
Abstract
Cardiovascular disease (CVD) is the leading cause of death and disability worldwide, and experiences of psychological trauma have been associated with subsequent CVD onset. Identifying key pathways connecting trauma with CVD has the potential to inform more targeted screening and intervention efforts to offset elevated cardiovascular risk. In this narrative review, we summarize the evidence for key psychological and biological mechanisms linking experiences of trauma with CVD risk. Additionally, we describe various methodologies for measuring these mechanisms in an effort to inform future research related to potential pathways. With regard to mechanisms involving posttraumatic psychopathology, the vast majority of research on psychological distress after trauma and CVD has focused on posttraumatic stress disorder (PTSD), even though posttraumatic psychopathology can manifest in other ways as well. Substantial evidence suggests that PTSD predicts the onset of a range of cardiovascular outcomes in trauma-exposed men and women, yet more research is needed to better understand posttraumatic psychopathology more comprehensively and how it may relate to CVD. Further, dysregulation of numerous biological systems may occur after trauma and in the presence of posttraumatic psychopathology; these processes of immune system dysregulation and elevated inflammation, oxidative stress, mitochondrial dysfunction, renin-angiotensin system dysregulation, and accelerated biological aging may all contribute to subsequent cardiovascular risk, although more research on these pathways in the context of traumatic stress is needed. Given that many of these mechanisms are closely intertwined, future research using a systems biology approach may prove fruitful for elucidating how processes unfold to contribute to CVD after trauma.
Similar content being viewed by others

Introduction
Despite advances in prevention and intervention, cardiovascular disease (CVD), encompassing a range of disorders of the heart and blood vessels, remains the leading cause of death and disability worldwide [1, 2]. Globally, CVD accounts for approximately one-third of all deaths; in the United States, CVD claims more lives each year than cancer and chronic lower respiratory disease combined [1, 2]. The vast majority of CVD events are preventable, but the burden of CVD has been growing faster than the ability to tackle it. Identifying novel targets for reducing CVD risk is a critical step toward reversing current CVD incidence trends.
Psychosocial factors have been increasingly recognized as risk factors for CVD [3]. Indeed, depression and anxiety symptoms have been found to predict cardiovascular conditions with similar effect sizes as more traditional risk factors like smoking and obesity [4]. Over the past few decades, research has documented links between experiences of trauma with CVD onset [5, 6]. Trauma (e.g., natural disasters, unwanted sexual contact) is highly prevalent; the majority of individuals will experience a psychological trauma in their lifetime [7, 8]. Lifetime trauma exposure has been linked to a range of cardiovascular outcomes across studies, including coronary heart disease (CHD), myocardial infarction, and stroke, even when accounting for potential confounders and pathway variables [5, 9,10,11,12] Furthermore, in recent years, the American Heart Association has drawn attention to these issues in Scientific Statements highlighting the literature linking childhood adversity [13] and traumatic stress [14] with cardiovascular risk.
Given evidence of associations between trauma with CVD, there have been numerous efforts to understand underlying mechanisms. Identifying pathways linking trauma with CVD can inform targeted screening and intervention efforts to offset elevated cardiovascular risk. In this narrative review, we summarize the evidence for psychological and biological mechanisms linking trauma with CVD risk. Although not a systematic review, we present a comprehensive examination of the empirical literature on mechanisms, focusing in particular on mechanisms that have not received as much attention in the literature. Further, despite a robust preclinical literature, we highlight research in humans in this review. To inform future research in this area, we also describe various methodologies for measuring mechanisms, especially potential biological pathways. Finally, we highlight remaining gaps in understanding and recommend future directions to advance research in this area.
Psychological mechanisms linking trauma with CVD risk
Adverse psychological responses to traumatic events have been posited as a key mechanism linking trauma with poor physical health [6]. Although many individuals are resilient after trauma, a sizeable proportion subsequently experience emotional difficulties [15]. Contingent upon trauma exposure, posttraumatic stress disorder (PTSD) is the quintessential trauma-related mental disorder [16]. However, PTSD is not the only mental disorder that can onset after trauma; other stress-related conditions (e.g., acute stress disorder) and mood, anxiety, and substance use disorders can all develop in response to a traumatic event [17,18,19,20,21]. Furthermore, PTSD is often comorbid with depression, anxiety, and substance misuse. Here, we summarize and evaluate the empirical evidence for manifestations of posttraumatic psychopathology as psychological mechanisms linking trauma with CVD risk.
PTSD
The vast majority of research on posttraumatic psychopathology and CVD has focused on PTSD, and PTSD has been increasingly recognized as a key psychological mechanism underlying elevated cardiovascular risk after trauma [22]. In an initial meta-analysis of six studies, PTSD was associated with a 55% higher rate of incident CHD; although attenuated when adjusting for depression, the pooled hazard ratio (HR) for the PTSD-CHD association still provided evidence of an effect (HR = 1.27, 95% CI: 1.08–1.49) [23]. A more recent meta-analysis of nine longitudinal studies estimated that PTSD was associated with a 61% higher rate of incident CHD; again, depression did not account for the PTSD-incident CHD relation entirely (HR = 1.46, 95% CI: 1.26–1.69) [24]. In addition to this meta-analytic evidence, methodologically rigorous, longitudinal research has demonstrated that PTSD precedes and predicts a range of cardiovascular conditions, including myocardial infarction, stroke, venous thromboembolism, heart failure, and atrial fibrillation [12, 25,26,27,28,29,30,31,32,33,34,35,36]. Furthermore, PTSD has been associated with numerous cardiometabolic risk factors (e.g., hyperlipidemia, hypertension, diabetes, obesity) [37,38,39,40]. In these studies, PTSD has been measured in various ways, including symptom questionnaires [12, 29], clinical interview-based diagnoses [27], and diagnostic codes in electronic health records [25, 35].
For years, PTSD and CVD was studied in predominantly male veteran samples. This limited the ability to draw conclusions about civilians and women—notable shortcomings given the wide-ranging nature of trauma and established sex differences in PTSD and CVD [7, 41]. However, prospective research in population-based health registry samples has addressed this gap [25, 26]. In addition, longitudinal research has demonstrated associations between PTSD and incident CVD in community-dwelling women and women veterans, with effect sizes similar to those observed in men [12, 28, 35].
Together, this work provides substantial epidemiologic support that PTSD may increase risk of CVD. Indeed, in light of this evidence—and the potential public health implications—the National Heart, Lung, and Blood Institute held a workshop in 2018 to outline important directions for future research [42]. One such direction was to extend the research in observational cohorts and improve causal inference with Mendelian randomization (MR) using results from large-scale genome-wide association studies (GWAS) of PTSD and CVD. MR is an instrumental variable method that uses genetic instruments (which can be derived from publicly available GWAS summary statistics) to address the causal relation between a risk factor and health outcome [43]. Two MR studies on PTSD and cardiovascular risk have been conducted, and they suggest that genetically determined PTSD predicts hypertension [44] and CHD [45]. In contrast, no evidence for genetic predisposition for hypertension predicting PTSD was observed [44], and genetic predisposition for CHD was associated with reduced PTSD symptom severity [45]. The latter finding is contrary to evidence from cohort studies suggesting that cardiovascular events can serve as an index trauma for PTSD [22].
Other posttraumatic psychopathology
Even though posttraumatic psychopathology can manifest in various ways, research that comprehensively considers a constellation of posttraumatic psychopathology as predictors of CVD is lacking. Research in national health registry samples has considered other stress-related disorders that—like PTSD—are contingent upon experiencing a severely stressful event (e.g., acute stress reaction, adjustment disorder); these stress-related mental disorders were associated with elevated risk of incident cardiovascular events and conditions [25, 26]. However, research in trauma-exposed samples has generally treated other mental disorders (e.g., depression) as confounders when evaluating PTSD and CVD, rather than investigating these other expressions of posttraumatic psychopathology as CVD predictors themselves [12, 28, 29]. This gap in the literature is notable, because depression, anxiety, and substance misuse are associated with CVD in non-trauma-exposed samples [46,47,48].
The predominant focus on PTSD has thus resulted in an incomplete characterization of the psychopathological effects of trauma exposure on subsequent CVD risk. Moreover, initial evidence suggests that considering psychiatric comorbidities after trauma may help identify those most at-risk for adverse physical health outcomes. Compared to women without trauma or depression, trauma-exposed women with high PTSD and depressive symptom levels had a nearly four-fold greater risk of all-cause mortality, plus higher rates of death from CVD [49]. These findings highlight the importance of considering co-occurrence of mental disorders when studying cardiovascular risk after trauma.
Downstream pathways and shared genetic risk
Numerous behavioral and biological changes associated with posttraumatic psychopathology may subsequently contribute to cardiovascular risk. Here, we highlight some of these pathways, often focusing on ones associated with PTSD in particular as it has received the most attention within the literature. We describe additional biological mechanisms relevant to trauma and related psychopathology in more detail in Section 3.0. We also describe evidence for shared genetic factors that may increase risk of psychopathology and CVD after trauma.
Meta-analytic evidence has linked PTSD to numerous poor health behaviors and conditions, including physical inactivity, unhealthy diet, smoking, and obesity [50]. Additionally, medication nonadherence and greater substance use and abuse have been observed in individuals with PTSD [51, 52]. Other psychopathology such as depression and anxiety have also been linked to unhealthy behaviors [53,54,55]. Although numerous studies detect an association between PTSD and CVD even when accounting for these behaviors, effect sizes are often attenuated, suggesting these factors explain some of the excess cardiovascular risk in individuals with posttraumatic psychopathology [12, 27,28,29]. For example, unhealthy behaviors and obesity may contribute to metabolic dysregulation and related conditions (e.g., insulin resistance, metabolic syndrome, diabetes) that, in turn, increase vulnerability to CVD [56]. Furthermore, some research suggests that PTSD may contribute to psychopathology like depression, which—in turn—may contribute to unhealthy behaviors like physical inactivity and smoking, thereby increasing CVD risk [57]. This research demonstrates that considering various psychological consequences of trauma may help elucidate drivers of CVD-relevant pathophysiology.
A substantial literature has also described a strong connection between PTSD and dysregulation of the biological stress response, including the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic-adrenal-medullary (SAM) system; changes in these systems may produce a cascade of effects conducive to poor cardiovascular health. Studies of individuals with PTSD have reported decreased cortisol levels, increased sensitivity of glucocorticoid receptors, and enhanced negative feedback of the HPA axis, although inconsistencies in findings across studies exist and may reflect differences in trauma exposure, manifestations and/or duration of posttraumatic psychopathology, and methodological factors [58,59,60]. In addition, PTSD has been characterized by hyperarousal of the sympathetic nervous system (SNS; e.g., elevated heart rate, blood pressure, skin conductance) and diminished parasympathetic activity (e.g., lower heart rate variability reflecting reduced vagal tone) [59,60,61]. Changes in HPA and autonomic functioning have implications for cardiovascular health (e.g., excessive catecholamines from SNS hyperreactivity can induce cardiac injury [62]), and they can influence other cardio-relevant biological mechanisms described in Section 3.0 (e.g., inflammation, oxidative stress) [63]. Other psychopathology such as depression has been characterized by HPA axis and autonomic dysfunction as well, although often in distinct ways from PTSD (e.g., depression is characterized by reduced feedback inhibition of the HPA axis, whereas PTSD is characterized by enhanced negative feedback) [58, 64], again suggesting a need to examine comprehensively the psychological sequelae of trauma when delineating these pathways.
Finally, shared genetic risk factors may also contribute to poor mental and cardiovascular health after trauma. For example, one study found associations between candidate genes for PTSD and CVD; of the 87 PTSD candidate risk genes, 37 were also risk genes for CVD, with many implicated in pathways related to immune function [65]. Research using results from GWAS of PTSD, depression, and cardiovascular outcomes has also pointed to positive genetic correlations between these mental and cardiovascular conditions [44, 66]. These results thus provide initial evidence for genetic overlap between PTSD and depression with cardiovascular risk, situating these conditions in a shared genetic milieu.
Biological mechanisms linking trauma with CVD risk
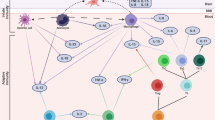
Both trauma and posttraumatic psychopathology can contribute to downstream processes that increase risk of CVD. For example, as described in Section 2.3, PTSD is characterized by dysregulation of the HPA axis and SAM system [58,59,60], and trauma exposure—even without trauma-related psychopathology—is associated with similar dysfunction, although generally to a lesser degree than PTSD [67, 68]. These physiological changes can contribute to alterations in biological processes relevant to cardiovascular health, including immune dysregulation and elevated inflammation, oxidative stress, mitochondrial dysfunction, dysregulation of the renin-angiotensin system, and accelerated biological aging (Fig. 1). For each of these processes, we briefly introduce the underlying biology, describe links to CVD and other biological mechanisms, and review different measurement approaches to support the incorporation of these mechanisms in future studies. We then summarize and evaluate the empirical evidence for these biological mechanisms, first focusing on links with trauma and then with PTSD. Although some of these mechanisms are related to other psychopathology (e.g., depression [69, 70]), we focus on PTSD given the predominance of this disorder in the traumatic stress-CVD literature to date. Additionally, we provide details for some exemplar studies for each biological mechanism to convey further information about notable methods and key findings (Table 1).

Experiences of trauma and severe stress precede manifestations of posttraumatic psychopathology, such as posttraumatic stress disorder (PTSD) and depression. Subsequent dysregulation of biological stress response systems, including the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic-adrenal-medullary (SAM) system, can contribute to further dysregulation in several interconnected biological systems, potentially leading to immune dysregulation and elevated inflammation, oxidative stress, mitochondrial dysfunction, and dysregulation of the renin-angiotensin system (RAS). Not only can these biological processes further influence one another (as indicated by the recursive arrows), but they can also contribute to accelerated biological aging. Together, these biological alterations can lead to the accumulation of intermediary cardiovascular risk factors, such as hypertension, endothelial dysfunction, and atherosclerosis, which—in turn—increase risk of developing CVD. Furthermore, these psychological and biological processes may unfold after trauma within a milieu of shared genetic risk.
Immune dysregulation and elevated inflammation
The immune system encompasses cells, chemicals, and processes that defend the body from noxious stimuli, and inflammation is a key component of the immune response [71]. Inflammatory responses involve a complex cascade of signaling molecules; although acute increases in inflammation in response to injury or infection are critical for health, a chronic state of inflammation can contribute to disease. Inflammation has been implicated in cardiovascular event onset, disease progression, and adverse prognosis [72]. For example, epidemiologic studies have demonstrated that elevated inflammatory biomarkers predict CVD [73], and atherosclerosis—the narrowing of arteries due to plaque accumulation, a major CVD risk factor—is now conceptualized as an inflammatory condition [72].
Immune- and inflammation-related processes can be measured in research in various ways. Quantifying levels of peripheral inflammation-related biomarkers, such as C-reactive protein (CRP) and interleukin-6 (IL-6), are common in the field of traumatic stress [74, 75], although imaging-based approaches that capture cellular glycolysis (e.g., fluorodeoxyglucose positron emission tomography) have begun to be used to detect vascular inflammation [76, 77]. Immune function can also be captured, for example, by considering in vitro stimulated measures of inflammatory cytokine- and/or chemokine-producing immune cells [78].
Trauma, PTSD, and inflammation-related biomarkers have been studied extensively, with several meta-analyses on these relations. A meta-analysis of 36 independent samples found moderate positive correlations between trauma exposure and several inflammation-related biomarkers [CRP, IL-1β, IL-6, tumor necrosis factor-α (TNF-α)]; no associations were observed for fibrinogen, IL-2, IL-4, IL-8, or IL-10 [75]. In addition, childhood trauma was associated with elevated CRP, IL-6, and TNF-α levels with small effects in a meta-analysis of 25 studies [79]. Several systematic reviews and meta-analyses examining PTSD and inflammatory markers have also been conducted [74, 80,81,82]. In the most recent meta-analysis of 54 studies examining over 15 inflammatory markers, elevated levels of CRP (moderate effect), IL-6 (large effect), and TNF-α (large effect) were observed in individuals with PTSD compared to controls; there was also weak evidence of a small effect for PTSD and IL-1β [82]. Together, these results suggest that trauma and PTSD are associated with elevations in peripheral inflammatory markers. However, most studies have been cross-sectional, which limits the ability to draw directional and causal conclusions. Indeed, bidirectional associations between PTSD and inflammation have been postulated, and the relatively few existing longitudinal studies have yet to provide robust evidence that a proinflammatory milieu leads to PTSD and/or that trauma and PTSD promote inflammation [83]. In addition to studies of peripheral biomarkers, preliminary research in small samples of individuals with PTSD has examined vascular inflammation using imaging-based approaches, although findings have been mixed [76, 77].
Nevertheless, support for links between trauma, PTSD, and inflammation has been observed across omics studies. For example, hypothesis-free genetic, epigenetic, and gene expression studies of PTSD have identified genes related to the immune system [e.g., genes in the HLA region and those encoding inflammatory cytokines (IL-8, IL-16)] [84,85,86], which parallels work with candidate genes [65]. Additionally, some studies have found that variation in and/or methylation of immune-relevant genes underlie associations of PTSD and inflammation. For instance, in some (but not all) studies of military veterans, methylation of the Absent in Melanoma 2 (AIM2) gene, which has been implicated in the inflammatory response, partially accounted for associations of PTSD with elevated inflammatory markers [87,88,89]. Furthermore, MR research has begun to address causality in the PTSD-inflammation and inflammation-PTSD relations, with an initial study documenting small bidirectional associations between PTSD and CRP [90]. Finally, several metabolomics studies have identified metabolites related to inflammation and immune function (e.g., sphingolipids, glycerophospholipids) in profiles distinguishing individuals with and without PTSD [91,92,93].
Oxidative stress
Oxidative stress may also underlie associations of trauma and PTSD with CVD. Cellular energy production generates pro-oxidants [e.g., reactive oxygen species (ROS)] as naturally occurring byproducts, and regulation by an antioxidant defense system is key for cellular functioning [94, 95]. Oxidative stress arises when there is an imbalance between ROS and other pro-oxidant molecules and their neutralization by antioxidants [94, 95]. A prolonged state of oxidative stress can result in cellular damage and death. Oxidative stress and inflammation often co-occur and influence one another; inflammatory signaling can trigger ROS production, and oxidative stress stimulates the immune response [94, 95]. Furthermore, oxidative stress has been linked to CVD [96]. Traditional cardiovascular risk factors like hyperlipidemia and hypertension contribute to ROS production [97], and oxidative stress can contribute to endothelial dysfunction [98]—an early indicator of reduced capacity of blood vessels to respond to cardiovascular demand [99]—and atherosclerosis [97].
Although it is challenging to measure pro-oxidant molecules like ROS directly due to short half-lives and low concentrations, approaches have been developed to quantify oxidative stress [100]. For example, biomarkers indicative of damage induced by oxidative stress to lipids, proteins, and DNA (e.g., F2-isoprostanes, protein carbonyls, 8-hydroxy-deoxyguanosine, respectively) can be measured using immunoassays or mass spectrometry [100, 101]. Alternatively, antioxidant-based measures can be used to capture oxidative stress. Such approaches include quantifying antioxidant enzyme levels and activity, as well as the antioxidant capacity in bodily fluids—an in vitro metric that uses color or fluorescence changes to quantify the extent to which pro-oxidants are counteracted by antioxidants [101, 102].
Although some research has linked life stress (e.g., chronic caregiving, examination stress) with greater oxidative stress [103, 104], few studies have considered traumatic experiences, with all focused on early-life adversity. In two small studies of adolescents, those with a history of early-life adversity had elevated markers of oxidative damage to lipids (F2-isoprostanes) and proteins (protein carbonyls), and an increased enzymatic pro-oxidant/antioxidant defense ratio and lower non-enzymatic antioxidant capacity [105, 106]. Furthermore, one small study of postpartum women found that more severe childhood maltreatment was associated with serum metabolites associated with greater oxidative stress and lower antioxidant capacity [107]. However, some studies in children [108] and adults [109] have failed to detect associations between early-life adversity and oxidative stress markers. Notably, all studies employed retrospective reports of early-life adversity in relatively small samples, and research on lifetime experiences of trauma is lacking.
Whereas the research on trauma and oxidative stress has been limited, numerous studies have assessed oxidative stress in individuals with PTSD. A recent meta-analysis did not find an association between PTSD with malondialdehyde (a product of lipid peroxidation) or two antioxidant enzymes (catalase, paraoxonase-1) [82], although only five studies were included and three drew participants from the same sample [110,111,112,113,114]. Overall, additional studies of PTSD and oxidative stress—most conducted among combat veterans [115,116,117,118]—have had mixed results [119,120,121]. Furthermore, studies have varied widely in measures of oxidative stress, which complicates comparisons. Some additional evidence for a link between PTSD and oxidative stress comes from omics studies. For example, a variant in the retinoic acid orphan receptor (RORA) gene, involved in oxidative stress-related biology, was identified in the first GWAS of PTSD [122], and findings from epigenetic and gene expression studies of PTSD have implicated genes related to oxidative stress [123,124,125]. Additionally, metabolite profiling studies comparing individuals with and without PTSD have identified differences in metabolites related to oxidative stress (e.g., proline, hydroxyproline, 4Z,15E-bilirubin IXa) [91, 93, 126].
Mitochondrial dysfunction
Given their critical role in energy production, mitochondria are key producers of ROS that are closely connected with immune modulation and inflammation-related processes [127]. Mitochondria have their own DNA—mitochondrial DNA (mtDNA)—with critical genes for energy production. In addition, mitochondria can change in structure and function as a result of environmental signals they detect; some of these changes can contribute to mtDNA damage, which can lead to mitochondrial dysfunction and adversely impact energy metabolism, ROS production, and signaling [127, 128]. Changes in mitochondrial structure and function can contribute to cardiometabolic dysregulation, including obesity, hypertension, and hyperlipidemia [129], and mitochondrial dysfunction can lead to endothelial dysfunction and atherosclerosis [130].
Multiple mitochondria-relevant metrics exist; measuring aspects of mitochondria function and content provides the most comprehensive assessment [127]. Functional measures include quantifying oxygen consumption rate in living cells and respiratory chain enzymatic activity in frozen cells [131]. Additionally, mitochondria morphology can be assessed with electron microscopy [132], and activity of the mitochondrial enzyme citrate synthase can be quantified to measure the density of the mitochondrial network per cell [133,134,135]. Several mtDNA-related measures [e.g., mtDNA copy number (mtDNAcn) per cell, mutations, deletions] can also be assessed as potential indicators of mitochondrial dysfunction [127, 136]. Additionally, circulating cell-free mtDNA (ccf-mtDNA) is a pro-inflammatory mitochondria-derived signaling molecule detectable in peripheral samples [127].
As with research on oxidative stress, most research has examined stress and mitochondrial dysfunction [127, 131, 133], but some studies have examined early-life adversity. One small sample of postpartum women found that more severe experiences of childhood maltreatment were associated with greater mitochondrial routine physiological activity and augmented energy and ROS production, but not with citrate synthase activity [107]. These findings were partially replicated in a larger sample of mother-newborn dyads in the My Childhood-Your Childhood study from the same research group; mothers with vs. without a history of childhood maltreatment had higher mitochondrial routine physiological activity, augmented energy production, and higher citrate synthase activity [137]. However, no associations between maternal childhood maltreatment and newborns’ mitochondrial measures were observed. My Childhood-Your Childhood study investigators also examined whether maternal childhood maltreatment was related to differential change in mitochondrial metrics within the first year after birth [138]. At the 1-year follow-up, childhood maltreatment-related differences in mitochondrial function and intracellular density at baseline (reported in Gumpp et al. [137]) were no longer observed, suggesting that these mitochondrial changes may have only been detectable after the physiological demands of childbirth.
Whereas links between early-life adversity and mitochondrial function have been examined predominantly in postpartum women, associations between early-life adversity and mtDNAcn have been investigated in more diverse samples [136]. For example, history of childhood maltreatment was associated with greater mtDNAcn in adults [139] and children [140], however, in one additional study, the positive association between childhood sexual abuse and mtDNAcn was only observed in individuals with depression [141]. In contrast, maternal lifetime trauma exposure was negatively associated with mtDNAcn in placenta, but not cord blood [142]. Additionally, no significant differences in mtDNAcn were observed in Holocaust survivors or their descendants compared to age-matched controls [143]. With respect to circulating mitochondrial-related markers, two studies of traumatic injury patients observed higher concentrations of mtDNA in plasma compared to controls [144, 145], and women who experienced sexual trauma during adolescence had significantly more ccf-mtDNA than women with no sexual trauma or who experienced sexual trauma during childhood or adulthood [146].
Studies of PTSD and mitochondria are even fewer than those investigating trauma and mitochondrial dysfunction. In male combat veterans, individuals with vs. without a current PTSD diagnosis had lower mtDNAcn; this was driven by individuals with mild or severe symptoms [147]. In a prospective pregnancy cohort, greater prenatal PTSD symptoms were associated with lower mtDNAcn in placenta [148]. However, in trauma-exposed women, there was no significant correlation between current PTSD symptoms and ccf-mtDNA [146]. Indirect support for a link between PTSD and mitochondrial function also comes from mitochondrial GWAS [149], along with epigenetic [150], gene expression [125], and metabolomics studies [93].
Renin-angiotensin system (RAS) dysregulation
Dysregulation of the RAS, which interacts with the HPA axis and SAM system, represents another biological mechanism potentially linking traumatic stress with CVD risk [151,152,153]. The RAS is a key regulator of blood pressure, along with fluid and salt balance [152, 154]. The enzyme renin leads to the production of angiotensin I and II in blood and tissues. As the main effector molecule of the RAS, angiotensin II has numerous functions, including constricting blood vessels, stimulating sodium reabsorption and triggering the adrenal cortex to release aldosterone, and promoting norepinephrine release [152]. RAS activation can contribute to cardiovascular risk via elevated SNS activity, blood pressure, inflammation, oxidative stress, and endothelial dysfunction [63, 154], and elevated renin predicts myocardial infarction [155, 156]. Furthermore, elements of the RAS have become cardiovascular pharmacotherapy targets; angiotensin converting enzyme inhibitors (ACE-Is) and angiotensin receptor blockers (ARBs) are RAS blockers commonly used to manage hypertension and have been shown to reduce SNS activity [63, 154].
RAS components can be assessed from peripheral samples. For example, plasma renin activity, a measure of renin’s capacity to generate angiotensin I, can be determined by activity assay; immunoassays can also be used to estimate renin concentration in plasma [157, 158]. Additionally, immunoassays can be used to quantify endogenous angiotensins (e.g., angiotensin II) and the steroid hormone aldosterone [158, 159].
Although acute stress has been associated experimentally with increases in RAS components (renin, aldosterone) [153], only three studies have examined trauma, PTSD, and the RAS. In a large general population sample from the Study of Health in Pomerania, greater childhood trauma was associated with higher plasma concentrations of aldosterone—but not renin—and greater adulthood trauma exposure was associated with higher plasma renin—but not aldosterone—concentrations [160]. In a second Study of Health in Pomerania investigation, individuals with trauma but without PTSD and individuals with PTSD had elevated renin (but not aldosterone) levels compared to individuals without trauma; those with PTSD showed the most pronounced renin elevations [161]. Additionally, middle-aged women with chronic PTSD had lower aldosterone levels compared to women without trauma [162]. Although findings have been mixed from the few studies examining traumatic stress and RAS components, further indirect evidence for a PTSD-RAS link comes from ACE-I/ARB studies. Specifically, use of ACE-Is/ARBs (but not other antihypertensive medications) has been associated with lower PTSD symptoms [163, 164], with some evidence of moderation by genetic variation or sex [164, 165]. However, there was no evidence for clinical benefit of ARB use for PTSD in a 10-week randomized, placebo-controlled trial [166].
Accelerated biological aging
Many of the biological processes described above, including elevated inflammation and oxidative stress, may contribute to CVD via accelerated biological aging (BA). Indeed, trauma and PTSD have been linked to early-onset CVD [12, 34, 35], prompting interest in whether the pace of cellular aging may be hastened after trauma and contribute to premature disease [167]. Cellular markers of BA associated with aging-related process and CVD include telomeres, epigenetic clocks, and composite biomarker-based estimates. For example, telomeres—noncoding DNA sequences at the ends of chromosomes—shorten with age, although telomere attrition can accelerate due to environmental factors [168]. Cellular senescence is triggered when telomeres shorten to a particular length, and shortened telomeres predict mortality and incident CVD [169, 170]. Further, MR research suggests a causal link between telomere length and CVD [171]. Numerous “epigenetic clocks” have also been developed that estimate age based on DNA methylation (DNAm) data across the genome [172]. The extent to which these DNAm age estimates are accelerated relative to chronological age predicts mortality and CVD [173, 174]. In addition, BA estimators that integrate information from clinical biomarkers across multiple physiological systems have been developed that predict mortality and morbidity [175,176,177].
Multiple methods can be leveraged to quantify these BA measures. Telomere length can be measured in several specimens, including blood and saliva, using various methods (e.g., terminal restriction fragment length analysis by Southern blot, quantitative polymerase chain reaction with DNA analytes [178, 179]); however, tissue type and analytic approach have been found to influence reliability [179,180,181]. Recently, a DNAm estimator of telomere length was also developed [182]. Additionally, multiple epigenetic clocks are available, including first-generation clocks calibrated to predict chronological age (the pan-tissue Horvath clock [183] and blood-based Hannum clock [184]) and second-generation clocks trained to predict morbidity- and mortality-related outcomes (PhenoAge [176] and GrimAge [185]). Most of these clocks can be applied to DNAm from blood samples in adults, although the Horvath clock was calibrated across various tissues and in youths and adults [177, 183]. In addition, individual variability in the pace of biological aging was recently distilled into a single timepoint DNAm measure (DunedinPACE) [186]. Finally, composite biomarker estimates of BA can be calculated from various biomarkers (e.g., CRP, creatinine) often collected in clinical settings; several computational methods are available (e.g., Klemera-Doubal method, Phenotypic Age algorithm [175, 176]). Calculation of various DNAm age estimates and composite biomarker estimates is facilitated by the availability of online calculators and statistical packages [183, 187]. Interestingly, relatively low agreement has been found between various BA measures, suggesting that they capture different aspects of aging-related processes [188].
Trauma, PTSD, and telomeres have been studied extensively, as summarized in several systematic reviews and meta-analyses. Early-life adversity has generally been linked to shorter telomere length, despite heterogeneity across investigations [189,190,191,192,193,194]; in the largest meta-analysis of 41 studies in youths and adults, there was a small-to-medium overall association between early-life adversity and reduced telomere length [190]. Furthermore, there is some meta-analytic evidence that early adverse experiences characterized by threat—not deprivation or socioeconomic disadvantage—are associated with accelerated BA in youths (measured across telomere and DNAm age metrics) [195]. In several studies of early-life adversity and telomeres published since these review papers, most—but not all—have demonstrated negative associations between early-life adversity and telomere length [196,197,198,199,200,201,202,203], yet there has been mixed evidence for intergenerational transmission of trauma via telomere length [204,205,206]. Fewer studies have examined traumatic experiences in adulthood (e.g., military service, solitary confinement during war captivity, lifetime trauma) and telomere length, with mixed results [203, 207,208,209]. Research on PTSD and telomeres has also been summarized in reviews. One meta-analysis of five studies found a small overall effect of PTSD on shorter telomere length; as with research on early-life adversity, substantial heterogeneity was detected [191]. A more recent systematic review included 13 studies of PTSD and telomeres, with six finding a negative association, three (all in military samples) not detecting an association, one finding a positive association, and three finding mixed results [210]. More recent studies have continued to have inconsistencies or nuance in results, with some observing a negative PTSD-telomere length relation only for older individuals [211] or for re-experiencing symptoms [212], and others detecting a positive association between PTSD and telomere length [213, 214]. A few studies of telomeres in trauma-exposed individuals have also considered manifestations of posttraumatic psychopathology beyond PTSD, detecting shorter telomeres in former prisoners of war with greater depressive symptoms [215] and in women with comorbid PTSD and depressive symptoms [216].
Although fewer studies have examined associations between trauma, PTSD, and DNAm age compared to the telomere literature, this is an area of growing interest. A recent systematic review identified 10 studies of traumatic stress and DNAm age in adults; four of these examined early-life adversity, with one finding an association with advanced DNAm age relative to chronological age (i.e., epigenetic age acceleration) [217]. In a large meta-analysis of individuals across nine cohorts, childhood trauma was associated with Hannum (but not Horvath) epigenetic age acceleration, however only when measured with the Childhood Trauma Questionnaire [218]. Greater experiences of early-life adversity were also linked to DNAm age acceleration based on several epigenetic clocks in two large longitudinal cohorts [219, 220]. In youths, early-life adversity has been linked to epigenetic age acceleration, with nuances in the findings detected. For example, in the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort, early-life adversity during early and middle childhood was associated with Hannum (but not Horvath) epigenetic age acceleration—suggesting sensitive periods, rather than cumulative or recency effects—when it comes to early-life adversity and BA [221]. Additional work in ALSPAC found that greater early-life adversity was associated with Horvath (but not Hannum) epigenetic age acceleration in adolescent girls and not boys [222]. In contrast, as noted in the Lim and colleagues [217] systematic review, relatively few studies have detected a link between lifetime trauma and epigenetic age acceleration, a finding echoed in more recent investigations [218, 223]. Research on PTSD and DNAm age has more consistently demonstrated evidence for epigenetic age acceleration, although there is variability in which epigenetic clocks have effects detected. The Lim and colleagues [217] systematic review and a recent review [224] reported that 5 of 7 studies and 7 of 11 studies, respectively, observed an association between PTSD and accelerated epigenetic age. Recent studies have a similar overall finding, although again which epigenetic clock is accelerated varies [225,226,227,228]. There is also initial evidence that other posttraumatic psychopathology may be relevant to DNAm age. For example, current alcohol use disorder—but not depression or generalized anxiety disorder—was associated with a faster pace of the Horvath epigenetic clock in a longitudinal study of veterans [229]. Additionally, although PTSD diagnosis was not related to pace of the epigenetic clock, avoidance and numbing symptoms of PTSD were predictors.
To date, only a handful of studies have investigated early-life adversity and composite biomarker-based BA measures. Across three studies in large longitudinal cohorts, early-life adversity was associated with more advanced BA based on the Klemera-Doubal method, Phenotypic Age algorithm, and a multi-biomarker indicator of pace of BA [230,231,232].
Recommendations and future directions
There is now an extensive literature suggesting that CVD risk is elevated after trauma. The psychological response to trauma appears to be a key mechanism linking these experiences with adverse cardiovascular health. Not only is dysregulation of cardio-relevant biological processes generally more pronounced in those with posttraumatic psychopathology than with trauma alone [67, 68, 233], but studies that directly compare risk of incident CVD in trauma-exposed individuals with and without posttraumatic psychopathology generally detect larger effects in individuals with psychological distress after trauma [12, 34]. These findings have been observed in a variety of trauma-exposed samples using rigorous longitudinal designs that account for numerous potential confounders, and this work has been complemented by MR studies that further suggest a causal association. However, despite the many ways in which posttraumatic psychopathology manifests, the vast majority of research has focused on PTSD. This is a notable limitation, especially as the few studies considering multiple forms of posttraumatic psychopathology have often found more pronounced health risks in individuals with comorbid symptoms, suggesting the value of looking beyond just PTSD [49, 216].
Going forward, it is critical to consider whether other posttraumatic psychopathology has an etiologic effect on cardiovascular health. For example, it is unclear whether there are combinations and/or sequences of psychopathology after trauma that are particularly cardiotoxic, and whether these combinations vary for different groups (e.g., men vs. women), trauma types (e.g., interpersonal vs. non-interpersonal trauma), and cardiovascular outcomes. Furthermore, given racial and ethnic inequities in trauma exposure, risk of trauma-related psychopathology, access to treatment, and CVD risk, studies in diverse populations are critical [1, 234]. In addition, research is needed that extends beyond traditional diagnostic categories and investigates how transdiagnostic symptom dimensions relate to cardiovascular risk after trauma. Such an approach may identify key posttraumatic symptoms that may be targeted to reduce cardiovascular risk. For example, initial research suggests that fear-related symptoms after trauma may be particularly associated with cardiovascular risk [235,236,237].
Extensive research also suggests dysregulation of a variety of inter-related biological processes that may contribute to elevated cardiovascular risk both after trauma and in individuals with PTSD. Immune- and inflammation-related processes are some of the most well-studied and supported mechanisms to date, with evidence from meta-analyses and multiple omics studies. However, most investigations have been cross-sectional, and longitudinal research is needed to better understand risk processes after trauma. There is also considerable support for accelerated BA after trauma and in those with posttraumatic psychopathology, particularly for telomeres. Despite clear connections between stress-related biological systems that are dysregulated after trauma and in PTSD and the other biological mechanisms highlighted in this review (e.g., oxidative stress, mitochondrial dysfunction, RAS dysregulation), the latter processes have yet to be the subject of ample empirical study. Further, most studies have focused on early-life adversity—typically reported retrospectively—and sample sizes have generally been small and methods varied.
More comprehensive research on how experiences of trauma over the lifespan, and a range of posttraumatic psychopathology, relate to downstream biological processes is thus needed. Furthermore, although some research has considered potential sex differences in the traumatic stress-CVD relation [25, 26], research that extends this consideration to biological mechanisms is needed. For example, gonadal hormones (e.g., estradiol, testosterone) have documented effects on many of the biological mechanisms discussed in this review (e.g., inflammation, the RAS) [238, 239], and an important future direction is to consider sex differences in these mechanisms after trauma and potential consequences for CVD risk. Additionally, given the interconnections between processes, an integrative systems biology approach is likely to be valuable for understanding mechanisms contributing to cardiovascular risk. Preclinical experimental studies of trauma and PTSD offer elegant examples for studying processes across multiple levels of analysis and their impact on cardiovascular metrics [240, 241]. Although systems biology research is of growing interest in the traumatic stress field [242], this approach has yet to be implemented in trauma and CVD research. Additionally, examining dynamic changes in biological mechanisms in response to stress and trauma-related stimuli may shed light on how risk processes unfold—and how interventions may affect these processes [243, 244].
Ultimately, a more refined understanding of psychological and biological mechanisms can inform CVD intervention efforts. Initial evidence suggests that PTSD treatment may attenuate cardiovascular risk [245, 246], and a trial of gold-standard treatment for PTSD is underway to investigate potential impact on cardiovascular risk markers [247]. Although treating PTSD to improve mental health itself is an important goal, it is of interest to examine whether trauma-focused psychotherapies for PTSD may improve cardiovascular risk markers and related biological mechanisms directly or via reductions in posttraumatic psychopathology. Nevertheless, a longitudinal study in veterans did not find that clinically meaningful reductions in PTSD symptoms were associated with reduced incidence of CVD [248]. Investigating interventions that engage behavioral and/or biological mechanisms linking trauma with adverse cardiovascular health (e.g., anti-inflammatory or antioxidant treatments [95], physical activity interventions [249]) may also prove fruitful for developing multi-modal treatment approaches for reducing cardiovascular risk after trauma.
References
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–639.
Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25.
Everson-Rose SA, Lewis TT. Psychosocial factors and cardiovascular diseases. Annu Rev Public Health. 2005;26:469–500.
Niles AN, O’Donovan A. Comparing anxiety and depression to obesity and smoking as predictors of major medical illnesses and somatic symptoms. Health Psychol. 2019;38:172–81.
Scott KM, Koenen KC, Aguilar-Gaxiola S, Alonso J, Angermeyer MC, Benjet C, et al. Associations between lifetime traumatic events and subsequent chronic physical conditions: a cross-national, cross-sectional study. PLoS One. 2013;8:e80573.
Schnurr PP, Wachen JS, Green BL, Kaltman S. Trauma exposure, PTSD, and physical health. In Handbook of PTSD: Science and Practice (eds Friedman MJ, Keane TM, Resick PA) pp. 502–21 (Guilford Press, New York, 2014).
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–60.
Benjet C, Bromet E, Karam E, Kessler R, McLaughlin K, Ruscio A, et al. The epidemiology of traumatic event exposure worldwide: results from the World Mental Health Survey Consortium. Psychol Med. 2016;46:327–43.
Sledjeski EM, Speisman B, Dierker LC. Does number of lifetime traumas explain the relationship between PTSD and chronic medical conditions? Answers from the National Comorbidity Survey-Replication (NCS-R). J Behav Med. 2008;31:341–9.
Atwoli L, Platt JM, Basu A, Williams DR, Stein DJ, Koenen KC. Associations between lifetime potentially traumatic events and chronic physical conditions in the South African Stress and Health Survey: a cross-sectional study. BMC Psychiatry. 2016;16:214.
Husarewycz MN, El-Gabalawy R, Logsetty S, Sareen J. The association between number and type of traumatic life experiences and physical conditions in a nationally representative sample. Gen Hosp Psychiatry. 2014;36:26–32.
Sumner JA, Kubzansky LD, Elkind MS, Roberts AL, Agnew-Blais J, Chen Q, et al. Trauma exposure and posttraumatic stress disorder symptoms predict onset of cardiovascular events in women. Circulation. 2015;132:251–9.
Suglia SF, Koenen KC, Boynton-Jarrett R, Chan PS, Clark CJ, Danese A, et al. Childhood and adolescent adversity and cardiometabolic outcomes: a scientific statement from the American Heart Association. Circulation. 2018;137:e15–e28.
Levine GN, Cohen BE, Commodore-Mensah Y, Fleury J, Huffman JC, Khalid U, et al. Psychological health, well-being, and the mind-heart-body connection: a scientific statement from the American Heart Association. Circulation. 2021;143:763–83.
Galatzer-Levy IR, Huang SH, Bonanno GA. Trajectories of resilience and dysfunction following potential trauma: a review and statistical evaluation. Clin Psychol Rev. 2018;63:41–55.
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders 5th edn (American Psychiatric Association, Washington, DC, 2013).
Bryant RA. The current evidence for acute stress disorder. Curr Psychiatry Rep. 2018;20:111.
O’Donnell ML, Bryant RA, Creamer M, Carty J. Mental health following traumatic injury: toward a health system model of early psychological intervention. Clin Psychol Rev. 2008;28:387–406.
Goldmann E, Galea S. Mental health consequences of disasters. Annu Rev Public Health. 2014;35:169–83.
Asselmann E, Wittchen H-U, Lieb R, Perkonigg A, Beesdo-Baum K. Incident mental disorders in the aftermath of traumatic events: a prospective-longitudinal community study. J Affect Disord. 2018;227:82–89.
Walsh K, McLaughlin KA, Hamilton A, Keyes KM. Trauma exposure, incident psychiatric disorders, and disorder transitions in a longitudinal population representative sample. J Psychiatr Res. 2017;92:212–8.
Edmondson D, von Känel R. Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatry. 2017;4:320–9.
Edmondson D, Kronish IM, Shaffer JA, Falzon L, Burg MM. Posttraumatic stress disorder and risk for coronary heart disease: a meta-analytic review. Am Heart J. 2013;166:806–14.
Akosile W, Colquhoun D, Young R, Lawford B, Voisey J. The association between post-traumatic stress disorder and coronary artery disease: a meta-analysis. Australas Psychiatry. 2018;26:524–30.
Gradus JL, Farkas DK, Svensson E, Ehrenstein V, Lash TL, Milstein A, et al. Associations between stress disorders and cardiovascular disease events in the Danish population. BMJ Open. 2015;5:e009334.
Song H, Fang F, Arnberg FK, Mataix-Cols D, de la Cruz LF, Almqvist C, et al. Stress related disorders and risk of cardiovascular disease: population based, sibling controlled cohort study. BMJ. 2019;365:l1255.
Vaccarino V, Goldberg J, Rooks C, Shah AJ, Veledar E, Faber TL, et al. Post-traumatic stress disorder and incidence of coronary heart disease: a twin study. J Am Coll Cardiol. 2013;62:970–8.
Kubzansky LD, Koenen KC, Jones C, Eaton WW. A prospective study of posttraumatic stress disorder symptoms and coronary heart disease in women. Health Psychol. 2009;28:125–30.
Kubzansky LD, Koenen KC, Spiro A, Vokonas PS, Sparrow D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch Gen Psychiatry. 2007;64:109–16.
Boscarino JA. A prospective study of PTSD and early-age heart disease mortality among Vietnam veterans: implications for surveillance and prevention. Psychosom Med. 2008;70:668–76.
Rosman L, Lampert R, Ramsey CM, Dziura J, Chui PW, Brandt C, et al. Posttraumatic stress disorder and risk for early incident atrial fibrillation: a prospective cohort study of 1.1 million young adults. J Am Heart Assoc. 2019;8:e013741.
Rosman L, Sico JJ, Lampert R, Gaffey AE, Ramsey CM, Dziura J, et al. Posttraumatic stress disorder and risk for stroke in young and middle-aged adults: a 13-year cohort study. Stroke. 2019;50:2996–3003.
Roy SS, Foraker RE, Girton RA, Mansfield AJ. Posttraumatic stress disorder and incident heart failure among a community-based sample of US veterans. Am J Public Health. 2015;105:757–63.
Sumner JA, Kubzansky LD, Kabrhel C, Roberts AL, Chen Q, Winning A, et al. Associations of trauma exposure and posttraumatic stress symptoms with venous thromboembolism over 22 years in women. J Am Heart Assoc. 2016;5:e003197.
Ebrahimi R, Lynch KE, Beckham JC, Dennis PA, Viernes B, Tseng C-H, et al. Association of posttraumatic stress disorder and incident ischemic heart disease in women veterans. JAMA Cardiol. 2021;6:642–51.
Jordan HT, Miller-Archie SA, Cone JE, Morabia A, Stellman SD. Heart disease among adults exposed to the September 11, 2001 World Trade Center disaster: results from the World Trade Center Health Registry. Prev Med. 2011;53:370–6.
Sumner JA, Kubzansky LD, Roberts AL, Gilsanz P, Chen Q, Winning A, et al. Posttraumatic stress disorder symptoms and risk of hypertension over 22 years in a large cohort of younger and middle-aged women. Psychol Med. 2016;46:3105–16.
Suliman S, Anthonissen L, Carr J, du Plessis S, Emsley R, Hemmings SM, et al. Posttraumatic stress disorder, overweight, and obesity: a systematic review and meta-analysis. Harv Rev Psychiatry. 2016;24:271–93.
Vancampfort D, Rosenbaum S, Ward PB, Steel Z, Lederman O, Lamwaka AV, et al. Type 2 diabetes among people with posttraumatic stress disorder: systematic review and meta-analysis. Psychosom Med. 2016;78:465–73.
Džubur Kulenović A, Kučukalić A, Maleč D. Changes in plasma lipid concentrations and risk of coronary artery disease in army veterans suffering from chronic posttraumatic stress disorder. Croat Med J. 2008;49:506–14.
Appelman Y, van Rijn BB, Ten Haaf ME, Boersma E, Peters SA. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis. 2015;241:211–8.
O’Donnell CJ, Longacre LS, Cohen BE, Fayad ZA, Gillespie CF, Liberzon I, et al. Posttraumatic stress disorder and cardiovascular disease: state of the science, knowledge gaps, and research opportunities. JAMA Cardiol. 2021;6:1207–16.
Davies NM, Holmes MV, Smith GD. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601.
Seligowski AV, Misganaw B, Duffy LA, Ressler K, Guffanti G. Leveraging large-scale genetics of PTSD and cardiovascular disease demonstrates robust shared risk and improves risk prediction accuracy. Am J Psychiatry. 2022;179:814–23.
Polimanti R, Wendt FR, Pathak GA, Tylee DS, Tcheandjieu C, Hilliard AT, et al. Understanding the comorbidity between posttraumatic stress severity and coronary artery disease using genome-wide information and electronic health records. Mol Psychiatry. 2022;27:3961–9.
Nicholson A, Kuper H, Hemingway H. Depression as an aetiologic and prognostic factor in coronary heart disease: a meta-analysis of 6362 events among 146 538 participants in 54 observational studies. Eur Heart J. 2006;27:2763–74.
Batelaan NM, Seldenrijk A, Bot M, van Balkom AJ, Penninx BW. Anxiety and new onset of cardiovascular disease: critical review and meta-analysis. Br J Psychiatry. 2016;208:223–31.
Roerecke M, Rehm J. Chronic heavy drinking and ischaemic heart disease: a systematic review and meta-analysis. Open Heart. 2014;1:e000135.
Roberts AL, Kubzansky LD, Chibnik LB, Rimm EB, Koenen KC. Association of posttraumatic stress and depressive symptoms with mortality in women. JAMA Netw Open. 2020;3:e2027935.
van den Berk-Clark C, Secrest S, Walls J, Hallberg E, Lustman PJ, Schneider FD, et al. Association between posttraumatic stress disorder and lack of exercise, poor diet, obesity, and co-occuring smoking: a systematic review and meta-analysis. Health Psychol. 2018;37:407–16.
Zen AL, Whooley MA, Zhao S, Cohen BE. Post-traumatic stress disorder is associated with poor health behaviors: findings from the heart and soul study. Health Psychol. 2012;31:194–201.
McDevitt-Murphy ME, Murphy JG, Monahan CM, Flood AM, Weathers FW. Unique patterns of substance misuse associated with PTSD, depression, and social phobia. J Dual Diagn. 2010;6:94–110.
Bonnet F, Irving K, Terra J-L, Nony P, Berthezène F, Moulin P. Anxiety and depression are associated with unhealthy lifestyle in patients at risk of cardiovascular disease. Atherosclerosis. 2005;178:339–44.
Lasser K, Boyd JW, Woolhandler S, Himmelstein DU, McCormick D, Bor DH. Smoking and mental illness: a population-based prevalence study. JAMA. 2000;284:2606–10.
Roshanaei-Moghaddam B, Katon WJ, Russo J. The longitudinal effects of depression on physical activity. Gen Hosp Psychiatry. 2009;31:306–15.
Farr OM, Sloan DM, Keane TM, Mantzoros CS. Stress- and PTSD-associated obesity and metabolic dysfunction: a growing problem requiring further research and novel treatments. Metabolism. 2014;63:1463–8.
Hoerster KD, Campbell S, Dolan M, Stappenbeck CA, Yard S, Simpson T, et al. PTSD is associated with poor health behavior and greater body mass index through depression, increasing cardiovascular disease and diabetes risk among US veterans. Prev Med Rep. 2019;15:100930.
Yehuda R. Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann N. Y Acad Sci. 2006;1071:137–66.
Sherin JE, Nemeroff CB. Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci. 2011;13:263–78.
Pitman RK, Rasmusson AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, et al. Biological studies of post-traumatic stress disorder. Nat Rev Neurosci. 2012;13:769–87.
Schneider M, Schwedtfeger A. Autonomic dysfunction in posttraumatic stress disorder indexed by heart rate variability: a meta-analysis. Psychol Med. 2020;50:1937–48.
Haft JI. Cardiovascular injury induced by sympathetic catecholamines. Prog Cardiovasc Dis. 1974;17:73–86.
Brudey C, Park J, Wiaderkiewicz J, Kobayashi I, Mellman TA, Marvar PJ. Autonomic and inflammatory consequences of posttraumatic stress disorder and the link to cardiovascular disease. Am J Physiol Regul Integr Comp Physiol. 2015;309:R315–21.
Jovanovic T, Norrholm SD, Blanding NQ, Davis M, Duncan E, Bradley B, et al. Impaired fear inhibition is a biomarker of PTSD but not depression. Depress Anxiety. 2010;27:244–51.
Pollard HB, Shivakumar C, Starr J, Eidelman O, Jacobowitz DM, Dalgard CL, et al. “Soldier’s heart”: a genetic basis for elevated cardiovascular disease risk associated with post-traumatic stress disorder. Front Mol Neurosci. 2016;9:1–12.
Sumner JA, Duncan LE, Wolf EJ, Amstadter AB, Baker DG, Beckham JC, et al. Posttraumatic stress disorder has genetic overlap with cardiometabolic traits. Psychol Med. 2017;47:2036–9.
D’Andrea W, Sharma R, Zelechoski AD, Spinazzola J. Physical health problems after single trauma exposure: when stress takes root in the body. J Am Psychiatr Nurses Assoc. 2011;17:378–92.
Yehuda R, Flory JD, Pratchett LC, Buxbaum J, Ising M, Holsboer F. Putative biological mechanisms for the association between early life adversity and the subsequent development of PTSD. Psychopharmacology. 2010;212:405–17.
Beurel E, Toups M, Nemeroff CB. The bidirectional relationship of depression and inflammation: double trouble. Neuron. 2020;107:234–56.
Palta P, Samuel LJ, Miller ER III, Szanton SL. Depression and oxidative stress: results from a meta-analysis of observational studies. Psychosom Med. 2014;76:12–19.
Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol. 2017;18:826–31.
Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43.
Emerging Risk Factors Collaboration. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N. Engl J Med. 2012;367:1310–20.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2:1002–12.
Tursich M, Neufeld R, Frewen P, Harricharan S, Kibler J, Rhind S, et al. Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry. 2014;4:e413.
Tawakol A, Ishai A, Takx RA, Figueroa AL, Ali A, Kaiser Y, et al. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet. 2017;389:834–45.
Toczek J, Hillmer AT, Han J, Liu C, Peters D, Emami H, et al. FDG PET imaging of vascular inflammation in post-traumatic stress disorder: a pilot case–control study. J Nucl Cardiol. 2021;28:688–94.
Gola H, Engler H, Sommershof A, Adenauer H, Kolassa S, Schedlowski M, et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry. 2013;13:40.
Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol Psychiatry. 2016;21:642–9.
Speer K, Upton D, Semple S, McKune A. Systemic low-grade inflammation in post-traumatic stress disorder: a systematic review. J Inflamm Res. 2018;11:111–21.
Yang J-J, Jiang W. Immune biomarkers alterations in post-traumatic stress disorder: a systematic review and meta-analysis. J Affect Disord. 2020;268:39–46.
Peruzzolo TL, Pinto JV, Roza TH, Shintani AO, Anzolin AP, Gnielka V, et al. Inflammatory and oxidative stress markers in post-traumatic stress disorder: a systematic review and meta-analysis. Mol Psychiatry. 2022;27:3150–63.
Sumner JA, Nishimi KM, Koenen KC, Roberts AL, Kubzansky LD. Posttraumatic stress disorder and inflammation: untangling issues of bidirectionality. Biol Psychiatry. 2020;87:885–97.
Zass LJ, Hart SA, Seedat S, Hemmings SM, Malan-Müller S. Neuroinflammatory genes associated with post-traumatic stress disorder: implications for comorbidity. Psychiatr Genet. 2017;27:1–16.
Mehta D, Miller O, Bruenig D, David G, Shakespeare‐Finch J. A systematic review of DNA methylation and gene expression studies in posttraumatic stress disorder, posttraumatic growth, and resilience. J Trauma Stress. 2020;33:171–80.
Katrinli S, Zheng Y, Gautam A, Hammamieh R, Yang R, Venkateswaran S, et al. PTSD is associated with increased DNA methylation across regions of HLA-DPB1 and SPATC1L. Brain Behav Immun. 2021;91:429–36.
Miller M, Maniates H, Wolf E, Logue M, Schichman S, Stone A, et al. CRP polymorphisms and DNA methylation of the AIM2 gene influence associations between trauma exposure, PTSD, and C-reactive protein. Brain Behav Immun. 2018;67:194–202.
Hawn SE, Neale Z, Wolf EJ, Zhao X, Pierce M, Fein‐Schaffer D, et al. Methylation of the AIM2 gene: an epigenetic mediator of PTSD‐related inflammation and neuropathology plasma biomarkers. Depress Anxiety. 2022;39:323–33.
Young RM, Lawford B, Mellor R, Morris CP, Voisey J, McLeay S, et al. Investigation of C-reactive protein and AIM2 methylation as a marker for PTSD in Australian Vietnam veterans. Gene. 2021;803:145898.
Carvalho CM, Wendt FR, Maihofer AX, Stein DJ, Stein MB, Sumner JA, et al. Dissecting the genetic association of C-reactive protein with PTSD, traumatic events, and social support. Neuropsychopharmacology. 2021;46:1071–7.
Karabatsiakis A, Hamuni G, Wilker S, Kolassa S, Renu D, Kadereit S, et al. Metabolite profiling in posttraumatic stress disorder. J Mol Psychiatry. 2015;3:2.
Kuan P-F, Yang X, Kotov R, Clouston S, Bromet E, Luft BJ. Metabolomics analysis of post-traumatic stress disorder symptoms in World Trade Center responders. Transl Psychiatry. 2022;12:174.
Konjevod M, Erjavec GN, Perkovic MN, Sáiz J, Tudor L, Uzun S, et al. Metabolomics in posttraumatic stress disorder: untargeted metabolomic analysis of plasma samples from Croatian war veterans. Free Radic Biol Med. 2021;162:636–41.
Karanikas E, Daskalakis NP, Agorastos A. Oxidative dysregulation in early life stress and posttraumatic stress disorder: a comprehensive review. Brain Sci. 2021;11:723.
Miller MW, Lin AP, Wolf EJ, Miller DR. Oxidative stress, inflammation, and neuroprogression in chronic PTSD. Harv Rev Psychiatry. 2018;26:57–69.
Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47.
Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713–35.
Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med. 2014;24:165–9.
Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, et al. The assessment of endothelial function: from research into clinical practice. Circulation. 2012;126:753–67.
Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D, et al. Clinical relevance of biomarkers of oxidative stress. Antioxid Redox Signal. 2015;23:1144–70.
Marrocco I, Altieri F, Peluso I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid Med Cell Longev. 2017;2017:6501046.
Miller MW, Sadeh N. Traumatic stress, oxidative stress and post-traumatic stress disorder: neurodegeneration and the accelerated-aging hypothesis. Mol Psychiatry. 2014;19:1156–62.
Aschbacher K, O’Donovan A, Wolkowitz OM, Dhabhar FS, Su Y, Epel E. Good stress, bad stress and oxidative stress: insights from anticipatory cortisol reactivity. Psychoneuroendocrinology. 2013;38:1698–708.
Nakhaee A, Shahabizadeh F, Erfani M. Protein and lipid oxidative damage in healthy students during and after exam stress. Physiol Behav. 2013;118:118–21.
Horn SR, Leve LD, Levitt P, Fisher PA. Childhood adversity, mental health, and oxidative stress: a pilot study. PLoS One. 2019;14:e0215085.
do Prado CH, Grassi-Oliveira R, Wieck A, Zaparte A, Daruy Filho L, da Silva, et al. The impact of childhood maltreatment on redox state: relationship with oxidative damage and antioxidant defenses in adolescents with no psychiatric disorder. Neurosci Lett. 2016;617:173–7.
Boeck C, Koenig AM, Schury K, Geiger ML, Karabatsiakis A, Wilker S, et al. Inflammation in adult women with a history of child maltreatment: the involvement of mitochondrial alterations and oxidative stress. Mitochondrion. 2016;30:197–207.
Şimşek Ş, Kaplan I, Uysal C, Yüksel T, Alaca R. The levels of cortisol, oxidative stress, and DNA damage in the victims of childhood sexual abuse: a preliminary study. J Child Sex Abus. 2016;25:175–84.
Fanning JR, Lee R, Gozal D, Coussons-Read M, Coccaro EF. Childhood trauma and parental style: relationship with markers of inflammation, oxidative stress, and aggression in healthy and personality disordered subjects. Biol Psychol. 2015;112:56–65.
Atli A, Bulut M, Bez Y, Kaplan I, Özdemir PG, Uysal C, et al. Altered lipid peroxidation markers are related to post-traumatic stress disorder (PTSD) and not trauma itself in earthquake survivors. Eur Arch Psychiatry Clin Neurosci. 2016;266:329–36.
Tezcan E, Atmaca M, Kuloglu M, Ustundag B. Free radicals in patients with post-traumatic stress disorder. Eur Arch Psychiatry Clin Neurosci. 2003;253:89–91.
Ogłodek EA. Evaluation of ADMA, carbonyl groups, CAT and NKA in depressed patients with and without posttraumatic stress disorder. Pharm Rep. 2017;69:730–7.
Ogłodek EA. The role of PON-1, GR, IL-18, and OxLDL in depression with and without posttraumatic stress disorder. Pharm Rep. 2017;69:837–45.
Ogłodek EA. Changes in the concentrations of inflammatory and oxidative status biomediators (MIP-1 α, PMN elastase, MDA, and IL-12) in depressed patients with and without posttraumatic stress disorder. Pharm Rep. 2018;70:110–8.
Borovac Štefanović L, Kalinić D, Mimica N, Beer Ljubić B, Aladrović J, Mandelsamen Perica M, et al. Oxidative status and the severity of clinical symptoms in patients with post-traumatic stress disorder. Ann Clin Biochem. 2015;52:95–104.
Čeprnja M, Đerek L, Unić A, Blažev M, Fistonić M, Kozarić-Kovačić D, et al. Oxidative stress markers in patients with post-traumatic stress disorder. Coll Antropol. 2011;35:1155–60.
Perković MN, Milković L, Uzun S, Mimica N, Pivac N, Waeg G, et al. Association of lipid peroxidation product 4-hydroxynonenal with post-traumatic stress disorder. Biomolecules. 2021;11:1365.
Bersani FS, Wolkowitz OM, Lindqvist D, Yehuda R, Flory J, Bierer LM, et al. Global arginine bioavailability, a marker of nitric oxide synthetic capacity, is decreased in PTSD and correlated with symptom severity and markers of inflammation. Brain Behav Immun. 2016;52:153–60.
Michels L, Schulte-Vels T, Schick M, O’Gorman RL, Zeffiro T, Hasler G, et al. Prefrontal GABA and glutathione imbalance in posttraumatic stress disorder: preliminary findings. Psychiatry Res Neuroimaging. 2014;224:288–95.
Ozdemir PG, Kaplan I, Uysal C, Bulut M, Atli A, Bez Y, et al. Serum total oxidant and antioxidant status in earthquake survivors with post-traumatic stress disorder. Acta Neuropsychiatr. 2015;27:153–8.
Şimşek Ş, Yüksel T, Kaplan I, Uysal C, Aktaş H. The levels of cortisol and oxidative stress and DNA damage in child and adolescent victims of sexual abuse with or without post-traumatic stress disorder. Psychiatry Investig. 2016;13:616–21.
Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, Reardon AF, et al. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol Psychiatry. 2013;18:937–42.
Katrinli S, Maihofer AX, Wani AH, Pfeiffer JR, Ketema E, Ratanatharathorn A, et al. Epigenome-wide meta-analysis of PTSD symptom severity in three military cohorts implicates DNA methylation changes in genes involved in immune system and oxidative stress. Mol Psychiatry. 2022;27:1720–8.
Zieker J, Zieker D, Jatzko A, Dietzsch J, Nieselt K, Schmitt A, et al. Differential gene expression in peripheral blood of patients suffering from post-traumatic stress disorder. Mol Psychiatry. 2007;12:116–8.
Su YA, Wu J, Zhang L, Zhang Q, Su DM, He P, et al. Dysregulated mitochondrial genes and networks with drug targets in postmortem brain of patients with posttraumatic stress disorder (PTSD) revealed by human mitochondria-focused cDNA microarrays. Int J Biol Sci. 2008;4:223–35.
Mellon SH, Bersani FS, Lindqvist D, Hammamieh R, Donohue D, Dean K, et al. Metabolomic analysis of male combat veterans with post traumatic stress disorder. PLoS One. 2019;14:e0213839.
Picard M, McEwen BS. Psychological stress and mitochondria: a conceptual framework. Psychosom Med. 2018;80:126–40.
Zhunina OA, Yabbarov NG, Grechko AV, Starodubova AV, Ivanova E, Nikiforov NG, et al. The role of mitochondrial dysfunction in vascular disease, tumorigenesis, and diabetes. Front Mol Biosci. 2021;8:671908.
Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, et al. Mitochondria and cardiovascular diseases—from pathophysiology to treatment. Ann Transl Med. 2018;6:256.
Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol. 2013;305:459–76.
Picard M, McEwen BS. Psychological stress and mitochondria: a systematic review. Psychosom Med. 2018;80:141–53.
Sasaki S. Determination of altered mitochondria ultrastructure by electron microscopy. Methods Mol Biol. 2010;648:279–90.
Picard M, Prather AA, Puterman E, Cuillerier A, Coccia M, Aschbacher K, et al. A mitochondrial health index sensitive to mood and caregiving stress. Biol Psychiatry. 2018;84:9–17.
Eigentler A, Draxl A, Wiethüchter A, Kuznetsov A, Lassing B, Gnaiger E Laboratory protocol: citrate synthase, a mitochondrial marker enzyme. Mitochondrial Physiol Network http://wiki.oroboros.at/index.php/MiPNet17.04_CitrateSynthase (2020).
Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–60.
Ridout KK, Khan M, Ridout SJ. Adverse childhood experiences run deep: toxic early life stress, telomeres, and mitochondrial DNA copy number, the biological markers of cumulative stress. Bioessays. 2018;40:e1800077.
Gumpp AM, Boeck C, Behnke A, Bach AM, Ramo-Fernández L, Welz T, et al. Childhood maltreatment is associated with changes in mitochondrial bioenergetics in maternal, but not in neonatal immune cells. Proc Natl Acad Sci USA. 2020;117:24778–84.
Gumpp AM, Behnke A, Ramo-Fernández L, Radermacher P, Gündel H, Ziegenhain U et al. Investigating mitochondrial bioenergetics in peripheral blood mononuclear cells of women with childhood maltreatment from post-parturition period to one-year follow-up. Psychol Med. https://doi.org/10.1017/S0033291722000411 (2022).
Tyrka AR, Parade SH, Price LH, Kao H-T, Porton B, Philip NS, et al. Alterations of mitochondrial DNA copy number and telomere length with early adversity and psychopathology. Biol Psychiatry. 2016;79:78–86.
Ridout KK, Parade SH, Kao H-T, Magnan S, Seifer R, Porton B, et al. Childhood maltreatment, behavioral adjustment, and molecular markers of cellular aging in preschool-aged children: a cohort study. Psychoneuroendocrinology. 2019;107:261–9.
Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, et al. Molecular signatures of major depression. Curr Biol. 2015;25:1146–56.
Brunst KJ, Sanchez-Guerra M, Chiu Y-HM, Wilson A, Coull BA, Kloog I, et al. Prenatal particulate matter exposure and mitochondrial dysfunction at the maternal-fetal interface: effect modification by maternal lifetime trauma and child sex. Environ Int. 2018;112:49–58.
Cai N, Fňašková M, Konečná K, Fojtová M, Fajkus J, Coomber E, et al. No evidence of persistence or inheritance of mitochondrial DNA copy number in holocaust survivors and their descendants. Front Genet. 2020;11:87.
Lam NY, Rainer TH, Chiu RW, Joynt GM, Lo YD. Plasma mitochondrial DNA concentrations after trauma. Clin Chem. 2004;50:213–6.
Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS One. 2013;8:e72834.
Morrison KE, Stenson AF, Marx-Rattner R, Carter S, Michopoulos V, Gillespie CF, et al. Developmental timing of trauma in women predicts unique extracellular vesicle proteome signatures. Biol Psychiatry. 2022;91:273–82.
Bersani FS, Morley C, Lindqvist D, Epel ES, Picard M, Yehuda R, et al. Mitochondrial DNA copy number is reduced in male combat veterans with PTSD. Prog Neuropsychopharmacol Biol Psychiatry. 2016;64:10–17.
Brunst KJ, Sanchez Guerra M, Gennings C, Hacker M, Jara C, Bosquet Enlow M, et al. Maternal lifetime stress and prenatal psychological functioning and decreased placental mitochondrial DNA copy number in the PRISM study. Am J Epidemiol. 2017;186:1227–36.
Flaquer A, Baumbach C, Ladwig K, Kriebel J, Waldenberger M, Grallert H, et al. Mitochondrial genetic variants identified to be associated with posttraumatic stress disorder. Transl Psychiatry. 2015;5:e524.
Hammamieh R, Chakraborty N, Gautam A, Muhie S, Yang R, Donohue D, et al. Whole-genome DNA methylation status associated with clinical PTSD measures of OIF/OEF veterans. Transl Psychiatry. 2017;7:e1169.
Seligowski AV, Webber TK, Marvar PJ, Ressler KJ, Philip NS. Involvement of the brain–heart axis in the link between PTSD and cardiovascular disease. Depress Anxiety. 2022;39:663–74.
Patel S, Rauf A, Khan H, Abu-Izneid T. Renin-angiotensin-aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. 2017;94:317–25.
Gideon A, Sauter C, Fieres J, Berger T, Renner B, Wirtz PH. Kinetics and interrelations of the renin aldosterone response to acute psychosocial stress: a neglected stress system. J Clin Endocrinol Metab. 2020;105:762–73.
Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369:1208–19.
Alderman MH, Madhavan S, Ooi WL, Cohen H, Sealey JE, Laragh JH. Association of the renin-sodium profile with the risk of myocardial infarction in patients with hypertension. N. Engl J Med. 1991;324:1098–104.
Campbell DJ, Woodward M, Chalmers JP, Colman SA, Jenkins AJ, Kemp BE, et al. Prediction of myocardial infarction by N-terminal-pro-B-type natriuretic peptide, C-reactive protein, and renin in subjects with cerebrovascular disease. Circulation. 2005;112:110–6.
Campbell DJ, Nussberger J, Stowasser M, Danser AJ, Morganti A, Frandsen E, et al. Activity assays and immunoassays for plasma renin and prorenin: information provided and precautions necessary for accurate measurement. Clin Chem. 2009;55:867–77.
Cartledge S, Lawson N. Aldosterone and renin measurements. Ann Clin Biochem. 2000;37:262–78.
Chappell MC, Pirro NT, South AM, Gwathmey TM. Concerns on the specificity of commercial ELISAs for the measurement of angiotensin (1–7) and angiotensin II in human plasma. Hypertension. 2021;77:29–31.
Terock J, Hannemann A, Janowitz D, Van der Auwera S, Bahls M, Völzke H, et al. Differential activation of the renin-angiotensin-aldosterone-system in response to childhood and adulthood trauma. Psychoneuroendocrinology. 2019;107:232–40.
Terock J, Hannemann A, Janowitz D, Freyberger HJ, Felix SB, Dörr M, et al. Associations of trauma exposure and post-traumatic stress disorder with the activity of the renin–angiotensin–aldosterone-system in the general population. Psychol Med. 2019;49:843–51.
Nishimi K, Adler GK, Roberts AL, Sumner JA, Jung SJ, Chen Q, et al. Associations of trauma and posttraumatic stress disorder with aldosterone in women. Psychoneuroendocrinology. 2021;132:105341.
Khoury NM, Marvar PJ, Gillespie CF, Wingo A, Schwartz A, Bradley B, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73:849–55.
Seligowski AV, Duffy LA, Merker JB, Michopoulos V, Gillespie CF, Marvar PJ, et al. The renin–angiotensin system in PTSD: a replication and extension. Neuropsychopharmacology. 2021;46:750–5.
Nylocks K, Michopoulos V, Rothbaum AO, Almli L, Gillespie C, Wingo A, et al. An angiotensin‐converting enzyme (ACE) polymorphism may mitigate the effects of angiotensin‐pathway medications on posttraumatic stress symptoms. Am J Med Genet B Neuropsychiatr Genet. 2015;168:307–15.
Stein MB, Jain S, Simon NM, West JC, Marvar PJ, Bui E, et al. Randomized, placebo-controlled trial of the angiotensin receptor antagonist losartan for posttraumatic stress disorder. Biol Psychiatry. 2021;90:473–81.
Wolf EJ, Schnurr PP. Posttraumatic stress disorder-related cardiovascular disease and accelerated cellular aging. Psychiatr Ann. 2016;46:527–32.
Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–8.
Rode L, Nordestgaard BG, Bojesen SE. Peripheral blood leukocyte telomere length and mortality among 64 637 individuals from the general population. J Natl Cancer Inst. 2015;107:djv074.
Willeit P, Willeit J, Brandstätter A, Ehrlenbach S, Mayr A, Gasperi A, et al. Cellular aging reflected by leukocyte telomere length predicts advanced atherosclerosis and cardiovascular disease risk. Arterioscler Thromb Vasc Biol. 2010;30:1649–56.
Scheller Madrid A, Rode L, Nordestgaard BG, Bojesen SE. Short telomere length and ischemic heart disease: observational and genetic studies in 290 022 individuals. Clin Chem. 2016;62:1140–9.
Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–84.
Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai P-C, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging. 2016;8:1844–59.
Roetker NS, Pankow JS, Bressler J, Morrison AC, Boerwinkle E. Prospective study of epigenetic age acceleration and incidence of cardiovascular disease outcomes in the ARIC study (Atherosclerosis Risk in Communities). Circ Genom Precis Med. 2018;11:e001937.
Levine ME. Modeling the rate of senescence: can estimated biological age predict mortality more accurately than chronological age? J Gerontol A Biol Sci Med Sci. 2013;68:667–74.
Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10:573–91.
Liu Z, Kuo P-L, Horvath S, Crimmins E, Ferrucci L, Levine M. A new aging measure captures morbidity and mortality risk across diverse subpopulations from NHANES IV: a cohort study. PLoS Med. 2018;15:e1002718.
Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, et al. Telomere length: a review of methods for measurement. Nurs Res. 2014;63:289–99.
Lin J, Smith DL, Esteves K, Drury S. Telomere length measurement by qPCR–Summary of critical factors and recommendations for assay design. Psychoneuroendocrinology. 2019;99:271–8.
Martin-Ruiz CM, Baird D, Roger L, Boukamp P, Krunic D, Cawthon R, et al. Reproducibility of telomere length assessment: an international collaborative study. Int J Epidemiol. 2015;44:1673–83.
Aubert G, Hills M, Lansdorp PM. Telomere length measurement—caveats and a critical assessment of the available technologies and tools. Mutat Res. 2012;730:59–67.
Lu AT, Seeboth A, Tsai P-C, Sun D, Quach A, Reiner AP, et al. DNA methylation-based estimator of telomere length. Aging. 2019;11:5895–923.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:115.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67.
Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11:303–27.
Belsky DW, Caspi A, Corcoran DL, Sugden K, Poulton R, Arseneault L, et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife. 2022;11:1–26.
Kwon D, Belsky DW. A toolkit for quantification of biological age from blood chemistry and organ function test data: BioAge. GeroScience. 2021;43:2795–808.
Belsky DW, Moffitt TE, Cohen AA, Corcoran DL, Levine ME, Prinz JA, et al. Eleven telomere, epigenetic clock, and biomarker-composite quantifications of biological aging: do they measure the same thing? Am J Epidemiol. 2018;187:1220–30.
Coimbra BM, Carvalho CM, Moretti PN, Mello MF, Belangero SI. Stress-related telomere length in children: a systematic review. J Psychiatr Res. 2017;92:47–54.
Ridout KK, Levandowski M, Ridout SJ, Gantz L, Goonan K, Palermo D, et al. Early life adversity and telomere length: a meta-analysis. Mol Psychiatry. 2018;23:858–71.
Li X, Wang J, Zhou J, Huang P, Li J. The association between post-traumatic stress disorder and shorter telomere length: a systematic review and meta-analysis. J Affect Disord. 2017;218:322–6.
Nelles-McGee T, Khoury J, Kenny M, Joshi D, Gonzalez A. Biological embedding of child maltreatment: a systematic review of biomarkers and resilience in children and youth. Psychol Trauma. 2022;14:S50–62.
Bürgin D, O’Donovan A, d’Huart D, Di Gallo A, Eckert A, Fegert J, et al. Adverse childhood experiences and telomere length a look into the heterogeneity of findings—a narrative review. Front Neurosci. 2019;13:490.
Hanssen LM, Schutte NS, Malouff JM, Epel ES. The relationship between childhood psychosocial stressor level and telomere length: a meta-analysis. Health Psychol Res. 2017;5:6378.
Colich NL, Rosen ML, Williams ES, McLaughlin KA. Biological aging in childhood and adolescence following experiences of threat and deprivation: a systematic review and meta-analysis. Psychol Bull. 2020;146:721–64.
Aas M, Elvsåshagen T, Westlye LT, Kaufmann T, Athanasiu L, Djurovic S, et al. Telomere length is associated with childhood trauma in patients with severe mental disorders. Transl Psychiatry. 2019;9:97.
Boeck C, Krause S, Karabatsiakis A, Schury K, Gündel H, Waller C, et al. History of child maltreatment and telomere length in immune cell subsets: associations with stress-and attachment-related hormones. Dev Psychopathol. 2018;30:539–51.
Edmonds GW, Hampson SE, Côté HC, Hill PL, Klest B. Childhood personality, betrayal trauma, and leukocyte telomere length in adulthood: a lifespan perspective on conscientiousness and betrayal traumas as predictors of a biomarker of cellular ageing. Eur J Pers. 2016;30:426–37.
Robakis TK, Zhang S, Rasgon NL, Li T, Wang T, Roth MC, et al. Epigenetic signatures of attachment insecurity and childhood adversity provide evidence for role transition in the pathogenesis of perinatal depression. Transl Psychiatry. 2020;10:48.
Vincent J, Hovatta I, Frissa S, Goodwin L, Hotopf M, Hatch SL, et al. Assessing the contributions of childhood maltreatment subtypes and depression case-control status on telomere length reveals a specific role of physical neglect. J Affect Disord. 2017;213:16–22.
Elam KK, Johnson SL, Ruof A, Eisenberg DT, Rej PH, Sandler I, et al. Examining the influence of adversity, family contexts, and a family-based intervention on parent and child telomere length. Eur J Psychotraumatol. 2022;13:2088935.
Kuehl L, De Punder K, Deuter C, Martens D, Heim C, Otte C, et al. Telomere length in individuals with and without major depression and adverse childhood experiences. Psychoneuroendocrinology. 2022;142:105762.
Rungnirundorn T, Krusong K, Kalayasiri R, Maes M. Leukocyte telomere length is not shortened in methamphetamine dependence or methamphetamine-induced psychosis but is increased following traumatic events. World J Biol Psychiatry. 2022;23:613–21.
Jones CW, Esteves KC, Gray SA, Clarke TN, Callerame K, Theall KP, et al. The transgenerational transmission of maternal adverse childhood experiences (ACEs): insights from placental aging and infant autonomic nervous system reactivity. Psychoneuroendocrinology. 2019;106:20–27.
Etzel L, Hastings WJ, Mattern BC, Oxford ML, Heim C, Putnam FW, et al. Intergenerational transmission of childhood trauma? Testing cellular aging in mothers exposed to sexual abuse and their children. Psychoneuroendocrinology. 2020;120:104781.
Izano MA, Cushing LJ, Lin J, Eick SM, Goin DE, Epel E, et al. The association of maternal psychosocial stress with newborn telomere length. PLoS One. 2020;15:e0242064.
Boks MP, van Mierlo HC, Rutten BP, Radstake TR, De Witte L, Geuze E, et al. Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology. 2015;51:506–12.
Stein JY, Levin Y, Uziel O, Abumock H, Solomon Z. Traumatic stress and cellular senescence: the role of war-captivity and homecoming stressors in later life telomere length. J Affect Disord. 2018;238:129–35.
Howard JT, Janak JC, Santos-Lozada AR, McEvilla S, Ansley SD, Walker LE, et al. Telomere shortening and accelerated aging in US military veterans. Int J Environ Res Public Health. 2021;18:1743.
Pousa PA, Souza RM, Melo PHM, Correa BH, Mendonça TS, Simões-e-Silva AC, et al. Telomere shortening and psychiatric disorders: a systematic review. Cells. 2021;10:1423.
Connolly SL, Stoop TB, Logue MW, Orr EH, De Vivo I, Miller MW, et al. Posttraumatic stress disorder symptoms, temperament, and the pathway to cellular senescence. J Trauma Stress. 2018;31:676–86.
Carvalho CM, Coimbra BM, Xavier G, Bugiga AV, Fonseca T, Olff M, et al. Shorter telomeres related to posttraumatic stress disorder re-experiencing symptoms in sexually assaulted civilian women. Front Psychiatry. 2022;13:835783.
Küffer AL, O’Donovan A, Burri A, Maercker A. Posttraumatic stress disorder, adverse childhood events, and buccal cell telomere length in elderly Swiss former indentured child laborers. Front Psychiatry. 2016;7:147.
Womersley JS, Xulu KR, Sommer J, Hinsberger M, Kidd M, Elbert T, et al. Associations between telomere length and symptoms of posttraumatic stress disorder and appetitive aggression in trauma-exposed men. Neurosci Lett. 2022;769:136388.
Lahav Y, Avidor S, Stein JY, Zhou X, Solomon Z. Telomere length and depression among ex-prisoners of war: the role of subjective age. J Gerontol B Psychol Sci Soc Sci. 2020;75:21–29.
Ratanatharathorn A, Roberts AL, Chibnik L, Choi K, De Vivo I, Kim Y et al. PTSD, depression and accelerated aging: leukocyte telomere length in the Nurses’ Health Study 2. Biol Psychiatry Glob Open Sci. https://doi.org/10.1016/j.bpsgos.2022.05.006 (2022).
Lim S, Nzegwu D, Wright ML. The impact of psychosocial stress from life trauma and racial discrimination on epigenetic aging—a systematic review. Biol Res Nurs. 2022;24:202–15.
Wolf EJ, Maniates H, Nugent N, Maihofer AX, Armstrong D, Ratanatharathorn A, et al. Traumatic stress and accelerated DNA methylation age: a meta-analysis. Psychoneuroendocrinology. 2018;92:123–34.
McCrory C, Fiorito G, O’Halloran AM, Polidoro S, Vineis P, Kenny RA. Early life adversity and age acceleration at mid-life and older ages indexed using the next-generation GrimAge and Pace of Aging epigenetic clocks. Psychoneuroendocrinology. 2022;137:105643.
Copeland WE, Shanahan L, McGinnis EW, Aberg KA, van den Oord EJ. Early adversities accelerate epigenetic aging into adulthood: a 10‐year, within‐subject analysis. J Child Psychol Psychiatry. 2022;63:1308–15.
Marini S, Davis KA, Soare TW, Zhu Y, Suderman MJ, Simpkin AJ, et al. Adversity exposure during sensitive periods predicts accelerated epigenetic aging in children. Psychoneuroendocrinology. 2020;113:104484.
Tang R, Howe LD, Suderman M, Relton CL, Crawford AA, Houtepen LC. Adverse childhood experiences, DNA methylation age acceleration, and cortisol in UK children: a prospective population-based cohort study. Clin Epigenetics. 2020;12:55.
McKenna B, Mekawi Y, Katrinli S, Carter S, Stevens JS, Powers A, et al. When anger remains unspoken: anger and accelerated epigenetic aging among stress-exposed black Americans. Psychosom Med. 2021;83:949–58.
Reed K, Cleveland S, Thomas J, Hsu A, Jeong A, Nguyen J et al. in Epigenetics of Stress and Stress Disorders (ed Youssef N) Ch 10 (Academic Press, San Diego, 2022).
Kuan P-F, Ren X, Clouston S, Yang X, Jonas K, Kotov R, et al. PTSD is associated with accelerated transcriptional aging in World Trade Center responders. Transl Psychiatry. 2021;11:311.
Mehta D, Bruenig D, Pierce J, Sathyanarayanan A, Stringfellow R, Miller O, et al. Recalibrating the epigenetic clock after exposure to trauma: the role of risk and protective psychosocial factors. J Psychiatr Res. 2022;149:374–81.
Shenk CE, O’Donnell KJ, Pokhvisneva I, Kobor MS, Meaney MJ, Bensman HE, et al. Epigenetic age acceleration and risk for posttraumatic stress disorder following exposure to substantiated child maltreatment. J Clin Child Adolesc Psychol. 2022;51:651–61.
Wang Z, Hui Q, Goldberg J, Smith N, Kaseer B, Murrah N, et al. Association between posttraumatic stress disorder and epigenetic age acceleration in a sample of twins. Psychosom Med. 2022;84:151–8.
Wolf EJ, Logue MW, Morrison FG, Wilcox ES, Stone A, Schichman SA, et al. Posttraumatic psychopathology and the pace of the epigenetic clock: a longitudinal investigation. Psychol Med. 2019;49:791–800.
Graf GH-J, Li X, Kwon D, Belsky D, Widom CS. Biological aging in maltreated children followed up into middle adulthood. Psychoneuroendocrinology. 2022;143:105848.
Belsky DW, Caspi A, Cohen HJ, Kraus WE, Ramrakha S, Poulton R, et al. Impact of early personal‐history characteristics on the Pace of Aging: implications for clinical trials of therapies to slow aging and extend healthspan. Aging Cell. 2017;16:644–51.
Mian O, Belsky DW, Cohen AA, Anderson LN, Gonzalez A, Ma J, et al. Associations between exposure to adverse childhood experiences and biological aging: evidence from the Canadian Longitudinal Study on Aging. Psychoneuroendocrinology. 2022;142:105821.
Sumner JA, Chen Q, Roberts AL, Winning A, Rimm EB, Gilsanz P, et al. Cross-sectional and longitudinal associations of chronic posttraumatic stress disorder with inflammatory and endothelial function markers in women. Biol Psychiatry. 2017;82:875–84.
Roberts AL, Gilman SE, Breslau J, Breslau N, Koenen KC. Race/ethnic differences in exposure to traumatic events, development of post-traumatic stress disorder, and treatment-seeking for post-traumatic stress disorder in the United States. Psychol Med. 2011;41:71–83.
Ho GW, Karatzias T, Vallières F, Bondjers K, Shevlin M, Cloitre M, et al. Complex PTSD symptoms mediate the association between childhood trauma and physical health problems. J Psychosom Res. 2021;142:110358.
Sumner JA, Kubzansky LD, Roberts AL, Chen Q, Rimm EB, Koenen KC. Not all posttraumatic stress disorder symptoms are equal: fear, dysphoria, and risk of developing hypertension in trauma-exposed women. Psychol Med. 2020;50:38–47.
von Känel R, Hepp U, Traber R, Kraemer B, Mica L, Keel M, et al. Measures of endothelial dysfunction in plasma of patients with posttraumatic stress disorder. Psychiatry Res. 2008;158:363–73.
Bereshchenko O, Bruscoli S, Riccardi C. Glucorticoids, sex hormones, and immunity. Front Immunol. 2018;9:1332.
Medina D, Mehay D, Arnold AC. Sex differences in cardiovascular actions of the renin-angiotensin system. Auton Res. 2020;30:393–408.
Manukhina EB, Tseilikman VE, Komelkova MV, Lapshin MS, Goryacheva AV, Kondashevskaya MV, et al. Сardiac injury in rats with experimental posttraumatic stress disorder and mechanisms of its limitation in experimental posttraumatic stress disorder-resistant rats. J Appl Physiol. 2021;130:759–71.
Sahafi E, Peeri M, Hosseini M-J, Azarbyjani MA. Cardiac oxidative stress following maternal separation stress was mitigated following adolescent voluntary exercise in adult male rat. Physiol Behav. 2018;183:39–45.
Dalvie S, Chatzinakos C, Al Zoubi O, Georgiadis F, Lancashire L, Daskalakis NP. From genetics to systems biology of stress-related mental disorders. Neurobiol Stress. 2021;15:100393.
Lima BB, Hammadah M, Wilmot K, Pearce BD, Shah A, Levantsevych O, et al. Posttraumatic stress disorder is associated with enhanced interleukin-6 response to mental stress in subjects with a recent myocardial infarction. Brain Behav Immun. 2019;75:26–33.
Bremner D, Gurel NZ, Jia Y, Wittbrodt MT, Levantsevych OM, Huang M, et al. Transcutaneous vagal nerve stimulation blocks stress-induced activation of Interleukin-6 and interferon-γ in posttraumatic stress disorder: a double-blind, randomized, sham-controlled trial. Brain Behav Immun Health. 2020;9:100138.
Burg MM, Brandt C, Buta E, Schwartz J, Bathulapalli H, Dziura J, et al. Risk for incident hypertension associated with PTSD in military veterans, and the effect of PTSD treatment. Psychosom Med. 2017;79:181–8.
van den Berk Clark C, Kansara V, Fedorova M, Ju T, Renirie T, Lee J, et al. How does PTSD treatment affect cardiovascular, diabetes and metabolic disease risk factors and outcomes? A systematic review. J Psychosom Res. 2022;157:110793.
LoSavio ST, Beckham JC, Wells SY, Resick PA, Sherwood A, Coffman CJ, et al. The effect of reducing posttraumatic stress disorder symptoms on cardiovascular risk: design and methodology of a randomized clinical trial. Contemp Clin Trials. 2021;102:106269.
Scherrer JF, Salas J, Schneider FD, Friedman MJ, van den Berk-Clark C, Norman SB, et al. PTSD improvement and incident cardiovascular disease in more than 1000 veterans. J Psychosom Res. 2020;134:110128.
Hall KS, Morey MC, Beckham JC, Bosworth HB, Sloane R, Pieper CF, et al. Warrior wellness: a randomized controlled pilot trial of the effects of exercise on physical function and clinical health risk factors in older military veterans with PTSD. J Gerontol A Biol Sci Med Sci. 2020;75:2130–8.
Funding
This work was funded by grants from the National Heart, Lung, and Blood Institute (R01HL139614 to JAS, R01HL160850 to JAS, JLG).
Author information
Authors and Affiliations
Contributions
JAS conceptualized the paper and its organization, designed the review structure, and wrote the manuscript. SC and TC contributed to sections on psychological mechanisms linking trauma with cardiovascular disease risk, assisted with creation of the table and figure, and reviewed and edited other sections of the paper. JLG assisted with conceptualization of the review, comprehensively reviewed all sections, and provided critical edits and revisions on all drafts.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sumner, J.A., Cleveland, S., Chen, T. et al. Psychological and biological mechanisms linking trauma with cardiovascular disease risk. Transl Psychiatry 13, 25 (2023). https://doi.org/10.1038/s41398-023-02330-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-023-02330-8
This article is cited by
-
Resting heart rate associations with violence exposure and posttraumatic stress symptoms: sex differences in children
Biology of Sex Differences (2024)
-
Childhood maltreatment and health in the UK Biobank: triangulation of outcome-wide and polygenic risk score analyses
BMC Medicine (2024)
-
Psychological Health and Ischemic Heart Disease in Women: A Review of Current Evidence and Clinical Considerations across the Healthspan
Current Atherosclerosis Reports (2024)
