novel therapeutic targets for HCC.
The highly conserved molecular chaperone Hsp90 is an ATP-dependent chaperone protein that controls the maturation, function and stability of various oncogenic proteins [5,6,7,8]. Hsp90 clients include Akt, C-Raf, Erk and epidermal growth factor receptor (EGFR), which play crucial roles in tumor proliferation, survival and signaling [9,10,11]. Hsp90 is overexpressed in cancer cells at levels 2–10-fold higher than those in normal cells [12, 13]. Thus, Hsp90 may be a critical protein associated with tumor growth and proliferation and an emerging target for cancer treatment [14, 15]. Therefore, Hsp90 inhibitors have considerable potential as antitumor agents. Inhibiting Hsp90 activity can promote the degradation of Hsp90 client proteins and trigger apoptosis in different cancer types [16, 17]. It was reported that Hsp90 is overexpressed in HCC [18]. However, it is still important to investigate the mechanisms by which Hsp90 promotes hepatocellular carcinoma and the efficacy of Hsp90 inhibitors against HCC.
Curcumin is a polyphenol derived from turmeric plants (Curcuma longa). Previous studies have revealed that curcumin has numerous pharmacological properties, including inflammatory, anticancer, and antioxidant activities [19,20,21]. It was reported that curcumin may be a lead compound for new types of Hsp90 inhibitors through the downregulation of Hsp90 clients and the co-chaperone p23 [22, 23]. Therefore, we synthesized a series of novel curcumin analogs that have been shown to exhibit lead-like activities and are more potent than curcumin in inhibiting Hsp90 and exerting anticancer effects [24,25,26,27].
C0818 [3,5-(E)-bis(3-methoxy-4-hydroxybenzal)-4-piperidinonehydrochloride], a novel structural derivative of curcumin, was synthesized in our laboratory. This analog contains the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore, which was reported to show excellent cytotoxic properties, particularly in the presence of secondary amino groups [28,29,30]. In our previous work, C0818 was shown to inhibit Hsp90 function by interacting with the C-terminal dimerization domain of Hsp90 and inhibiting its ATPase activity [31].
The current study investigated the antitumor effect of C0818 on human hepatocellular carcinoma cell lines in vitro. Our results demonstrated that C0818 significantly inhibited proliferation and induced G2/M phase arrest and apoptosis in HCC cells, which may be associated with C0818-induced proteasomal degradation of Hsp90 clients and downstream RAS/RAF/MEK/ERK and PI3K/AKT pathways. These findings indicate that C0818 is a potent Hsp90 inhibitor to protect against HCC.
Materials and methods
Regents and antibodies
C0818 was synthesized in our lab and prepared as a 20 mmol/L stock solution in dimethylsulfoxide (DMSO). Antibodies against Ras, C-Raf, P-C-Raf, Erk, P-Erk, Akt, P-Akt, Mek, P-Mek, β-Actin, Bax, Bcl-2, caspase-3, cleaved caspase-3, caspase-7, cleaved caspase-7, caspase-9, cleaved caspase-9, PARP, cleaved PARP, PI3K, mTOR, P-mTOR, Cdc2, P-cdc2, cyclin B1, Cdc25c and P21 were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against Hsp90 was purchased from Abcam (Cambridge, UK). All other chemical reagents were of analytical reagent grade.
Cell culture
Human HepG2 and Sk-Hep-1 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 25 µg/mL amphotericin B, 10,000 U/mL penicillin and 10,000 µg/mL streptomycin at 37 °C and 5% CO2 in a humidified incubator.
MTT assay
Cells were seeded in 96-well culture plates in triplicate at a density of 4 × 103/well and treated with vehicle (DMSO) or various doses of curcumin and C0818 for 48 h. To examine cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was used. In each well, 20 μL of (5 mg/mL) MTT solution was added and incubated for 4 h. The medium was then removed, and 150 μL of dimethylsulfoxide was used to dissolve the formazan crystals. Using a 96-well microplate reader, the optical density values were measured at 570 nm. To determine the inhibitory effect of curcumin and C0818 on cell growth, the half-maximal inhibitory concentration (IC50) was calculated.
Colony formation assay
Cells were seeded in 12-well plates (500/well) and treated with vehicle (DMSO) or various concentrations of C0818. C0818-containing medium was removed after 48 h, and the cells were allowed to grow in complete medium without C0818. After 14 days, the medium was removed, and the colonies were fixed for 20 min with methanol, stained for 30 min with 0.1% crystal violet and counted by light microscopy.
Apoptosis assay
To measure apoptosis, the Annexin-V: APC Apoptosis Detection Kit Ι (BD Biosciences) was used according to the manufacturer’s instructions. Briefly, cells were treated for 48 h with vehicle (DMSO) or different concentrations of C0818. Then, the cells were collected, centrifuged and washed once with phosphate-buffered saline (PBS). The cells were subsequently resuspended in 100 μL of binding buffer, stained for 20 min at room temperature in the dark with annexin V-APC and PI and analyzed by flow cytometry (BD FACSCanto TM II). FlowJo software was used to analyze the data.
EDU Click-iT proliferation assay
Cells were seeded in 6-well plates and treated for 48 h with vehicle (DMSO) or various concentrations of C0818. Then, the cells were stimulated for 12 h with fresh medium containing 20 μM EdU, harvested, fixed and permeabilized. The cells were then stained with the Click-iT Plus reaction cocktail and incubated for 30 min in the dark at room temperature. The cells were then washed with PBS and analyzed by flow cytometry.
Cell cycle analysis
Cells were treated for 48 h with vehicle (DMSO) or different concentrations of C0818. After that, the cells were harvested, resuspended in PBS, fixed with ice-cold 70% ethanol and stored at 4 °C overnight. The cells were then washed with PBS and incubated with 25 μg/mL RNase and propidium iodide (PI) for 30 min in the dark. Using flow cytometry, the percentage of cells in each cell cycle phase was measured.
Measurement of mitochondrial membrane potential (MMP)
Mitochondrial membrane potential (MMP) was determined using JC-1 assay kit (KeyGen Biotech). Cells were treated for 48 h with vehicle (DMSO) or various concentrations of C0818. The cells were then harvested, resuspended in staining solution containing JC-1 dye and incubated for 30 min in the dark at 37 °C. The cells were subsequently centrifuged, resuspended in PBS and analyzed by flow cytometry. A reduction in MMP is expressed as the percentage of JC-1 monomer fluorescence- expressing cells.
Detection of cellular reactive oxygen species (ROS)
Cells were treated for 48 h with vehicle (DMSO) or different concentrations of C0818 in the presence or absence of NAC (a ROS scavenger). Cells were treated with 50 μg/mL Rosup as a positive control for 2 h. The cells were then harvested, resuspended in culture medium (without FBS) containing 10 μM DCFH-DA (Beyotime) and incubated for 30 min in the dark at 37 °C. The relative ROS level was determined by flow cytometry.
Proteins extraction and Western blot analysis
Cells were harvested by trypsinization, washed with PBS, lysed with NP-40 lysis buffer (50 mmol/L Tris pH 8.0, 150 mmol/L NaCl, and 1% NP-40) on ice, and centrifuged at 12000 r/min for 15 min. The supernatants were collected, and the protein concentration was determined using a bicinchoninic acid (BCA) assay. Proteins (40 μg/lane) were separated on 10%, 12% or 15% SDS-polyacrylamide gels and transferred to 0.2 μm polyvinylidene fluoride membranes. The membranes were blocked for 2 h at room temperature with 5% skimmed milk in TBST [Tris-buffered saline (TBS) containing 0.1% Tween 20] and incubated overnight at 4 °C with specific antibodies. The membranes were washed three times in TBST for 5 min each and then incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 h. The signals were measured using an enhanced chemiluminescence detection kit and visualized using a ChemiDocTM imaging system. Anti-β-actin was used as a loading control. All Western blots analyses were repeated at least three times.
Small interfering RNA (siRNA) transfection
Cells were allowed to grow in antibiotic-free normal growth medium supplemented with FBS until they reached 50%–60% confluence. Control siRNA (sc-37007, Santa Cruz Biotechnology) and single siRNA oligonucleotides targeting human Hsp90α/β (sc-35608) were transfected according to the manufacturer’s protocol using siRNA transfection reagent (sc-29528). Six hours after transfection, the medium containing the transfection reagents was removed, and the cells were incubated in fresh medium for 48 h. The cells were then treated with vehicle (DMSO) or C0818 for 24 h. The cells were allowed to grow for an additional 72 h to determine cell proliferation, and the inhibition rate was calculated by comparing the cell number in the siRNA- treated group with that in the control group. Western blotting was used to confirm the knockdown of Hsp90.
Immunoprecipitation
HepG2 cells were treated for 12 h with vehicle (DMSO) or C0818 (8 μmol/L). The cells were then harvested, lysed in RIPA buffer and centrifuged for 15 min at 12,000 rpm. The protein concentration was determined using a BCA assay. Then, 400 μg of lysate was incubated with 2 μg of anti-HSP90 antibody overnight at 4 °C. After that, 20 μL of protein A Mag Sepharose™ (GE Healthcare, UK) was added to the mixture and incubated for 2 h at room temperature. The immunoprecipitated protein complexes were washed in lysis buffer five times, resuspended in SDS gel loading buffer and boiled for 5 min. The proteins were then separated by SDS–PAGE and transferred to polyvinylidene fluoride membranes. The membranes were incubated with anti-Hsp90, anti-C-Raf and anti-Akt antibodies overnight at 4 °C, washed in TBST and then incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h. The blots were detected by enhanced chemiluminescence.
Quantitative real-time PCR
Cells were treated for 48 h with vehicle (DMSO) or various concentrations of C0818. The cells were harvested, and total RNA was extracted from control and treated cells with TRIzol reagent (Life Technologies Corporation). The reverse transcription (RT) step was performed using GoScriptTM reverse transcriptase (Promega) according to the manufacturer’s protocol. Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) was carried out using the following primers: C-Raf (forward 5′-CACCTCCAGTCCCTCATCTG-3′ and reverse 5′-CTCAATCATCCTGCTGCTCA-3′), Akt (forward 5′-TTGAGAGAAGCCACGCTGT-3′ and reverse 5′-CGGAGAACAAACTGGATGAA-3′) and GAPDH (forward 5′-AGAAGGCTGGGGCTCATTTG-3′ and reverse 5′-AGGGGCCATCCACAGTCTTC-3′).
Statistical analysis
Using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA), all data were analyzed by one-way or two-way ANOVA for multiple comparisons. All experiments were performed in triplicate. The values are expressed as the mean ± standard deviation (SD). P < 0.05 was considered to be statistically significant.
Results
C0818 inhibits proliferation and induces apoptosis in HCC cells
Various concentrations of C0818 and curcumin were used to evaluate the antiproliferative effect on HCC cells using the MTT assay. The IC50 values of C0818 and curcumin at 48 h were 2.1 μmol/L and 19.3 μmol/L and were 1.9 μmol/L and 19 μmol/L for HepG2 and Sk-Hep-1 cells, respectively (Fig. 1a). Different in vitro experiments were performed based on these concentrations. The inhibition of colony formation by C0818 was observed in both HepG2 and Sk-Hep-1 cells in a dose-dependent manner, and a nearly complete inhibition in colony formation was observed at the highest dose of C0818 in both cell lines (Fig. 1b). To further explore whether C0818 could promote cell death through the induction of apoptosis in HCC, HepG2 and Sk-Hep-1 cells were used to investigate the effect of C0818 on apoptosis using annexin V–APC/PI staining. C0818 induced an increase in the percentages of both early and late apoptotic cells in a dose-dependent manner in both HepG2 and Sk-Hep-1 cells (Fig. 1c). Taken together, these findings suggested that C0818 could significantly reduce HCC cells viability by inhibiting the proliferation and induction of apoptosis and its antiproliferative effects were more potent than those of curcumin.

a The viability of HepG2 and Sk-Hep-1 cells was analyzed by MTT assay. b The results of the colony formation assay in HepG2 and Sk-Hep-1 cells are shown. c The percentages of apoptotic HepG2 and Sk-Hep-1 cells were analyzed by flow cytometry. The data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001 versus the control.
C0818 inhibits DNA synthesis and induces G2/M phase arrest in HCC cells
To further investigate the effect of C0818 on cell proliferation by targeting DNA synthesis, Click-iT EdU proliferation assay was carried out. C0818 significantly inhibited DNA synthesis in both HepG2 and Sk-Hep-1 cells in a dose-dependent manner (Fig. 2a). To analyze the stage at which C0818 induced HCC cells growth inhibition, we investigated the effects of C0818 on the cell cycle using flow cytometry. C0818 induced obvious G2/M phase arrest in a dose-dependent manner in both HepG2 and Sk-Hep-1 cells. The increase in percentage of cells in the G2/M phase was consistent with the decrease in the percentage of cells in both the S and G0/G1 phases (Fig. 2b). In addition, Western blot analysis was used to examine the expression levels of proteins regulating the cell cycle. The expression levels of Cdc2, P-Cdc2 (Thr161), Cyclin B1 and Cdc25c were downregulated, and P21 was upregulated by C0818 (Fig. 2c). These data indicated that C0818 promoted G2/M phase arrest by regulating cell cycle-related proteins.

a Cell proliferation was determined by EdU incorporation assay using flow cytometry. b The cell cycle distribution percentages were determined by flow cytometry. c The expression of cell cycle-regulated proteins was measured by Western blotting. The data are presented as the mean ± SD (n = 3). **P < 0.01 and ***P < 0.001 versus the control.
C0818 induces ROS and caspase-dependent apoptosis in HCC cells through the mitochondrial-mediated pathway
To analyze the molecular mechanism by which C0818 induces cell death, we examined the molecular changes associated with apoptosis. Caspases have been reported to be the central signaling proteins in various types of apoptosis [32]. Western blot analysis showed that C0818 induced an increase in the cleavage of caspase-3, -7, -8, and -9 and PARP in a dose- and time-dependent manner in both HepG2 and Sk-Hep-1 cells (Fig. 3a). To confirm the significance of caspase activation in C0818-induced cell death, HepG2 and Sk-Hep-1 cells were pretreated with the general caspase inhibitor z-VAD-fmk. The C0818-induced cleavage of PARP and caspase-3 was significantly inhibited by z-VAD-fmk (Fig. 3b). In addition, C0818-induced cell death and apoptosis were prevented by z-VAD-fmk (Fig. 3c, d). Previous studies have shown that Bcl-2 family proteins regulate the mitochondrial apoptotic pathway by controlling mitochondrial permeability [33, 34]. Moreover, mitochondria are the major source of ROS, which could determine the fate of cancer cells by regulating various signaling pathways, and excessive ROS production affects mitochondrial permeability and cytochrome c release [35,36,37]. We investigated whether mitochondrial function was affected by C0818. C0818 upregulated the Bax/Bcl-2 ratio in a dose- and time-dependent manner in both HepG2 and Sk-Hep-1 cells (Fig. 3e). We analyzed disruptions in MMP using JC-1 staining. After C0818 treatment of HepG2 and Sk-Hep-1 cells for 48 h, it was found that C0818 led to a significant reduction in MMP in a dose-dependent manner in both cell lines, as demonstrated by increased JC-1 fluorescence (Fig. 3f). DCFH-DA probe was used to detect intracellular ROS production in HepG2 and Sk-Hep-1 cells. After C0818 treatment for 48 h, flow cytometry showed a significant increase in ROS production in a dose-dependent manner in both cell lines (Fig. 4a). To confirm the role of ROS, HepG2 and Sk-Hep-1 cells were pretreated with the ROS scavenger N-Acetyl-L-cysteine (NAC) in the presence or absence of C0818 to inhibit the accumulation of ROS (Fig. 4b). The annexin V–APC/PI staining results showed that the percentage of apoptotic cells was significantly attenuated by pretreatment with NAC (Fig. 4c). In addition, changes in the expression levels of Bax, Bcl-2, cleaved PARP and cleaved caspase 9 were partially reversed by pretreatment with NAC (Fig. 4d). MTT assay was carried out to investigate cell viability, and C0818-induced cell growth inhibition was markedly decreased by pretreatment with NAC (Fig. 4e). These findings suggest that C0818 can upregulate the ratio of Bax/Bcl-2 and promote mitochondrial dysfunction, resulting in ROS production, MMP reduction, mitochondrial permeability enhancement, and caspase-dependent and mitochondrial-mediated apoptosis activation.

a The expression levels of apoptosis-related proteins were measured by Western blotting. b The expression levels of cleaved caspase-3 and cleaved PARP in cells pretreated with z-VAD-fmk were measured by Western blotting. c Quantification of viability (MTT) in cells pretreated with z-VAD-fmk. d Flow cytometric quantification of apoptosis (annexin V/PI staining) in cells pretreated with z-VAD-fmk. e The expression levels of Bax and Bcl-2 were measured by Western blotting. f The mitochondrial membrane potential was measured by flow cytometry. The data are presented as the mean ± SD (n = 3). **P < 0.01 and ***P < 0.001 versus the control, ##P < 0.01 and ###P < 0.001 versus C0818 treatment.

a Flow cytometric quantification of ROS levels in cells (Rosup as a positive control). b Flow cytometric quantification of ROS in cells pretreated with NAC. c Flow cytometric quantification of apoptosis (annexin V/PI staining) in cells pretreated with NAC. d The expression levels of Bax, Bcl-2, cleaved PARP and cleaved caspase-9 in cells pretreated with NAC were measured by Western blotting. e Cell viability was measured using MTT in cells pretreated with NAC. The data are presented as the mean ± SD (n = 3). **P < 0.01 and ***P < 0.001 versus the control. ##P < 0.01 and ###P < 0.001 versus C0818 treatment.
C0818 downregulates Hsp90 client proteins and the associated RAS/RAF/MEK/ERK and PI3K/AKT pathways in HCC cells
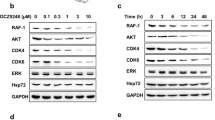
The RAS/RAF/MEK/ERK and PI3K/AKT pathways are known to play important roles in cancer cell transformation and the maintenance of malignant phenotype. Therefore, downregulation of Hsp90 client proteins, which are key molecules in these pathways, can inhibit the signal transduction of these pathways and produce antitumor effects. To evaluate the role of Hsp90 in HCC, HepG2 and Sk-Hep-1 cells were transfected with Hsp90 siRNA. Hsp90 knockdown in both cell lines was confirmed by Western blot analysis (Fig. 5a). Hsp90 knockdown significantly reduced the viability of HCC cells (Fig. 5b). Next, we measured the expression of Hsp90 client proteins after C0818 treatment by Western blot analysis. C0818 reduced the levels of RAS, C-Raf, P-C-Raf, Erk, P-ERK, MEK and P-MEK in a dose- and time-dependent manner in both HepG2 and Sk-Hep-1 cells (Fig. 5c). In addition, the levels of PI3K, AKT, P-AKT, mTOR and P-mTOR were also decreased in the same manner (Fig. 5d). These data suggest that C0818 downregulates Hsp90 client proteins and inhibits their associated pathways, including RAS/RAF/MEK/ERK and PI3K/AKT, which may explain C0818-mediated inhibition of HCC cells growth.

a SiRNA reduced the expression level of Hsp90 in HepG2 and Sk-Hep-1 cells. b Hsp90 knockdown reduced the viability of HepG2 and Sk-Hep-1 cells. c The expression levels of Hsp90 clients were measured by Western blotting. d The expression levels of PI3K, AKT, P-AKT, mTOR and P-mTOR were measured by Western blotting. The data are presented as the mean ± SD (n = 3). *P < 0.05 versus the control.
C0818 disrupts the molecular chaperone functions of Hsp90 in HCC cells
It was reported that both the proteasome and the lysosome are responsible for the degradation of Hsp90 client proteins [38, 39]. To examine the molecular mechanism underlying the reduction in Hsp90 client protein levels by C0818, we first examined whether C0818 treatment affected the stability of the protein C-Raf. HepG2 and Sk-Hep-1 cells were treated with the protein synthesis inhibitor cycloheximide (CHX), and the level of C-Raf was measured at the indicated times in the absence or presence of C0818. C0818 induced degradation of C-Raf much faster when new protein synthesis was inhibited by CHX in HepG2 and Sk-Hep-1 cells. These findings suggest that C0818 induced a reduction in C-Raf protein levels by enhancing C-Raf degradation instead of inhibiting protein synthesis (Fig. 6a). HepG2 and Sk-Hep-1 cells were used to investigate whether C0818 mediated Hsp90 client proteins degradation through the proteasome and/or the lysosome. Treatment with the proteasome inhibitor MG132 completely blocked the C0818-induced degradation of C-Raf and AKT (Fig. 6b). In addition, degradation was not affected by treatment with the lysosome inhibitor NH4Cl in either cell lines (Fig. 6c). Furthermore, after downregulating the expression of Hsp90 using siRNA, C0818 could not promote C-Raf or AKT protein degradation, suggesting that Hsp90 is a direct target of C0818 (Fig. 6d). Immunoprecipitation (IP) experiments were carried out to investigate the effects of C0818 on the chaperone functions of Hsp90. IP analysis showed that C0818 inhibited the binding of C-Raf and AKT with Hsp90 and induced proteasomal degradation of these clients (Fig. 6e). In addition, C0818 had no obvious effect on the transcription of Hsp90 client proteins such as C-Raf and AKT (Fig. 6f).

a C-Raf was analyzed by Western blotting in cells treated with cycloheximide in the absence or presence of C0818. b, c The expression levels of C-Raf and AKT proteins were measured by Western blotting in the cells treated with MG132 or NH4Cl. d C-Raf and AKT protein levels were analyzed by Western blotting in cells transfected with Hsp90 siRNA in the absence or presence of C0818. e Immunoprecipitation was used to evaluate the interaction of Hsp90 with its client proteins C-Raf and AKT. f C-Raf and AKT mRNA levels in HepG2 cells were measured by quantitative real-time PCR; GAPDH was used as an internal control. The results from three independent experiments are shown.
These data indicate that C0818 is a promising Hsp90 inhibitor that has the ability to disrupt the molecular chaperone functions of Hsp90, resulting in the degradation of Hsp90 client proteins.
Discussion
Liver cancer has become the most common malignancy in China. Due to the limitations of the current treatment options available, novel approaches are needed to lower the occurrence and mortality associated with liver cancer [40,41,42].
In cancer treatment, the molecular chaperone Hsp90 has emerged as an attractive therapeutic target. Hsp90 includes four homologs: HSP90α, HSP90β, glucose-regulated protein (Grp94) in the endoplasmic reticulum and mitochondrial tumor necrosis factor (TNF) receptor-associated protein 1 (Trap1) [43, 44]. Under stress conditions, the level of Hsp90 increases by ~2–10 -fold in cells, and its mRNA levels have been shown to be upregulated in different types of tumors [45]. Hsp90 plays a crucial role in numerous cellular functions and pathophysiological processes in tumors, including the folding, maturation and stability of various oncoproteins [46,47,48]. The mechanisms of anti-Hsp90 treatments include the degradation of Hsp90 clients, which activate multiple signaling pathways that regulate tumor growth and proliferation [11, 49].
It was reported that most Hsp90 inhibitors have been designed to inhibit Hsp90 chaperone functions by binding to Hsp90 [50]. The ansamycin antibiotic geldanamycin (GA) and its derivatives 17-allylamino-geldanamycin (17AAG) and 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) were first developed as Hsp90 inhibitors and evaluated in clinical trials. However, several drawbacks have been reported in clinical applications, including hepatotoxicity [8, 51]. Therefore, great effort has been directed to the identification of compounds with improved safety profiles.
Curcumin exerts protective effects against liver injury by improving hepatic functions [52, 53]. In addition, numerous studies have shown the curative effects of curcumin in HCC based on its potent anti-inflammatory and antioxidant properties [54, 55]. Therefore, many strategies have been created to improve the therapeutic efficacy of curcumin, including the design and synthesis of novel analogs. We synthesized the novel curcumin analoge 3,5-(E)-bis(3-methoxy-4-hydroxybenzal)-4-piperidinone hydrochloride (C0818). In our previous study, we demonstrated the ability of C0818 to interact with Hsp90 and its inhibitory effects on the ATPase activity of Hsp90 [31].
In this study, we investigated whether C0818 potently inhibits the proliferation of HCC cells by promoting apoptosis and cell cycle arrest via the degradation of Hsp90 client proteins. In addition, we observed that the curcumin derivative C0818 has more potent antiproliferative effects than curcumin on HCC cells.
It is now widely accepted that inhibiting Hsp90 triggers the degradation of Hsp90 client proteins that are critical for tumor growth and proliferation [56]. Hsp90 inhibitors can promote the degradation of numerous Hsp90 client proteins. Proteasome-dependent and lysosome-dependent mechanisms are involved in protein degradation [57, 58]. Both pathways were reported to promote the degradation of Hsp90 clients [38, 39]. In this study, we showed that C0818 induced the degradation of RAS, C-Raf, P-C-Raf, Erk, P-ERK, MEK and P-MEK in a proteasome-dependent and lysosome-independent manner in both HepG2 and Sk-Hep-1 cells. It was reported that both the RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways are interesting molecular targets in the treatment of HCC [59]. C0818 showed obvious effects on the PI3K/AKT and Raf/MEK/ERK signaling pathways by downregulating the levels of C-Raf, the key activator of MEK/ERK signaling, and AKT, the downstream target of PI3K. These results suggested that the mechanism of the antitumor effect of C0818 on HCC cells has been investigated at both the molecular and cellular levels.
Apoptosis is a well-known form of programmed cell death. Previous studies have reported that apoptosis is regulated through two main apoptotic pathways: the intrinsic or mitochondrial pathway and the extrinsic or death receptor pathway [60]. Hsp90 inhibitors can activate both pathways [61]. In the extrinsic apoptotic pathway, caspase-8 is first activated to trigger subsequent downstream cascade reactions. The mitochondria-mediated intrinsic pathway functions in response to diverse stimuli [62]. MMP induction and cytochrome c release promote the activation of molecules such as caspase-9, -3, -6 and -7 that in turn cleave nuclear proteins, such as PARP, and trigger apoptosis [63]. In this study, C0818 treatment induced mitochondrial dysfunction and a sharp reduction in MMP and led to the activation of caspase-3, caspase-8, and caspase-9 and cleaved-PARP. Taken together, these findings demonstrate that C0818 can promote apoptosis by both the extrinsic and intrinsic pathways in HCC cells.
Bcl-2 family proteins play a critical role in the regulation of mitochondrial outer membrane permeability. The Bcl-2 protein family is composed of different pro- and antiapoptotic molecules that form heterodimers to activate or inhibit each other [64]. Antiapoptotic members, including Bcl-2 and Bcl-xL, are often overexpressed in many tumors and induce protection against apoptotic stimuli. The proapoptotic members Bax, Bid, Bad and Bak promote the reduction in MMP to facilitate the release of cytochrome c into the cytosol, caspase activation, and ultimately apoptosis [65]. A variety of chemotherapeutics induce mitochondria-mediated apoptosis in susceptible cells through the upregulation of Bax/Bid/Bad and/or the downregulation of Bcl-2/Bcl-xL. In this study, C0818 treatment induced significant Bax upregulation and Bcl-2 downregulation. These findings suggest that C0818 promotes mitochondrial-mediated apoptosis in HCC cells via the upregulation of the Bax/Bcl-2 ratio. Multiple stimuli, including mitochondrial dysfunction and DNA damage trigger an increase in ROS levels, which are involved in the regulation of apoptosis [66]. Increasing ROS levels have been reported to induce cell death in HCC cells [67]. In the present study, it was demonstrated that C0818 markedly increased ROS production in HCC cells and that the accumulation of ROS triggered apoptosis in C0818-treated cells.
In conclusion, C0818 inhibits proliferation of HCC cells by inhibiting the PI3K/AKT and RAS/RAF/MEK/ERK signaling pathways in HCC cells. C0818 promotes the degradation of Hsp90 client proteins, including RAS, C-Raf, P-C-Raf, Erk, P-ERK, MEK and P-MEK, via a proteasome-dependent pathway.
Additionally, C0818 potentiates both the mitochondrial and death receptor pathways of apoptosis and promotes Bax upregulation, Bcl-2 downregulation, activation of caspase-8 and cleavage of caspase-3, caspase-7, caspase-9 and PARP. The loss of MMP and excessive ROS production are important in inhibiting the growth and proliferation of HCC cells. Our findings provide a basis for further development of C0818 as a promising Hsp90-targeted therapy in the treatment of HCC.
References
Tang ZY. Hepatocellular carcinoma–cause, treatment and metastasis. World J Gastroenterol. 2001;7:445–54.
Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108.
Bartlett A, Heaton N. Hepatocellular carcinoma: defining the place of surgery in an era of organ shortage. World J Gastroenterol. 2008;14:4445–53.
Xu Q, Liu X, Zheng X, Yao Y, Wang M, Liu Q. The transcriptional activity of Gli1 is negatively regulated by AMPK through Hedgehog partial agonism in hepatocellular carcinoma. Int J Mol Med. 2014;34:733–41.
Esfahani K, Cohen V. HSP90 as a novel molecular target in non-small-cell lung cancer. Lung Cancer. 2016;7:11–7.
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72.
Richter K, Hendershot LM, Freeman BC. The cellular world according to Hsp90. Nat Struct Mol Biol. 2007;14:90–4.
Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49.
Pearl LH, Prodromou C. Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv Protein Chem. 2001;59:157–86.
Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin Drug Discov. 2011;6:559–87.
Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med. 2004;82:488–99.
Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–7.
Chiosis G, Neckers L. Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol. 2006;1:279–84.
Neckers L, Mimnaugh E, Schulte TW. Hsp90 as an anti-cancer target. Drug Resist Updat. 1999;2:165–72.
Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–30.
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10.
Hertlein E, Wagner AJ, Jones J, Lin TS, Maddocks KJ, Towns WH, et al. 17-DMAG targets the nuclear factor-kappaB family of proteins to induce apoptosis in chronic lymphocytic leukemia: clinical implications of HSP90 inhibition. Blood. 2010;116:45–53.
Pascale RM, Simile MM, Calvisi DF, Frau M, Muroni MR, Seddaiu MA, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology. 2005;42:1310–9.
Joe B, Vijaykumar M, Lokesh BR. Biological properties of curcumin-cellular and molecular mechanisms of action. Crit Rev Food Sci Nut. 2004;44:97–111.
Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res. 2003;23:363–98.
Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225.
Wu LX, Xu JH, Huang XW, Zhang KZ, Wen CX, Chen YZ. Down-regulation of p210(bcr/abl) by curcumin involves disrupting molecular chaperone functions of Hsp90. Acta Pharmacol Sin. 2006;27:694–9.
Jung Y, Xu W, Kim H, Ha N, Neckers L. Curcumin-induced degradation of ErbB2: a role for the E3 ubiquitin ligase CHIP and the Michael reaction acceptor activity of curcumin. Biochim Biophys Acta. 2007;1773:383–90.
Fan YJ, Zhou YX, Zhang LR, Lin QF, Gao PZ, Cai F, et al. C1206, a novel curcumin derivative, potently inhibits Hsp90 and human chronic myeloid leukemia cells in vitro. Acta Pharmacol Sin. 2018;39:649–58.
Ye M, Huang W, Wu WW, Liu Y, Ye SN, Xu JH. FM807, a curcumin analogue, shows potent antitumor effects in nasopharyngeal carcinoma cells by heat shock protein 90 inhibition. Oncotarget. 2017;8:15364–76.
Chen C, Liu Y, Chen Y, Xu J. C086, a novel analog of curcumin, induces growth inhibition and down-regulation of NFκB in colon cancer cells and xenograft tumors. Cancer Biol Ther. 2011;12:797–807.
Wu L, Yu J, Chen R, Liu Y, Lou L, Wu Y, et al. Dual inhibition of Bcr-Abl and Hsp90 by C086 potently inhibits the proliferation of imatinib-resistant CML cells. Clin Cancer Res. 2015;21:833–43.
Das U, Sharma RK, Dimmock JR. 1,5-diaryl-3-oxo-1,4-pentadienes: a case for antineoplastics with multiple targets. Curr Med Chem. 2009;16:2001–20.
Dimmock JR, Padmanilayam MP, Puthucode RN, Nazarali AJ, Motaganahalli NL, Zello GA, et al. A conformational and structure-activity relationship study of cytotoxic 3,5-bis(arylidene)-4-piperidones and related N-acryloyl analogues. J Med Chem. 2001;44:586–93.
Dimmock JR, Arora VK, Wonko SL, Hamon NW, Quail JW, Jia Z, et al. 3,5-Bis-benzylidene-4-piperidones and related compounds with high activity towards P388 leukemia cells. Drug Des Deliv. 1990;6:183–94.
Fan Y, Liu Y, Zhang L, Cai F, Zhu L, Xu J. C0818, a novel curcumin derivative, interacts with Hsp90 and inhibits Hsp90 ATPase activity. Acta Pharm Sin B. 2017;7:91–6.
Utz PJ, Anderson P. Life and death decisions: regulation of apoptosis by proteolysis of signaling molecules. Cell Death Differ. 2000;7:589–602.
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–32.
Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65.
Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–90.
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62.
Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J Biol Chem. 2005;280:18558–61.
Shen S, Zhang P, Lovchik MA, Li Y, Tang L, Chen Z, et al. Cyclodepsipeptide toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J Cell Biol. 2009;185:629–39.
Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 2006;16:895–901.
Gu D, Kelly TN, Wu X, Chen J, Samet JM, Huang JF, et al. Mortality attributable to smoking in China. N Engl J Med. 2009;360:150–9.
Okuno M, Kojima S, Moriwaki H. Chemoprevention of hepatocellular carcinoma: concept, progress and perspectives. J Gastroenterol Hepatol. 2001;16:1329–35.
Kensler TW, Qian GS, Chen JG, Groopman JD. Translational strategies for cancer prevention in liver. Nat Rev Cancer. 2003;3:321–9.
Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28.
Sreedhar AS, Kalmár E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562:11–5.
Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann NY Acad Sci. 2007;1113:202–16.
Falsone SF, Gesslbauer B, Tirk F, Piccinini AM, Kungl AJ. A proteomic snapshot of the human heat shock protein 90 interactome. FEBS Lett. 2005;579:6350–4.
Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10:283–90.
Zhao R, Davey M, Hsu YC, Kaplanek P, Tong A, Parsons AB, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–27.
Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, et al. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem. 2009;9:1479–92.
Neckers L. Development of small molecule Hsp90 inhibitors: utilizing both forward and reverse chemical genomics for drug identification. Curr Med Chem. 2003;10:733–9.
Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–15.
Rivera-Espinoza Y, Muriel P. Pharmacological actions of curcumin in liver diseases or damage. Liver Int. 2009;29:1457–66.
Shao W, Yu Z, Chiang Y, Yang Y, Chai T, Foltz W, et al. Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS One. 2012;7:e28784.
Wang WZ, Cheng J, Luo J, Zhuang SM. Abrogation of G2/M arrest sensitizes curcumin-resistant hepatoma cells to apoptosis. FEBS Lett. 2008;582:2689–95.
Cao J, Jia L, Zhou HM, Liu Y, Zhong LF. Mitochondrial and nuclear DNA damage induced by curcumin in human hepatoma G2 cells. Toxicol Sci. 2006;91:476–83.
Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–53.
Mortimore GE, Pösö AR. Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr. 1987;7:539–64.
Fuertes G, Villarroya A, Knecht E. Role of proteasomes in the degradation of short-lived proteins in human fibroblasts under various growth conditions. Int J Biochem Cell Biol. 2003;35:651–64.
Dimri M, Humphries A, Laknaur A, Elattar S, Lee TJ, Sharma A, et al. NAD(P)H quinone dehydrogenase 1 ablation inhibits activation of the phosphoinositide 3-kinase/Akt serine/threonine kinase and mitogen-activated protein kinase/extracellular signal-regulated kinase pathways and blocks metabolic adaptation in hepatocellular carcinoma. Hepatology. 2020;71:549–68.
Matthews GM, Newbold A, Johnstone RW. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv Cancer Res. 2012;116:165–97.
Kim YJ, Lee SA, Myung SC, Kim W, Lee CS. Radicicol, an inhibitor of Hsp90, enhances TRAIL-induced apoptosis in human epithelial ovarian carcinoma cells by promoting activation of apoptosis-related proteins. Mol Cell Biochem. 2012;359:33–43.
Liang H, Salinas RA, Leal BZ, Kosakowska-Cholody T, Michejda CJ, Waters SJ, et al. Caspase-mediated apoptosis and caspase-independent cell death induced by irofulven in prostate cancer cells. Mol Cancer Ther. 2004;3:1385–96.
Sánchez-Alcázar JA, Khodjakov A, Schneider E. Anticancer drugs induce increased mitochondrial cytochrome c expression that precedes cell death. Cancer Res. 2001;61:1038–44.
Rudner J, Elsaesser SJ, Müller AC, Belka C, Jendrossek V. Differential effects of anti-apoptotic Bcl-2 family members Mcl-1, Bcl-2, and Bcl-xL on celecoxib-induced apoptosis. Biochem Pharmacol. 2010;79:10–20.
Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–607.
Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schüssel K, et al. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer’s disease. Biochem Pharmacol. 2003;66:1627–34.
Wang B, Zhou TY, Nie CH, Wan DL, Zheng SS. Bigelovin, a sesquiterpene lactone, suppresses tumor growth through inducing apoptosis and autophagy via the inhibition of mTOR pathway regulated by ROS generation in liver cancer. Biochem Biophys Res Commun. 2018;499:156–63.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (81973364), the Joint Funds for the Innovation of Science and Technology, Fujian province, China (2019Y9131), the external cooperation project of Fujian Provincial Department of Science and Technology (2020I0016) and the National Science and Technology Foundation of China for Key Projects of “Major New Drugs Innovation and Development” (2012ZX09103-101-028).
Author information
Authors and Affiliations
Contributions
JHX, SNY provided funding, designed the research and revised the paper; AAAA performed the research and wrote the paper; PZ, WL and YJF performed a part of the research and provided technical support.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abdelmoaty, A.A.A., Zhang, P., Lin, W. et al. C0818, a novel curcumin derivative, induces ROS-dependent cytotoxicity in human hepatocellular carcinoma cells in vitro via disruption of Hsp90 function. Acta Pharmacol Sin 43, 446–456 (2022). https://doi.org/10.1038/s41401-021-00642-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00642-3
Keywords
This article is cited by
-
HSP90 mediates the connection of multiple programmed cell death in diseases
Cell Death & Disease (2022)
