
Abstract
Rape is associated with a high risk for posttraumatic stress disorder (PTSD). DNA methylation changes may confer risk or protection for PTSD following rape by regulating the expression of genes implicated in pathways affected by PTSD. We aimed to: (1) identify epigenome-wide differences in methylation profiles between rape-exposed women with and without PTSD at 3-months post-rape, in a demographically and ethnically similar group, drawn from a low-income setting; (2) validate and replicate the findings of the epigenome-wide analysis in selected genes (BRSK2 and ADCYAP1); and (3) investigate baseline and longitudinal changes in BRSK2 and ADCYAP1 methylation over six months in relation to change in PTSD symptom scores over 6 months, in the combined discovery/validation and replication samples (n = 96). Rape-exposed women (n = 852) were recruited from rape clinics in the Rape Impact Cohort Evaluation (RICE) umbrella study. Epigenome-wide differentially methylated CpG sites between rape-exposed women with (n = 24) and without (n = 24) PTSD at 3-months post-rape were investigated using the Illumina EPIC BeadChip in a discovery cohort (n = 48). Validation (n = 47) and replication (n = 49) of BRSK2 and ADCYAP1 methylation findings were investigated using EpiTYPER technology. Longitudinal change in BRSK2 and ADCYAP1 was also investigated using EpiTYPER technology in the combined sample (n = 96). In the discovery sample, after adjustment for multiple comparisons, one differentially methylated CpG site (chr10: 61385771/ cg01700569, p = 0.049) and thirty-four differentially methylated regions were associated with PTSD status at 3-months post-rape. Decreased BRSK2 and ADCYAP1 methylation at 3-months and 6-months post-rape were associated with increased PTSD scores at the same time points, but these findings did not remain significant in adjusted models. In conclusion, decreased methylation of BRSK2 may result in abnormal neuronal polarization, synaptic development, vesicle formation, and disrupted neurotransmission in individuals with PTSD. PTSD symptoms may also be mediated by differential methylation of the ADCYAP1 gene which is involved in stress regulation. Replication of these findings is required to determine whether ADCYAP1 and BRSK2 are biomarkers of PTSD and potential therapeutic targets.
Similar content being viewed by others


Introduction
Rape and sexual assault are associated with a high risk for the development of posttraumatic stress disorder (PTSD) compared to other trauma types [1, 2]. Prospective studies have reported PTSD prevalence rates ranging between 35% and 45% at 3-months post-rape, with many survivors of sexual assault continuing to experience PTSD symptoms at 6-months and 12-months post-rape [3,4,5,6]. PTSD is a complex, multifactorial disorder and an array of environmental and genetic putative risk and protective factors mediate or contribute to the development of the disorder [3, 5, 7]. Epigenetic mechanisms, including DNA methylation, are known to respond to environmental exposures such as trauma, leading to stable changes in gene expression [8, 9]. DNA methylation responses may confer risk or protection for PTSD, as they may alter the ability to adapt to traumatic events on a molecular level [10]. Using a hypothesis-neutral, genome-wide approach to study epigenome-wide signatures (while accounting for potential environmental and biological confounding factors), and validating and replicating these findings, may bring us closer to uncovering the complexity of the disorder [10].
To date, twelve epigenome-wide association studies (EWASs) of blood DNA methylation differences in PTSD cases and controls have been published (see Table 1 for details). In sum, the majority of genes identified as differentially methylated in PTSD are linked to central nervous system functioning (e.g., neuron development, axonal outgrowth, synaptic connectivity, neurotransmitter release, neuroinflammation, and apoptosis) [11,12,13,14,15,16,17] and the immune response (T cell expression, cytokine and interferon release, phagocytosis) [13, 14, 18, 19].
A meta-analysis of three North American mixed-gender civilian EWASs [13, 17, 18, 20] found that PTSD was associated with the neuregulin1 (NRG1) and hepatocyte growth factor-regulated tyrosine kinase substrate (HGS), both of which are related to central nervous system functioning [21]. The largest EWAS meta-analysis to date included 796 participants with PTSD and 1100 healthy controls [22]. North American and European male and female participants were drawn from three civilian cohorts [13, 17, 18, 20] and seven combat-exposed cohorts [15, 16] were included. Associations with PTSD were observed at four CpG sites of the human aryl hydrocarbon receptor repressor (AHRR) gene, which has been linked to both pro-inflammatory and anti-inflammatory immune regulation [22, 23]. Ring finger protein 6 (RNF6) associated with immune function, ATPase phospholipid transporting 9A (ATP9A), associated with glucose metabolism, family with sequence similarity 75-like protein FLJ46321 (FLJ46321), associated with cell differentiation; microRNA 3170 (MIR3170), and the long intergenic non-protein coding RNA 599 (LINC00599) genes were also associated with PTSD [22].
None of the gene-specific findings have been replicated across EWASs. Heterogeneity between and within EWASs may explain the lack of consistent findings. The majority of EWASs have been cross-sectional studies [11,12,13,14,15, 17,18,19,20, 22, 24, 25] and have investigated differential methylation in combat-exposed populations and first responders [12, 14,15,16, 20, 24, 25]. PTSD symptoms may manifest differently in combat-exposed samples (increased hypervigilance and compulsive behavior) compared to civilian samples [26, 27]. In civilians, PTSD symptom presentation, severity and recovery rates also differ depending on trauma type [26, 28, 29]. Civilian EWASs have investigated a mixture of traumas and none have investigated rape exclusively [30]. Civilian EWASs have also been predominantly conducted in mixed-gender [11, 13, 18, 19, 25, 31], North American samples [11,12,13, 17,18,19,20, 25].
Ethnicity-specific and sex-specific characteristics may influence methylation profiles [32,33,34]. Women have a two-fold increased risk of developing PTSD compared to men [34]. Increased risk for PTSD in women may be X-chromosome linked, given that PTSD heritability is considerably higher among women compared to men [35, 36]. Sex-specific expression of reproductive genes may also mediate the increased risk for PTSD in women, for example, estrogen levels have been associated with an altered hypothalamic-pituitary-adrenal (HPA) axis stress response in women [17, 37, 38] and differential methylation of estrogen response elements (EREs) in genes associated with HPA-axis functioning has been reported [17, 39].
We sought to address the design shortcomings and demographic differences in prior EWASs by conducting a cross-sectional EWAS study, complemented by validation of the results, replication, and longitudinal investigation of a demographically similar group of rape-exposed African black women in a low-income setting. Specific aims were to: (1) identify genome-wide differentially methylated CpG sites/regions associated with PTSD status at 3-months post-rape using an EWAS approach in a discovery sample; (2) validate the significant EWAS results in selected genes using an alternate methodology; (3) replicate the findings in 2 using a larger sample; (4) determine whether methylation levels of selected genes at baseline predict PTSD status change over 6-months; and (5) determine whether methylation changes in selected genes covary with PTSD symptom scores over 6 months.
Methods
Participant recruitment and setting
Participants were recruited through the Rape Impact Cohort Evaluation (RICE) study conducted in South Africa (n = 852). A detailed description of the methods of the RICE study has been published elsewhere [40]. In short, female survivors of rape were recruited from rape clinics. Interested participants were invited to the study site to enrol in the study following informed consent procedures. Recruitment was restricted to female participants between 18 and 40 years who reported rape in the preceding 20 days of the baseline visit. In this study, we excluded women who: (1) were pregnant or lactating during the course of the study; (2) met criteria for PTSD at the baseline visit, as this would be indicative of PTSD due to a past traumatic event other than the rape; and (3) had HIV-seroconverted. Samples from 48 participants comprised the “discovery” sample, i.e., those that were included in the epigenome-wide DNA methylation analysis. These samples were subsequently utilized to technically validate the results from the EWAS study using EpiTYPER Sequenom MassARRAY technology (Agena Bioscience, California, United States). The “replication” sample comprised 96 participants, 47 from the discovery sample and 49 additional samples.
Ethical approval for the RICE parent study was obtained from the Human Research Ethics Committee at the South African Medical Research Council (SAMRC; EC019-10/2013) and approval to conduct the sub-study was obtained from the Health Research Ethics Committee at Stellenbosch University (S16/08/146).
Clinical measures
At the baseline visit, a research assistant supervised by a registered trauma counselor or registered nurse assessed for PTSD (in relation to prior criterion A traumas other than the rape) on the Mini-International Neuropsychiatric Interview (MINI) version 7.0.0 [41]. An HIV rapid test, pregnancy test, blood collection for DNA analysis, and assessment of body mass index (BMI) were undertaken by a nurse at all time points (baseline, 3-months, and 6-months post-rape).
A research assistant administered a demographic questionnaire, a modified version of the Childhood Trauma Questionnaire-Short Form (CTQ-SF) [42], and a modified version of the Life Events Checklist (LEC) [43, 44] at baseline. The Davidson Trauma Scale (DTS) [45], the Alcohol Use Disorders Identification Test, alcohol consumption subscale (AUDIT-C) [46], and the Center for Epidemiologic Studies Depression Scale (CES-D) [47] was administered at all time points. The DTS was used to measure PTSD symptoms with a cut-off score of forty or more considered indicative of PTSD [45]. This cut-off was used to group participants into PTSD cases and controls at 3-months post-rape (see supplementary material for more details) [45]. All assessments were completed face-to-face and responses were recorded and electronically captured in real-time on a secure server. Item-level missing values were imputed using a multiple imputation model whilst maintaining a multivariate normal distribution.
Demographic and clinical characteristics of the sample
The baseline demographic and clinical characteristics of the sample were investigated using descriptive statistics. Differences in baseline demographic and clinical characteristics between the discovery/validation sample and the replication sample were investigated using non-parametric tests since most of the variables did not conform to a normal distribution. Mann–Whitney U tests were used to compare groups on continuous variables, i.e., age, body mass index (BMI), childhood trauma score, number of childhood traumas endorsed, number of lifetime traumas endorsed, alcohol use, and depression symptom scores. Chi-square statistics were used to compare groups on several categorical variables (completed secondary education, relationship status, smoking status, HIV status, medication use, childhood neglect, witnessed domestic violence in the childhood home, childhood emotional abuse, childhood physical abuse, childhood sexual abuse, imprisonment, civil unrest or war, serious injury, being close to death, murder of a family member or friend, unnatural death of a family member or friend, murder of a stranger, robbed at gun/knifepoint, kidnaped, hazardous alcohol use and depression status).
The same variables and methods used to investigate baseline demographic and clinical differences between the discovery/validation and replication samples were used to investigate differences between those with and without PTSD at 3-months post-rape.
Cross-sectional analyses (3 months post-rape)
Discovery sample
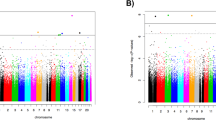
Forty-eight participants, 24 with PTSD and 24 without PTSD at 3-months post-rape, were included in the discovery sample. We selected the 3-months post-rape time point since it was the first time point, in the parent study, at which a PTSD diagnosis could be made, based on DSM-5 criteria [48] We implemented a cross-sectional, case-control design to identify genome-wide differentially methylated positions (DMPs) and differentially methylated regions (DMRs) between individuals with and without PTSD. Consecutive cases of PTSD at 3-months post-rape were identified until the target number was reached. Controls were perfectly matched to cases, based on HIV status and as closely as possible (in descending hierarchical order of importance) on age, childhood trauma scores, lifetime trauma exposure, BMI, smoking, education, and income. DNA was extracted from peripheral blood samples and assayed using the Human Illumina EPIC BeadChip array (Illumina, California, United States) [49].
Raw probe intensity data (iDAT) files produced by Illumina GenomeStudio were decompressed and parsed into text format using the meffil R package [50] in R statistics version 3.6.2 [51]. All EWAS analyses, including quality control measures and beta normalization, were completed using the meffil R package [50].
All samples passed the quality control checks (see Supplementary Material for more details). Probes not passing the quality control checks (n = 29936) were excluded from the downstream analyses. Previously identified cross-reactive probes for 43254 CpG sites were also excluded [52]. Probes targeting CpG sites on the X chromosome were retained since all participants included in the study were female.
The percentage of methylated alleles for each CpG site in each sample was calculated as β = M/(M + U + 100) where M and U symbolize raw probe fluorescent intensities for methylated and unmethylated signals, respectively [53]. Technical bias and batch effects were corrected for using functional normalization (Supplementary Material, Supplementary Figs. 1–5, Supplementary Tables 1 and 2) [54]. Any residual effects were handled by including surrogate variables as covariates in the EWAS models. These were estimated following functional normalization using surrogate variable analysis (SVA) [55]. Cell type composition was estimated by applying the Houseman algorithm to the normalized DNA methylation profiles and a publicly available blood cell type reference dataset (Gene Expression Omnibus accession number GSE35069) [56]. Cell type composition was included in the final EWAS models (Supplementary Fig. 6). Epigenome-wide associations were investigated using logistic regression models to identify DMPs associated with PTSD status. A Bonferroni correction was applied to correct for multiple testing with an adjusted p-value < 0.05 indicating genome-wide significance [57].
The dmrff R package was applied to EWAS summary statistics to identify DMRs [58]. DMRs were defined as a region covering two or more CpG sites with less than 100 bp between consecutive sites showing the same direction of effect with an uncorrected p-value < 0.05 (see supplementary material for more details) [58]. A DMR was considered significant on an epigenome-wide level if a Bonferroni-adjusted p < 0.05 was observed. Coordinates resulting from the DMP and DMR analyses were annotated using the Illumina EPIC_v-1-0_B4 manifest [53]. Co-variation in methylation levels between blood and brain tissue was explored using the online Blood-Brain DNA Methylation Comparison Tool [59]. Prior findings reporting a link between any exposure or phenotype and the CpG sites identified from the EWAS were identified using the Medical Research Council Integrative Epidemiology Unit (MRC-IEU) catalog of epigenome-wide association studies [60] and the China National Center for Bioinformation National Genomics Data Center epigenome-wide association studies atlas [61]. Prior findings reporting a link between mood, anxiety, or trauma-related disorders and any CpG site in the genes identified from the EWAS were identified using the aforementioned databases for EWAS studies, and the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) genome-wide association study (GWAS) Catalog for GWAS studies [62]. Prior findings reporting a link between PTSD and any CpG site in the genes identified from the EWAS were identified through a literature search in PubMed [63]. All genomic coordinates reported in this study are in reference to the Hg19/GRCh37 human genome assembly (see Supplementary Material for more details).
Validation analysis
A candidate gene approach was used to validate the findings of the EWAS in 47 of the 48 participants included in the discovery sample. One participant was excluded from the validation sample due to incomplete data at the time of validation analysis. Samples were assayed using EpiTYPER. DNA methylation was investigated at CpG sites in two selected regions at 3-months post-rape. Brain-specific serine/threonine-protein kinase 2 (BRSK2) and adenylate cyclase-activating polypeptide 1 (ADCYAP1) were selected for validation since they contained CpG sites found to be differentially methylated (prior to correction for multiple testing) between cases and controls in the EWAS. Both genes were also found to contain differentially methylated regions. A CpG site in BRSK1, a paralog of BRSK2, was found to be differentially methylated in a prior PTSD EWAS study [14] and ADCYAP1 receptor 1 (ADCYAP1R1) has been linked to the development of PTSD in several prior studies [64,65,66].
DNA methylation percentages were exported using the EpiTYPER Analyzer software. The validation analyses were completed using IBM SPSS Statistics 27.0. Logistic regression models were used to determine if differential methylation of BRSK2 and ADCYAP1 at 3-months post-rape was associated with PTSD status at 3-months post-rape.
The relationship between baseline confounding variables, PTSD status at 3-months post-rape, BRSK2 methylation at 3-months post-rape, and ADCYAP1 methylation at 3-months post-rape was investigated using Mann−Whitney U tests, Chi-square tests, and Spearman’s correlations. Potential confounders included continuous variables (age, BMI, childhood trauma score, number of lifetime traumas endorsed, alcohol use, and depression) and categorical variables (HIV status, smoking, and medication use). Confounding variables significantly associated with PTSD or BRSK2/ADCYAP1 methylation were entered in logistic regression models as covariates, in a stepwise manner.
Replication analysis
To replicate the validation analysis, an additional 49 consecutively selected participants from the parent study were included in the DNA methylation replication analyses. These participants were not matched on PTSD status or potential methylation covariates. Samples were assayed using EpiTYPER.
Logistic regression models, including potential confounding variables, were used to determine if differential methylation of BRSK2 and ADCYAP1 at 3-months post-rape was associated with PTSD status at 3-months post-rape in the replication sample, following the same procedure applied in the validation analyses.
Comparison of previous findings from candidate gene studies and EWASs
Candidate gene studies and EWASs investigating the relationship between methylation and PTSD were identified from published literature. For EWASs, the Illumina CpG identification number for significant findings was manually recorded and cross-checked against the findings of the current EWAS. For candidate gene studies, the genomic coordinates of the sites were identified from the publications and converted to Hg19/GRCh37 positions using the BLAT function of the University of California, Santa Cruz (UCSC) genome browser (if not already indicated as Hg19/GrCh37 positions). The genomic locations were manually recorded and cross-checked with the Illumina EPIC_v-1-0_B4 manifest to determine if the sites were included on the Illumina EPIC array. Significant CpG sites resulting from the current EWAS and corresponding to prior findings are reported in the results.
Agreement between the Illumina EPIC array and EpiTYPER
Spearman’s correlation coefficients were used to investigate the level of agreement between methylation levels resulting from the Illumina EPIC array at 3-months post-rape and methylation levels resulting from EpiTYPER at 3-months post-rape.
Longitudinal investigation (baseline, 3-months, and 6-months post-rape)
Combined sample
The validation and replication samples were combined and methylation data from the baseline and 6-month post-rape samples were added to the dataset, for the same combined group. The group consisted of 96 participants with methylation data at all time points (baseline, 3-months, and 6-months). The samples were assayed using EpiTYPER. We investigated the same BRSK2 and ADCYAP1 CpG sites investigated in the validation and replication samples but followed a longitudinal cohort design with PTSD symptom scores as the outcome, instead of a cross-sectional case-control design with PTSD status at 3-months as the outcome.
PTSD scores at each time point were compared between the discovery/validation sample and the replication sample using Mann-Whitney U tests. The relationship between PTSD, BRSK2 methylation, ADCYAP1 methylation (at all time points), and potential baseline confounders (age, BMI, childhood trauma, lifetime traumas, alcohol use, depression, HIV status, smoking, and medication use) was investigated using Mann-Whitney U tests, Chi-square tests and Spearman’s correlations.
Baseline ADCYAP1 and BRSK2 methylation levels were investigated as predictors of change in PTSD symptom scores over six months, in the first set of mixed regression models. In the second set of mixed regression models, we investigated change in BRSK2 and ADCYAP1 methylation levels over six months in relation to change in PTSD symptom scores over six months. Confounding variables significantly associated with PTSD or BRSK2/ADCYAP1 methylation at any time point were entered in the mixed regression models as covariates, in a stepwise manner.
Results
Baseline demographic and clinical characteristics of the sample
Table 2 presents the baseline demographic and clinical characteristics of the discovery/validation and replication samples. The samples were similar with regard to demographic and clinical characteristics. The only variable that differed between the samples was the prevalence of lifetime exposure to the murder of a family member or friend, which was more frequently endorsed in the discovery/validation sample compared to the replication sample (25.5% vs. 8.2%, respectively; χ2 = 5.2, p = 0.022).
Comparison of baseline demographic and clinical characteristics between the PTSD groups at 3-months post-rape
Table 3 presents group comparisons by PTSD status (at 3-months post-rape) in the discovery/validation sample and the replication sample, consecutively. Participants with and without PTSD had similar baseline demographic and clinical characteristics in the discovery/validation and replication samples. However, in the discovery/validation sample, those with PTSD were more likely to endorse being robbed with a gun or knife compared to those without PTSD (50% and 21.7%, respectively; z = 4.1, p = 0.044). In the replication sample, those with PTSD endorsed less lifetime traumas (M = 0.5, SD = 0.7) compared to those without PTSD (M = 1.4, SD = 1.3, z = −2.5, p = 0.014).
Discovery sample: genome-wide differentially methylated genes associated with PTSD status at 3-months post-rape
Table 4 presents selected findings from the top twenty DMPs that were associated with PTSD before correction for multiple comparisons (p < 0.05) (see Supplementary Table 3 and Supplementary Figs. 7–9 for more details). Only one DMP, cg01700569, remained significant after correcting for multiple testing (adjusted p < 0.05). This intergenic site (cg01700569) is located 24694 bases downstream of solute carrier family 16 member 9 (SLC16A9). Other genes previously linked to mood, anxiety, or trauma-related disorders included protein zeta-1 (FEZ1), ADCYAP1, BRSK2, catenin alpha 3 (CTNNA3), and par-3 family cell polarity regulator (PARD3).
Thirty-four DMRs were identified from the regional analysis after Bonferroni correction for multiple testing. The regions previously linked to mood, anxiety, or trauma-related disorders included coiled-coil and C2 domain-containing protein 2 A (CC2D2A), BRSK2, and ADCYAP1. The findings related to these genes are also presented in Table 4.
Validation and replication sample: differential methylation of BRSK2 in relation to PTSD status at 3-months post-rape
The BRSK2 region (chr11:1463541-1463670; adjusted p < 0.05) identified from the EWAS included five CpG sites (CpG1-cg12186219, CpG2-cg14064268, CpG3-cg10590925, CpG4-cg17429870, CpG5-cg18651858) that showed decreased methylation in participants with PTSD (see Fig. 1). Based on prior findings, DNA methylation of these CpG sites in blood was highly correlated with DNA methylation in the prefrontal cortex, superior temporal gyrus, and the cerebellum (see Supplementary Fig. 10a–d) [59] Three of the five CpG sites (CpG3, CpG4, and CpG5) were investigated in the validation and replication sample. We could not investigate CpG1 or CpG2, as the mass of CpG1 was too low to be measured by the EpiTYPER mass spectrometer, and CpG2 contained a silent peak that overlapped with the non-methylated peak for this site (see Supplementary Table 4 for the genomic coordinates and sequence for CpG3, CpG4, and CpG5).

Boxplots indicating methylation levels between participants with and without PTSD for the five CpG sites in the BRSK2 region found to be associated with PTSD at 3-months post-rape in the epigenome-wide association study.
Baseline age, HIV status, BMI, smoking status, childhood trauma score, lifetime trauma, alcohol use, depression, and medication use were not associated with BRSK2 methylation at 3-months post-rape in either the validation or replication samples. PTSD status at 3-months post-rape was associated with lifetime trauma (z = −2.47, p = 0.014) in the replication sample only (see Supplementary Tables 5 and 6).
In the validation analysis, methylation levels of BRSK2 CpG3 (β = −0.04, p = 0.050, OR 0.96) and CpG4 (β = −0.04, p = 0.052, OR 0.96) at 3-months post-rape were not significantly associated with PTSD status at 3-months post-rape. Decreased methylation of BRSK2 CpG5 (β = −0.04, p = 0.048, OR 0.96) at 3-months post-rape was significantly associated with PTSD status at 3-months post-rape, but the association was no longer significant when lifetime trauma was added as a covariate to the model (see Supplementary Tables 7). In the replication analysis, methylation levels of BRSK2 CpG3 (β = −0.00, p = 0.889, OR 1.00), CpG4 (β = −0.01, p = 0.667, OR 0.99) and CpG5 (β = 0.00, p = 0.866, OR 1.00) were not significantly associated with PTSD status at 3-months post-rape (see Supplementary Table 8).
Validation and replication samples: differential methylation of ADCYAP1 in relation to PTSD status at 3-months post-rape
The ADCYAP1 region (chr18:905177-905180) identified from the EWAS included only two differentially methylated CpG sites (CpG1 – cg22388954, CpG2 – cg11773720) which both showed increased methylation in participants with PTSD (see Fig. 2). Based on prior findings, DNA methylation of these CpG sites in blood was not correlated with DNA methylation in brain tissue (Supplementary Fig. 11a, b) [59]. EpiTYPER signals for ADCYAP1 CpG1 and CpG2 were combined for analysis, due to their proximity to each other (see supplementary Table 9 for the genomic coordinates and sequence of CpG1 and CpG2).

Boxplots indicating methylation levels between participants with and without PTSD for the two CpG sites in the ADCYAP1 region were found to be associated with PTSD in the epigenome-wide methylation study.
Baseline age, HIV status, BMI, smoking status, childhood trauma score, lifetime trauma, alcohol use, depression, and medication use were not associated with ADCYAP1 methylation at 3-months post-rape in the validation or replication samples (see Supplementary Tables 5 and 6). In the validation analysis, methylation levels of ADCYAP1 CpG1&2 (β = −0.09, p = 0.382, OR 0.92) were not significantly associated with PTSD status at 3-months post-rape (see supplementary Tables 7). In the replication sample, methylation levels of ADCYAP1 CpG1&2 (β = −0.06, p = 0.639, OR 0.94) were also not significantly associated with PTSD status at 3-months post-rape (see supplementary Table 8).
Agreement between the Illumina EPIC array and EpiTYPER
Large positive correlations were found when comparing the Illumina EPIC array and EpiTYPER methylation levels for BRSK2 CpG3 (r = 0.881, p < 0.000), CpG4 (r = 0.900, p < 0.000), and CpG5 (r = 0.831, p = 0.831) at 3-months post-rape (see Supplementary Table 10). Small, non-significant correlations were found when comparing the Illumina EPIC array and EpiTYPER methylation levels for ADCYAP1 CpG1&2 (r = 0.254, p > 0.05; see Supplementary Table 11).
Replication of previous candidate gene and EWAS findings
Differential methylation of five CpG sites previously investigated was replicated in this EWAS study, prior to correction for multiple testing (see Supplementary Table 12 and 13). These sites were located in the HTR3A (chr11:113846004, cg20621129, p = 0.028) [67], AHRR (two CpG sites: chr5:373378, cg05575921, p = 0.033; chr5:377358, cg26703534, p = 0.031) [22], DUSP22 (chr6:291882, cg21548813, p = 0.032) [15] and TPR (chr1:186344558, cg24577137, p = 0.0008) genes [13]. Since decreased methylation of AHRR is strongly linked to smoking, [22] we investigated the link between smoking and AHRR methylation (based on the values obtained from our EWAS) and found decreased AHRR methylation levels in smokers (M = 78.91, SD = 14.95, n = 5) compared to non-smokers (M = 93.88, SD = 1.45, n = 42) at cg05575921 (z = −2.92, p = 0.001).
Combined sample: longitudinal relationship between BRSK2, ADCYAP1, PTSD scores, and confounding variables
Baseline childhood trauma, alcohol use, and depression were associated with PTSD scores at one or more time points. Baseline childhood and lifetime trauma scores were associated with BRSK2 methylation at one or more time points. Baseline HIV status was associated with ADCYAP1 methylation at 3-months post-rape (see supplementary Table 14).
Combined sample: longitudinal change in PTSD symptom scores
The mean PTSD scores at baseline, 3-months, and 6-months, stratified by sample (discovery/validation, replication, combined), are presented in Fig. 3. There were no significant differences between the discovery/validation samples and the replication sample for either baseline (z = −0.79, p = 0.431), 3-month (z = −1.37, p = 0.172), or 6-month (z = −0.15, p = 0.883) PTSD scores. There was a significant decline in PTSD scores from baseline to 3-months (p < 0.000) and from 3-months to 6-months (p = 0.021), in the combined sample.

Symptoms over 6 months for the discovery/validation samples, replication sample, and combined sample.
Combined sample: baseline BRSK2 and ADCYAP1 methylation levels and longitudinal change in PTSD scores
Table 5 presents the results of the mixed regression models investigating baseline BRSK2 and ADCYAP1 methylation as predictors of change in PTSD symptom scores over time. Decreased baseline BRSK2 CpG3, CpG4, and CpG5 methylation levels were significant predictors of increased PTSD symptom scores at 3-months (CpG3 ß = −0.39, p < 0.001, CpG4 ß = −0.33, p = 0.005, CpG5 ß = −0.27, p = 0.009) and 6-months (CpG3 ß = −0.49, p < 0.001, CpG4 ß = −0.44, p < 0.001, CpG5 ß = −0.38, p < 0.001) post-rape. However, the relationships between BRSK2 CpG3, CpG4, and CpG5 methylation levels and PTSD scores at 3-and 6-months post-rape were no longer significant when childhood trauma, alcohol consumption, depression, and lifetime trauma were added to the models as covariates.
Increased baseline ADCYAP1 CpG1&2 methylation was a significant predictor of increased PTSD scores at baseline (ß = 5.34, p < 0.001) and decreased PTSD scores at 6-months (ß = −3.52, p = 0.004) post-rape, but the associations were no longer significant when covariates were added to the model.
Combined sample: longitudinal change in ADCYAP1 and BRSK2 methylation levels in relation to longitudinal change in PTSD scores
Table 6 presents the results of the mixed regression models investigating change in BRSK2 and ADCYAP1 methylation over time as predictors of change in PTSD symptom scores over time. Decreased BRSK2 CpG3 (ß = −0.39, p < 0.001), CpG4 (ß = −0.36, p = 0.001), and CpG5 (ß = −0.32, p = 0.001) methylation at 3-months post-rape was associated with increased PTSD scores at 3-months post-rape. Decreased BRSK2 CpG3 (ß = −0.49, p < 0.001), CpG4 (ß = −0.46, p < 0.001), and CpG5 (ß = −0.43, p < 0.001) methylation at 6-months post-rape was also associated with increased PTSD scores at 6-months post-rape. The relationship between PTSD score at 3-month post-rape and methylation of BRSK2 CpG3 (ß = −0.30, p = 0.049) was the only association that remained significant after the addition of covariates to the models.
Increased baseline ADCYAP1 CpG1&2 methylation was associated with increased PTSD scores at baseline (ß = 4.67, p < 0.001), while decreased ADCYAP1 CpG1&2 methylation at 3-months (ß = −2.61, p = 0.001) and 6-months (ß = −5.01, p < 0.001) was associated with increased PTSD scores at 3-months and 6-months post-rape. The associations were no longer significant when covariates were added to the model.
Discussion
In this study, we identified one DMP (cg01700569) and thirty-four DMRs associated with PTSD at 3-months post-rape on an epigenome-wide level. The gene closest to the aforementioned DMP is SLC16A9. Although investigating this DMP further may have been of value, little is known about it in the context of mental health. The site (cg01700569) is located in an intergenic region, which further complicates the interpretation of the clinical significance of the finding.
We investigated two DMRs in the BRSK2 and ADCYAP1 genes further. We were able to validate, but not replicate, the BRSK2 CpG5 finding, confirming decreased BRSK2 methylation in rape-exposed participants with PTSD at 3-months post-rape, compared to those without PTSD. We also found that decreased baseline BRSK2 CpG3, CpG4, and CpG5 methylation was associated with increased PTSD scores at 3-months and 6-months post-rape. Decreased BRSK2 methylation at 3-months and 6-months post-rape was associated with increased PTSD scores at the same time points. However, the associations between decreased BRSK2 CpG3 methylation at 3-months post-rape and increased PTSD scores at 3-months post-rape were the only ones that remained significant after childhood trauma, alcohol consumption, depression, and lifetime trauma were added as covariates to the models.
We were unable to validate or replicate our ADCYAP1 CpG1&2 findings. We found that decreased baseline ADCYAP1 CpG1&2 methylation was associated with increased PTSD scores at 6-months post-rape. Decreased ADCYAP1 methylation at 3-and 6-months post-rape was also associated with increased PTSD scores at the same time points, while decreased baseline ADCYAP1 CpG1&2 methylation was associated with decreased PTSD scores at baseline. The findings did not remain significant after PTSD covariates were added to the models.
Decreased methylation of the BRSK2 paralog, BRSK1 [68], has been associated with a PTSD diagnosis in a prior EWAS [14]. BRSK1 and BRSK2 share a 68% overlap in genetic sequence, both are highly expressed in the brain, and decreased expression of both has been linked to disorganized presynaptic vesicle formation, uncoordinated release and reuptake of neurotransmitters, altered axonal development, and abnormal neuronal polarization in animal studies [68,69,70,71,72,73]. In human studies, a BRSK2 polymorphism (rs1881509) has been associated with heroin dependence [69], and functional variants of BRSK2 have been associated with autism spectrum disorder, cognitive impairment, intellectual disability, and speech delays [74, 75].
BRSK1 and BRSK2 are expressed most strongly in the cerebellum and the hippocampus [69]. The hippocampus is closely linked to PTSD since it is involved in memory consolidation [76]. When memories are not consolidated into autobiographical memory networks, they may involuntarily resurface (e.g., flashbacks, intrusions, nightmares, and dissociation) and activate the limbic system, which induces the fight-or-flight response [77]. Differential methylation and expression of BRSK2 may also alter the expression of neurotransmitters previously found to be associated with PTSD (norepinephrine, epinephrine, dopamine, and serotonin) through altered presynaptic vesicle and synaptic cleft development [78, 79].
In addition to their functions in the brain, BRSK1 and BRSK2 have been linked to metabolic processes and glucose homeostasis [80, 81]. Animal studies have found increased expression of BRSK1 and BRSK2 in pancreatic cells and knockdown of BRSK2 resulted in a significant increase in serum insulin levels [80, 81]. In a human study, BRSK2 was found to be highly expressed in human pancreatic insulin-producing B cells, and activation of BRSK2 was linked to reduced insulin secretion [81]. Moreover an EWAS found that participants with type 1 diabetes and neuropathy showed decreased methylation at four CpG sites in the BRSK2 gene compared to participants with type 1 diabetes without neuropathy [82].
The BRSK2 CpG sites investigated in this study were located in intron 4 of the gene. The function of methylation in gene bodies is not well established, but methylation is abundant in these regions and is generally positively correlated with expression [83]. Assuming the latter, we can hypothesize that decreased methylation of BRSK2 may contribute to adverse neuronal development, neuronal maintenance, and dysregulated blood glucose levels which may explain the increased risk for diabetes and cardiovascular disease observed in prior PTSD studies [84, 85]. The relationship between BRSK2 methylation and adverse neuronal development and maintenance is further supported by prior findings of a high correlation between BRSK2 blood methylation and methylation in brain tissue [59].
We investigated ADCYAP1 further, since its protein product, PACAP, has been identified as a master regulator of the HPA-axis and the stress response [86]. The highest concentration of PACAP in the brain is found in the hypothalamus [87]. PACAP binding in the hypothalamus triggers the release of corticotrophin-releasing hormone (CRH) and signals the activation of the stress response [86]. In the adrenal medulla, PACAP binding to PAC1R (product of ADCYAP1R1) stimulates the release of catecholamines as part of the sympathetic nervous system (SNS) [88]. PACAP binding to PACR1 in preganglionic neurons triggers the release of phenylethanolamine-N-methyltransferase (PNMT) and tyrosine hydroxylase (TH) in effector organs of the SNS. PNMT and TH are catecholamine-synthesizing enzymes and sustain the release of catecholamines in the effector organs during the stress response [88].
Researchers investigating PACAP/ADCYAP1 and PACR1/ADCYAP1R1 in relation to PTSD in a predominantly African− American sample with a mixture of trauma types found that, in women more than men, increased PACAP blood levels were associated with increased PTSD symptom severity and an increased acoustic startle reflex response [64, 89]. They also found that women carrying the ADCYAP1R1 rs2267735 CC genotype showed decreased ADCYAP1R1 mRNA expression, increased PTSD symptom severity, increased dark-enhanced startle response, and increased amygdala and hippocampal activity in response to viewing threatening face stimuli [64,65,66, 89]. In both men and women, increased methylation of ADCYAP1R1 was associated with decreased cortical mRNA expression and increased PTSD symptom severity [64, 90]. However, the functional effects of ADCYAP1 and ADCYAP1R1 seem to be more pronounced in women compared to men [64,65,66], due to the presence of several estrogen response elements (EREs) in the ADCYAP1R1 promoter. The CC genotype of rs2267735 has been associated with decreased binding of estrogen receptor alpha to the EREs and decreased expression of ADCYAP1R1 [91]. The role of estrogen in ADCYAP1R1 and HPA-axis activity may in part explain why women have an increased risk of PTSD compared to men [35, 92].
The two ADCYAP1 CpG sites investigated in this study are located in a CpG island spanning the 1st intron of the gene. Methylation in CpG islands and in the 1st intron of a gene is generally associated with decreased expression of the gene [93,94,95]. Our longitudinal findings, therefore, correspond with prior findings since decreased methylation of ADCYAP1 is likely to result in increased expression of PACAP and increased PTSD symptom severity [65, 66, 91, 96]. Decreased PACAP is also likely to result in decreased binding to PAC1 and reduced activation of the HPA-axis [86, 88].
Based on prior findings, ADCYAP1 CpG1&2 DNA methylation in blood was not significantly correlated with DNA methylation at the same sites in brain tissue [59]. However, the brain regions investigated did not specifically focus on the region where PACAP is most abundantly expressed i.e., the paraventricular nucleus of the hypothalamus, and investigating blood-brain methylation in this region may show different results [37]. It is also likely that the expression of PACAP in the endocrine system has a more profound effect on the regulation of the HPA-axis compared to PACAP expression in the brain [37].
We found that, before correction for multiple testing, CpG sites in HTR3A [67], AHRR [22], DUSP22 [15], and TPR [13] were associated with PTSD. The results from our study are in line with recent results from the largest EWAS meta-analysis of PTSD published to date [22], where AHRR cg05575921 and cg26703534 were found to exhibit reduced DNA methylation in individuals with PTSD. Decreased AHRR methylation at these CpG sites was also associated with decreased kynurenine and kynurenic acid in the same study [22]. Kynurenine ligand binding to aryl hydrocarbon receptors has been associated with the expression of anti-inflammatory genes which may be disrupted by decreased methylation of AHRR [22, 25]. This may result in increased levels of proinflammatory cytokines and the low-grade inflammatory state often observed in PTSD [97, 98]. Upregulation in kynurenine to restore the imbalance between pro-inflammatory and anti-inflammatory cytokines may also result in reduced levels of serotonin since both kynurenine and serotonin are synthesized from tryptophan [99]. A strong link between decreased AHRR methylation and smoking has also been reported in previous studies although some studies have reported a significant relationship between AHRR methylation and PTSD independent of the effect of smoking [22, 100,101,102].
Our findings should be interpreted in light of a number of limitations. First, the EWAS was conducted in a small sample of participants. However, the study was well designed to limit variation between groups. Second, we used DNA extracted from whole blood to measure methylation levels while differential methylation in brain tissue is a more direct approximation of PTSD pathophysiology. However, based on prior findings, we observed that blood-brain methylation was highly correlated at the BRSK2 CpG sites investigated in this study, but not at the ADCYAP1 CpG sites. Blood is easily accessible and blood biomarkers of PTSD risk may be a more pragmatic approach for personalized treatment of individuals at high risk of developing PTSD following trauma exposure [103]. Third, we may have overcorrected for confounding variables in the EWAS given that SVA was used along with the inclusion of cell-type composition as a covariate in the final models. Fourth, we did not investigate methylation quantitative trait loci (meQTL) located in the BRSK2 and ADCYAP1 genes. SNPs located in these genes may predict or mediate the methylation profiles observed in relation to PTSD status and symptom scores. Finally, DNA methylation in relation to gene expression and/or protein levels was not objectively measured and conclusions related to the functional effects of methylation are speculative.
The study has many strengths. First, all participants were rape-exposed women from similar sociodemographic backgrounds and from the same ethnicity group thus making the sample relatively homogenous. Second, the analyses were robust with a variety of confounding factors controlled for i.e., participants who were pregnant/lactating were excluded, none of the participants were on psychotropic medication and participants were of similar age. Baseline measures of age, HIV status, BMI, smoking, childhood trauma, lifetime trauma, alcohol use, and depression were controlled for by matching participants on these variables in the cross-sectional EWAS and including these factors as covariates/confounders in the longitudinal analyses. Third, we attempted to expand the findings of the EWAS by including longitudinal data which allowed us to investigate changes in methylation in relation to change in PTSD symptom scores over time. Fourth, investigating the agreement between the results obtained from the two different laboratory methods used (Illumina EPIC array and EpiTYPER) also allowed identification of potential bias/variation introduced by the different procedures involved in each method.
In summary, this study provides evidence that differential methylation of genes related to neurogenesis/development, glucose homeostasis, and HPA-axis regulation may be involved in PTSD development following rape. Our findings are supported by previous research implicating ADCYAP1/ADCYAP1R1 (especially in women) and BRSK1/BRSK2 in the development of PTSD. However, replication of these findings is required to determine whether the differentially methylated regions identified in this study are consistently linked to the development of PTSD.
References
Breslau N, Troost JP, Bohnert K, Luo Z. Influence of predispositions on post-traumatic stress disorder: Does it vary by trauma severity? Psychol Med. 2013;43:381–90.
Yehuda R, Mcfarlane A, Vermetten E, Lanius R. Post-traumatic stress disorder. Nat Rev. 2015;1:1–22.
Elklit A, Christiansen DM. Risk factors for posttraumatic stress disorder in female help-seeking victims of sexual assault. Violence Vict. 2013;28:552–68.
Elklit A, Christiansen DM. ASD and PTSD in rape victims. J Interpers Violence. 2010;25:1470–88.
Möller AT, Bäckström T, Söndergaard HP, Helström L. Identifying risk factors for PTSD in women seeking medical help after rape. PLoS ONE. 2014;9:1–9.
Nickerson A, Steenkamp M, Aerka IM, Salters-Pedneault K, Carper TL, Barnes JB, et al. Prospective investigation of mental health following sexual assault. Depress Anxiety. 2013;30:444–50.
Mgoqi-Mbalo N, Zhang M, Ntuli S. Risk factors for PTSD and depression in female survivors of rape. Psychol Trama. 2017;9:301–8.
Ryan J, Chaudieu I, Saffery R. Biological underpinnings of trauma and post-traumatic stress disorder: focusing on genetics and epigenetics. Epigenomics. 2016;8:1553–69.
Nievergelt CM, Ashley-Koch AE, Dalvie S, Hauser MA, Morey RA, Smith AK, et al. Genomic approaches to posttraumatic stress disorder: The Psychiatric Genomic Consortium Initiative. Biol Psychiatry. 2018;83:831–9.
Morrison FG, Miller MW, Logue MW, Assef M, Wolf EJ. DNA methylation correlates of PTSD: recent findings and technical challenges. Prog Neuro-Psychopharmacol Biol Psychiatry. 2019;90:223–34.
Mehta D, Klengel T, Conneely KN, Smith AK, Altmann A, Pace TW, et al. Childhood maltreatment is associated with distinct genomic and epigenetic profiles in posttraumatic stress disorder. Proc Natl Acad Sci USA. 2013;110:8302–7.
Hammamieh R, Chakraborty N, Gautam A, Muhie S, Yang R, Donohue D, et al. Whole-genome DNA methylation status associated with clinical PTSD measures of OIF/OEF veterans. Transl Psychiatry. 2017;7:e1169.
Smith AK, Conneely KN, Kilaru V, Mercer KB, Weiss TE, Bradley B, et al. Differential immune system DNA methylation and cytokine regulation in Post-Traumatic Stress Disorder. Am J Med Genet Part B. 2011;156B:700–8.
Mehta D, Bruenig D, Carrillo-Roa T, Lawford B, Harvey W, Morris CP, et al. Genomewide DNA methylation analysis in combat veterans reveals a novel locus for PTSD. Acta Psychiatr Scand. 2017;136:493–505.
Rutten BPF, Vermetten E, Vinkers CH, Ursini G, Daskalakis NP, Pishva E, et al. Longitudinal analyses of the DNA methylome in deployed military servicemen identify susceptibility loci for post-traumatic stress disorder. Mol Psychiatry. 2018;23:1145–56.
Snijders C, Maihofer AX, Ratanatharathorn A, Baker DG, Boks MP, Geuze E, et al. Longitudinal epigenome-wide association studies of three male military cohorts reveal multiple CpG sites associated with post-traumatic stress disorder. Clin Epigenetics. 2020;12:1–13.
Maddox SA, Kilaru V, Shin J, Jovanovic T, Almli LM, Dias BG, et al. Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD. Mol Psychiatry. 2018;23:658–65.
Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, et al. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci USA. 2010;107:9470–5.
Chen Y, Li X, Kobayashi I, Tsao D, Mellman T. Expression and methylation in Posttraumatic Stress Disorder and resilience; evidence of the role of odorant receptors. Psychiatry Res. 2016;245:36–44.
Kuan PF, Waszczuk MA, Kotov R, Marsit CJ, Guffanti G, Gonzalez A, et al. An epigenome-wide DNA methylation study of PTSD and depression in World Trade Center responders. Transl Psychiatry. 2017;7:e1158.
Uddin M, Ratanatharathorn A, Armstrong D, Kuan P, Allison E. Epigenetic meta-analysis across three civilian cohorts identifies NRG1 and HGS as blood-based biomarkers for post-traumatic stress disorder. Epigenomics. 2018;10:1585–601.
Smith A, Ratanatharathorn A, Maihofer AX, Naviaux RK, Aiello AE, Amstadter AB, et al. Epigenome-wide meta-analysis of PTSD across 10 military and civilian cohorts identifies novel methylation loci. bioRxiv. https://doi.org/10.1101/585109. 2019.
Yang SY, Ahmed S, Satheesh SV, Matthews J. Genome-wide mapping and analysis of aryl hydrocarbon receptor (AHR)‑ and aryl hydrocarbon receptor repressor (AHRR)‑ binding sites in human breast cancer cells. Arch Toxicol. 2018;92:225–40.
Krzyzewska IM, Ensink J, Nawijn L, Mul AN, Koch SB, Venema A, et al. Genetic variant in CACNA1C is associated with PTSD in traumatized police officers. Eur J Hum Genet. 2018;26:247–57.
Logue MW, Miller MW, Wolf EJ, Huber BR, Morrison FG, Zhou Z, et al. An epigenome-wide association study of posttraumatic stress disorder in US veterans implicates several new DNA methylation loci. Clin Epigenetics. 2020;12:1–14.
Vincenzes KA. Comparison of civilian trauma and combat trauma. VISTAS Online. 2013;45:1–8.
Tuerk PW, Grubaugh AL, Hamner MB, Foa EB. Diagnosis and treatment of PTSD-related compulsive checking behaviors in veterans of the iraq war: the influence of military context on the expression of PTSD symptoms. Am J Psychiatry. 2009;166:762–67.
Hagenaars MA, Fisch I, van Minnen A. The effect of trauma onset and frequency on PTSD-associated symptoms. J Affect Disord. 2011;132:192–9.
Kessler RC, Aguilar-Gaxiola S, Alonso J, Benjet C, Bromet EJ, Cardoso G, et al. Trauma and PTSD in the WHO World Mental Health Surveys. Eur J Psychotraumatol. 2017;8:1353383.
Howie H, Rijal CM, Ressler KJ. A review of epigenetic contributions to post-traumatic stress disorder. Dialogues Clin Neurosci. 2019;21:417–28.
Nawijn L, Krzyzewska IM, van Zuiden M, Henneman P, Koch S, Mul AN, et al. Oxytocin receptor gene methylation in male and female PTSD patients and trauma-exposed controls. Eur Neuropsychopharmacol. 2019;29:147–55.
Galanter JM, Gignoux CR, Oh SS, Torgerson D, Pino-Yanes M, Thakur N, et al. Differential methylation between ethnic sub-groups reflects the effect of genetic ancestry and environmental exposures. Elife. 2017;6:e20532.
Yuan V, Price EM, Del Gobbo G, Mostafavi S, Cox B, Binder AM, et al. Accurate ethnicity prediction from placental DNA methylation data. Epigene Chromatin. 2019;12:1–14.
Merikangas AK, Almasy L. Using the tools of genetic epidemiology to understand sex differences in neuropsychiatric disorders. Genes Brain Behav. 2020;19:e12660.
Sartor CE, McCutcheon VV, Pommer NE, Nelson EC, Grant JD, Duncan AE, et al. Common genetic and environmental contributions to posttraumatic stress disorder and alcohol dependence in young women. Psychol Med. 2011;41:1497–505.
Duncan LE, Ratanatharathorn A, Aiello AE, Almli LM, Amstadter AB, Ashley-Koch AE, et al. Largest GWAS of PTSD (N = 20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry. 2018;23:666–73.
Goldstein JM, Jerram M, Abbs B, Whitfield-gabrieli S, Makris N. Sex differences in stress response circuitry activation dependent on female hormonal cycle. J Neurosci. 2010;30:431–8.
Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychol. 2005;69:113–32.
Ramikie TS, Ressler KJ. Stress-related disorders, pituitary adenylate cyclase-activating peptide (PACAP)ergic system, and sex differences. Dialogues Clin Neurosci. 2016;18:403–13.
Abrahams N, Seedat S, Lombard C, Kengne AP, Myers B, Sewnath A, et al. Study protocol for a longitudinal study evaluating the impact of rape on women’s health and their use of health services in South Africa. BMJ Open. 2017;7:1–13.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59:34–57.
Bernstein D, Fink L. Childhood Trauma Questionnaire: a retrospective self-report manual. San Antonio: The Psychological Corporation; 1998.
Jewkes R, Nduna M, Levin J, Jama N, Dunkle K, Khuzwayo N, et al. A cluster randomized-controlled trial to determine the effectiveness of Stepping Stones in preventing HIV infections and promoting safer sexual behaviour amongst youth in the rural Eastern Cape, South Africa: Trial design, methods and baseline findings. Trop Med Int Heal. 2006;11:3–16.
Weathers F, et al. The Life Events Checklist for DSM-5 (LEC-5). USA: National Center for PTSD. 2013.
Davidson JR, Book SW, Colket JT, Tupler LA, Roth S, David D, et al. Assessment of a new self-rating scale for post-traumatic stress disorder. Psychol Med. 1997;27:153–60.
Saunders J, Aasland O, Babor T, De La Fuente J, Grant M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption-II. Addiction. 1993;88:791–804.
Radloff L. The CES-D Scale: a self-report depression scale for research in the general population. Appl Psychol Meas. 1977;1:385–401.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th edn. Arlington, 2013.
Illumina. Infinium® MethylationEPIC BeadChip. Illumina Mark. 2015;4.
Min JL, Hemani G, Smith G, Relton C, Suderman M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics. 2018;34:3983–9.
R Core Team. R: a language and environment for statistical computing. https://www.r-project.org/. 2018.
Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:1–17.
Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. Genomics High density DNA methylation array with single CpG site resolution Unmethylated locus Methylated locus Unmethylated locus Methylated locus. Genomics. 2011;98:288–95.
Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014;15:1–17.
Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161–35.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13. https://doi.org/10.1186/1471-2105-13-86.
Bonferroni C. Teoria statistica delle classi e calcolo delle probabilita. R Instituto Superiore de Scienze Economiche e Commerciali: Firenze, Italia, 1936.
Suderman M, Staley JR, French R, Arathimos R, Simpkin A, Tilling K. Dmrff: Identifying Differentially Methylated Regions Efficiently With Power and Control. bioRxiv. 2018;508556.
Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: Implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–32.
Staley J, et al. The MRC-IEU catalog of epigenome-wide association studies. EWAS Cat. http://ewascatalog.org/. 2017.
Li M, Zou D, Li Z, Gao R, Sang J, Zhang Y, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019;47:983–8.
Buniello, A, MacArthur J A L, Cerezo M, Harris L W, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics. Nucleic Acids Res. 2019; 47.
NCBI Resource C. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018;46:D8–D13.
Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and PAC1 receptor. Nature. 2011;470:492–7.
Almli LM, Mercer KB, Kerley K, Feng H, Bradley B, Conneely KN, et al. ADCYAP1R1 genotype associates with post-traumatic stress symptoms in highly traumatized African-American females. Am J Med Genet Part B. 2013;162:262–72.
Stevens JS, Almli LM, Fani N, Gutman DA, Bradley B, Norrholm SD, et al. PACAP receptor gene polymorphism impacts fear responses in the amygdala and hippocampus. Proc Natl Acad Sci USA. 2014;111:3158–63.
Schechter DS, Moser DA, Pointet VC, Aue T, Stenz L, Paoloni-Giacobino A, et al. The association of serotonin receptor 3A methylation with maternal violence exposure, neural activity, and child aggression. Behav Brain Res. 2017;325:268–77.
Tamir TY, Bowman BM, Agajanian MJ, Goldfarb D, Schrank TP, Stohrer T, et al. Gain-of-function genetic screen of the kinome reveals BRSK2 as an inhibitor of the NRF2 transcription factor. J Cell Sci. 2020;133:jcs241356.
Kalsi G, et al. Genome-Wide Association of Heroin Dependence in Han Chinese. 2016:1–18.
Kim JSM, Lilley BN, Zhang C, Shokat KM, Sanes JR, Zhen M. A chemical-genetic strategy reveals distinct temporal requirements for SAD-1 kinase in neuronal polarization and synapse formation. Neural Dev. 2008;3:1–14.
Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–32.
Sample V, Ramamurthy S, Gorshkov K, Ronnett G V, Chernoff J. Polarized activities of AMPK and BRSK in primary hippocampal neurons. Mol Biol Cell. 2015;26:1935–46.
Inoue E, Mochida S, Takagi H, Higa S, Deguchi-Tawarada M, Takao-Rikitsu E, et al. SAD: a presynaptic kinase associated with synaptic vesicles and the active zone cytomatrix that regulates neurotransmitter release. Neuron. 2006;50:261–75.
Hiatt SM, Thompson ML, Prokop JW, Lawlor J, Gray DE, Bebin EM, et al. Deleterious variation in BRSK2 associates with neurodevelopmental disorder. Am J Hum Genet. 2019;104:701–8.
Feliciano P, Zhou X, Astrovskaya I, Turner T N, Wang T, Brueggeman L, et al. Exome sequencing of 457 autism families recruited online provides evidence for novel ASD genes. mpj Genomic Med. 2019; 4.
Ross DA, Arbuckle MR, Travis MJ, Dwyer JB, van Schalkwyk GI, Ressler KJ. An integrated neuroscience perspective on formulation and treatment planning for posttraumatic stress disorder an educational review. JAMA Psychiatry. 2017;74:407–15.
van Marle H. PTSD as a memory disorder. Eur J Psychotraumatol. 2015;6:27633.
Krystal J, Neumeister A. Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Res. 2009;1293:13–23.
Sherin JE, Nemeroff CB. Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci. 2011;13:263–78.
Bright NJ, Carling D, Thornton C. Investigating the Regulation of Brain-specific Kinases 1 and 2 by Phosphorylation. 2008;283:14946–54.
Chen XY, Gu XT, Saiyin H, Wan B, Zhang YJ, Li J, et al. Brain-selective Kinase 2 (BRSK2) phosphorylation on PCTAIRE1 negatively regulates glucose-stimulated insulin secretion in pancreatic β -Cells. J Biol Chem 2012;287:30368–75.
Gastoł J, Kapusta P, Polus A, Pitera E, Biela M, Wołkow P, et al. Epigenetic mechanism in search for the pathomechanism of diabetic neuropathy development in diabetes mellitus type 1 (T1DM). Endocrine. 2020;68:235–40.
Jjingo D, Conley AB, Yi SV, Lunyak VV, King J,I. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3:462–74.
Edmondson D, Kronish IM, Shaffer JA, Falzon L, Burg MM. Posttraumatic stress disorder and risk for coronary heart disease: a meta-analytic review. Am Heart J. 2013;166:806–14.
Ryder AL, Azcarate PM, Cohen BE. PTSD and physical health. Curr Psychiatry Rep. 2018;20. https://doi.org/10.1007/s11920-018-0977-9.
Mustafa T. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP). Adv Pharm. 2013;68:445–57.
Hammack SE, Cheung J, Rhodes KM, Schutz KC, Falls WA, Braas KM, et al. Chronic stress increases pituitary adenylate cyclase-activating peptide (PACAP) and brain-derived neurotrophic factor (BDNF) mRNA expression in the bed nucleus of the stria terminalis (BNST): roles for PACAP in anxiety-like behavior. Psychoneuroendocrinology. 2009;34:833–43.
Rudecki AP, Gray SL. PACAP in the defense of energy homeostasis. Trends Endocrinol Metab. 2016;27:620–32.
Mcmahon FJ, Akula N, Schulze TG, Muglia P, Tozzi F, Detera-Wadleigh SD, et al. Meta-analysis of genome-wide association data detects a risk locus for major mood disorders on chromosome 3p21.1. Genome. 2010;42:128–31.
Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–9.
Mercer KB, et al. Functional evaluation of a PTSD-associated genetic variant: estradiol regulation and ADCYAP1R1. Transl Psychiatry. 2016;6:1–7.
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the national comorbidity survey. Arch Gen Psychiatry. 1995;52:1048–60.
Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7.
Anastasiadi D, Esteve-Codina A, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin. 2018;11:1–17.
Dias BG, Ressler KJ. PACAP and the PAC1 receptor in post-traumatic stress disorder. Neuropsychopharmacology. 2013;38:245–7.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2:1002–12.
Speer K, Upton D, Semple S, McKune A. Systemic low-grade inflammation in post-traumatic stress disorder: a systematic review. J Inflamm Res. 2018;11:111–21.
Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J. 2012;279:1375–85.
Wilson R, Wahl S, Pfeiffer L, Ward-Caviness CK, Kunze S, Kretschmer A, et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics. 2017;18:1–15.
Philibert R, Dogan M, Noel A, Miller S, Krukow B, Papworth E, et al. Dose response and prediction characteristics of a methylation sensitive digital PCR assay for cigarette consumption in adults. Front Genet. 2018;9:1–9.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–47.
Ratanatharathorn A, Aiello A, Boks M, Hauser M, Fen Kuan P, Logue M, et al. Epigenetic biomarkers Of PTSD: updates from the EWAS Working Group of the PTSD PGC. Eur Neuropsychopharmacol. 2019;29:S750.
Acknowledgements
This research is supported by: (i) The South African Research Chair in PTSD hosted by Stellenbosch University, funded by the Department of Science and Technology (DST) and administered by the South African National Research Foundation (NRF) and (ii) South African Medical Research Council in terms of the SAMRC’s Flagships Awards Project SAMRC-RFA-IFSP-01-2013/RAPE COHORT. The first author was supported by: (i) the SAMRC through its Division of Research Capacity Development under the National Health Scholarship Programme from funding received from the Public Health Enhancement Fund/South African National Department of Health and (2) under the SAMRC Intra-Mural Postdoctoral Fellowship Programme from funding received from the South African National Treasury. The content hereof is the sole responsibility of the authors and does not necessarily represent the official views of the SAMRC.
Author information
Authors and Affiliations
Contributions
JN, NA, CL, SS, and SMJH contributed to the conception and design of the study. JN, ST, MS, SM, CL, and SMJH analyzed the data. All authors contributed to the interpretation of the data. NA was involved in the acquisition of the primary data. MS developed the statistical package used in the epigenome-wide analysis. JN drafted the paper and all other authors revised it. All authors read and approved the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nöthling, J., Abrahams, N., Toikumo, S. et al. Genome-wide differentially methylated genes associated with posttraumatic stress disorder and longitudinal change in methylation in rape survivors. Transl Psychiatry 11, 594 (2021). https://doi.org/10.1038/s41398-021-01608-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01608-z
