
Abstract
Electroconvulsive therapy (ECT) is a quick-acting and powerful antidepressant treatment considered to be effective in treating severe and pharmacotherapy-resistant forms of depression. Recent studies have suggested that epigenetic mechanisms can mediate treatment response and investigations about the relationship between the effects of ECT and DNA methylation have so far largely taken candidate approaches. In the present study, we examined the effects of ECT on the methylome associated with response in depressed patients (n = 34), testing for differentially methylated CpG sites before the first and after the last ECT treatment. We identified one differentially methylated CpG site associated with the effect of ECT response (defined as >50% decrease in Hamilton Depression Rating Scale score, HDRS), TNKS (q < 0.05; p = 7.15 × 10−8). When defining response continuously (ΔHDRS), the top suggestive differentially methylated CpG site was in FKBP5 (p = 3.94 × 10−7). Regional analyses identified two differentially methylated regions on chromosomes 8 (Šídák’s p = 0.0031) and 20 (Šídák’s p = 4.2 × 10−5) associated with ΔHDRS. Functional pathway analysis did not identify any significant pathways. A confirmatory look at candidates previously proposed to be involved in ECT mechanisms found CpG sites associated with response only at the nominally significant level (p < 0.05). Despite the limited sample size, the present study was able to identify epigenetic change associated with ECT response suggesting that this approach, especially when involving larger samples, has the potential to inform the study of mechanisms involved in ECT and severe and treatment-resistant depression.
Similar content being viewed by others

Introduction
Depression is a major contributor to global burden of disease and despite worldwide research efforts, the heterogeneous nature of the disorder makes it difficult to definitively unravel its underlying etiology and the factors influencing treatment response1,2. Electroconvulsive therapy (ECT) is an intervention with rapid and striking antidepressant effects and is the treatment of choice for patients with severe and treatment-resistant depression3,4. Studying biological changes associated with response to ECT in such a subgroup of severely depressed patients is a promising approach to gain insights into the underlying mechanisms of depression and treatment response.
DNA methylation is thought to be involved in disease pathology through its influence on gene expression and cellular function5,6. There is evidence that pre-treatment methylation profiles may predict likelihood of achieving remission7 and the literature on DNA methylation in depression has pointed to several possible genes of interest (e.g., BDNF, SLC6A4, NR3C1, FKBP5, etc. for reviews, see7,8,9,10) as being related to antidepressant response.
Investigating change of methylation levels during treatment may inform the biological processes underlying both depression and antidepressant response. Examining these changes in ECT patients offers an optimal research setting as: (1) treatment effects are substantial and occur soon after the intervention, and (2) ECT patients represent a subgroup of patients with the most severe form of depression. It is likely that this subgroup is not only clinically but also genetically more homogenous, especially as these patients tend to show a higher genetic burden for major depression than those with less severe forms11.
The few studies exploring differences in methylation related to ECT are only beginning to give insight into the factors involved. A translational study observed that the response to electroconvulsive stimulation (ECS) in rats was predicted by higher p11 promoter methylation and found this also to be the case in two human samples (n = 11; n = 65) in response to ECT12. Another candidate gene is brain-derived neurotrophic factor (BDNF) which is involved in neuroplastic changes associated with stress and depression13,14. Methylation differences of the promoter region of BDNF have been observed in many psychiatric disorders15, and also after use of antidepressant medication16. Studies in animals and humans also propose an involvement of BDNF in response to ECT: after ECS/ECT, alterations of BDNF levels have been found in rats17, and in humans, differences in expression of BDNF have been described18,19. Furthermore, associations between pre-treatment BDNF levels and ECT outcome have been described, but whether BDNF levels are indicative of response remains to be determined20,21.
Research on ECT-related changes in methylation levels has largely focused on candidate genes. However, as ECT is a non-specific treatment with widespread, yet uncertain effects on biological processes, the selection of predefined candidates, relying on prior knowledge may not give the full picture. As such, an approach investigating the whole methylome is expected to yield new and relevant insights. Few studies have made use of these technologies in ECT samples to date. Moschny et al.22 examined longitudinal DNA methylation before and after ECT in a small group of patients (8 responders and 4 non-responders). They did not find any significant differences in global DNA methylation, but identified eight genes potentially implicated in either ECT response or its mechanism through analysis of single probe variance, and two genes whose methylation changed during treatment course.
In the present study, we aimed to identify changes in methylation levels associated with the effects of ECT and to find potential biomarkers for antidepressant response. We obtained and compared epigenome-wide DNA methylation levels of ECT patients (n = 34) before and after ECT. Differentially methylated CpG sites and regions associated with response were examined. Pathway analyses were employed to search for functional pathways affected by ECT. Finally, we took a targeted look at methylation in genes which have been previously implicated in ECT response and depression-related studies.
Materials and methods
This study was approved by the Ethics Committee (II), Medical Faculty Mannheim, University of Heidelberg. All patients provided written consent after a detailed explanation of the content of the study. All experimental procedures were performed in accordance with the Declaration of Helsinki.
Participants
Patients (n = 34) diagnosed with a major unipolar depressive episode (based on International Classification of Diseases version 10, ICD-10), older than 18 years, and assigned to ECT treatment were recruited at the Department of Psychiatry and Psychotherapy at the Central Institute of Mental Health (CIMH) in Mannheim, Germany, between 2014 and 2016. Criteria for assignment to ECT were treatment-resistant depression (i.e., failure of two adequate dose-duration antidepressants from different classes, or psychotherapy in the current episode), positive experience of ECT from a previous episode, or severe depression with (a) psychotic symptoms, (b) severe suicidality, or (c) the refusal of food or fluid intake. Patients were excluded if they had any substance-use-related disorders (other than tobacco and alcohol use disorders) or a lifetime diagnosis of schizophrenia. All participants were of Caucasian descent. The patients kept the same medication regimens throughout ECT treatment. This sample is a subset of a sample reported in a previous genetic study in ECT patients11. Descriptive and clinical characteristics of the participants are described in Table 1.
Depression and DNA methylation assessment
The 21-item version of the Hamilton Depression Rating Scale (HDRS) was administered and blood was collected from patients prior to the first (T1) and after the last (T2) session of ECT (average sessions: 10.82, range: 5–25 sessions). T2 was between 1 and 7 days after the last ECT session. Time of collection was kept constant between 8:30 and 9:30 a.m. at both timepoints to keep it close to the clinical interview conducted around the same time, as well as preclude any potential confounding effects arising from ward routines and circadian fluctuations23,24. Standard procedures for extraction and processing were followed. DNA extraction was performed using the Chemagic Magnetic Separation Module 1 (Chemagen Biopolymer-Technologie AG; Baesweiler, Germany). All genomic DNA samples were stored at −20 °C prior to analysis. Epigenome-wide DNA methylation was measured using the Illumina Infinium Methylation EPIC array (>850,000 CpG sites). The arrays were processed at the Genome Analysis Center at the HelmholtzZentrum München and Max-Planck-Institute for Psychiatry in Munich, Germany.
ECT treatment
ECT was conducted with a Thymatron IV device (Somatics, LLC. Lake Bluff, IL, USA). Anesthetic drugs used were: s-ketamine (~1.0 mg/kg)25,26 and succinylcholine for muscle relaxation (~1.0 mg/kg). Seizure thresholds were titrated at the initial session; dosing in subsequent sessions was given at >2.5 above this threshold. If patients did not show a clinical improvement or seizures were insufficient, energy used was increased. Patients received 2–3 ECT sessions per week. All patients started with unilateral stimulation with the possibility to change to bilateral stimulation at the discretion of the ECT supervisor.
Statistical analysis
Data processing, quality control (QC) and other statistical analyses were performed using R (versions 3.4.4 and 3.6.3) analysis software (https://cran.r-project.org/).
Data preprocessing, QC, and filtering
Methylation values were extracted using an updated version of the pipeline indicated in27, adapted in-house for use with the Illumina Infinium EPIC array. Illumina background correction was applied to all intensity values. A detection p-value threshold of p < 10−16 was used and intensity values with detection p ≥ 10−16 were designated as missing data. The proportion of missing data points was determined, allowing the calculation of sample and CpG site-specific call rates. Samples with insufficient DNA quality as denoted by a call rate of <95% were excluded. Intensity values were quantile normalized for each of the six probe types present on the array separately. Intensity values were converted to methylation beta values according to the manufacturer’s recommendation. White blood cell fractions were estimated according to28. Five of the six resulting estimates were subsequently included as covariates in downstream analyses to control for influences of cell type distribution on DNA methylation. The estimate for granulocytes showed the highest variance inflation factor (VIF) and was omitted to avoid collinearity issues. CpG sites were filtered by removing cross-hybridizing probes, probes with high missing rate (>0.02), and probes linked to X- and Y-chromosomes. Correction for batch effects and other technical parameters was done by performing a principal component analysis on internal control probe intensity values and including the first 10 extracted principal components (PCs) as covariates in the downstream analyses. Prior to analysis, all methylation beta values were logit transformed (base 2) to M-values, which were used in downstream analysis as recommended in29.
Differentially methylated single CpG sites
Association testing of methylation M-values for each CpG site was done using a mixed linear model approach as implemented in the Limma R package. Participant ID was used as a blocking factor, and estimated cell fractions, 10 control probe PCs, age, sex, and smoking status were included as additional covariates to adjust for confounding factors. It was observed that sex was highly correlated with the 5th control probe PC, thus this PC was not included in the statistical models to avoid collinearity issues.
The main effects of interest specified included: (1) response (responders vs. non-responders), (2) timepoint (change after intervention), and (3) the interaction between timepoint and response (the difference in change between response groups). Models were calculated with response specified both as a binary (>50% decrease in HDRS score) and continuous variable (i.e., change in HDRS score, ΔHDRS).
In addition, we examined the relationship between baseline (T1) methylation and response (both binary and continuous) in additional models.
Significance was defined as false discovery rate (FDR) q < 0.05. Results at a suggestive threshold of p < 10−5 were also reported.
Differentially methylated regions
Differentially methylated region (DMR) analysis was performed on the results of the above analyses using the comb-p package30. Comb-p parameters were specified as: seed p-value = 0.001 and a maximum distance between probes of 500 base pairs. These parameters follow those used in previous studies in the field31,32 and results from simulation experiments33.
Pathway analysis
A Gene Ontology (GO) enrichment analysis was performed on the results of the different models using the missMethyl (v1.12.0) R package. We examined CpG sites at the suggestive threshold of 1 × 10−5.
Targeted examination of methylation change in candidate CpG sites
Change in methylation of candidate genes from the literature was examined in an exploratory search. First, we selected: (1) candidates implicated in reviews of DNA methylation and antidepressant medication, i.e., BDNF, MAOA, SLC6A2, SLC6A4, HTR1A, HTR1B, IL6, IL117; SLC6A4, NR3C1, FKBP5, and OXTR8; and (2) candidates specific to ECT, i.e., S100A10 (p11)12, RNF175, RNF213, TBC1D14, TMC5, WSCD1, AC018685.2, AC098617.1, CLCN3P1, AQP10, and TRERF122.
Autosomal CpG sites which were annotated to these candidate genes in the UCSC Genome Browser NCBI curated RefSeq (retrieved: August 10, 2018) were extracted from the results of the single site analyses above (for each variable of interest in both binary and continuous models). Furthermore, to examine their predictive value in our sample, association between baseline methylation and response was also examined. A secondary FDR correction was applied to the list of all candidate CpG sites to control for false positives.
Results
Descriptive statistics of the sample are shown in Table 1. In the sample analyzed, using binary criteria, (defined as decrease of HDRS score of more than 50%) 25 were responders and 9 were non-responders to ECT. Levene’s test found no significant differences between group variances. Briefly, as also reported for the total sample in11, binary response to ECT was positively correlated with sex (being male) while continuous response (ΔHDRS score) was also associated with male sex and positively correlated with increased age.
Single CpG site analysis
Binary response
In the binary response model, one significantly differentially methylated site (q < 0.05), cg10005358, mapped to TNKS, was observed as an effect of response. Eight sites reached a suggestive threshold of p < 1 × 10−5 (see Table 2). No significantly differentially methylated CpG sites were observed for the effect of timepoint (at p < 1 × 10−5; 22 CpG sites), or interaction effect (at p < 1 × 10−5; 12 CpG sites). Several CpG sites annotated to the same gene appeared among the top hits of these effects of interest (see Tables 2 and S1.1–S1.3, e.g., TNKS, PCM1, RAPGEF2, RAB21; all suggestive at p < 1 × 10−5).
Continuous response (ΔHDRS)
In the continuous response model, no effects yielded significantly differentially methylated CpG sites at q < 0.05. At a suggestive threshold of p < 1 × 10−5, 7, 9, and 5 differentially methylated CpG sites were observed for the effects of ΔHDRS, timepoint, and ΔHDRS × timepoint, respectively (see Table 2 and Tables S2.1–S2.3). FKBP5 (CHR 6: cg01294490) was the top hit for both effect of ΔHDRS (p = 4.46 × 10−7) and effect of interaction of ΔHDRS × timepoint (p < 3.94 × 10−7). FXR2 (CHR 17: cg02936535) was also observed among the top hits for all effects of interest (ΔHDRS p = 3.79 × 10−6; timepoint p = 6.78 × 10−6; ΔHDRS × timepoint p = 7.38 × 10−6).
Baseline methylation and response
Methylation at baseline was not significantly associated with either binary or continuous response. At a suggestive threshold of p < 1 × 10−5 baseline methylation was associated with binary response at 9 CpG sites and with ΔHDRS at 6 CpG sites (see Tables S3.1 and S3.2).
Differentially methylated region analysis
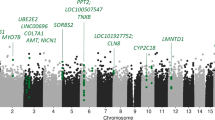
Two DMRs were identified as associated with effect of ΔHDRS in the continuous response model. One significant DMR on chromosome 8 was identified (3 probes, Šídák’s corrected p = 0.0031) and another on chromosome 20 (13 probes, Šídák’s corrected p = 4.2 × 10−5). The DMR on chromosome 8 (CHR 8: 127568854-127569023) is located in the LRATD2 (FAM84B) gene, while the chromosome 20 DMR (CHR 20: 36148620-36148861) is located in the BLCAP gene and in the promoter region of NNAT (791 base pairs upstream of the transcription start site, TSS). Two other regions on chromosomes 14 and 19 were nominally significant but did not remain significant after Šídák correction (see Table 3 and Fig. 1).

Significantly differentially methylated regions associated with the effect of ΔHDRS are indicated in red (chromosomes 8 and 20).
Analysis of the results from timepoint and timepoint × ΔHDRS models, as well as all binary response models, did not identify significant DMRs.
Pathway analysis
No significant pathways were observed in any of the models.
Candidate analysis
In the binary model, at nominal significance (p < 0.05 uncorrected), 43, 37, and 36 CpG sites were associated with response, timepoint, and response × timepoint interaction, respectively. The top 3 candidate CpG sites associated with binary response were: FKBP5 (cg01294490, p = 8.74 × 10−5), BDNF-AS (cg02386995, p = 1.51 × 10−4), and NR3C1 (cg23273257, p = 0.0012), but no CpG site tested survived secondary FDR correction for multiple testing.
In the continuous response model, 41, 43, and 42 CpG sites were nominally associated with ΔHDRS, timepoint and ΔHDRS × timepoint interaction, respectively. The top 3 candidate CpG sites associated with continuous response were, FKBP5 (cg01294490, p = 4.46 × 10−7), BDNF (cg15710245, p = 0.0033), and BDNF-AS (cg09878183, p = 0.0034). The site annotated to FKBP5 survived the secondary correction for multiple testing (q = 0.0004).
Discussion
The present study examined treatment-associated changes of DNA methylation levels in 34 patients in an epigenome-wide manner. By investigating the relationship between response status and change in methylation levels, this study identified several potential CpG sites involved in ECT response and outlines potential differences between response groups.
The top CpG site associated with binary response is located in TNKS, which is a protein-coding gene associated with blood pressure, alcohol consumption, implicated in cancer pathology, and involved in various processes such as the Wnt signaling pathway, telomere length, and vesicle trafficking34. Telomere length, a marker associated with aging is also known to be associated with psychiatric disorders35 including major depressive disorder36, as well as depressive symptoms37. In several genome-wide association studies (GWASs) of depression-related traits, TNKS was found to be associated with (p = 7.68 × 10−10)38, bipolar disorder (p = 3 × 10−6)39, and positive affect (p = 0.0003)40.
Among the top 5 CpG sites suggestively associated with binary response, genes associated with processes such as cell adhesion, cell growth, apoptosis in malignant tumors, protein metabolism (RAB21)41, and signaling in glucose metabolism (AQR)42 were found. RAB21 was previously reported to be associated with remission (p = 0.0103) in a GWAS of selective serotonin reuptake inhibitors in MDD patients43 and AQR as related to neuroticism (p = 9.58 × 10−8) and worry (p = 2.06 × 10−6)44, a well-known symptom in depression. RAPGEF2, a protein-coding gene suggested to be involved in signal transmission, in BDNF receptor pathway signaling45, in schizophrenia46, and is found to be a target for regulated miRNAs in MDD47. PCM1, located in a chromosomal region on 8p, which has been implicated in various neuropsychiatric disorders including schizophrenia and depression48, is a protein-coding gene critical for cell division, and is involved in the proliferation and neurogenesis of neuroprecursors49.
The top site in the continuous response analysis was located in FKBP5, a gene which is known to be an important endogenous regulator of the stress hormone system possibly linked to stress-related psychiatric disorders such as depression50. FKBP5 demethylation resulting from childhood trauma has been linked to long-term stress hormone system deregulation and effects on immune function and brain areas associated with stress regulation51. Depressive phenotypes are shown to be associated with the age-related decrease in FKBP5 methylation52. Altered epigenetic and genetic FKBP5 regulation may contribute to stress-related disease risk. Findings related to FKBP5 have pointed to it as important in the interaction with environment in stress-related disorders such as major depression53. The present study found an association between methylation in a CpG site in FKBP5 and the reduction in patients’ HDRS scores. Although the CpG site identified here has not been associated with antidepressant response in previous studies, the findings support FKBP5 as an important gene requiring further investigation in the present context.
Among the top 5 CpG sites suggestively associated with continuous response were CpG sites annotated to D2HGDH and FXR2. D2HGDH encodes for the enzyme D-2-hydroxyglutarate dehydrogenase, and is suggested to be downregulated in depressed patients during remission54. Proteins of the FXR family have commonly been reported in autism spectrum disorders, and evidence from GWASs in mood disorders and schizophrenia suggests that Fragile X mental retardation syndrome-related proteins are involved in the development of mental disorders55.
Two DMRs associated with continuous response were identified. The DMR on chromosome 8 lies in LRATD2 (FAM84B), which is known to be involved in gastric and prostate cancer56,57. In a recent large genome-wide gene-environment analysis, its paralog, LRATD1, was observed to be associated with unipolar depression and response to trauma exposure58. The DMR on chromosome 20 is located in BLCAP and in the promoter region of NNAT. BLCAP encodes a protein that regulates cell proliferation and reduces cell growth by stimulating apoptosis59, and NNAT is involved in brain development and neuronal differentiation60. Together with the single site results, these findings are in line with previous works in the field of ECT; alterations in mechanisms such as neurogenesis and neuroinflammatory immune response are proposed to be among the mechanisms of ECT action61,62,63.
Several candidate genes proposed in the literature were found harboring CpG sites with nominally significant changes between T1 and T2, and T1 methylation values for a number of them were also nominally associated with response (both binary and continuous) (see Tables S4.1–S6.2); the roles they play remain unclear. The present results appear to lend support to previous research which has suggested the importance of these candidates but these results are preliminary and further investigation is warranted. Also, the identification of FKBP5 in the present study suggests that future research should assess and control for factors such as childhood trauma and stress51,64.
This study had several limitations. Although the largest study to date, the present sample size was limited, and it is expected that future studies using a similar approach in larger samples will be able to further clarify our results. Sample size notwithstanding, we identified a single significantly differentially methylated CpG site, as well as some suggestive ones which need further investigation. While we assessed methylation levels in whole blood, ECT is applied to the brain; both central and peripheral mechanisms may be affected by the global nature of the treatment and care should be taken with the interpretation of these findings65. The possible effect of anesthesia and pharmacotherapy is a potential confounding factor in methylation studies. However, medication in each patient was kept constant during the ECT course, and there were no differences between patients regarding anesthesia administration or treatment dosage. Therefore, the observed changes are unlikely to have resulted from these medications.
The genes implicated in our findings have been previously involved in the etiology of depression and treatment response, but confirmation in larger samples is needed. Multi-center approaches and collaborative efforts could help in obtaining the sample sizes required to allow a more robust characterization of ECT response and give insights into the biological processes underlying the striking antidepressant effects of ECT.
References
World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates, 2017 (WHO, accessed 11 February 2021); https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf?sequence=1.
Krishnan, V. & Nestler, E. J. The molecular neurobiology of depression. Nature 455, 894–902 (2008).
Jaffe, R. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging: a task force report of the American Psychiatric Association. Am. J. Psychiatry 159, 331 (2002).
Singh, A. & Kar, S. K. How electroconvulsive therapy works? Understanding the neurobiological mechanisms. Clin. Psychopharmacol. Neurosci. 15, 210–221 (2017).
Jones, P. A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492 (2012).
Meissner, A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 (2008).
Webb, L.M., Phillips, K.E., Ho, M.C., Veldic, M. & Blacker, C.J. The relationship between DNA methylation and antidepressant medications: a systematic review.Int. J. Mol. Sci. 21, 826 (2020).
Chen, D., Meng, L., Pei, F., Zheng, Y. & Leng, J. A review of DNA methylation in depression. J. Clin. Neurosci. 43, 39–46 (2017).
Lisoway, A. J., Zai, C. C., Tiwari, A. K. & Kennedy, J. L. DNA methylation and clinical response to antidepressant medication in major depressive disorder: a review and recommendations. Neurosci. Lett. 669, 14–23 (2018).
Menke, A., Klengel, T. & Binder, E. B. Epigenetics, depression and antidepressant treatment. Curr. Pharm. Des. 18, 5879–5889 (2012).
Foo, J. C. et al. Evidence for increased genetic risk load for major depression in patients assigned to electroconvulsive therapy. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 180, 35–45 (2019).
Neyazi, A. et al. P11 promoter methylation predicts the antidepressant effect of electroconvulsive therapy. Transl. Psychiatry 8, 25 (2018).
Levy, M. et al. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacology 235, 2195–2220 (2018).
Pittenger, C. & Duman, R. S. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33, 88–109 (2008).
Zheleznyakova, G. Y., Cao, H. & Schioth, H. B. BDNF DNA methylation changes as a biomarker of psychiatric disorders: literature review and open access database analysis. Behav. Brain Funct. 12, 17 (2016).
Tadic, A. et al. Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 19, 281–283 (2014).
Sartorius, A. et al. Correlations and discrepancies between serum and brain tissue levels of neurotrophins after electroconvulsive treatment in rats. Pharmacopsychiatry 42, 270–276 (2009).
Kleimann, A. et al. BDNF serum levels and promoter methylation of BDNF exon I, IV and VI in depressed patients receiving electroconvulsive therapy. J. Neural Transm. 122, 925–928 (2014).
Stelzhammer, V. et al. Electroconvulsive therapy exerts mainly acute molecular changes in serum of major depressive disorder patients. Eur. Neuropsychopharmacol. 23, 1199–1207 (2013).
Rocha, R. B. et al. Increased BDNF levels after electroconvulsive therapy in patients with major depressive disorder: a meta-analysis study. J. Psychiatr. Res. 83, 47–53 (2016).
van Zutphen, E. M. et al. Brain-derived neurotrophic factor as a possible predictor of electroconvulsive therapy outcome. Transl. Psychiatry 9, 155 (2019).
Moschny, N. et al. Novel candidate genes for ECT response prediction - a pilot study analyzing the DNA methylome of depressed patients receiving electroconvulsive therapy. Clin. Epigenetics 12, 114 (2020).
Rijo-Ferreira, F. & Takahashi, J. S. Genomics of circadian rhythms in health and disease. Genome Med. 11, 82 (2019).
Liu, C. & Chung, M. Genetics and epigenetics of circadian rhythms and their potential roles in neuropsychiatric disorders. Neurosci. Bull. 31, 141–159 (2015).
Hoyer, C., Kranaster, L., Janke, C. & Sartorius, A. Impact of the anesthetic agents ketamine, etomidate, thiopental, and propofol on seizure parameters and seizure quality in electroconvulsive therapy: a retrospective study. Eur. Arch. Psychiatry Clin. Neurosci. 264, 255–261 (2014).
Kranaster, L., Kammerer-Ciernioch, J., Hoyer, C. & Sartorius, A. Clinically favourable effects of ketamine as an anaesthetic for electroconvulsive therapy: a retrospective study. Eur. Arch. Psychiatry Clin. Neurosci. 261, 575–582 (2011).
Lehne, B. et al. A coherent approach for analysis of the Illumina HumanMethylation450 BeadChip improves data quality and performance in epigenome-wide association studies. Genome Biol. 16, 37 (2015).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587 (2010).
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Mooney, M. A. et al. Large epigenome-wide association study of childhood ADHD identifies peripheral DNA methylation associated with disease and polygenic risk burden. Transl. Psychiatry 10, 8 (2020).
Roberts, S. et al. Longitudinal investigation of DNA methylation changes preceding adolescent psychotic experiences. Transl. Psychiatry 9, 69 (2019).
Mallik, S. et al. An evaluation of supervised methods for identifying differentially methylated regions in Illumina methylation arrays. Brief. Bioinform. 20, 2224–2235 (2018).
GeneCards. TNKS Gene-GeneCards | TNKS1 Protein | TNKS1 Antibody (accessed 22 January 2021); https://www.genecards.org/cgi-bin/carddisp.pl?gene=TNKS.
Darrow, S. M. et al. The association between psychiatric disorders and telomere length: a meta-analysis involving 14,827 persons. Psychosom. Med. 78, 776–787 (2016).
Ridout, K. K., Ridout, S. J., Price, L. H., Sen, S. & Tyrka, A. R. Depression and telomere length: a meta-analysis. J. Affect. Disord. 191, 237–247 (2016).
Humphreys, K. L., Sisk, L. M., Manczak, E. M., Lin, J. & Gotlib, I. H. Depressive symptoms predict change in telomere length and mitochondrial DNA copy number across adolescence. J. Am. Acad. Child Adolesc. Psychiatry 59, 1364–1370 (2020).
Jones, S. E. et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat. Commun. 10, 343 (2019).
Stahl, E. A. et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803 (2019).
Baselmans, B. M. et al. Multivariate genome-wide analyses of the well-being spectrum. Nat. Genet. 51, 445–451 (2019).
GeneCards. RAB21 Gene-Genecards | RAB21 Protein | RAB21 Antibody (accessed 22 January 2021); https://www.genecards.org/cgi-bin/carddisp.pl?gene=RAB21&keywords=RAB21.
Song, C. et al. AQR is a novel type 2 diabetes-associated gene that regulates signaling pathways critical for glucose metabolism. J. Genet. Genom. 45, 111–120 (2018).
Ji, Y. et al. Pharmacogenomics of selective serotonin reuptake inhibitor treatment for major depressive disorder: genome-wide associations and functional genomics. Pharmacogenomics J. 13, 456–463 (2013).
Nagel, M., Watanabe, K., Stringer, S., Posthuma, D. & van der Sluis, S. Item-level analyses reveal genetic heterogeneity in neuroticism. Nat. Commun. 9, 905 (2018).
GeneCards. RAPGEF2 Gene-Genecards | RPGF2 Protein | RPGF2 Antibody (accessed 22 January 2021); https://www.genecards.org/cgi-bin/carddisp.pl?gene=RAPGEF2&keywords=RAPGEF2.
Han, J. & Gage, F.H. A role for miR-19 in the migration of adult-born neurons and schizophrenia.Neurogenesis 3, e1251873 (2016).
Garbett, K. A. et al. Coordinated messenger RNA/microRNA changes in fibroblasts of patients with major depression. Biol. Psychiatry 77, 256–265 (2015).
Tabares-Seisdedos, R. & Rubenstein, J. L. Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol. Psychiatry 14, 563–589 (2009).
Zhang, W. et al. MiRNA-128 regulates the proliferation and neurogenesis of neural precursors by targeting PCM1 in the developing cortex. eLife 5, e11324 (2016).
Binder, E. B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 34, 186–195 (2009).
Klengel, T. et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 16, 33–41 (2013).
Zannas, A. S. et al. Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-kappaB-driven inflammation and cardiovascular risk. Proc. Natl Acad. Sci. USA 116, 11370–11379 (2019).
Zannas, A. S., Wiechmann, T., Gassen, N. C. & Binder, E. B. Gene-stress-epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology 41, 261–274 (2016).
Scifo, E. et al. Sustained molecular pathology across episodes and remission in major depressive disorder. Biol. Psychiatry 83, 81–89 (2018).
Khlghatyan, J. & Beaulieu, J. M. Are FXR family proteins integrators of dopamine signaling and glutamatergic neurotransmission in mental illnesses? Front. Synaptic Neurosci. 10, 22 (2018).
Zhang, Y. et al. Long non-coding RNA FAM84B-AS promotes resistance of gastric cancer to platinum drugs through inhibition of FAM84B expression. Biochem. Biophys. Res. Commun. 509, 753–762 (2019).
Wong, N. et al. Upregulation of FAM84B during prostate cancer progression. Oncotarget 8, 19218–19235 (2017).
Coleman, J. R. I. et al. Genome-wide gene-environment analyses of major depressive disorder and reported lifetime traumatic experiences in UK Biobank. Mol. Psychiatry 25, 1430–1446 (2020).
NCBI. BLCAP BLCAP Apoptosis Inducing Factor – Gene (accessed 22 January 2021); https://www.ncbi.nlm.nih.gov/gene/10904.
NCBI. NNAT Neuronatin-Gene (accessed 22 Jan 2021); https://www.ncbi.nlm.nih.gov/gene/4826.
Nakamura, K. et al. Effects of single and repeated electroconvulsive stimulation on hippocampal cell proliferation and spontaneous behaviors in the rat. Brain Res. 1491, 88–97 (2013).
van Buel, E.M. et al. Immune and neurotrophin stimulation by electroconvulsive therapy: is some inflammation needed after all?. Transl. Psychiatry 5, e609 (2015).
Mindt, S., Neumaier, M., Hoyer, C., Sartorius, A. & Kranaster, L. Cytokine-mediated cellular immune activation in electroconvulsive therapy: a CSF study in patients with treatment-resistant depression. World J. Biol. Psychiatry 21, 139–147 (2019).
Weder, N. et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child. Adolesc. Psychiatry 53, 417–424 (2014).
Hannon, E., Lunnon, K., Schalkwyk, L. & Mill, J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 10, 1024–1032 (2015).
Acknowledgements
The authors wish to thank Susann Sauer and Maik Ködel at the Genome Analysis Center at the HelmholtzZentrum München and Max-Planck-Institute for Psychiatry in Munich, Germany, for running the methylation arrays. This work was supported by the German Research Foundation [DFG; grant FOR2107; RI908/11-2 and WI3429/3-2], the German Federal Ministry of Education and Research (BMBF) through the Integrated Network IntegraMent, under the auspices of the e:Med Programme [01ZX1314G; 01ZX1614G] through grants 01EE1406C, 01EE1409C, Target-OXY [031L0190A] and through ERA-NET NEURON, “SynSchiz-linking synaptic dysfunction to disease mechanisms in schizophrenia-a multilevel investigation” [01EW1810], through ERA-NET NEURON “EMBED-impact of Early life MetaBolic and psychosocial strEss on susceptibility to mental Disorders; from converging epigenetic signatures to novel targets for therapeutic intervention” [01EW1904], and by a grant of the Dietmar-Hopp Foundation. The funders had no role in the design of the study nor in its execution, analyses, interpretation of results, and decision to prepare and submit the manuscript for publication.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sirignano, L., Frank, J., Kranaster, L. et al. Methylome-wide change associated with response to electroconvulsive therapy in depressed patients. Transl Psychiatry 11, 347 (2021). https://doi.org/10.1038/s41398-021-01474-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01474-9
This article is cited by
-
Nrf2 activation rescues stress-induced depression-like behaviour and inflammatory responses in male but not female rats
Biology of Sex Differences (2024)
-
Hippocampal neuroplasticity, major depression and, not to forget: ECT
Molecular Psychiatry (2022)
