
Abstract
Increasing evidence suggests that the use of potent neuroprotective agents featured with novel pharmacological mechanism would offer a promising strategy to delay or prevent the progression of neurodegeneration. Here, we provide the first demonstration that the chiral nonracemic isochroman-2H-chromene conjugate JE-133, a novel synthetic 1,3-disubstituted isochroman derivative, possesses superior neuroprotective effect against oxidative injuries. Pretreatment with JE-133 (1–10 μM) concentration-dependently prevented H2O2-induced cell death in SH-SY5Y neuroblastoma cells and rat primary cortical neurons. Pretreatment with JE-133 significantly alleviated H2O2-induced apoptotic changes. These protective effects could not be simply attributed to the direct free radical scavenging as JE-133 had moderate activity in reducing DPPH free radical. Further study revealed that pretreatment with JE-133 (10 μM) significantly decreased the phosphorylation of MAPK pathway proteins, especially ERK and P38, in the neuronal cells. In addition, blocking PI3K/Akt pathway using LY294002 partially counteracted the cell viability-enhancing effect of JE-133. We conclude that JE-133 exerts neuroprotection associated with dual regulative mechanisms and consequently activating cell survival and inhibiting apoptotic changes, which may provide important clues for the development of effective neuroprotective drug lead/candidate.
Similar content being viewed by others


Introduction
In recent years, the utilization of antioxidants in the treatment of Alzheimer’s disease (AD) has been a popular research topic [1,2,3]. Oxidative stress results from the accumulation of reactive oxygen species (ROS) or an excess of free radicals and is involved in multiple pathological processes of neurodegenerative diseases, including AD [4,5,6]. As a consequence, a series of clinical trials have been conducted with the aim of treating or preventing AD using antioxidants [7, 8], such as a mixture of vitamin C and vitamin E [9], the natural polyphenolic compound curcumin [10] and Ginkgo biloba tree leaf extract (EGb761) [11]. Many of the abovementioned compounds have shown promising antioxidative and cognitive enhancing effects in animal experiments; however, there is currently no curative therapy for AD [12, 13]. Hence, the discovery of new lead compounds with novel antioxidative pharmacological mechanisms is promising for achieving better therapeutic benefits.
Antioxidative interventions aimed at directly scavenging free radicals have not yet exhibited satisfactory clinical efficacy. Alternatively, accumulating studies have indicated that various small molecules can significantly ameliorate neuronal oxidative damage by activating prosurvival pathways or inhibiting the pathogenic pathways [14,15,16,17]. Evidence has shown that many complex signaling pathways are involved in the process of oxidative stress, with the mitogen-activated protein kinase (MAPK) [18, 19] and phosphatidylinositol 3-kinase (PI3K)/Akt [16, 20, 21] pathways being of great importance. The activation of the MAPK signaling pathway, including the phosphorylated proteins c-Jun N-terminal kinase (JNK), P38 and extracellular signal-regulated kinase 1/2 (ERK1/2), triggers the cleavage of apoptosis-related proteins, such as caspase-3, and consequently leads to apoptosis [18, 22,23,24]. In contrast, the PI3K/Akt pathway is critical for neuronal survival through the promotion of cell survival and the inhibition of apoptosis induced by deleterious stimuli [20, 21, 25,26,27]. The above findings provide an alternative and promising therapeutic strategy for resisting oxidative insults.
Our laboratory is engaged in identifying potent antioxidants through cell-based phenotypic screening [28, 29], which is believed to well simulate complex pathologic cellular environments [30] of interacting proteins and signaling pathways and to possess distinct advantages over single molecular target-based screening for drug discovery and development [30,31,32]. Meanwhile, this method maintains the capacity for high-throughput screening (HTS) [30]. Employing cell-based phenotypic screening, we discovered that the chiral nonracemic isochroman-2H-chromene conjugate JE-133 (structure shown in Fig. 1a), a novel 1,3-disubstituted isochroman derivative, shows potent antioxidative effects on SH-SY5Y neuroblastoma cells. To better understand the protective potential of JE-133, the present study was designed to further verify the antioxidant effects of JE-133 in SH-SY5Y cells and rat primary cortical neurons, and to elucidate whether the MAPK and PI3K/Akt signaling pathways are involved in the abovementioned neuroprotective effects.

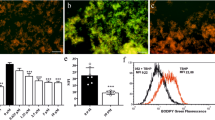
Synthesis and stereochemical analysis of the compound JE-133. a Synthesis of the compound JE-133. b Twenty computed low-energy overlapping solution conformers (level of DFT optimization: CAM-B3LYP/TZVP PCM/MeCN) of (1R,3R,2’S)-JE-133 with an overall Boltzmann population of 87.4%. c Experimental ECD spectrum of JE-133 (black curve) compared with the Boltzmann-weighted PBE0/TZVP (PCM/MeCN) ECD spectrum (red curve) of (1R,3R,2’S)-JE-133 computed for the B97D/TZVP PCM/MeCN conformers. The bars represent the rotational strengths of the lowest-energy conformer
Materials and methods
Preparation of compounds
JE-133 and N-acetylcysteine (NAC, Sigma-Aldrich, St. Louis, MO, USA) were dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM to generate stock solutions, stored at −20 °C and diluted with culture medium before use.
SH-SY5Y cell culture
Human SH-SY5Y neuroblastoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). SH-SY5Y cells were cultured in MEM/F12 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 60 mg/L penicillin and 50 mg/L streptomycin in a humid atmosphere of 5% CO2 at 37 °C. SH-SY5Y cells were seeded into 96- or 6-well plates at a density of 3 × 105 cells/mL.
Primary cortical neuron culture
Primary cortical neuron cultures were prepared from 17-day-old embryos of Sprague-Dawley rats as previously described [33]. Briefly, the cortices were rapidly dissected and minced on ice and then dissociated in 0.125% trypsin with 0.2 mg/mL DNase I at 37 °C for 15 min. Next, the tissues were triturated with a pipette in HG-DMEM medium with 10% FBS and filtered through sieves (300/400 mesh). The cells were plated in plates coated with poly-L-lysine (Sangon Biotech, Shanghai, China). The culture medium was replaced with neurobasal medium (Invitrogen, Carlsbad, CA, USA) containing 0.5 mM L-glutamine, 2% B27 supplement, penicillin (60 mg/L) and streptomycin (50 mg/L) after 4 h. Half of the culture medium was refreshed every 3 days. The neurons were allowed to grow for 9 days before further treatment.
Cell viability measurement
Morphological changes in SH-SY5Y cells and primary cortical neurons were observed with an inverted optical microscope (Nikon TE2000, Melville, NY, USA), and cell viability was evaluated by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT, Sigma-Aldrich) assay. Briefly, SH-SY5Y cells or primary cortical neurons (3 × 104 cells/well in 96-well plates) were incubated with 100 μM or 300 μM H2O2 at 37 °C for 24 h after being pretreated with or without JE-133 (or NAC) and then incubated with MTT at a final concentration of 0.5 mg/mL for 3 h. The medium was removed, and 100 μL DMSO was added. The cell plates were shaken for 5 min, and the absorbance of each well was read at 490 nm using a microplate reader (DTX 800 Multimode Detector, Beckman Coulter, Fullerton, CA). The optical density (OD) values of all groups were normalized to the values of the control group, and cell viability is presented as the percentage of surviving cells relative to that in the control group.
Apoptosis assay
Apoptosis was detected by Annexin V-FITC and PI staining following the procedures described in the apoptosis detection kit (Rainbio, Shanghai, China). Annexin V is used to detect early apoptotic cells, and propidium iodide (PI) can penetrate into the nuclei of cells undergoing late-stage apoptosis, while intact cells cannot be stained by Annexin V or PI. SH-SY5Y cells (6 × 105 cells/well in 6-well plates) were incubated with 100 μM H2O2 for 24 h after being pretreated with or without JE-133 (or NAC). The cells were then collected, suspended in 500 μL binding buffer, and stained with 5 μL Annexin V-FITC and 5 μL PI. After 15 min of reaction in the dark, the apoptosis rates of the different groups (10,000 cells for each group) were analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA). The FL-1 and FL-3 channels were chosen for the detection of Annexin V-FITC- and PI-labeled cells, respectively.
DPPH free radical scavenging assay
The free radical scavenging ability of JE-133 was analyzed using stable 1,1-diphenyl-2-picrylhydrazyl (DPPH, Sigma-Aldrich) dissolved in 95% ethanol. Briefly, various concentrations of JE-133 (or NAC) in 20 μL solution were added to 180 μL DPPH radical solution (80 μM) and incubated for 60 min in the dark at room temperature. The absorbance of the mixture was measured with a microplate reader (DTX 800 Multimode Detector, Beckman Coulter, Fullerton, CA) at 517 nm. The absorbance value was then converted to the DPPH free radical scavenging rate according to the following equation: DPPH scavenging rate (IP%) = [1−(Ai−Aj)/Ac] × 100%, with Ai representing the absorbance value of the compounds upon reaction with DPPH; Aj representing the absorbance value of the compounds upon reaction with 95% ethanol; and Ac representing the absorbance value of DPPH solution.
Western blot analysis
After exposure to 100 μM (SH-SY5Y cells) or 300 μM (primary neurons) H2O2 for 15 min (6 h or 24 h) following pretreatment with or without JE-133 (or LY294002, purchased from Selleck, Shanghai, China), the cells were lysed in RIPA buffer [50 mM Tris 50, 150 mM NaCl, 0.5% (v/v) sodium-deoxycholate, 1% (v/v) Triton X-100, 0.1% (v/v) SDS 0.1%, and 2 mM EDTA, pH 7.4] plus protease inhibitor cocktail (Sigma-Aldrich) for 30 min on ice and then centrifuged at 12,000 × g for 15 min at 4 °C. The supernatants were collected, and the protein concentrations were determined using a BCA assay kit (Pierce, Rockford, IL, USA). The proteins were separated by 10% or 12% SDS-PAGE and transferred to 0.2-μm nitrocellulose membranes. After blocking with 5% nonfat milk for 1 h at room temperature, the membranes were incubated with the following primary antibodies at 4 °C overnight: anti-cleaved caspase-3 (1:500, Cell Signaling, Beverly, MA, USA); anti-caspase-3 (1:2000, Cell Signaling); anti-p-Akt (1:1000, Cell Signaling); anti-Akt (1:3000, Cell Signaling); anti-p-JNK (1:800, Cell Signaling); anti-JNK (1:1000, Cell Signaling); anti-p-P38 (1:1000, Cell Signaling); anti-P38 (1:2000, Cell Signaling); anti-p-ERK1/2 (1:1000, Cell Signaling); anti-ERK1/2 (1:2000, Cell Signaling); and anti-β-Actin (1:10,0000, Sigma-Aldrich). Then, they were incubated with HRP-conjugated secondary antibodies (1:5,000; Kangcheng, Shanghai, China) for 2 h at room temperature. The bands were visualized by autoradiography after development by an ECL plus kit (Millipore, Billerica, MA, USA), and the intensity of each band was quantified with ImageJ software.
Statistical analysis
All data are presented as the mean ± SEM and were analyzed by Student’s t-test for single comparisons or by one-way ANOVA followed by Dunnett’s test for multiple comparisons. P-values below 0.05 were considered statistically significant.
Results
Synthesis and stereochemical analysis of JE-133
JE-133 was obtained by the diastereoselective oxa-Pictet-Spengler cyclization of 4-[(R)-2-hydroxypropyl]-benzene-1,2-diol (3) with ethyl (2S)-3-formyl-6-methoxy-2H-chromene-2-carboxylate (4) (Fig. 1a). Optically active 3 [34] was prepared in two steps; 1,2-bis-(benzyloxy)-4-bromobenzene (1) was lithiated with n-butyllithium and reacted with (R)-propylene oxide in the presence of BF3·OEt2, and then the benzyl protective groups were removed by catalytic hydrogenation (2 → 3). Optically active 4 was obtained from 2-hydroxy-5-methoxybenzaldehyde through an enantioselective organocatalytic oxa-Michael/aldol condensation domino reaction according to the procedure of Córdova et al. [35] with 93% enantiomeric excess. The oxa-Pictet-Spengler cyclization of 3 with 4 retained the (R) and (S) absolute configuration of the reactants, and a new chirality center was established at C-1 of the isochroman unit diastereoselectively with (R) absolute configuration. The (1R,3R) absolute configuration of the isochroman moiety afforded the low-energy equatorial arrangement of the C-1 and C-3 substituents, which was confirmed by the NOE correlation between the axial 1-H and 3-H oriented toward the same face of the molecule.
Since the three-dimensional shape of chiral nonracemic JE-133 is believed to be an important factor for its activity, the solution conformation and absolute configuration of JE-133 were studied by electronic circular dichroism (ECD) measurements supported by the solution time-dependent density functional theory-electronic circular dichroism (TDDFT-ECD) calculation protocol [36, 37]. In all the computed low-energy conformers, the isochroman and 2H-chromene subunits had the same well-defined relative arrangement, even though they were attached by a rotatable single bond (Fig. 1b). The C-1 and C-3 substituents adopted equatorial orientation, while the C-2′ ethoxycarbonyl group had axial orientation. The Boltzmann-weighted computed ECD spectra gave good agreement with the experimental ECD spectrum of (1R,3R,2′S)-JE-133, which confirmed the (1R,3R,2’S) absolute configuration and determined the preferred solution conformation.
The best agreement was produced by the PBE0/TZVP PCM/MeCN ECD computed for the CAM-B3LYP/TZVP PCM/MeCN conformers, which is shown in Fig. 1c.
The spectroscopic characterization and additional stereochemical results are shown in supplementary Figs. S1 –S7.
JE-133 attenuated H2O2-induced neurotoxicity
SH-SY5Y cells exposed to H2O2 for 24 h exhibited morphological cell damage characterized by dendrite fragmentation, body swelling and decreased cell number (Fig. 2a). In contrast, the cell morphological damage induced by H2O2 was markedly attenuated by pretreatment with 10 μM JE-133 (Fig. 2a). Consistent with the morphological observations, H2O2-induced cytotoxicity was also confirmed by a significant reduction in values in the MTT assay (50.62% ± 3.23%, P < 0.001) (Fig. 2b) and excessive release of LDH (34.28% ± 1.84%, P < 0.001) (supplementary Fig. S8) compared to those of the control group. However, pretreatment with JE-133 markedly attenuated cytotoxicity induced by H2O2 in a concentration-dependent manner, with the maximum protective effect at a concentration of 10 μM (cell viability restored to 95.88% ± 3.06%, P < 0.001 vs the H2O2 group) (Fig. 2b). Similarly, in cells pretreated with 3 and 10 μM JE-133, intracellular LDH release was significantly inhibited (25.25% ± 2.37%, P < 0.05; 12.97% ± 1.44%, P < 0.001, respectively) (Supplementary Fig. S8). The antioxidant NAC, the positive control, significantly attenuated the decline in cell viability in the MTT and LDH assays (78.25% ± 2.14%, P < 0.001; 16.21% ± 2.48%, P < 0.001, respectively) (Fig. 2b and supplementary Fig. S8). To verify whether the neuroprotective effects of JE-133 against H2O2 are limited to SH-SY5Y cells, we also evaluated its protective effects on primary cultured cortical neurons. Similar as in SH-SY5Y cells, JE-133 markedly attenuated H2O2-induced cell morphological damage (Fig. 2c) and alleviated the H2O2-induced reduction in cell viability in a concentration-dependent manner in primary cultured cortical neurons (Fig. 2d).

Effects of JE-133 on H2O2-induced cytotoxicity in neuronal cells. Cells were pretreated with different concentrations of JE-133 (1, 3 or 10 μM) or NAC (100 μM) for 2 h and then exposed to H2O2 for 24 h. The morphology of SH-SY5Y cells (a) and primary cortical neurons (c) observed under a microscope at 100× magnification after different treatments. Scale bar = 100 μm. The viability of SH-SY5Y cells (b) and primary cortical neurons (d) was determined using the MTT assay, n = 4. The data are presented as the mean ± SEM. ###P < 0.001 vs the control group; *P < 0.05, **P < 0.01, ***P < 0.001 vs the H2O2 group
JE-133 pretreatment protected SH-SY5Y cells against H2O2-induced apoptosis
To explore whether the neuroprotective effects of JE-133 in SH-SY5Y cells are associated with the amelioration of apoptotic changes, flow cytometry with Annexin V-FITC and PI double staining was performed. SH-SY5Y cells in the upper and lower right quadrants represented cells that underwent the late and early stages of apoptosis, respectively. As shown in Fig. 3a, b, after exposure to H2O2 for 24 h, the percentage of SH-SY5Y cells in the early and late stages of apoptosis was greatly increased from 24.45% ± 3.89% to 49.25% ± 1.60% (P < 0.01 vs the control group). However, pretreatment with 3 and 10 μM JE-133 suppressed the apoptosis ratio to 30.83% ± 4.35% (P < 0.05 vs the H2O2 group) and 25.79% ± 5.13% (P < 0.01 vs the H2O2 group), respectively. Similarly, 100 μM NAC significantly decreased the rate of apoptosis to 29.12% ± 5.46% (P < 0.05 vs the H2O2 group). The cleavage of caspase-3 was examined by Western blot analysis. As shown in Fig. 3c, H2O2 caused a robust increase in the level of cleaved caspase-3 compared to that in the control group (9.86 ± 1.09-fold of the control group, P < 0.01). In contrast, the activation of caspase-3 was markedly alleviated in SH-SY5Y cells upon preincubation with JE-133 (10 μM) for 2 h followed by exposure to H2O2 for 24 h, as shown by a significant reduction in cleaved caspase-3 levels (1.60 ± 0.26-fold of the control group, P < 0.01 vs the H2O2 group) (Fig. 3c).

Effects of JE-133 on H2O2-induced apoptosis in SH-SY5Y cells and on free radical scavenging. Cells were pretreated with various concentrations of JE-133 or 100 μM NAC for 2 h and then exposed to H2O2 for 24 h. a Representative FACS analyses of apoptosis. Q1: necrotic cells; Q2: late apoptotic cells; Q3: early apoptotic cells; Q4: live cells. The apoptosis rate is equal to the sum of Q2 and Q3. b A histogram of the percentage of apoptotic cells, n = 4. c Statistical results of cleaved caspase-3. The protein levels of caspase-3 and cleaved caspase-3 were detected by Western blotting using β-actin as a loading control, n = 3. d The direct radical scavenging capacity of FAN was determined using the DPPH free radical scavenging assay, n = 3. The data are presented as the mean ± SEM. ##P < 0.01 vs the control group; *P < 0.05, **P < 0.01 vs the H2O2 group
JE-133 (at its working concentration) had a weak ability to scavenge DPPH free radicals against H2O2-induced oxidative cytotoxicity
We next examined the DPPH radical scavenging capacity of JE-133 to further understand its antioxidative mechanism using the antioxidant NAC as a positive control. The scavenging ratio of 100 μM NAC, the working concentration that was effective against H2O2-induced oxidative cytotoxicity, on DPPH free radicals was 98.67%. In contrast, the clearance ratio of DPPH free radicals by 10 μM JE-133, the working concentration against H2O2-induced oxidative cytotoxicity, was 15.00% (Fig. 3d).
JE-133 prevented the upregulation of MAPK pathway protein phosphorylation induced by H2O2 in neuronal cells
To reveal the potential molecular mechanism involved in the antioxidative effect of JE-133, its influence on ERK1/2, P38 and JNK phosphorylation was measured. The exposure of SH-SY5Y cells to H2O2 for 6 h significantly increased the levels of phosphorylated ERK1/2, P38 and JNK (3.14 ± 0.34-fold of the control group, P < 0.01; 12.19 ± 1.76-fold of the control group, P < 0.01; and 2.28 ± 0.06-fold of the control group, P < 0.001 vs the control group, respectively) (Fig. 4a–c). In contrast, upon pretreatment with 10 μM JE-133, the H2O2-induced increases in ERK1/2, P38 and JNK phosphorylation were markedly decreased (1.48 ± 0.09-, 4.96 ± 1.10-, and 1.40 ± 0.18-fold of the control group, respectively, P < 0.05 for all vs the H2O2 group) (Fig. 4a–c). We also evaluated changes in the MAPK pathway in primary cortical neurons exposed to H2O2 after pretreatment with or without JE-133. As shown in Fig. 4d–f, the exposure of primary cortical neurons to H2O2 for 6 h significantly increased the levels of phosphorylated ERK1/2 and P38 (1.64 ± 0.08-fold of control group, P < 0.001; 1.80 ± 0.11-fold of the control group, P < 0.01, respectively). Upon pretreatment with 10 μM JE-133, the H2O2-induced increases in ERK1/2 and P38 phosphorylation were markedly decreased (1.27 ± 0.11- and 1.38 ± 0.12-fold of the control group, respectively, P < 0.05 for both vs the H2O2 group) (Fig. 4d, e). H2O2 treatment with or without JE-133 had no effect on JNK phosphorylation (Fig. 4f).

Effects of JE-133 on the phosphorylation of MAPK pathway proteins in H2O2-exposed neuronal cells. Cells were pretreated with JE-133 (10 μM) for 2 h and then exposed to H2O2 for 6 h. The protein levels of p-ERK1/2 (a), p-P38 (b), and p-JNK (c) in SH-SY5Y cells, n = 3. The protein levels of p-ERK1/2 (d), p-P38 (e), and p-JNK (f) in primary cortical neurons, n = 4. The above protein levels were measured by Western blotting using β-actin as a loading control. The data are presented as the mean ± SEM. ##P < 0.01, ###P < 0.001 vs the control group; *P < 0.05 vs the H2O2 group
PI3K/Akt pathway partially mediated the neuroprotective effects of JE-133
As the PI3K/Akt signaling pathway plays a key role in cell survival, we further evaluated whether the neuroprotective effect of JE-133 against H2O2-induced neurotoxicity is related to the PI3K/Akt pathway. The exposure of SH-SY5Y cells to H2O2 for 5–60 min increased the phosphorylation of Akt, and the maximal phosphorylation level of Akt was reached 15 min after H2O2 exposure (data not shown). In contrast, pretreatment with JE-133 (10 μM) for 2 h significantly enhanced the phosphorylation of Akt (P < 0.05 vs the H2O2 group) in H2O2-exposed SH-SY5Y cells (Fig. 5a). We then used LY294002 (an inhibitor of PI3K/Akt activation) to further evaluate how the PI3K/Akt signaling pathway is involved in the antioxidative neuroprotective effects of JE-133 in SH-SY5Y cells. As shown in Fig. 5b, the neuroprotective effects of JE-133 against H2O2-induced SH-SY5Y cell injury (95.58% ± 0.43%, P < 0.001 vs the H2O2 group) were effectively antagonized by 5 μM LY294002 (80.24% ± 0.30%, P < 0.05 vs the H2O2 + JE-133 group). In contrast, pretreatment with 5 μM LY294002 for 2 h exerted no influence on the viability of SH-SY5Y cells incubated with or without H2O2. As in SH-SY5Y cells, the protective effect of JE-133 against H2O2-induced reduction in cell viability was inhibited by LY294002 in primary cortical neurons (Fig. 5c). JE-133 pretreatment had no effect on the phosphorylation of Akt in H2O2-exposed primary neurons (supplementary Fig. S9).

The PI3K/Akt pathway mediated the neuroprotective effect of JE-133 in H2O2-exposed neuronal cells. a The level of p-AKT detected by Western blotting, n = 3. b The effect of LY294002 on the H2O2-induced reduction in viability of SH-SY5Y cells, n = 3. c The effect of LY294002 on the H2O2-induced reduction in viability of primary cortical neurons, n = 4. Cell viability was detected by the MTT assay. The data are presented as the mean ± SEM. #P < 0.05, ###P < 0.001 vs the control group; *P < 0.05, ***P <0.001 vs the H2O2 group; &P < 0.05 vs the H2O2 + JE-133 group
Blocking the PI3K/Akt pathway influenced the suppressive effects of JE-133 on the phosphorylation of MAPK pathway proteins in H2O2-exposed SH-SY5Y cells
We further investigated the relationship between the activation of PI3K/AKT signaling and the inhibition of MAPK regulation induced by JE-133 treatment. The cells were pretreated with LY294002 and/or JE-133 (10 μM) for 2 h and then exposed to H2O2 for 6 h, and the phosphorylation of MAPK pathway proteins was then measured. Pretreatment with LY294002 significantly reversed the inhibitory effects of JE-133 on P38 phosphorylation (P < 0.01 vs the H2O2 + JE-133 group, Fig. 6a) and ERK1/2 phosphorylation (P < 0.05 vs the H2O2 + JE-133 group, Fig. 6b) but did not reverse the inhibitory effects of JE-133 on JNK phosphorylation (Fig. 6c) in H2O2-exposed SH-SY5Y cells. In addition, pretreatment with LY294002 had no influence on P38 and ERK1/2 phosphorylation in cells not exposed to H2O2 (Fig. 6a, b).

Effect of LY294002 on suppressive effect of JE-133 on the phosphorylation of MAPK pathway proteins in H2O2-exposed SH-SY5Y cells. a The protein levels of p-P38 and P38. b The protein levels of p-ERK1/2 and ERK1/2. c The protein levels of p-JNK and JNK. The above protein levels were analyzed by Western blotting using β-actin as a loading control. A histogram of the protein expression ratio of p-P38 (p-ERK1/2, p-JNK)/β-actin based on the results of six independent experiments. The data are presented as the mean ± SEM. #P < 0.05, ##P < 0.01 vs the control group; *P < 0.05, ***P < 0.001 vs the H2O2 group; &P < 0.05, &&P < 0.01 vs the H2O2 + JE-133 group
Discussion
In the current study, we demonstrated for the first time that (i) JE-133, a novel optically active isochroman-2H-chromene conjugate, exhibited potent neuroprotective effects against H2O2-induced aberrant changes in SH-SY5Y cells and primary cortical neurons and (ii) the MAPK and PI3K/Akt pathways were both closely associated with the neuroprotective effects mediated by JE-133.
Hydrogen peroxide (H2O2), a major form of ROS produced during oxidative stress, plays an important role in the development and progression of neurodegenerative diseases, including AD. Exogenous H2O2 can penetrate the cell membrane, generate various free radicals and cause toxicity to neurons in the AD brain [38]. H2O2 is widely used as an inducer of oxidative stress to establish cellular oxidative damage models [39,40,41,42]. In our study, an H2O2-stimulated SH-SY5Y neuronal injury model was used for cell-based phenotypic screening to identify effective lead compounds that protect against oxidative neuronal injury. Consistent with previous studies [40, 43,44,45], the addition of H2O2 to SH-SY5Y cells induced obvious neuronal damage, as indicated by cell morphology observations and MTT and LDH measurements (Fig. 2a, b and Supplementary Fig. S8). These aberrant changes were significantly ameliorated by JE-133 in a concentration-dependent manner to a greater extent than the positive control (Fig. 2a, b and Supplementary Fig. S8). Moreover, similar neuroprotective effects of JE-133 were also verified in H2O2-stimulated primary cortical neurons (Fig. 2c, d). The above results prove that JE-133 is a novel small molecule with potent antioxidant effects and the capacity to attenuate oxidative stress-induced neuronal injury.
A large number of studies have shown that oxidative stress injury eventually leads to apoptosis. Under oxidative stress insult, excessive ROS accumulation can activate caspase-3 in various cells, including neurons [22, 41, 46,47,48]. By double staining SH-SY5Y cells with Annexin V-FITC and PI, we confirmed that the apoptosis rate of the H2O2-exposed group was significantly increased and that this increase was effectively suppressed by JE-133 treatment (Fig. 3a, b). Taken together, these results suggest that the ability of JE-133 to downregulate caspase-3 cleavage in H2O2-exposed SH-SY5Y cells (Fig. 3c) and the neuroprotective effects afforded by JE-133 may be partially attributed to its anti-apoptotic ability.
The imbalance between the generation and scavenging of intracellular free radicals is considered the key factor in triggering oxidative stress. Free radicals damage the plasma membrane, impair mitochondrial function, result in insufficient cell energy supply, and give rise to DNA fragmentation [49]. Antioxidants such as NAC [50], a classic antioxidant used as a positive control in our study, directly react with free radicals. The antioxidative capacity of JE-133 was estimated using DPPH free radicals, which have excellent stability and are widely used for testing free radical scavenging activity [51]. The results showed that JE-133 had moderate free radical scavenging activity (Fig. 3d), indicating that the neuroprotective effect of JE-133 is not mainly attributed to its free radical scavenging ability.
Numerous studies have indicated that the MAPK signaling pathway plays a critical role in oxidative stress-induced cell damage [52, 53]. MAPK signaling initiates the mitochondrial apoptotic pathway through enhanced pro-apoptotic protein activation [54, 55]. Consistent with previous reports [24, 56], H2O2 stimulated all three MAPK signaling proteins in SH-SY5Y cells in our study. Similar stimulation of the ERK and P38 pathways but not the JNK pathway was detected in H2O2-treated primary cortical neurons. Compared with H2O2 treatment alone, JE-133 treatment significantly attenuated the upregulation of MAPK signaling proteins, indicating the potential involvement of MAPK signaling, especially ERK and P38, in the neuroprotective effects of JE-133 against oxidative stress-evoked cell damage.
On the other hand, the PI3K/Akt pathway is believed to activate and trigger cell survival signals, which play a crucial role in neuroprotection against oxidative stress and apoptosis [14, 16, 21]. In the present study, the PI3K/Akt pathway was activated at the early stage (15 min), and this acute activation of Akt may have protected cells against oxidative damage as a survival-facilitating compensation for H2O2 stimulation. Treatment with JE-133 upregulated the phosphorylation level of Akt in SH-SY5Y cells, indicating that JE-133 may protect cells by strengthening this compensatory effect. In contrast, JE-133 pretreatment had no effect on the phosphorylation of Akt in H2O2-exposed primary neurons. However, the inhibition of the PI3K/Akt pathway (by LY294002) [20] significantly reduced the cell viability-enhancing efficacy of JE-133 in both SH-SY5Y cells and primary neurons (Fig. 5b, c). These results suggest that the PI3K/Akt pathway is responsible, at least partially, for the neuroprotective effects of JE-133 against oxidative damage. Interestingly, LY294002 reduced the levels of p-P38 and p-ERK1/2 but not the p-JNK level in SH-SY5Y cells (Fig. 6), suggesting that the regulatory effects of JE-133 on P38 and ERK phosphorylation may be a downstream effect of its ability to trigger Akt signaling. In contrast, our results indicate that the inhibitory effect of JE-133 on JNK phosphorylation is independent of its regulation of Akt signaling, which is in line with the incomplete inhibitory effect of LY294002 on the JE-133-induced enhancement of cell survival. This dual regulatory effect of JE-133 on the MAPK and PI3K/Akt pathways may provide the first insight into the molecular mechanisms underlying the neuroprotective effect of this novel small molecule. Overall, as a novel synthetic 1,3-disubstituted isochroman derivative obtained by diastereoselective oxa-Pictet-Spengler cyclization, JE-133 affords superior neuroprotective efficacy against H2O2-induced oxidative neuronal injuries associated with the regulation of the MAPK and PI3K/Akt signaling pathways (Fig. 7). Our study might provide important clues for the development of effective drug leads/candidates against oxidative stress-associated neurodegeneration. However, further animal studies are needed to determine the in vivo efficacy of JE-133.

Schematic diagram of the beneficial effects of JE-133 against H2O2-induced neurotoxicity and the potential mechanisms. H2O2 exposure induces intracellular oxidative stress, which triggers the overphosphorylation of the MAPK pathway proteins ERK1/2, P38 and JNK, and induces the upregulation of Akt phosphorylation, consequently activating neuronal apoptosis and ultimately leading to neuronal damage. JE-133 prevents H2O2-induced neuronal damage, which might be mediated by the downregulation of ERK1/2 and P38 phosphorylation and associated with the PI3K/Akt pathway. The dual regulatory effects on the PI3K/Akt and MAPK pathways might synergistically contribute to the neuroprotective effects of JE-133
References
Zhao Y, Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev. 2013;2013:316523.
Poprac P, Jomova K, Simunkova M, Kollar V, Rhodes CJ, Valko M. Targeting free radicals in oxidative stress-related human diseases. Trends Pharmacol Sci. 2017;38:592–607.
Jiang T, Sun Q, Chen S. Oxidative stress: a major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog Neurobiol 2016;147:1–19.
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842:1240–7.
Li J, O W, Li W, Jiang ZG, Ghanbari HA. Oxidative stress and neurodegenerative disorders. Int J Mol Sci. 2013;14:24438–75.
Pohanka M. Oxidative stress in Alzheimer disease as a target for therapy. Bratisl Lek Listy. 2018;119:535–43.
Di Domenico F, Barone E, Perluigi M, Butterfield DA. Strategy to reduce free radical species in Alzheimer’s disease: an update of selected antioxidants. Expert Rev Neurother. 2015;15:19–40.
Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932.
Arlt S, Muller-Thomsen T, Beisiegel U, Kontush A. Effect of one-year vitamin C- and E-supplementation on cerebrospinal fluid oxidation parameters and clinical course in Alzheimer’s disease. Neurochem Res. 2012;37:2706–14.
Baum L, Lam CW, Cheung SK, Kwok T, Lui V, Tsoh J, et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J Clin Psychopharmacol. 2008;28:110–3.
Snitz BE, O’Meara ES, Carlson MC, Arnold AM, Ives DG, Rapp SR, et al. Ginkgo biloba for preventing cognitive decline in older adults: a randomized trial. JAMA 2009;302:2663–70.
Persson T, Popescu BO, Cedazo-Minguez A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid Med Cell Longev. 2014;2014:427318.
Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, et al. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275:251–83.
Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192:1–15.
Azmi NH, Ismail N, Imam MU, Ismail M. Ethyl acetate extract of germinated brown rice attenuates hydrogen peroxide-induced oxidative stress in human SH-SY5Y neuroblastoma cells: role of anti-apoptotic, pro-survival and antioxidant genes. BMC Complement Alter Med. 2013;13:177.
Fu XY, Yang MF, Cao MZ, Li DW, Yang XY, Sun JY, et al. Strategy to suppress oxidative damage-induced neurotoxicity in PC12 cells by curcumin: the role of ROS-mediated DNA damage and the MAPK and AKT pathways. Mol Neurobiol. 2016;53:369–78.
Zhao X, Zeng Z, Gaur U, Fang J, Peng T, Li S, et al. Metformin protects PC12 cells and hippocampal neurons from H2O2-induced oxidative damage through activation of AMPK pathway. J Cell Physiol. 2019. https://doi.org/10.1002/jcp.28337
Liu X, Zhang J, Wang S, Qiu J, Yu C. Astragaloside IV attenuates the H2O2-induced apoptosis of neuronal cells by inhibiting alpha-synuclein expression via the p38 MAPK pathway. Int J Mol Med. 2017;40:1772–80.
Kwon SH, Hong SI, Ma SX, Lee SY, Jang CG. 3’,4’,7-Trihydroxyflavone prevents apoptotic cell death in neuronal cells from hydrogen peroxide-induced oxidative stress. Food Chem Toxicol. 2015;80:41–51.
Sadidi M, Lentz SI, Feldman EL. Hydrogen peroxide-induced Akt phosphorylation regulates Bax activation. Biochimie. 2009;91:577–85.
Wang S, Huang L, Zhang Y, Peng Y, Wang X, Peng Y. Protective effects of L-3-n-butylphthalide against H2O2-induced injury in neural stem cells by activation of PI3K/Akt and Mash1 pathway. Neuroscience. 2018;393:164–74.
Clementi ME, Pani G, Sampaolese B, Tringali G. Punicalagin reduces H2O2-induced cytotoxicity and apoptosis in PC12 cells by modulating the levels of reactive oxygen species. Nutr Neurosci. 2018;21:447–54.
Jantas D, Krawczyk S, Lason W. The predominant protective effect of tianeptine over other antidepressants in models of neuronal apoptosis: the effect blocked by inhibitors of MAPK/ERK1/2 and PI3-K/Akt pathways. Neurotox Res. 2014;25:208–25.
Zhao ZY, Luan P, Huang SX, Xiao SH, Zhao J, Zhang B, et al. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci Ther. 2013;19:163–9.
Feng HX, Li CP, Shu SJ, Liu H, Zhang HY. A11, a novel diaryl acylhydrazone derivative, exerts neuroprotection against ischemic injury in vitro and in vivo. Acta Pharmacol Sin. 2019;40:160–9.
Li L, Yao Y, Jiang Z, Zhao J, Cao J, Ma H. Dehydroepiandrosterone prevents H2O2-Induced BRL-3A cell oxidative damage through activation of PI3K/Akt pathways rather than MAPK pathways. Oxid Med Cell Longev. 2019;2019:2985956.
Yi J, Chen B, Yao X, Lei Y, Ou F, Huang F. Upregulation of the lncRNA MEG3 improves cognitive impairment, alleviates neuronal damage, and inhibits activation of astrocytes in hippocampus tissues in Alzheimer’s disease through inactivating the PI3K/Akt signaling pathway. J Cell Biochem. 2019;120:18053–65.
Guo X, Chen Y, Liu Q, Wu J, Wang L, Tang X, et al. Ac-cel, a novel antioxidant, protects against hydrogen peroxide-induced injury in PC12 cells via attenuation of mitochondrial dysfunction. J Mol Neurosci. 2013;50:453–61.
Wang XJ, Wang LY, Fu Y, Wu J, Tang XC, Zhao WM, et al. Promising effects on ameliorating mitochondrial function and enhancing Akt signaling in SH-SY5Y cells by (M)-bicelaphanol A, a novel dimeric podocarpane type trinorditerpene isolated from Celastrus orbiculatus. Phytomedicine. 2013;20:1064–70.
Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18:1067–73.
Costello JC, Stolovitzky G. Seeking the wisdom of crowds through challenge-based competitions in biomedical research. Clin Pharmacol Ther. 2013;93:396–8.
Prior M, Chiruta C, Currais A, Goldberg J, Ramsey J, Dargusch R, et al. Back to the future with phenotypic screening. ACS Chem Neurosci. 2014;5:503–13.
Tao LX, Huang XT, Chen YT, Tang XC, Zhang HY. Acetylcholinesterase-independent protective effects of huperzine A against iron overload-induced oxidative damage and aberrant iron metabolism signaling in rat cortical neurons. Acta Pharmacol Sin. 2016;37:1391–400.
Brozda D. The synthesis of 1-(3,4-dihydroxyphenyl)-2-propanone and 1-(3,4-dihydroxyphenyl)-2-propanol. Pharmazie. 1994;49:573–5.
Sunden H, Ibrahem I, Zhao GL, Eriksson L, Cordova A. Catalytic enantioselective domino oxa-michael/aldol condensations: asymmetric synthesis of benzopyran derivatives. Chemistry. 2007;13:574–81.
Mandi A, Kurtan T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat Prod Rep. 2019;36:889–918.
Pescitelli G, Bruhn T. Good computational practice in the assignment of absolute configurations by TDDFT calculations of ECD spectra. Chirality. 2016;28:466–74.
Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–54.
Park HR, Lee H, Park H, Jeon JW, Cho WK, Ma JY. Neuroprotective effects of Liriope platyphylla extract against hydrogen peroxide-induced cytotoxicity in human neuroblastoma SH-SY5Y cells. BMC Complement Alter Med. 2015;15:171.
Zhu A, Wu Z, Meng J, McGeer PL, Zhu Y, Nakanishi H, et al. The neuroprotective effects of ratanasampil on oxidative stress-mediated neuronal damage in human neuronal SH-SY5Y cells. Oxid Med Cell Longev. 2015;2015:792342.
Li P, Li Z. Neuroprotective effect of paeoniflorin on H2O2-induced apoptosis in PC12 cells by modulation of reactive oxygen species and the inflammatory response. Exp Ther Med. 2015;9:1768–72.
Bhaskaran N, Srivastava JK, Shukla S, Gupta S. Chamomile confers protection against hydrogen peroxide-induced toxicity through activation of Nrf2-mediated defense response. Phytother Res. 2013;27:118–25.
Ismail N, Ismail M, Fathy SF, Musa SN, Imam MU, Foo JB, et al. Neuroprotective effects of germinated brown rice against hydrogen peroxide induced cell death in human SH-SY5Y cells. Int J Mol Sci. 2012;13:9692–708.
Gu J, Chi M, Sun X, Wang G, Li M, Liu L, et al. Propofol-induced protection of SH-SY5Y cells against hydrogen peroxide is associated with the HO-1 via the ERK pathway. Int J Med Sci. 2013;10:599–606.
Wang W, Sun F, An Y, Ai H, Zhang L, Huang W, et al. Morroniside protects human neuroblastoma SH-SY5Y cells against hydrogen peroxide-induced cytotoxicity. Eur J Pharmacol. 2009;613:19–23.
Kang SS, Lee JY, Choi YK, Kim GS, Han BH. Neuroprotective effects of flavones on hydrogen peroxide-induced apoptosis in SH-SY5Y neuroblostoma cells. Bioorg Med Chem Lett. 2004;14:2261–4.
Annunziato L, Amoroso S, Pannaccione A, Cataldi M, Pignataro G, D’Alessio A, et al. Apoptosis induced in neuronal cells by oxidative stress: role played by caspases and intracellular calcium ions. Toxicol Lett. 2003;139:125–33.
Wang Y, Liu Q, Xu Y, Zhang Y, Lv Y, Tan Y, et al. Ginsenoside Rg1 protects against oxidative stress-induced neuronal apoptosis through myosin IIA-actin related cytoskeletal reorganization. Int J Biol Sci. 2016;12:1341–56.
Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med. 2000;29:323–33.
Lan A, Liao X, Mo L, Yang C, Yang Z, Wang X, et al. Hydrogen sulfide protects against chemical hypoxia-induced injury by inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in PC12 cells. PLoS ONE. 2011;6:e25921.
Sharma OP, Bhat TK. DPPH antioxidant assay revisited. Food Chem. 2009;113:1202–5.
Runchel C, Matsuzawa A, Ichijo H. Mitogen-activated protein kinases in mammalian oxidative stress responses. Antioxid Redox Signal. 2011;15:205–18.
Spencer JP. The interactions of flavonoids within neuronal signalling pathways. Genes Nutr. 2007;2:257–73.
Kwon SH, Hong SI, Kim JA, Jung YH, Kim SY, Kim HC, et al. The neuroprotective effects of Lonicera japonica THUNB. against hydrogen peroxide-induced apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH-SY5Y cells. Food Chem Toxicol. 2011;49:1011–9.
Cho ES, Jang YJ, Hwang MK, Kang NJ, Lee KW, Lee HJ. Attenuation of oxidative neuronal cell death by coffee phenolic phytochemicals. Mutat Res. 2009;661:18–24.
Hu XL, Niu YX, Zhang Q, Tian X, Gao LY, Guo LP, et al. Neuroprotective effects of Kukoamine B against hydrogen peroxide-induced apoptosis and potential mechanisms in SH-SY5Y cells. Environ Toxicol Pharmacol. 2015;40:230–40.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant numbers. 81703507, 81872859, 81522045). The research of the Hungarian authors was supported by the EU and cofinanced by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008 and the National Research Development and Innovation Office (grant number: K-120181).
Author information
Authors and Affiliations
Contributions
LXT and HYZ designed the pharmacological experiments; LXT, SSJ, and XD performed the pharmacological assays and analyzed the data; DS, TK, AM, and SA synthesized JE-133; TK instructed the chemical synthesis and wrote the part of the manuscript involving the chemical synthesis; LXT, SSJ, and HYZ wrote the manuscript with input from all of the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Tao, Lx., Ji, Ss., Szalóki, D. et al. An optically active isochroman-2H-chromene conjugate potently suppresses neuronal oxidative injuries associated with the PI3K/Akt and MAPK signaling pathways. Acta Pharmacol Sin 42, 36–44 (2021). https://doi.org/10.1038/s41401-020-0391-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0391-9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}