
Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disease associated with a complex genetic etiology. Besides the apolipoprotein E ε4 (APOE ε4) allele, a few dozen other genetic loci associated with AD have been identified through genome-wide association studies (GWAS) conducted mainly in individuals of European ancestry. Recently, several GWAS performed in other ethnic groups have shown the importance of replicating studies that identify previously established risk loci and searching for novel risk loci. APOE-stratified GWAS have yielded novel AD risk loci that might be masked by, or be dependent on, APOE alleles. We performed whole-genome sequencing (WGS) on DNA from blood samples of 331 AD patients and 169 elderly controls of Korean ethnicity who were APOE ε4 carriers. Based on WGS data, we designed a customized AD chip (cAD chip) for further analysis on an independent set of 543 AD patients and 894 elderly controls of the same ethnicity, regardless of their APOE ε4 allele status. Combined analysis of WGS and cAD chip data revealed that SNPs rs1890078 (P = 6.64E−07) and rs12594991 (P = 2.03E−07) in SORCS1 and CHD2 genes, respectively, are novel genetic variants among APOE ε4 carriers in the Korean population. In addition, nine possible novel variants that were rare in individuals of European ancestry but common in East Asia were identified. This study demonstrates that APOE-stratified analysis is important for understanding the genetic background of AD in different populations.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the most common form of dementia, accounting for 60–70% of all cases1. In the United States, the prevalence of AD in people aged 65 years and older is forecast to increase from 4.7% in 2010 to 13.8% in 20502,3. Likewise, in South Korea, the prevalence of AD in people aged 65 years and older is forecast to increase from 9.95% in 2017 to 16.09% in 20504.
Late-Onset Alzheimer’s disease (LOAD), the non-Mendelian form of AD, accounts for >99% of AD and is highly heritable (estimates of 58–79%), with complex genetic etiology5,6. The APOE ε4 allele is the strongest genetic risk factor for LOAD, with a population attributable fraction of approximately 30–35%7. The search for additional genetic risk factors through genome-wide association studies (GWAS) conducted mainly in European populations have yielded a few dozen genetic loci, beyond APOE ε4, associated with LOAD8,9,10,11,12. However, the genetic liability of AD by APOE and GWAS findings is estimated to be only 24–33%, which is not enough to explain the 58–79% heritability for AD revealed by twin studies5,6.
The effects of many risk loci differ across ethnic groups. For example, variants in ABCA7 have greater effects on AD risk for individuals of African–American ancestry than on those of European ancestry13. One main reason for this is differences in frequencies of risk alleles in different ethnic groups, which affects calculations of odds ratios and statistical significance. Many AD loci can be revealed by focusing on ethnic groups other than European. For example, GWAS in non-European populations have identified novel genetic risk factors associated with AD, such as SORL1 from Japanese studies and ACE from Israeli–Arabs14,15. Morris et al. recently showed that racial differences are correlated with molecular biomarkers, such as the levels of t-tau and p-tau181 proteins in cerebrospinal fluid of AD patients16. Several GWAS where individuals were stratified based on APOE status have also led to the identification of novel loci17,18. For example, KANSL1 region on chromosome 17 near MAPT was identified using only APOE ε4 negative (APOE ε4-) samples in the International Genomics of Alzheimer’s Project (IGAP)17. Novel associations were also identified with variants in ISYNA1, OR8G5, IGHV3-7, and SLC24A3, among APOE ε4 carriers19. We therefore rationalized that we would be more likely to find novel genetic variants by focusing on a subgroup of individuals of Korean ethnicity that was stratified based on APOE ε4 allele status.
The goal of the present study was to identify genetic markers that are significantly associated with AD among APOE ε4 carriers in the Korean population by conducting the following analyses: (1) identifying possible candidate markers from a dataset of 500 whole-genome sequencing (WGS) comprised of 331 AD cases and 169 controls; (2) designing a customized genotyping AD chip (cAD chip) containing our WGS candidates and previously known genetic variants associated with AD; and (3) validating significant genetic markers in independent datasets from the Korean population by using the cAD chip.
Materials and methods
Sample information
In total, 331 AD patients and 169 control participants aged 70 years or older, with normal cognitive ability, were recruited as a discovery set. All 500 participants used in the discovery set were of Korean descent and bore the APOE ε4 positive (APOE ε4+) allele. A separate set of 1915 participants, also of Korean descent, including 287 bearing the APOE ε4+ allele, were recruited as a validation set. All participants were enrolled on the basis of the medical records written by Geriatric neuropsychiatrists with expertise in dementia research. Each participant’s cognitive status was diagnosed based on a medical history assessment, neuropsychological test, and medical imaging with magnetic resonance imaging or amyloid positron emission tomography data. All study protocols were reviewed and approved by the relevant Institutional Review Boards at the Samsung Medical Center, Seoul, South Korea.
Whole-genome sequencing analysis
WGS data were generated using the Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA). Library construction and 150-bp paired-end sequencing were performed according to the manufacturer’s instructions. The average depth showed 18× coverage of the whole genome. Sequencing fastq data were aligned to the reference genome, hg19, with the decoy sequence using the BWA-mem algorithm implemented in BWA 0.7.1020. Duplicate reads were removed using Picard 1.1 (https://broadinstitute.github.io/picard/). Variant detection was performed using GenomeAnalysisTK-3.3–021. Variant annotation was conducted using ANNOVAR22 for refGene with dbSNP 147; population frequency was assessed with gnomAD23. In total, 22,526,987 variants (SNPs and indels) were obtained from the raw variant call set. We applied the following hard filter criteria: (1) exclude multi-allelic variants; (2) include variants with total read depth over six; (3) include variants with alternative read depth over three, (4) exclude segmental duplication variants, and (5) exclude very rare variants with minor allele frequency (MAF) less than 0.1%.
Design of the customized genotyping AD chip
To validate our candidates, we designed a cAD chip (based on an Axiom® myDesign GW genotyping array; Thermo Fisher Scientific, Waltham, MA, USA) that would allow precise genetic characterization of samples in the context of genetic variants associated with AD. Candidates available on the cAD chip were taken from three primary sources: variants in AD-related databases, variants reported in the literature, and candidates from the WGS analysis of the discovery set.
For known AD-related candidates, we referenced the AlzGene database (http://www.alzgene.org/), the NHGRI-EBI GWAS catalog (www.ebi.ac.uk/gwas/), the single nucleotide polymorphism database (dbSNP; https://www.ncbi.nlm.nih.gov/SNP/), the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/), as well as other candidates reported in the literature. From the AlzGene database, we selected 1448 genetic variants associated with AD24. A systematic review of published literature was performed to include variants known to be involved in AD. We used the NHGRI-EBI GWAS catalog curated from published GWAS since 200825. The GWAS catalog data were downloaded from the University of California, Santa Cruz table browser, and candidates were selected that included the word “Alzheimer” for disease and trait type. The AD chip also contains the IGAP data released from the 2013 meta-analysis results for AD8. We selected 4131 variants with P values less than 0.00001 in the stage 1 results of 17,008 AD cases and 37,154 controls. Meta-analysis results included an independent set of 8572 AD cases and 11,312 controls with combined P values. We also considered APOE-stratified GWAS results re-analyzed from IGAP consortium data for APOE ε4+ (10,352 cases and 9207 controls) and APOE ε4- (7184 cases and 26,968 controls) subgroups. An additional 1127 candidates were derived from the original article published by Jun et al.17. Exonic variants located in APP, PSEN1, and PSEN2, known to cause autosomal dominant early-onset AD, were included from dbSNP 147 and HGMD (Supplementary Table 1).
To prioritize variants from WGS analysis of the discovery set, we gathered five types of candidate groups: coding variants, non-coding variants, case-only, control-only, and expression quantitative trait loci (eQTL) groups. Supplementary Table 1 shows the summarized information of the candidate variants in the cAD chip. First, to identify association signals from coding and non-coding region candidates, we conducted an association analysis of whole-genome variants by comparing AD samples versus controls for 500 WGS samples. We selected candidates with P values less than 0.05 for variants within a coding region and less than 0.001 for variants within a non-coding region. This led to the inclusion of 1396 coding variants and 29,606 non-coding variants on the chip. Second, to include rare variants, we selected variants that appeared in the above two samples in AD cases or controls. We identified 2357 AD case-only variants and 327 control-only variants. Finally, we added 570 known eQTL variants expressed in brain tissues from the HaploReg v4.1 database that mapped to the non-coding variants from the 500 WGS data26.
Chip genotyping and QCs
We genotyped samples from 1915 participants using the cAD chip for replication of WGS analysis and performance evaluation of the customized chip. All DNA samples were extracted from whole blood. Our genotype calling workflow on the Affymetrix® GeneTitan® platform with the Axiom® myDesign GW genotyping array was performed according to the Axiom® 2.0 reagent kit protocol for 384 samples (Affymetrix®/Thermo Fisher Scientific). Fifty samples previously used for WGS analysis were also included to evaluate the cAD chip performance in order to compare the WGS results for the corresponding SNPs that overlapped both datasets. One sample with experimental error was excluded from the genotyping analysis. Raw CEL data files generated by the GeneTitan® were imported into the Axiom® Analysis Suite Software (version 3.0, Affymetrix/Thermo Fisher Scientific), and analysis was performed using Affymetrix Best Practices Workflow with the default threshold settings. All five plates passed plate QC; 19 samples with failing dish QC < 0.82, 70 samples with calling QC call rates < 0.97, and five samples determined to be technical duplicates were excluded from the analysis. The average call rate for the passing samples was 99.57%. We also reviewed the individual clinical charts for each patient and adopted only AD patients and candidates with normal cognition as controls, excluding 255 samples from participants with mild cognitive impairment and samples from candidates less than 55 years old. We excluded 78 samples with discordant clinical information regarding APOE status and sex and computed results based on the cAD chip. After genotyping, we also applied high-accuracy variant QC that excluded missing genotype rates > 5%, significant deviations from Hardy–Weinberg equilibrium P < 0.000001 in controls, and monomorphic variants, and included the best-recommended variants from the Axiom® Analysis Suite Software. In total, 26,242 variants in 1437 samples, 543 AD cases, and 894 healthy controls including 190 AD cases and 97 controls from the APOE ε4 carriers, were used for the final analysis.
Statistical analysis
All statistical analyses, including association analysis, were performed using PLINK version 1.0927 and the statistical software R (R Foundation for Statistical Computing, Austria). LocusZoom software was used to depict candidate regions in detail28. Linkage disequilibrium (LD) information from the 1000 Genomes Project (1KGP) and functional annotation of non-coding variants were obtained from HaploReg v4.126.
Results
Identification of candidate variants by using WGS in a Korean population
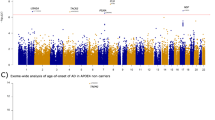
We conducted WGS of AD patients and matched controls in a Korean population who were APOE ε4 carriers, to find genetic variants dependent on APOE ε4 allele status, which is highly associated with AD pathogenesis. Our WGS analysis identified 22,526,987 variants from an APOE ε4 carrier dataset comprised of 331 AD patients and 169 controls. Results from all the samples confirmed that there was an ethnic overlap in genetic background with the East Asian population (JPT + CHB population) and that there was no population stratification in our discovery set (Supplementary Fig. 1). To overcome limitations in sample size and lack of power to reach genome-wide significance, we replicated 34,256 variants identified by WGS using a cAD chip (Supplementary Table 1). The chip included known variants associated with AD collected from various sources such as the IGAP. Clinical and demographic characteristics are shown in Supplementary Table 2. Our discovery and validation procedures are schematized in Fig. 1.

The schematic workflow shows the procedures for detecting novel genetic variants associated with Alzheimer’s Disease (AD) in a Korean population of APOE ε4 carriers (A) and for assessing the reproducibility of previously reported AD loci and suggestive novel loci shown to be population-specific (B). cAD customized genotyping AD chip, IGAP International Genomics of Alzheimer’s Project.
Characterization of cAD chip content and concordance with WGS
In total, the AD chip contains 41,735 autosomal variants and 745 X-chromosome variants. Of these, 40,395 (95.1%) variants were found in the single nucleotide polymorphism database (dbSNP) build 147. In addition, 2113 small insertion or deletion variants were also included in the cAD chip. Annotation using ANNOVAR showed that the cAD chip contained 4855 exonic, 14,155 intronic, and 20,416 intergenic variants (Supplementary Tables 3 and 4).
To evaluate the accuracy of the cAD chip, we compared 50 samples randomly selected from the WGS discovery set. All samples exhibited high call rates (average = 99.59%) and dish quality control (QC) (average = 0.97) in the results generated by the cAD chip analysis. The average concordance rate of 25,416 autosomal genotype calls that overlapped both the cAD chip and WGS genotypes in 50 samples was 99.29% (range = 98.47–99.69%). Considering SNPs exclusively, we observed high concordance rates ranging from 98.50% to 99.70% (average = 99.31%).
Known genetic variants associated with AD in cAD chip application set
We first examined the reproducibility of previously reported AD loci in 543 AD cases and 894 controls in the cAD chip application set (Table 1). We assessed 40 susceptibility loci previously reported to be associated with AD from four GWAS published by Andrews et. al.11. We identified 12 loci: CR1, BIN1, CD2AP, GPR141, PILRA, EPHA1, CLU, PICALM, SORL1, SLC24A2, ABCA7, and APOE, exhibiting significant association (P value < 0.05) in the cAD chip analysis. The APOE region in particular [best SNP: rs429358, P = 2.59 × 10-33, OR = 4.81 (95% CI: 3.76–6.16)] exhibited the highest significant association signal in the Korean population. APOE ε4 is the strongest genetic risk factor for AD, that has been identified in recent decades. The PICALM region [best SNP: rs3851179, P = 2.25 × 10−3, OR = 0.78 (95% CI: 0.67–0.92)] was also replicated in our data and showed an effect size similar to that observed in the IGAP study (Fig. 2). Our best SNP (rs3851179) in the PICALM region is in high LD (r2 = 1 based on the 1KGP phase 1 population) with the SNP previously reported as the best level of association (rs10792832) with AD in IGAP results8. Although the EPHA1 region showed a significant association [best SNP: rs11771145, P = 4.75 × 10−3, OR = 1.24 (95% CI: 1.06–1.44)], the allelic effect of the same SNP from a previous report was in the opposite direction [OR: 1.24 (95% CI: 1.06–1.44) vs. 0.90 (95% CI: 0.88–0.93)]. Our study corroborated the association of known AD variants reported by Lambert et al. in a Korean population8. BIN1 [rs6733839, P = 0.049, OR = 1.17 (95% CI: 1.00–1.36)], CLU [rs9331896, P = 0.021, OR = 0.81(95% CI: 0.68–0.97)], PICALM [rs10792832, P = 0.0029, OR = 0.79(95% CI: 0.67–0.92)], and ABCA7 [rs4147929, P = 0.045, OR = 1.18 (95% CI: 1.01–1.38)] showed significant association with the same risk allele and direction in cAD chip results. However, we observed that not all SNPs showing the best level of association in the IGAP study were not the most strongly associated SNPs in the loci analyzed in our dataset. One such example is rs4732729 [P = 0.013, OR = 0.80 (95% CI: 0.67–0.96)], located in CLU, which exhibits a more significant association than rs9331896. Notably, the allelic effect (minor allele) of rs4732729 was opposite to that of the IGAP dataset [OR: 0.80 (95% CI: 0.67–0.96) vs. OR: 1.12 (95% CI: 1.07–1.16)]. This discrepancy may be attributed to the different LD relationships between rs9331896 and rs4732729 (r2 = 0.96 based on 1KGP phase 1 ASN population and r2 = 0.33 based on the 1KGP phase 1 EUR population) in different population. This suggests that it is important to interpret the results while considering the population LD structure. Ethnic comparison of known variants reported from IGAP between Korean and European populations showed a low correlation coefficient (r = 0.216, P < 2.2E−16; Supplementary Fig. 2). Taken together, our results suggest that there are ethnic differences in associated SNPs at loci related to AD among populations, even when shared risk variants are associated with AD.

The figures show the regional association plot of known representative loci, (A) 11q14.2; PICALM and (B) 19q13.32; APOE, in the cAD chip application set (n = 1,437). The purple shaded diamond shape represents rs3851179 and rs429358, which are the most significant SNPs in 11q14.2 (A) and 19q13.32 (B), respectively. The blue line indicates the recombination rate, while filled color represents the linkage disequilibrium score based on r2 values estimated from the 1000 genome Nov 2014 ASN data.
Novel variants associated with AD in APOE ε4 carriers
We identified two novel AD-associated SNPs in APOE ε4 carriers with a discovery P value < 10−3, a replication P value of 0.05, and a combined P value < 10−6 (Table 2). One intergenic SNP between SORCS1 and LINC01435 [rs1890078; P = 6.64 × 10−7, OR = 0.43 (95% CI: 0.30–0.61)] showed higher MAF allele, C in control samples as a protective effect associated with reduced risk of AD (Fig. 3). Located in the intron of CHD2, SNP rs12594991, showed higher frequency of the minor allele, A in AD cases. The frequency of the A allele in rs12594991 in the European population (0.52) is much higher than in the Asian population (0.15). Two SNPs (rs1890078 and rs12594991) did not exhibit any significant association with AD in the APOE ε4 non-carriers (Supplementary Table 5).

The figures show the regional association plot of known representative loci, (A) 10q25.1; SORCS1 and (B) 15q26.1; CHD2, in the cAD chip application set with a combined P value < 0.000001. The purple shaded diamond shape represents rs1890078 and rs12594991, which are the most significant SNPs in 10q25.1 (A) and 15q26.1 (B), respectively. The blue line indicates the recombination rate, while filled color represents the linkage disequilibrium score based on r2 values estimated from the 1000 genome Nov 2014 ASN data.
Novel variants associated with AD in the cAD chip application set
Candidate novel variants in the cAD chip application set were selected based on the following criteria: (1) only P values less than 0.005 were accepted; (2) MAF < 1% were excluded; and (3) common variants in non-Finnish European populations with a MAF value above 0.05 from the GnomAD database (http://gnomad.broadinstitute.org/) were removed; and (4) only the variants with the same direction of allelic effect, estimated using the odds ratio between the discovery set and validation set, were accepted. Subsequently, we identified nine AD-associated SNPs in eight genes (CLIC4, PTPRN2, PSD3, SORCS1, LOC102724301, LINC01578, ABR, and USP32) (Table 3). Six of these SNPs were not included in the IGAP consortium data because they were either not genotyped or were filtered out because of low MAF in European populations. Significant associations were not observed for the other three SNPs (rs12063304, rs967326, and rs79919241) in IGAP data. Notably, two intergenic SNPs found between SORCS1 and LINC01435 [rs144835823; P = 8.40 × 10−4, OR = 0.32 (95% CI: 0.15–0.65) and rs78442236; P = 9.57 × 10−5, OR = 0.17 (95% CI: 0.06–0.47)] and one intronic SNP in USP32 [rs117665140; P = 8.17 × 10−4, OR = 1.62 (95% CI: 1.23–2.13)] were not found in European populations but were identified in our study. Taken together, we identified ethnic differences in associated single nucleotide polymorphisms (SNPs) at loci related to AD among different populations. We also identified 15 rare LOF variants such as frameshift, stop-gain, and stop-loss from the case-only or control-only categories of WGS analysis. However, none of these variants was significant in the cAD chip analysis.
rs1890078, previously observed in APOE ε4 carriers, was a common variant in SORCS1. However, rs144835823 and rs78442236 were very rare variants located approximately 24 kb and 55 kb away from the common variant, respectively. In the cAD chip application set, rs1890078 showed significant association in the APOE ε4 carriers but not in the APOE ε4- samples, whereas association of rs144835823 and rs78442236 with AD pathogenesis was observed, regardless of the APOE ε4 status. rs79919241, located in an intron of LINC01578, was approximately 82 kb away from rs12594991 in CHD2. The two variants were in low LD (r2 = 0.39) with each other, and were identified in APOE ε4 carriers (Table 2). rs12594991 (P = 2.03 × 10−7) exhibited a significantly higher association with APOE ε4 carriers than rs79919241 (P = 1.08 × 10−4). These results show the importance of performing APOE ε4 stratification analysis in case-control studies to enable the detection of significant variants related to the APOE ε4 allele in universal GWAS.
Discussion
Large-scale GWAS such as the IGAP have reported various variants associated with AD; however, these studies have mainly focused on non-Asian populations, and the genetic architecture of AD is less clear in Asian populations. However, small-scale studies on the genetic diversity in AD across various ethnic groups have suggested the importance of understanding the genetic background of AD in diverse populations29. Since no large-scale genetic study of AD has been conducted in Koreans to date, this study sought to identify genetic variants associated with AD in a Korean population by using a large sample size. We discovered and validated genetic candidates associated with AD by using WGS data and a cAD chip dataset. Our findings provide an important perspective on AD pathogenesis related to the APOE ε4+ allele. The APOE ε4 allele, a well-known stratifying risk factor for AD, has been considered to primarily elucidate the genetic effects associated with AD pathogenesis. We suggest that a WGS approach is imperative to maximize the detection probability of the existence of population-specific or rare variants.
We detected two novel variants associated with AD in the APOE ε4 stratified analysis. Chromodomain helicase DNA binding protein 2 (CHD2) is characterized by the presence of a chromodomain that is responsible for chromatin remodeling. CHD2 is a risk factor for photosensitivity in epilepsy30 and is related to neurodevelopmental disorders31,32,33. CHD2 has previously been reported to interact with repressor element 1-silencing transcription factor (REST), which plays an important role in cognitive decline associated with AD34. The minor allele (A) confers risk for AD, affecting the gene expression of CHD2 as cis-eQTL. According to the eQTL database35, one novel SNP (rs12594991) with minor allele (A) showing an association signal in the APOE ε4 carriers was significantly associated with high expression of the assigned gene (CHD2; FDR P < 0.001). The minor allele A on rs12594991 might be associated with high expression levels and increased susceptibility to AD. CHD2 protein expression is high in the cerebral cortex and cerebellum, that is, in brain regions rather than other organs36 (The Human Protein Atlas; www.proteinatlas.org). Since sortilin-related VPS10 domain-containing receptor 1 (SORCS1) is associated with sortilin, SORCS1 may be involved in amyloid precursor protein (APP) processing and trafficking across membranes, as is the sortilin-related receptor (SORL1) gene, which is associated with AD susceptibility37. Intronic genetic variations rs10884402 and rs950809 of SORCS1 associated with late-onset AD have been reported in the Chinese Han population38. Interestingly, the intronic variants rs12571141, rs17277986, and rs6584777 of SORCS1 only exhibited significant association in the APOE ε4 carriers39. However, the rs17277986 variants included in our cAD chip did not show significant associations in our dataset. Our novel variants in SORCS1 are located in intergenic regions and show lower allelic frequencies than those of previously reported intronic variants. These results indicate that functional variants with biological implications are not always consistent with one another, despite being localized in the same gene, SORCS1.
We also identified putative novel variants that were not detected in European populations due to low allele frequency. Although our putative novel variants were selected by WGS analysis in the discovery stage with the APOE ε4 carriers, they were replicated in our cAD chip application set which was not stratified based on the APOE ε4 genotype. When we considered the APOE ε4 carriers in our cAD chip application set, four of these novel variants also showed significant association (P < 0.05) (Table 3). It is not known whether the four genes (PTPRN2, SORCS1, LINC01578, and ABR) containing the putative novel variants in the cAD chip dataset are directly involved in the pathogenesis of AD.
PH and SEC7 domain-containing protein 3 (PSD3) includes the putative Pleckstrin domain, indicating that PSD3 is involved in intracellular signaling. PSD3 was included in 30 top-scoring SNPs identified by a Bayesian combinatorial method in an AD GWAS dataset that was shown to be differentially overexpressed in AD40. Receptor-type tyrosine-protein phosphatase N2 (PTPRN2) could be involved in amyloid processing, based on the fact that PTPRN2 is one of the substrates involved in beta-site APP-cleaving enzyme 141. The expression of PTPRN2 was significantly altered in the hippocampus of AD sufferers42. The active BCR-related gene (ABR), localized at the synapses of neurons, may be involved in synaptic signaling; the ABR gene is abundantly expressed in the brain43.
A number of limitations should be considered when interpreting our results. First, although we have conducted our association study with AD data from the largest Korean population size to date, it may still be inadequate. Therefore, novel variants identified in this study should be analyzed in a greater number of samples to achieve genome-wide significance. Second, the rare putative novel variants with low allele frequencies might not be replicated in European populations. We must validate these variants in other independent Asian populations.
In summary, our results highlight that novel germline variants associated with AD in APOE ε4 carriers sampled from a Korean population were identified using whole-genome sequencing and cAD chip genotyping. Our results suggest that genetic association studies must be performed in diverse ethnic populations.
Data availability
The complete dataset will not be made publicly available because of restrictions imposed by the ethics committees due to the sensitive nature of the personal data collected. Requests for data can be made to the corresponding author.
References
Holtzman D. M., Morris J. C. & Goate A. M. Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr71 (2011).
Hebert, L. E., Weuve, J., Scherr, P. A. & Evans, D. A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783 (2013).
Alzheimer’s Association Report. Alzheimer’s disease facts and figures. Alzheimers Dement. 16, 391–460 https://doi.org/10.1002/alz.12068 (2020).
Kim, K. W. et al. A nationwide survey on the prevalence of dementia and mild cognitive impairment in South Korea. J. Alzheimers Dis. 23, 281–291 (2011).
Cuyvers, E. & Sleegers, K. Genetic variations underlying Alzheimer’s disease: evidence from genome-wide association studies and beyond. Lancet Neurol. 15, 857–868 (2016).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Van Cauwenberghe, C., Van Broeckhoven, C. & Sleegers, K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet. Med. 18, 421–430 (2016).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Andrews, S. J., Fulton-Howard, B. & Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 19, 326–335 (2020).
Marioni, R. E. et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99 (2018).
Reitz, C. et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492 (2013).
Meng, Y. et al. Association of polymorphisms in the Angiotensin-converting enzyme gene with Alzheimer disease in an Israeli Arab community. Am. J. Hum. Genet. 78, 871–877 (2006).
Miyashita, A. et al. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE 8, e58618 (2013).
Morris, J. C. et al. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273 (2019).
Jun, G. et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117 (2016).
Wang, B. et al. A rare variant in MLKL confers susceptibility to ApoE varepsilon4-negative Alzheimer’s disease in Hong Kong Chinese population. Neurobiol Aging 68, 160.e1–160.e7 (2018).
Ma, Y. et al. Analysis of Whole-exome sequencing data for Alzheimer disease stratified by APOE genotype. JAMA Neurol. 76, 1099–1108 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D. & Tanzi, R. E. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23 (2007).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42, D1001–D1006 (2014).
Ward, L. D. & Kellis, M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Jun, G. R. et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 13, 727–738 (2017).
Galizia, E. C. et al. CHD2 variants are a risk factor for photosensitivity in epilepsy. Brain 138, 1198–1207 (2015).
Carvill, G., Helbig I. & Mefford, H. CHD2-related neurodevelopmental disorders. (ed Adam, M. P. et al.). GeneReviews(R) (1993).
Kim, Y. J. et al. Chd2 is necessary for neural circuit development and long-term memory. Neuron 100, 1180–1193 e1186 (2018).
Nieto-Estevez, V. & Hsieh, J. CHD2: one gene, many roles. Neuron 100, 1014–1016 (2018).
Shen, T. J., Ji, F., Yuan, Z. Q. & Jiao, J. W. CHD2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 33, 1794–1806 (2015).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Coulson, E. J. & Andersen, O. M. The A-B-C for SORting APP. J. Neurochemistry 135, 1–3 (2015).
Xu, W. et al. The genetic variation of SORCS1 is associated with late-onset Alzheimer’s disease in Chinese Han population. PLoS ONE 8, e63621 (2013).
Wang, H. F. et al. SORCS1 and APOE polymorphisms interact to confer risk for late-onset Alzheimer’s disease in a Northern Han Chinese population. Brain Res. 1448, 111–116 (2012).
Floudas, C. S., Um, N., Kamboh, M. I., Barmada, M. M. & Visweswaran, S. Identifying genetic interactions associated with late-onset Alzheimer’s disease. BioData Min. 7, 35 (2014).
Dislich, B. et al. Label-free quantitative proteomics of mouse cerebrospinal fluid detects beta-site APP Cleaving enzyme (BACE1) protease substrates in vivo. Mol. Cell Proteom. 14, 2550–2563 (2015).
Hokama, M. et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488 (2014).
Oh, D. et al. Regulation of synaptic Rac1 activity, long-term potentiation maintenance, and learning and memory by BCR and ABR Rac GTPase-activating proteins. J. Neurosci. 30, 14134–14144 (2010).
Acknowledgements
This study was supported by a grant from the National Research Foundation (NRF) of Korea funded by the Ministry of Science, ICT (2014M3C7A1046049, to J-.W.K.). Specimens and clinical information obtained from Seoul National University Hospital were supported by a grant from the Ministry of Science and ICT (NRF2014M3C7A1046042, to D.Y.L.). E.A.L was supported by NIA (K01AG051791), NIH/OD (DP2AG072437), and the Suh Kyungbae Foundation.
Author information
Authors and Affiliations
Contributions
J.-W.K. designed this study and had full access to all the data. D.Y.L., M.S.B., J.H.L., D.Y., S.J.C., K.W.P, N.C., S.Y.K., W.Y., H.A., K.W.K., S.H.C., J.H.J., E.-J.K., S.W.S., and D.L.N. provided clinical samples and data. J.-H.P., I.P., E.M.Y., S.L., June-Hee P., E.M.Y., Jongan Lee., E.A.L., H.K., Junehawk Lee, and Y.K. performed formal data analyses and data interpretation. J.-H.P., I.P., and S.L. conducted statistical and bioinformatics analyses. E.M.Y. and June-Hee P. conducted all experiments. J.-H.P., I.P., E.M.Y., S.L., and J.-W.K. wrote the manuscript. J.-H.P., I.P., and E.M.Y. contributed equally to the work as co-first authors. All authors approved the final version of the manuscript and agreed to its submission of publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, JH., Park, I., Youm, E.M. et al. Novel Alzheimer’s disease risk variants identified based on whole-genome sequencing of APOE ε4 carriers. Transl Psychiatry 11, 296 (2021). https://doi.org/10.1038/s41398-021-01412-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-021-01412-9
