
Abstract
Cancer immunotherapy, mainly including immune checkpoints-targeted therapy and the adoptive transfer of engineered immune cells, has revolutionized the oncology landscape as it utilizes patients’ own immune systems in combating the cancer cells. Cancer cells escape immune surveillance by hijacking the corresponding inhibitory pathways via overexpressing checkpoint genes. Phagocytosis checkpoints, such as CD47, CD24, MHC-I, PD-L1, STC-1 and GD2, have emerged as essential checkpoints for cancer immunotherapy by functioning as “don’t eat me” signals or interacting with “eat me” signals to suppress immune responses. Phagocytosis checkpoints link innate immunity and adaptive immunity in cancer immunotherapy. Genetic ablation of these phagocytosis checkpoints, as well as blockade of their signaling pathways, robustly augments phagocytosis and reduces tumor size. Among all phagocytosis checkpoints, CD47 is the most thoroughly studied and has emerged as a rising star among targets for cancer treatment. CD47-targeting antibodies and inhibitors have been investigated in various preclinical and clinical trials. However, anemia and thrombocytopenia appear to be formidable challenges since CD47 is ubiquitously expressed on erythrocytes. Here, we review the reported phagocytosis checkpoints by discussing their mechanisms and functions in cancer immunotherapy, highlight clinical progress in targeting these checkpoints and discuss challenges and potential solutions to smooth the way for combination immunotherapeutic strategies that involve both innate and adaptive immune responses.
Similar content being viewed by others

Introduction
Generally, cancer cells will be eradicated by the complex system in the human immune system, but they develop resistance to the antitumor immune response to evade the immune surveillance. Cancer immunotherapy has revolutionized the oncology landscape as it utilizes patients’ own immune systems in combating cancer cells. It can be realized in two broad manners: immune checkpoints-targeted therapy and the adoptive transfer of manipulated immune cells. Both manners manipulate the immune system to recognize and attack cancer cells.1 Immune checkpoint inhibitors, such as programmed cell death ligand 1 (PD-L1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibodies and agonists of costimulatory molecules that override the inhibitory pathways to unleash the immune function, have achieved success in various clinical trials but still face problems such as low response rates, high costs, and nonspecific toxicity.2,3,4 Adoptive transfer of cells basically includes genetically engineered cells including chimeric antigen receptor (CAR)-T cells and many other cells, e.g., multipotent mesenchymal stem cells engineered to express a cytokine and characteristics of other manipulated cells.5,6 In a word, cancer immunotherapy has experienced remarkable advances since the clinical success of immune checkpoint blockade and CAR-T-cell therapies in recent years. It has become an innovative treatment and a powerful clinical strategy due to its incomparable advantages over traditional antitumor therapy including surgery, radiotherapy, and chemotherapy.
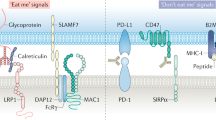
Most previously developed immunotherapies worked primarily by stimulating adaptive immunity, especially by revitalizing and boosting T cell responses. However, emerging studies have manifested that innate immune checkpoints expressed on the antigen-presenting cells (APCs) play a critical role in the immune evasion. These checkpoints detect and eliminate cancer cells by phagocytosis and inhibit the innate immune response. Innate immune cells that function as APCs, including macrophages, monocytes, dendritic cells (DCs), and natural killer (NK) cells are the first line of immune defense system. They establish proinflammatory responses to foreign invaders and repair damaged tissues. Cancer cells evade clearance by macrophages via overexpressing the anti-phagocytic membrane proteins termed “don’t eat me” signals, including cluster of differentiation 47 (CD47),7 cluster of differentiation 24 (CD24),8 PD-L1,9 the beta-2 microglobulin (β2M) subunit of the major histocompatibility class I complex (MHC-I),10 stanniocalcin 1 (STC-1),11 and GD212 (Figs. 1, 2). Phagocytosis is often facilitated by intrinsic “eat me” signals that function as ligands for phagocytic receptors, which can trigger extensive remodeling of the cytoskeleton and engulf the target.

Discovery of phagocytosis checkpoints

Phagocytosis checkpoints in cancer immunotherapy. Phagocytosis of tumor cells by macrophages is regulated by many “eat me” (pro-phagocytosis) and “don’t eat me” (anti-phagocytosis) signals. The expression of “don’t eat me” signals on tumor cells including CD47, CD24, PD-L1, MHC-I and STC-1 protect tumor cells from phagocytic clearance by interacting with their receptors on phagocytes. The working pathways are CD47-SIRPα, CD24-Siglec-10, MHC-1(B2M)-LILRB1, and PD-L1-PD-1. The high expression of tumor STC-1 traps the calreticulin in mitochondria and ER thus reducing the quantity of calreticulin on the cell surface, impairing phagocytosis and antigen processing and presentation, also leading to weak T cell response. Other anti-phagocytosis receptors such as SLAMF3, SLAMF4, FcγRIIB, and CLEC-1 facilitate the phagocytosis of tumor cells by phagocytes. The “eat me” signals such as calreticulin bind with the membrane glycans and are located on the cancer cell surface. It interacts with the lipoprotein receptor-related protein 1 (LRP1) receptor present on phagocytes. It seems that SLAMF7 expressed on tumor cells and MAC-1 on macrophages are both critical for inducing tumor phagocytosis, while the clear mechanism of SLAMF7-induced phagocytosis is under investigation
In this review, we summarize the phagocytosis checkpoints discovered to date, including basic knowledge, relevant pathways, and functions in cancers and the tumor microenvironment. We also discuss the expression and functions of these checkpoints in innate and adaptive immune responses. Finally, we highlight clinical progress in targeting these phagocytosis checkpoints, listing challenges and potential solutions for targeted cancer immunotherapy. We hope that this comprehensive review will not only help understand the current status of phagocytosis checkpoint research but also aid in the development of combinatorial treatment approaches, such as immunotherapy, that build on chemotherapy by targeting phagocytosis checkpoints.
Basic knowledge of phagocytosis checkpoints
CD47
The CD47-signal regulatory protein alpha (SIRPα) axis, identified in the late 2000s, is the first tumor phagocytosis checkpoint and is a typical myeloid-specific immune checkpoint that works directly via innate immunity.13 CD47, which serves as a “don’t eat me” signal on tumor cells, inhibits phagocytosis by macrophages in the immune system.14,15,16 Myriad CD47 inhibitors and antibodies are under investigation, and many of them are in clinical trials.17,18 In clinical trials, striking responses have been achieved for some solid tumors and hematologic malignancies upon CD47 inhibition.19,20 Moreover, CD47-SIRPα signaling relies on the phagocytic function of macrophages, which are the most abundant infiltrating leukocytes in tumors. Therefore, targeting CD47 likely represents a turning point in cancer immunotherapy. An elaborate discussion of CD47 regulation and its function in cancer immunotherapy will be presented in the following sections.
A brief history of CD47
CD47 was originally identified in 1987 on red blood cells (RBCs).21 Until 2000, CD47 was viewed as a “marker of self” on murine RBCs that binds to SIRPα on phagocytes.22,23 It was confirmed as a tumor phagocytosis checkpoint that delivers a “don’t eat me” signal during immune evasion in 2019, and CD47-targeting antibodies and inhibitors are currently in clinical trials.
Structure of CD47
In the immune system, CD47 is the only 5-transmembrane (5-TM) receptor.24 It contains three parts: a heavily glycosylated N-terminal extracellular domain (ECD), a 5-TM spanning domain and a short C-terminal domain (CTD).25 The ECD includes a V-set immunoglobulin superfamily domain binding to SIRPα. The CTD is alternatively spiced into 4 isoforms named from type I to type IV, which vary in expression in different cells.26 All the above structures and splicing isoforms are crucial for CD47 function.24
CD47 binding proteins
CD47 interacts with other extracellular proteins located on the membrane and inside cells. Most of its functions are attributed to its binding partners. The best-studied binding proteins of CD47 are thrombospondin 1 (TSP-1) and SIRPα. In addition to SIRPα, SIRPγ also binds to CD47 due to the similarity of its extracellular domain to SIRPα, but it has a tenfold lower affinity.27,28
TSP-1 was the first identified ligand for CD47.29,30 It interacts with CD47 via the RFYVVMWK sequence (4N1K) in the C-terminal of the CBD domain.31 The CD47-TSP-1 interaction inactivates the vascular endothelial growth factor receptor 2 (VEGFR2) and suppresses angiogenesis to inhibit tumor growth; thus, TSP-1 has also been viewed as a potent inhibitor of tumor growth and metastasis.32 The CD47-TSP-1 interaction also inhibits inflammatory responses such as cytokine secretion.33,34 TSP-1 deficiency in macrophages limits their phagocytic capacity.35 Furthermore, the interaction of CD47 and TSP-1 enhances the regeneration of stem cells by upregulating transcription factors of stem cells such as KLF4, Sox2, c-Myc and Oct4.36
SIRPα was identified as an endogenous ligand of CD47.37 It is also a transmembrane glycoprotein that is mainly expressed on macrophages, monocytes, and DCs. It contains one transmembrane domain, three lg-like domains, and four tyrosine phosphorylation sites. In the cytoplasmic tail, there are two immune receptor tyrosine-based inhibitor motifs (ITIMs).38 The interaction of SIRPα and CD47 is mediated by the N-terminal of SIRPα and the single lg-V domain of CD47.
The regulation of CD47
CD47 expression is regulated at different levels. First, transcription factors such as Myc,39 hypoxia-inducible factor-1 (HIF-1),40 and nuclear respiratory factor 1 (NRF-1)41 potentiate nuclear factor kappa B (NF-κB) CD47 expression.42 Moreover, cytokines, including tumor necrosis factor alpha (TNF-α),43,44 interferon-γ (IFN-γ)45 and interleukin,46,47 enhance CD47 expression. Conversely, various microRNAs and long noncoding RNAs (lncRNAs) negatively regulate CD47.48 At the posttranslational level, the pyroglutamylation and glycosylation of CD47 have been studied thoroughly.49 Lautenberg et al., Shana et al., and our group reported that CD47 is a substrate of QPCTL and that the N-terminal region of CD47 is pyroglutamylated. The pyroglutamylation of CD47 is catalyzed by QPCTL;50,51,52 this modification is critical for the recognition of CD47 by SIRPα and contributes to their interaction.49,51
Cellular function of CD47
CD47 plays a critical role in various biological and pathological processes. It either promotes or suppresses cell proliferation depending on cell status and type, and the expression of CD47 promotes cell proliferation in glioma cells but not in normal astrocytes.53 Moreover, CD47 enhances aerobic glycolysis, and CD47 activation contributes to the PI3K/Akt/mTOR oncogenic pathway.54
PD-L1
PD-L1, a ligand of programmed cell death protein 1 (PD-1), is a well-recognized immune checkpoint expressed on tumor cells. Antibodies targeting PD-1/PD-L1 have been widely used clinically for various types of tumors, and PD-1-PD-L1 blockade ushered in a new era of tumor treatment. Hence, it is a breakthrough of targeting the PD-1-PD-L1 pathway in tumor treatment.
In 1999, Chen’s team discovered a B7 homologous transmembrane protein, B7-H1 (now known as PD-L1).55 Later, it was found that PD-L1 is a ligand of PD-1, which clarified the negative immune regulation function of PD-L1 and highlighted its potential for application in tumor treatment.56 In 2002, PD-L1 was demonstrated to promote T cell apoptosis, and a B7-H1 antibody was applied to inhibit tumor growth, which demonstrated that PD-L1 functions in tumor immune escape for the first time.57,58 Since then, the effectiveness of PD-L1 antibody therapy has been witnessed by successive clinical trials.
Cd274 gene encodes PD-L1 protein, it is located on chromosome 9 of human and 19 of mouse. It is composed of a transmembrane region, typical immunoglobulin V-like plus C-like extracellular domains, and a short tail in cytosol.59 PD-L1 presents on a variety of hematopoietic cells, including DCs, macrophages, B cells and T cells, and other nonhematopoietic cells, such as vascular endothelial cells. Besides that, PD-L1 is also usually overexpressed in various types of cancer cells.60 PD-L1 expression on normal cells facilitates the regulation of immune responses in the periphery, but its overexpression on cancer cells protects cancer cells from immune surveillance.
PD-L1 expression is regulated by multiple factors at the genomic, transcriptional and posttranslational levels. For example, inflammatory signaling regulates PD-L1 expression. PD-L1 expression can be induced by both type I and type II interferons, TNF-α, and vascular endothelial growth factor (VEGF).60 Type I interferons, IFN-α and IFN-β stimulate PD-L1 expression.61 In prostate cancer and kidney cancer, TNF-α upregulates PD-L1 expression by activating NF-κB pathway.62,63 Type II interferon IFN-γ binds to IFNGR and triggers JAK-STAT1-IRF1 to modulate PD-L1 expression. Moreover, IL-6 activates the JAK-STAT3 or MEK/ERK signaling pathway to upregulate PD-L1 expression,64 and TGF-β also regulates the expression of PD-L1 in cancer cells.64 In addition to inflammatory factors, oncogenic pathways such as the epidermal growth factor receptor (EGFR), HIF-1, Myc, NF-κB, PTEN/PI3K-AKT, and mitogen-activated protein kinase (MAPK) pathways play vital roles in PD-L1 expression.65 The mechanisms by which PD-L1 expression is regulated were reviewed in another excellent review,66 and this article mainly focuses on phagocytosis and the PD-1-PD-L1 axis.
MHC-I
Major histocompatibility complex (MHC) is a cluster of closely linked genes that are highly polymorphic and located in a specific region of the mammalian chromosome. The molecules encoded by these genes are expressed on the surface of all nucleated cells and platelets but not on RBCs.67 They are involved in antigen presentation, governing intercellular recognition and the induction of immune responses. The basic function of MHC is to distinguish “self” and “non-self” and present the tumor-associated antigens (TAAs) to T cells to activate the adaptive immune response.68 Moreover, MHC-I on the surface of tumor cells binds to leukocyte immunoglobulin-like receptor subfamily B 1 (LILRB1) on the surface of macrophages to promote tumor cell escape from macrophage phagocytosis. Therefore, MHC-I-LILRB1 is another phagocytosis checkpoint in cancer immunotherapy.
A brief history of MHC-I
MHC genes were discovered in 1937. The key to successful transplantation is histocompatibility between the host and the donor, and the genes that mediate this recognition are called compatibility genes. They are closely linked on the same chromosome, and their product is the MHC, also known as the transplant antigen, which is the main determinant of transplant rejection.69 The MHC locus encodes classical MHC-I, MHC class II (MHC-II) and nonclassical MHC-I molecules. MHC-I, as the first human leukocyte antigen product, controls the immune response induced by protein antigens.70
Structure of MHC-I
In humans, MHC refers to human leukocyte antigen (HLA), which includes classical HLA-I, HLA-II, and nonclassical HLA-III molecules. HLA-I binds to and presents endogenous antigens. HLA is the most complex and polymorphically rich genetic system in humans by far,71 and it possesses a tremendous number of alleles to achieve the most appropriate immune response to pathogens and enable adaptation to a variable internal and external environment. Classical MHC-I is a heterodimer composed of an α heavy chain and a β2m light chain. The former chain contains three sites: three extracellular structural regions (α1, α2, and α3),72 a membrane-penetrating region and a cytoplasmic region. The α3 structural region is structurally homologous to the constant region of Ig and is the site of binding to CD8 on the surface of T cells.73 The α1 and α2 structural regions interact to form the antigen binding site of MHC-I. The binding groove is closed at both ends. The middle part of the antigenic peptide is generally elevated and recognized by the T-cell receptor (TCR) as a T-cell epitope. β2m is a soluble protein that cannot pass through the cell membrane. The sequence of amino acid for β2m is highly conserved, with minimal differences among species, and can be substituted for each other. The main function of β2m is to stabilize MHC-I molecules and facilitate their cell-surface expression.
MHC-I binds to LILR1 and LILRB2 on phagocytes to inhibit phagocytosis
MHC-I on tumor cells binds to LILRB1 and LILRB2, which are members of the LILR family,74 which belongs to the inhibitory class of the LIR receptor subfamily. LILRBs are overexpressed typically in immunosuppression-related cells, such as tolerogenic DCs and the immunosuppressive M2-type macrophages.75 LILRB1 expression is significantly increased after the differentiation of human monocytes into immature DCs. Subsequently, MHC-I molecules are upregulated for antigen-presenting functions, whereas LILRB1 is downregulated.76 Recently, it was found that the binding between β2m of MHC-I expressed on the surface of tumor cells and LILRB1 on the surface of tumor-associated macrophages (TAMs) inhibits the phagocytic activity of TAMs, leading to decreased immune surveillance and enhanced immune escape of tumor cells.10
Regulation of MHC-I
Dozens of genes have been reported to positively or negatively regulate MHC-I expression.77 The positive regulators include interferon signaling,78 mRNA processing and splicing,79 endoplasmic reticulum (ER) quality control,80 etc. The negative regulators include mammalian target of rapamycin (mTOR) regulation, mRNA capping and translation,81 polycomb repressive complex 2 (PRC2), the ubiquitin system,82 and a myriad of endo-lysosomal trafficking factors that are likely critical for internalizing MHC-I and its lysosomal degradation.83 MHC-I is removed from the cell surface when an HIV-1-encoded protein Nef is present.84 MIIP, CAMSAP3, SLC6A3 and KCTD19 were found to significantly inhibit Nef-induced MHC-I downregulation.85 Moreover, the 3’UTR of HLA-A2 mRNA has been found to bind the ubiquitin E3 ligase MEX-3C, which leads to its RING-dependent degradation.86
CD24
The CD24-sialic acid-binding immunoglobulin-like lectin-10 (Siglec-10) axis is known to protect the body from a lethal response involving pathological cell death.87 Recent studies indicated that blocking the binding of CD24 and Siglec-10 with a CD24 antibody significantly enhances the recognition of CD24-expressing tumor cells by macrophages, and after CD24 antibody treatment, the growth of murine orthotopic tumors was inhibited strikingly. Therefore, CD24 has been widely studied and explored as a new antitumor phagocytosis checkpoint.
A brief history of CD24
As a heat-stable antigen, CD24 was first found in 1978, and it was thought to be expressed on the membrane of immature B cells, T lymphocytes, and activated granulocytes as a marker of the differentiation and maturation of immune cells.88 In 2019, CD24, serving as a “don’t eat me” signal on tumor cells, was found to inhibit the phagocytosis of macrophages in the innate immune system.8 CD24 mediates adhesion between cells, cells and substrates and also functions in cell recognition, activation, signal transduction, proliferation, differentiation, extension and movement.89 Recently, increasing evidence has proven that the expression of CD24 on the surface of tumor cells, in contrast to that in adjacent tissue, is significantly elevated, which is positively associated with the occurrence and development of tumors.
Structure of CD24
The CD24 gene encodes a glycosylated protein and is located on chromosome 6q21. As a single-chain sialoglycoprotein, mature CD24 is a short peptide with only 30 amino acids. There are one or more O-linked glycosylation sites in the mature peptide backbone and four potential N-linked glycosylation sites in CD24.90 Thus, the glycosylation modifications of CD24 vary substantially among different cell types, resulting in molecular masses from 35 kDa to 45 kDa. Highly glycosylated CD24 requires anchoring on lipid rafts within the plasma membrane through a glycosyl-phosphatidyl-inositol (GPI) anchor protein.91
CD24 binding proteins and corresponding functions
Primarily, CD24 as a GPI-anchored protein, is located in the cell membrane in both normal and cancer cells, but is also distributed in the cytoplasm and nucleus in some cancer cells. The functions of CD24 on the membrane depend on its binding proteins. It binds to different proteins, such as Siglec10, Siglec E, platelet (P)-selectin, and L1-cell adhesion molecule (L1-CAM), to perform a variety of functions. Since only CD24-Signlec 10 is related to phagocytosis function, we focus on this binding protein in the following part.
CD24 binds to Siglec 10 on macrophages to avoid phagocytosis. Siglec10 is an immunosuppressive receptor, and the interaction between CD24 and Siglec10 significantly reduces the damage associated with damage-associated molecular pattern (DAMP)-related inflammatory responses, including liver injury87 and sepsis; this interaction also reduces antigen sensing at the cell surface or in the endosomal compartment and reduces the phagocytosis of tumor cells by tumor-associated macrophages, thus promoting tumor progression.92,93 Moreover, this interaction participates in the establishment of maternal immune tolerance in early pregnancy94 and is also involved in autoimmune diseases95 and graft-versus-host disease.96
The regulation of CD24
The expression of CD24 in tumors is regulated by a variety of factors. CD24 is upregulated by HIF1α in human bladder cancer,97 androgen receptor in urothelial carcinoma,98 DNA methyltransferase,99 estrogen receptor100 and truncated glioma-associated oncogene homolog 1101 in breast cancer. CD24 expression is negatively regulated by Twist in breast cancer,102 β-catenin/TCF in colorectal cancer,103 miR34a104 and miR-146a105 in oral squamous cell carcinoma, and histone deacetylase (HDAC)99 in breast cancer. As a highly glycosylated GPI-anchored protein, the localization of CD24 on the membrane is regulated by the proteins related to both the synthesis of N and O sugars and GPI assembly, such as PIGN, PIGP, and PGAP2.
Intracellular function of CD24
CD24 can be accumulated in the cytoplasm due to defects in the GPI system, such as loss of function of GPI assembly proteins, weak GPI anchor attachment, errors in the synthesis of CD24 in the ER, and the inclusion of CD24 in microvesicles.106 Localization of CD24 in cytosol also affects tumor cell development.107 CD24 in the cytoplasm of tumor cells inactivates and destabilizes p53 by disrupting the ARF-NPM interaction, which protects mutant p53 from degradation.108 The CD24-p53 axis also suppresses the tumorigenesis by maintaining intrahepatic macrophages, which can remove hepatocytes with DNA damage in hepatocellular carcinoma (HCC).107
The functions of cytoplasmic CD24 in tumor proliferation and metastasis are controversial. Mierke et al. reported in 2004 that CD24 enhances cell invasion through different pathways, such as increasing contractility and stimulating cell adhesion to fibronectin and collagen I and IV.109 However, a later study showed that intracellular CD24 suppresses tumor cell invasion and metastasis by influencing the posttranscriptional regulation of BART via G3BP RNase activity.110
STC-1
STC-1 was identified a phagocytosis checkpoint in 2021.11 STC-1 was first discovered in the corpuscles of the stannius of bony fishes,111 and its homologous genes in mammals, STC-1 and STC-2, were subsequently cloned.112 STC-1 is widely expressed in the ovary, prostate, bladder, kidney, adrenal gland, lung, heart, uterus, and pituitary gland in mammals,113 and its expression is upregulated in breast cancer, which potentiates invasiveness of breast cancer via JNK-/c-Jun pathway.114
STC-1, as a glycoprotein, functions in the regulation of serum calcium and phosphate homeostasis.112 It plays a more complex role in pregnancy, lactation, angiogenesis, organogenesis, proliferation, apoptosis, ischemia, and tumorigenesis.115,116 STC-1 acts as a SUMO E3 ligase in the SUMOylation cycle, and interacts with proteins located in the nucleus, endoplasmic reticulum, mitochondria, cytoplasm, membrane and secreted proteins.117 In diabetic nephropathy, STC-1 inhibits BNIP3 via AMPK/SIRT3 pathway and thus ameliorates renal injury.118 STC-1 also functions in the oxygen-induced retinopathy (OIR) stress response and development of pathologic vascular features in rodent OIR models by regulating VEGF levels.119 Emerging evidence has shown that STC-1 is present in various human cancer cells. It is closely associated with the efficacy of immunotherapy and is further related with patient survival negatively in various cancer types.11
GD2
Besides proteins, carbohydrates and lipids are also involved in the regulation of phagocytosis. GD2, a disialoganglioside, was identified as a tumor antigen of neuroblastoma in the 1980s; it is consistently overexpressed in neuroblastoma, sarcomas, gliomas, and neuroendocrine tumors and is regarded as the most promising tumor antigen.120 Anti-GD2 antibody has prolonged the survival of patients suffering from neuroblastoma.121,122 The role of GD2 as a cancer target has been reviewed elsewhere.123
GD2 is composed of five monosaccharides and contains glucose, galactose and two sialic acid residues linked to ceramide. GD2 is embedded in the outer plasma membrane via its ceramide tail, and the carbohydrate moiety is exposed to the extracellular space.124 GD2 expression is low in normal tissues and restricted to the brain, spinal cord, and skin melanocytes.125 The role of GD2 in normal development is thought to be involved in neural differentiation and repair,126 but clear mechanisms deserve further investigation.
As a complex ganglioside, GD2 regulates cell-cell recognition and signal transduction via specific binding lectins like Siglecs.127 GM2/GD2 synthase (B4GALNT1) deficient mice exhibit decreased central myelination, demyelination in peripheral nerves, and axonal degeneration in the nervous system, indicating the complex gangliosides role in the maintenance of the integrity of axons and myelin.128 Moreover, mice with GM2/GD2 synthase deficiency developed progressive behavioral neuropathies, indicating GM2/GD2 maintains the normal neural physiology.129 The function of GD2 in normal cellular physiology is not clearly illustrated, but GD2 augments cancer cell proliferation, adhesion, migration and invasion, and confers resistance to apoptosis.123
“Don’t eat me” receptors
In addition to the above “don’t eat me” signaling molecules that are highly expressed on cancer cells, there are many other “don’t eat me” receptors expressed on immune cells, including but not limited to SIRPα, Siglec-10, and LILRB1, which were mentioned in the previously described signaling pathways.
CD22
CD22 is expressed exclusively on B cells and is a cell surface sialoglycoprotein, it regulates the proliferation and function of B cells, acting as an inhibitory coreceptor of the B-cell antigen receptor (BCR).130 CD22 is present in the cytoplasm of progenitor and pre-B cells in early B-cell development and translocates to the surface of B cells as they mature.131 CD22 expression is highest in mature B cells. Therefore, it is an appealing therapeutic target for B-cell malignancies and autoimmune disorders. CD22 has been identified as an inhibitor of phagocytosis in microglia (Fig. 3a).132

The phagocytosis receptors CD22, Fc receptors, SLAMF3/4 and CLEC1. a CD22 binds α2,6-linked sialic acid and recruits tyrosine phosphatase SHP-1 to inhibit the phagocytic capacity of microglia. The anti-CD22 treatment enhanced clearance of injected oligomeric amyloid-β (Aβ), myelin debris and α-synuclein fibrils in aging brains. CMAS is a key synthase functioning in sialic acid synthesis, related to CD22 function. b FcγRIIb, FcγRI, FcγRIIIa, and FcγRIIa are expressed on macrophages. FcγRs crosslink IgG immune complex triggers phosphorylation of their ITAMs and activates kinases of SYK, SRC and PKC pathway, kinase activation leads to actin remodeling, which is crucial for phagocytosis of the IgG immune complex. FcγRIIB is the only phagocytosis-inhibitory receptor, and the other family members are phagocytosis-activating receptors within the human FcγR family. FcγRIIB contains an ITIM in its cytoplasmic region, and the phosphorylation and activation of the ITIM recruit the phosphatases SHP1 and SHP2 and inhibit downstream phagocytosis. c SLFRs are ubiquitously expressed in hematopoietic cells. SLAMF3 and SLAMF4 were identified as “don’t eat me” receptors on macrophages. They inhibit “eat me” signals, such as lipoprotein receptor-related protein 1 (LRP1) -mediated activation of mTOR and Syk to macrophages through SH2 domain-containing phosphatases and hematopoietic cells without SFRs are easily phagocyted by macrophages. d CLEC-1 is expressed primarily by myeloid cells, CLEC-1 on human DC dampens DC activation and restrains downstream Th17 responses, CLEC-1 is a novel myeloid immune checkpoint limiting tumor cells’ phagocytosis and tumor antigen presentation. CLRs binding to microbial surfaces influence phagocytosis by promoting inflammatory signals and triggering intracellular signaling to induce phagocytosis of microbes
The expression and function of CD22 are regulated by many molecules. Its synthase CMAS, a key enzyme in sialic acid synthesis, and PTPN6, which encodes SHP-1, are related to CD22 function,132 and spleen tyrosine kinase (Syk), phospholipase Cγ2 (PLCγ2), phosphoinositide 3-kinase (PI3K), Grb2, and Shc are the binding proteins of the CD22 cytoplasmic tail in response to BCR signaling.133
Ligands of CD22 have been identified on B cells, microglia,132 DCs and T cells.134 CD22 on B cells binds to α2,6-linked sialic acid on microglia and recruits the tyrosine phosphatase SHP-1 to inhibit the phagocytic capacity of microglia.132 Anti-CD22 treatment enhanced the clearance of injected oligomeric amyloid-β (Aβ), myelin debris and α-synuclein fibrils in the aging brain. Long-term CD22 blockade changes the transcriptional profile of microglia, including genes associated with microglial homeostasis, and improves cognitive function in aged mice.132 CD22-mediated phagocytosis in TAMs and in cancer immunotherapy requires further study. Moreover, DCs and bone marrow-derived immature DCs (iBMDCs) express glycan ligands of CD22, and iBMDCs induce strong inhibition of BCR-induced B-cell proliferation via a CD22-dependent mechanism.135 iBMDCs also suppress the proliferation and differentiation of B-cell subsets during Toll-like receptor (TLR) stimulation.136 Therefore, CD22 is a regulator of receptors that mediate both adaptive and innate immune responses. CD22 binds to ligands on T cells and affects T-cell activation. In addition, CD22 regulates B-cell responses to T-cell-independent type 2 antigens (TI-2 Ags). CD22 also negatively modulates TLR pathway, and CD22−/− B cells showed enhanced proliferative ability in response to TLR3, TLR7, and TLR9 agonists.137,138 Mechanistically, CD22 inhibits TLR signaling via intracellular signaling in B cells because the natural ligands for CD22 do not appear to affect proliferative responses to TLR agonists.138
CD22 plays a critical role in maintaining B-cell homeostasis in human immunity.139 The phosphorylated ITIMs of CD22 recruit the tyrosine phosphatase SHP-1 during antigen-mediated BCR crosslinking.140,141 CD22 knockout B cells induce responses, such as the intracellular calcium mobilization required for the proliferation and antibody production of B cells.142,143,144 CD22 also regulates the migration of recirculating B cells to the bone marrow,145 and CD22-deficient B cells inhibit homing to Peyer’s patches by reducing integrin expression via the CD22-Shp1 axis.146
CD22 is one of the most common antigens and is highly expressed in hematological malignancies, including human B-cell lymphomas and leukemias.147,148,149 Exon 12 depletion in infant B-precursor leukemia cells promotes their growth and survival.150 Moreover, CD22 conduces to protecting against pathogenic infection, and CD22 deficient mice are extremely sensitive to infection.151 In addition, CD22 expression is closely related to autoimmune disease, and CD22 levels are decreased in patients with systemic lupus erythematosus (SLE) and increased after effective treatment.152,153
Fc receptors
Fc receptors (FcRs) are cell-surface receptors present on several hematopoietic cells that specifically recognize the Fc region of immunoglobulin (Ig) to regulate phagocytosis and antibody-dependent cell-mediated cytotoxicity (ADCC).154 Generally, type I Fc common gamma receptors (FcγRs) are divided into activating or inhibitory subtypes. The activating FcγRs include FcγRI, FcγRIIa, FcγRIIc and FcγRIIIa, all of which contain immunoreceptor tyrosine activating motifs (ITAMs); FcγRIIB is the only phagocytosis-inhibitory receptor, and the others are phagocytosis-activating receptors within the human FcγR family.155 FcγRIIB comprises an ITIM in its cytoplasmic region,156 and the phosphorylation and activation of the ITIM recruit the phosphatases SHP1 and SHP2 and inhibit phagocytosis in their downstream (Fig.3b).
FcRs are present on different immune cells, such as monocytes, macrophages, DCs, and neutrophils, and the unique expression patterns of individuals or combinations of FcγRs balance cellular immune responses.156 FcγRIIb, FcγRI, FcγRIIIa, and FcγRIIa are expressed on macrophages. IgG immune complexes activate FcγR signaling for different subtypes of IgG, with complex binding specificity and affinity.157 After ligation of these immune complexes, ITAMs are phosphorylated by kinases of the SRC family, which recruits SYK-family kinases, followed by the activation of many downstream targets to activate the immune response, ADCC or phagocytosis.158 FcR function is important for the treatment of cancers especially when using the immune checkpoint-blocking drugs in cancer therapy.159,160,161 It may be possible to selectively exploit FcR activation or immune regulation function by engineering antibodies for different therapeutic environments.
Signaling lymphocytic activation molecule (SLAM) family receptors (SFRs)
Signaling lymphocyte activation molecules (SLAMs) are important immune regulatory receptors that have critical functions in immunity, cell survival, lymphocyte development, and cell adhesion.162 SLAM family receptors (SFRs) belong to an immunoglobulin superfamily that is expressed ubiquitously on hematopoietic cells, including macrophages, and modulate the activation and cytotoxicity of these cells. They recognize themselves as self-ligands and thus undergo homotypic interactions to constrain macrophage phagocytosis.163 Hematopoietic cells without SFRs are easily phagocytized by macrophages. The SFR members LAMF3 and SLAMF4 were identified as “don’t eat me” receptors on macrophages. They inhibit “eat me” signals in macrophages by SH2 domain-containing phosphatases (Fig. 3c). SFRs are markers that distinguish HSCs and their progenitors and prevent the inappropriate phagocytosis of self-HSCs. Mature RBCs express high levels of CD47 to avoid macrophage engulfment. SFRs can work in combination with the CD47 pathway but function independently of CD47 to mitigate macrophage phagocytosis.163 SLAMF3 is also expressed in cancer cells,164 but its function in phagocytosis in cancer immunotherapy remains unclear.
C-type lectin-like receptor-1 (CLEC-1)
C-type lectin-like receptors (CLRs) are a family of transmembrane receptors present on myeloid cells primarily. They recognize pathogen moieties for host defense and modify self-antigens. CLRs have at least one C-type lectin-like domain (CTLD) on the cell surface and either a transmembrane domain or a short intracellular signaling tail that boosts interaction with FcRγ that mediates signaling. CLRs binding to microbial surfaces influence phagocytosis by promoting inflammatory signals and triggering intracellular signaling to induce phagocytosis of microbes.165 C-type lectin-like receptor-1 (CLEC-1) is a prototypical CLR and an inhibitory receptor present on neutrophils, DCs and myeloid macrophages. CLEC-1 on human DCs dampens DC activation and restrains downstream Th17 responses.166 CLEC-1-deficient mice eradicate colorectal tumors by combining with cytotoxic and immunogenic chemotherapy, and CLEC-1 blocking antibodies augment the phagocytosis of CLEC-1 L-positive tumor cells by DCs and macrophages.167 CLEC-1 probably signifies a new therapeutic agent to regulate the immune response in transplantation, autoimmunity, and cancer. CLEC-1 is a novel myeloid immune checkpoint that limits tumor cell phagocytosis and tumor antigen presentation (Fig. 3d).167,168
“Eat me” signals
“Eat me” signals are molecules expressed on or released from cells to induce phagocytosis by a phagocyte. Most “eat me” signals are located on the cell surface, but some may be released extracellularly and bind back to the target cell. The lipid phosphatidylserine, the intracellular adhesion molecule ICAM-3, annexin I, calreticulin, cell surface-bound thrombospondin, complement factors, oxidized low-density lipoprotein, and other glycosylation alterations on apoptotic cells are “eat-me signals”.169 These signals have been reviewed previously.170
The phagocytosis process of tumor cells by macrophages or DCs is modulated by a large number of pro-phagocytosis (“eat me”) and anti-phagocytosis (“don’t eat me”) signals via the receptor-ligand axis. All the abovementioned checkpoints are antiphagocytosis proteins or signaling molecules. The “eat me” signals mainly include tumor-associated antigens generated in response to oncogenic stresses, the ER chaperone protein calreticulin and the glycoprotein SLAMF7.
Calreticulin
Calreticulin is an ER-resident protein and functions in various cellular processes, such as stress, and it functions as a chaperone and Ca2+ buffer to aid in appropriate protein folding and glycosylation.171 Calreticulin contributes highly to phagocytosis, the loss of wild-type calreticulin functions favors oncogenesis due to impaired cellular homeostasis in healthy cells and compromised natural and therapy-driven immunosurveillance.
Through binding with membrane glycans, calreticulin is anchored to the cancer cell surface, and it interacts with the low-density lipoprotein receptor-related protein 1 (LRP1) receptor present on phagocytes. LRP1 may recruit the adapter protein PTB domain-containing engulfment adapter protein 1 (GULP1) to regulate further phagocytic processes (Fig. 4a). Calreticulin translocates to the cell membrane and serves as an “eat me” signal to promote efferocytosis of apoptotic cells, including damaged, aged, and malignant cells, and leads to the elimination of these cells.172 Calreticulin has been demonstrated to be the dominant pro-phagocytic signal in a myriad of human cancers and is counterbalanced by CD47.

The “eat me” signals calreticulin and SLAMF7. a Stressed and dying tumor cells expose calreticulin on the surface of the cell from ER, and cell surface calreticulin binds to LRP1 on the phagocyte. LRP1 may recruit GULP1, an adapter protein LRP1 for regulating further phagocytic processes. b SLAMF7 on macrophage binds to MAC-1 on the macrophage, and MAC-1 interacts with FCRγ and DAP12 recruiting Src family Syk, and Btk kinases and promoting phagocytosis. SLAMF7 on macrophages combining with SIRPα on macrophage may affect the CD47-SIRPα axis, SLAMF7 in hematological cancers binds SLAMF7 on phagocytes and is necessary for phagocytosis
SLAMF7 synergizes with MAC-1 and promotes phagocytosis
SLAMF7, also known as CD319, CS1 or CRACC, is a member of the SLAM family of receptors that are present on both tumor cells173,174,175 and immune cells, including NK cells, B cells, DCs, and activated CD4 and CD8 T cells.162 SLAMF7 on macrophages recognizes homotypic SLAMF7 on hematopoietic cells to mediate phagocytosis. SLAMF7-deficient macrophages, but not macrophages deficient in other SFRs, have a defect in phagocytosis. SLAMF7 on macrophages interacts with integrin macrophage-1 antigen (MAC-1) on macrophages to promote the phagocytosis of cancer cells by macrophages. MAC-1 is a complement receptor (CR3) containing α-subunit CD11b (αm) and β-subunit CD18 (β2); it interacts with ITAM,175 FcRγ and DAP12 to mediate immune cell activation by Src, Syk, and Bruton’s tyrosine kinase (Btk) intrinsic signaling175 and enhance phagocytosis via the IgG-mediated FcR pathway (Fig. 4b).176 The expression of MAC-1 on macrophages is necessary for SLAMF7-dependent phagocytosis of cancer cells.175 Whether SLAMF7 is required for CD47-mediated phagocytosis is controversial. Chen et al. showed that during the CD47-SIRPα axis blockade, the phagocytosis of hematopoietic tumor cells was rigidly dependent on SLAMF7,175 but He et al. reported that SLAMF7 is not required for CD47-mediated phagocytosis.177 Given these controversial research results, the role of SLAMF7 in macrophage phagocytosis requires further investigation.
Signal pathways of phagocytosis checkpoints
The CD47-SIRPα signaling pathway
The mechanism of the CD47-SIRPα pathway
The intracellular region of SIRPα contains an ITIM, which is crucial for the inhibitory activity of the receptor.178,179 When an ITAM-containing receptor is triggered, the ITIM-containing receptor SIRPα counteracts cellular activation. The inhibition of this signaling pathway by SIRPα requires tyrosine residues’ phosphorylation in cytoplasmic ITIM sequences, which then recruits and activates the SH2-domain-containing protein tyrosine phosphatases SHP-1 and SHP-2.178,179 The recruitment of SHP-1 and SHP-2 phosphorylates myosin IIA and suppresses nonmuscle myosin IIA, which regulates phagolysosomal biogenesis in macrophages and functions in phagocytosis. Upon dephosphorylation of myosin IIA in macrophages, depolymerization of actin occurs, leading to a reduction in phagocytosis38,180 (Fig. 5a). The binding of CD47 on tumor cells and SIRPα on phagocytes promotes the phosphorylation of the ITIM in SIRPα by the Src family kinases SHP-1 and SHP-2 and thus contributes to the reduction of phagocytosis.181

Mechanisms of phagocytosis checkpoints. a CD47 on the surface of tumor cells binds to SIPRα on the membrane of the macrophage. This interaction promotes the phosphorylation of ITIM in SIRPα by Src family kinases SHP-1 and SHP-2. The recruitment of SHP-1 and SHP-2 phosphorylate myosin IIA, then suppresses the function of non-muscle myosin IIA, upon dephosphorylation of Myosin IIA in macrophages, the de-polymerization of actin occurs, resulting in the limitation of phagocytosis. b TP53 mutation increases the expression of PD-L1 on extracellular vesicles, leading to the block of phagocytosis of tumor cells by macrophages. c The β2M of MHC-1 binds to the extracellular region of LILRB1 to form a complex with the MHC- I heavy chain, this novel inhibitory MHC-I-LILRB1 axis inhibits the innate immune system. d The inhibitory receptor Siglec-10 on the macrophage surface binds to its ligand CD24 on cancer cells, resulting in an ITIM or ITIM-like motif in the cytoplasmic domain of Siglec-10 combing with Src family kinases. Then Src family kinases phosphorylate ITIM tyrosine in the cytoplasm, then recruit SHP-1/ SHP-2. SHP-1 can specifically bind to the intracellular phosphorylated ITIM domain to dephosphorylate it, leading to cytoskeleton remodeling and phagocytosis inhibition. e STC-1 interacts with the “eat me” signal calreticulin and abrogates the membrane calreticulin-directed phagocytosis by macrophages, thus impairing the antigen presentation from macrophages to T cells. Tumor STC-1 is crucial for intrinsic tumor resistance to tumor immunity, it traps calreticulin in mitochondria and ER to inhibit macrophage function and facilitate the tumor cell immune evasion and immunotherapy resistance. f GD2 (generated by the enzyme B4GALNT1) binding the Siglec-7 (the inhibitory immunoreceptor) on phagocyte triggers “don’t eat me” signals in the macrophages, calreticulin is an “eat me” signal on the surface of tumor cells, the ligation of GD2 leads to the upregulation of calreticulin, indicating GD2 may inhibit calreticulin signaling
The function of the CD47-SIRPα pathway
The best-studied function of CD47-SIRPα is the induction of tumor immune evasion during cancer immunotherapy (Fig. 6a, b). Cancer cells express CD47 highly, which binds to SIRPα on phagocytes, leading to the evasion from immune surveillance. CD47 inhibits the phagocytic function of macrophages, stimulates cell‒cell fusion, activates T cells and affects the migration of neutrophils.22,23,182,183,184 Moreover, CD47 is expressed highly on young RBCs and hematopoietic stem cells (HSCs) to protect them from phagocytosis,22 and damaged and senescent RBCs are phagocytosed by macrophages because their expression of CD47 is lower than that in younger RBCs. Targeting CD47 or inhibiting CD47-SIRPα signaling allows macrophages to engulf HSCs and RBCs (Fig. 6c, d). Besides its role in bulk tumor cells, CD47 also plays a crucial role in cancer stemness maintenance and the immunoresistance in cancer stem cells (CSCs).185 Furthermore, the CD47-SIRPα interaction also activates the Hedgehog/smoothened (SMO)/GLI family zinc finger 1 (Gli1) pathway in mesenchymal stem cell (MSC)-treated livers after ischemia/reperfusion (IR) stress, and activation of this pathway regulates cell growth, differentiation, and immune function.186

CD47-SIRPα pathway. a CD47 expressed on tumor cells interacts with SIRPα expressed on macrophages and other phagocytes to avoid immune surveillance. b Targeting CD47 or blocking the CD47-SIRPα axis interrupts their interaction and allows macrophages to phagocyte tumor cells. c CD47 expressed on hematopoietic cells or red blood cells interacts with SIRPα expressed on macrophages and other phagocytes to avoid phagocytosis. d Targeting CD47 or interrupting the CD47-SIRPα axis allows macrophages to phagocyte hematopoietic cells and thus brings the side effects such as anemia
The mechanism and function of the PD-1-PD-L1 axis in phagocytosis
As a T-cell immune checkpoint, the function of the PD-1-PD-L1 axis in T cells has been well elucidated; however, recent studies have shown that this axis also functions in the regulation of the phagocytic ability of TAMs.9 PD-1 is expressed not only in T cells in peripheral tissues but also in B cells, activated monocytes, DCs and NK cells.187,188 TAMs express high levels of PD-1 compared to splenic macrophages or circulating monocytes, and PD-1 expression increases with tumor volume after engraftment. Furthermore, PD-1 tends to promote the polarization of macrophages to M2 polarization, most PD-1+ TAMs are M2-like macrophages, which are regarded as the protumor population in the tumor microenvironment (TME).9,19,189 Bone marrow transplantation experiment shows that most PD-1+ TAMs originate from circulating leukocytes rather but not resident immune cells.9 And PD-1+ TAMs show a reduced capacity for phagocytosis in contrast to PD-1-TAMs, indicating that PD-1 on TAMs inhibits phagocytosis. PD-L1 deficiency increases phagocytosis by PD-1+ macrophages significantly but has no effect on phagocytosis by PD-1− macrophages. Blocking PD-1-PD-L1 signaling with either an anti-PD-1 blocker or a PD-L1 inhibitor (HAC, an engineered small protein lacking an Fc domain to eliminate interference with Fc-mediated phagocytosis) increases macrophage phagocytosis and increases the survival rate of NOD SCID gamma (NSG) mice lacking T cells, indicating the antitumor role of the PD-1-PD-L1 phagocytosis checkpoint.9 In addition, TP53-mutated tumor cells secrete more extracellular vesicles and show impaired macrophage phagocytosis, but blocking PD-L1 on the extracellular surface of TP53-mutant cells was able to restore the phagocytic capacity of macrophages, suggesting that the important role of PD-1-PD-L1 is in macrophage phagocytosis in TP53-mutated tumors (Fig. 6b.190 The PD-1-PD-L1 axis has direct effects on macrophages in tumors. This evidence implies that PD-1 inhibits not only cytotoxic T-cell activity but also macrophage phagocytosis, revealing a new mechanism of the PD-1-PD-L1 axis in macrophage-mediated phagocytosis. Furthermore, LPS stimulation of TLR4 signaling upregulates PD-1 in macrophages. Ligation of PD-1 in macrophages by PD-L1 potentiates the polarization of tolerogenic STAT6-dependent macrophages and subsequent tumor growth.191
In T cells, the tyrosines of the ITIM and the immune receptor tyrosine-based switch motif (ITSM) in the PD-1 intracellular domain are phosphorylated after PD-1 binding to its ligand, thereby recruiting the SH2 domain-containing tyrosine phosphatases SHP-1 and SHP-2 and downregulating TCR signaling to inhibit T-cell activation and proliferation.192 Therefore, PD-1 on macrophages may also trigger immunosuppressive signals to inhibit phagocytosis by macrophages; however, the detailed mechanism needs to be further studied.
The mechanism and function of the MHC-I–LILRB1 axis
The mechanism of the MHC-I–LILRB1 axis
The site of contact between LILRB1 on macrophages and MHC-I on tumor cells is located in the conserved α3 domain and β2M subunit rather than the highly polymorphic α1 and α2 domains of MHC-I and 1st and 2nd Ig domain of LILRB110 (Fig. 5c). LILRB1 contains an extracellular region with four Ig-like structural domains (D1-D4), the transmembrane structural domain, and a cytoplasmic tail containing four ITIMs that recruit SHP-1 tyrosine phosphatases(SHIP),193 LILRB1 triggers inhibitory signaling through the ITIM in the long cytoplasmic tail.194 Specifically, (1) the binding of LILRB1 and MHC-I results in the phosphorylation of ITIM; (2) then, the phosphorylated ITIM recruits phosphatases SHIP after tyrosine residues are phosphorylated by Src family protein tyrosine kinases.195 Two hydrophobic residues are symmetrically located at the N-terminal and C-terminal ends of the phosphorylated tyrosine residues in the ITIM of LILRB1, and they affect the ability of the ITIM to bind phosphatases. (3) The recruitment of SHIPs leads to the inactivation of ITAM tyrosine kinases, thereby inhibiting ITAM recruitment for the Syk/ZAP70 kinase family, leading to the activation of PI3K/AKT. (4) The above process promotes cancer cell proliferation and regulates immune cell function negatively, leading to the inhibition of phagocytosis by macrophages.196
The function of the MHC and MHC-I–LILRB1 pathways
The functions of MHC-I in organ transplantation were reported first. Later, more functions of MHC-I in immunity were explored. The main function of MHC-I in tumor immunotherapy is antigen presentation and induction of immune responses.
Antigen presentation
The basic function of MHC-I is to display antigens to CD8+ T cells and activate the acquired immune response. MHC-I binds to and presents endogenous antigenic peptides for recognition by CD8+ T cells. MHC-I is delivered to the cell surface by the Golgi apparatus to present tumor-associated peptides to CD8+ T cells, and CD8+ T cells recognize antigenic peptide fragments through the TCR as peptide-MHC-I complexes on transformed cells. Subsequently, CD8+ cells are stimulated to undergo clonal expansion and produce cytokines to enable cytolytic effector activity and the killing of tumor cells with antigen-secretion. Tumor cells have developed strategies such as downregulating MHC-I to inhibit HLA-I antigen expression and function to avoid the recognition and destruction by CD8+ T cells.197
Induction of the immune response
MHC-I protects tumor cells from phagocytosis by macrophages and killing by NK cells by binding to inhibitory receptors on the surface of macrophages and NK cells, respectively. Blocking MHC-I or inhibiting LILRB1 either in vitro or in vivo enhanced phagocytosis of tumor cells by macrophages, and tumor cells expressing β2m prevented phagocytosis by macrophages and enabled evasion of the immune response.10 This suggests that the MHC-I-LILRB1 signaling axis functions as an antiphagocytic signal. Tumor cells escape NK-cell killing via MHC-I module expression, which was introduced in the section on MHC-I binding proteins.
The mechanism and function of the CD24-Siglec-10 axis
The mechanism of the CD24-Siglec-10 pathway
The inhibitory receptor Siglec-10 on the macrophage surface binds to its ligand CD24 on cancer cells, resulting in the interaction of an ITIM or ITIM-like motif in the cytoplasmic domain of Siglec-10 with Src family kinases.198 Then, Src family kinases phosphorylate the ITIM tyrosine in the cytoplasm, thereby recruiting protein tyrosine phosphatases (PPPs), such as SHP-1 and SHP-2.199 SHP-1 specifically binds to the intracellular phosphorylated ITIM domain to dephosphorylate it, leading to cytoskeletal remodeling and phagocytosis inhibition (Fig. 5d). In addition, SHP-1 negatively regulates intracellular signal transduction involving cell adhesion molecules, extracellular matrix factors, hormones, cytokines, and growth factors.200 Hence, the interaction of CD24 with Siglec-10 inhibits phagocytosis by macrophages and promotes the immune escape of tumors. Blocking the expression of CD24 on tumor cells or Siglec-10 on macrophages genetically or via an antibody enhances the phagocytosis of macrophages and suppresses tumor growth in vivo.8,201
Furthermore, CD24 also binds to Siglec-10 on the surface of other immune cells, including T cells,202 DC cells,87,203 and NK cells,204 to inhibit their functions. The mechanisms are all dependent on Siglec10, which has an ITIM or an ITIM-like motif. The ITIM functions in the immunosuppression and tumor immune escape by blocking TLR-mediated inflammation and activating the following intracellular signaling pathways.205
The function of the CD24-Siglec-10 pathway
The highly expressed CD24 on tumor cells interacts with Siglec-10 on the surface of macrophages to inhibit phagocytosis by macrophages; thus, tumor cells cannot be cleared via phagocytosis by macrophages.8,201 Siglec-10, like other members of the Siglec family, preferentially binds to sialylated CD24 in tumor cells, and the sialylation of CD24 helps tumor cells escape engulfment by macrophages.8 The interaction between CD24 on tumor cells and Siglec-10 on NK cells helps tumor cells evade the killing effect of NK cells and promotes tumor immune escape.204 When interacting with Siglec-10 on the surface of T cells, CD24 blocks activation of the TCR by inhibiting T-cell receptor-related kinases such as Lck and ZAP-70,202 thereby promoting escape from killing by T cells. The binding of CD24 to Siglec-10 on the surface of B cells inhibits BCR-regulated signal transduction and promotes tumor escape. Furthermore, the interaction between CD24 and Siglec10 is involved in complex placental immunosuppressive responses.94
Mechanism of STC-1 in phagocytosis
STC-1 promotes tumor angiogenesis and metastasis by upregulating VEGF in a manner dependent on the activation of the PKCβII and ERK1/2 pathways in cancer cells.206 STC-1 has been demonstrated to be an intracellular “eat me” signal inhibitor and and unappreciated phagocytosis checkpoint previously. Mechanistically, STC-1 interacts with the “eat me” signal calreticulin in the cancer cell and abrogates membrane calreticulin-directed phagocytosis by APCs, including macrophages and DCs, thus impairing antigen presentation from APCs to T cells, meanwhile, macrophage phagocytosis of cancer cells is suppressed by this process. Tumor STC-1 is crucial for intrinsic tumor resistance to tumor immunity. It plays an essential role in the tumor immune evasion and immunotherapy resistance by trapping calreticulin in mitochondria and the ER to inhibit macrophage function (Fig. 5e). Targeting STC-1 and its interaction with calreticulin may be an approach to enable patients to be susceptible to cancer immunotherapy. Ovarian cancer cells treated with a neutralizing anti-STC-1 monoclonal antibody exhibit higher apoptosis rates than control cells.207 In a mouse model of human lung cancer, targeting STC-1-expressed tumor cells exhibits efficient antitumor effects.208
Mechanism of GD2 in phagocytosis
As a sialic acid-linked glycolipid (a sialoglycan), GD2 may be recognized by sialic acid-binding proteins such as Siglecs. GD2 binds to Siglec-7 specifically instead of other Siglecs in humans. Siglec-7 is an immunosuppressive molecule that contains a cytoplasmic ITIM domain and is present in human macrophages and NK cells.199 Anti-GD2 disrupts GD2-Siglec-7 interactions and upregulates calreticulin, an “eat me” signal, promoting phagocytosis (Fig. 5f). Anti-GD2 exhibits synergistic effects with anti-CD47 on phagocytosis. The combination of B6H12 (CD47 antibody) and dinutuximab (GD2 antibody) increases the phagocytosis of neuroblastoma cells by microglia significantly, substantially enhances antitumor responses and extends tumor-free survival in a syngeneic model in NSG mice. Monocytes are responsible for these synergistic responses to anti-GD2/anti-CD47.12 GD2 and CD47 blockade enhances macrophage phagocytosis by enhancing “eat me” signals and attenuating “don’t eat me” signals and recruits M1-like macrophages for an antitumor response, with potential for clinical application.12
Function of phagocytosis checkpoints in the immune system
Function of CD47 in immunity
Immune cells, such as monocytes, macrophages, DCs, T cells, and B cells express CD47. which is critical for both innate and adaptive immune responses. CD47 sends a potent potent “don’t eat me” signal to prevent phagocytosis and functions integrally plays in immune responses and autoimmunity.209
CD47 expression and function in the innate immune system
The CD47-binding protein SIRPα is expressed on macrophages, and the binding of CD47-SIRPα triggers a “don’t eat me” signal, protecting cancer cells from immune clearance.210 Another CD47 binding protein, TSP-1, is also expressed on macrophages, and CD47-TSP-1 contributes to the migration of monocytes and leads to nervous system inflammation and the occurrence of disease.211 Moreover, NK cells highly express CD47, which regulates the recruitment, activation and proliferation of NK cells.212 As a self-marker of DCs, CD47 not only regulates the activation, quantity, maturity, migration and apoptosis of DCs but also participates in the initiation of immune responses in DCs. The expression of SIRPα on DCs inhibits their phagocytosis,213 and blocking the CD47-SIRPα pathway activates DC cells to phagocytize tumor cells.214 CD47 inhibits the transformation of immature dendritic cells (iDCs) to mature cells in terms of both phenotype and function.213 CD47 also regulates DC migration to lymphatic organs. Under inflammatory conditions, CD47-SIRPα interactions are necessary for skin DC migration.215 Furthermore, CD47 expressed on neutrophils regulates their transepithelial migration and adhesion; it associates with leukocyte-specific integrin CD11b/CD18 in neutrophils’ membrane, and its loss results in impaired CD11b/CD18 activation. CD47 also regulates chemotaxis of human neutrophils, as SIRPα regulates neutrophil transmigration in vitro.183,216
CD47 expression and function in the adaptive immune system
CD47 expressed on T cells regulates the activation, proliferation, differentiation and apoptosis of T cells. CD47 is a costimulatory factor for T-cell activation, and the interaction of CD47 on T cells and SIRPα on DCs induces the activation of T lymphocytes by DCs and promotes the proliferation of T cells.217 Meanwhile, CD47 modulates T-cell differentiation by affecting both T cells and APCs. The CD47/TSP-1 interaction or blockade of CD47 induces T-cell apoptosis.218,219 CD47 limits TCR signaling and killing of irradiated target cells.220 The TSP-1/CD47 interaction inhibits TCR signal transduction and induces active T-cell anergy.221 In addition, CD47 expressed on Tregs regulates Treg cell generation, proliferation, and differentiation and contributes to Treg neuroprotection by binding to its receptors SIRPα or TSP-.222 Furthermore, CD47 is expressed on B cells and limits antibody-mediated phagocytosis and the growth of B cells.223,224 The interaction between CD47 on B cells and SIRPα on macrophages also plays a role in cell‒cell contact between B cells and macrophages, which is important for the differentiation of B lymphocytes.
The PD-1-PD-L1 phagocytosis checkpoint in immunity
The PD-1-PD-L1 phagocytosis checkpoint in innate immunity
PD-L1 expressed on DCs facilitates the migration of DCs from the skin to the lymph nodes and triggers intracellular signaling through the cytoplasmic tail of PD-L1. A mutated cytoplasmic domain of PD-L1 impairs CCR7 signaling, including G protein activation, extracellular signal-regulated kinase (ERK) phosphorylation, and F-actin polymerization.225 PD-L1 on DCs also regulates immunotherapy and reduces T-cell activation.226
PD-L1 expressed on macrophages exerts constitutive signaling effects, leading to suppressed activation and proliferation of macrophages via inhibition of the mTOR signaling pathway in macrophages. PD-L1−/− macrophages stimulate proliferation and activation, and PD-L1 antibody treatment upregulates the production of costimulatory molecules and spontaneous proinflammatory cytokines.227 Mechanistically, PD-L1 blockade upregulates costimulatory molecules’ (CD86 and MHC-II) and the secretion of the proinflammatory cytokines (TNFα and IL-12), consistent with the characteristics of M1-type macrophages.228,229 However, under the metabolic reprogramming, PD-L1 promotes M2 polarization via the Erk/Akt/mTOR signaling pathway.230 On the other hand, PD-1 regulates macrophage polarization to potentiate the inflammation, and PD-1 knockout promotes macrophage M1 instead of M2 polarization by potentiating STAT1, indicating that PD-1 expression is negatively associated with M1 polarization.231 PD-1 on macrophages reduces the phagocytic ability of macrophages for tumors and bacteria,9,232,233,234 suggesting that PD-1 affects tumor immunity by both innate and adaptive immune systems.9,235
The PD-1-PD-L1 axis plays important roles in protecting against pathogen infection via innate immunity. PD-1 and PD-L1 are expressed on CD4+ T cells and CD14+ monocytes but not on CD8+ T cells in patients with active tuberculosis infection. Blocking the PD-1-PD-L1 pathway increases the phagocytosis and intracellular killing of pathogens by macrophages,233 suggesting that the PD-1-PD-L1 pathway has an inhibitory effect on the function of macrophages in terms of phagocytosis of pathogens.
The PD-1-PD-L1 phagocytosis checkpoint in adaptive immunity
PD-L1 ligation of PD-1 limits immunogenic responses in T cells. PD-1 contains conserved ITIMs in its cytoplasmic tail, which recruit downstream phosphatases and attenuate activation signals, acting as an immune inhibitory receptor.236 PD-1 maintains immune homeostasis and tolerance to prevent immunopathology under physiological conditions, and PD-1 deficiency leads to autoimmune diseases. The PD-1-PD-L1 axis inhibits T-cell activation through a series of signals, eventually leading to a reduction in the activation of transcription factors, such as nuclear factor of activated T cells (NFAT), activator protein 1 (AP-1), and NF-κB, which are critical for T-cell proliferation, activation, survival and effector functions. Furthermore, PD-1 upregulates transcription factors such as basic leucine zipper transcription factor ATF-like (BATF), which can further antagonize effector transcriptional programs to inhibit T-cell functions.192
PD-L1 is also expressed in T cells, and PD-L1 blockade reduces the numbers of effector CD8+ T cells during the contraction phase of an immune response. Activated CD8+ T cells deficient in PD-L1 are more susceptible to Ca-dependent and Fas ligand-dependent killing by cytotoxic T cells, leading to a lower Bcl-xL. PD-L1 on primed T cells helps effector T cells survive in the contraction phase and thereby elicits optimal protective immunity.237 Moreover, PD-L1 deficiency results in increased activation of p38 MAPK, which results in the apoptosis of T-cells, indicating that PD-L1 suppresses p38 MAPK activation to preserve T-cell survival.238
MHC-I expression and function in the immune system
MHC-I is a cell surface recognition element expressed on all somatic cells, including all immune cells. It is primarily involved in T-cell-mediated adaptive immune responses but also functions in the innate immune system.
MHC-I is present on the surface of APCs, including both DCs and macrophages. The APCs load endogenous antigenic peptides in the ER onto MHC-I to form a correctly folded trimeric complex (pMHC/β2m), which is modified post-translationally in the Golgi complex, and finally, the complexes are transported to the cell surface, where they are presented to CD8+ T cells;239 thus, MHC-I bridges the innate and adaptive immunity via antigen-presenting cells.
MHC-I on DCs binds to TCRs on T cells and regulates T-cell differentiation and maturation. T cells that cannot bind to MHC-I are scheduled for apoptosis.240 T cells that pass positive selection should not have a strong affinity for MHC; otherwise, they will easily attack themselves. Therefore, only T cells that can bind to MHC via appropriate TCRs with low affinity successfully enter tissues through the blood circulation and exhibit immune surveillance and immune attack abilities. In addition, T cells and B cells also express MHC-1, but its function is rarely studied.241
Function of CD24 in immunity
CD24 is expressed on the surface of a variety of immune cells, including B cells, T cells, DCs, and neutrophils. CD24 interacts with Siglec10 on the surface of various immune cells to exert an immunosuppressive effect. All the above studies demonstrated that CD24 is a critical molecule in the immune system.
CD24 expression and function in the innate immune system
CD24 is expressed in all innate immune cells, such as macrophages and DCs, and its main function is endogenous antigen presentation. CD24 on DCs negatively regulates T-cell homeostatic proliferation.242 Moreover, CD24 on DCs interacts with Siglec-10 in humans or Siglec-G in mice on the surface of damaged cells; then, SHP-1 binds to the ITIM of Siglec10 and inhibits the activation of NF-kB, which inhibits the release of HMGB1 in turn and negatively regulates damage-associated molecular patterns.87 CD24 on the surface of DCs also interacts with Siglec10 in other cell types, which inhibits host inflammatory and immune responses triggered by damage-related molecules,87 but it also allows RNA viruses to evade host immunity.203 In addition, CD24 on microglia contributes to the activation and proliferation of pathogenic T cells since the costimulatory activity of microglia is reduced in CD24-deficient mice.243
CD24 expression and function in the adaptive immune system
CD24 was thought to be a marker of B cells originally; it is present highest on B-cell progenitors and is not expressed on terminally differentiated plasma cells because it disappears as B cells mature.244 CD24 knock-out leads to a reduction in the numbers of advanced pre-B cells and immature B cells in the bone marrow.245 CD24 on activated B cells serves as a CD4 T-cell costimulator for clonal expansion.246
As in B cells, CD24 is expressed on peripheral T cells weakly while present expressed on peripheral T cells highly. The difference in CD24 expression between T cells and B cells is that CD24 is upregulated on activated T cells.247 CD24 deficiency and CD28 deficiency synergistically suppress CD4 and CD8 T-cell responses.248 In addition, highly expressed CD24 on tumor cells binds to Siglec-10 on the surface of T cells and B cells, inhibiting TCR and BCR-related kinases to block activation of the TCR and BCR and ultimately promote tumor immune escape.
Phagocytosis checkpoints in diseases and the tumor microenvironment (TME)
CD47 in diseases and the TME
CD47 in cancer and the TME
For many types of malignancies, the low early detection rate is an obstacle to improved cancer control;249,250 therefore, efforts to identify novel diagnostic markers are valuable.251 CD47 has been demonstrated as a diagnostic biomarker for a variety of cancers. It is an innate immune checkpoint and is closely related to the survival in different cancers. High expression of CD47 contributes to tumor cell proliferation and tumor metastasis.
CD47 is overexpressed in a host of hematological malignancies, and its interaction with SIRPα on phagocytes prevents phagocytosis of tumor cells and promotes tumor evasion of immune surveillance.209,252 CD47 is expressed highly in both small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). In EGFR-mutant NSCLC, the augmented CD47 expression is closely related to the off-target resistance to the tyrosine kinase inhibitor (TKI) gefitinib.253 In glioblastoma multiforme (GBM), GBM cells with higher CD47 expression possess the characteristics of stem cells and have poor clinical results,254 and irradiation or temozolomide (TMZ) significantly enhances anti-CD47-mediated phagocytosis of GBM cells in vivo and in vitro. Specific inhibition of the TSP-1/CD47 interaction with a peptide antagonist decreases GBM cell invasion.255 CD47 is also expressed highly in ovarian cancer, HCC, cholangiocarcinoma (CCA), etc. High expression of CD47 may contribute to the resistance of CSCs to chemotherapy.256 In HER2-expressing cells, CD47 is upregulated preferentially, and the interaction between CD47 and HER2 is reflected in the significant difference in the expression levels of CD47 in HER2+ versus HER2− breast cancer cells.
CD47 in the TME
The TME affects immunotherapy efficacy and patient outcomes in various types of cancer. CD47 functions in immune homeostasis related to cancer prognosis, and its expression is closely related to immune infiltration. TAMs are key components in the TME that participate in the regulation of various biological behaviors and influence tumor growth and progression.257,258,259 Their phagocytic function has been demonstrated to be a key determinant of tumor metastasis and is closely related to the TME.260 The blockade of CD47 or SIRPα with blocking antibodies increases the phagocytic activity of TAMs and decreases tumor growth in different tumor models, including models of glioblastoma,261 melanoma,262 lymphoma,263 breast cancer,264 and colorectal cancer.265 Blocking CD47 promotes antitumor immunity through CD103 + DC-NK cell axis in murine HCC model.266 CD47 may also induce T-cell exhaustion by working with T-cell exhaustion markers such as PD-1 and CTLA-4, thus remodeling the TME.267
TSP-1, the binding protein of CD47, restricts antitumor immunity via CD47-dependent regulation of innate and adaptive immune cells by regulating angiogenesis and perfusion of the tumor vasculature. Moreover, The TSP-1/CD47 expression and interaction increase under hypoxia to promote tumor growth.268
CD47 in other diseases
In addition to playing critical roles in cancer and the TME, CD47 also functions in many other diseases. For example, in pulmonary arterial hypertension (PAH), the levels of both CD47 and TSP-1 are increased and promote hypoxia and ROS production in the environment.269 In addition, activated CD47 promotes acute kidney injury (AKI) by limiting autophagy, and CD47 has been demonstrated to be a target for preserving renal function following injury.270 TSP-1 expression is increased in response to AKI, and blocking TSP-1-CD47 signaling restricts tissue injury caused by ischemic stress in tissues.271,272 Furthermore, targeting CD47 attenuates fibrosis induced by various diseases. CD47 mediates immune escape in infectious diseases caused by parasites, bacteria, and viruses, including SARS-CoV-2 in COVID pathogenesis,273 and it interferes with the host immune response by binding to SIRPα on immune cells. The disruption of CD47-SIRPα increases the phagocytosis of P. falciparum-infected RBCs.274
PD-L1 in diseases and the TME
PD-L1 in cancer and the TME
Most tumors, including solid tumors such as melanoma, clear cell carcinoma, NSCLC, and breast cancer, as well as hematological tumors,65,275 overexpress PD-L1, whose expression is closely associated with poor prognosis. Tumors evade immune clearance by suppressing T-cell activation via overexpression of PD-L1. Under normal physiological conditions, the PD-1-PD-L1 interaction maintains T-cell immune homeostasis, thereby preventing T-cell hyperactivation and avoiding autoimmune diseases.59 However, tumors use PD-1-PD-L1 checkpoint inhibitory signals to evade the immune system, mainly by upregulating PD-L1 expression to suppress T cells, leading to T-cell inactivation and triggering T-cell dysfunction.66 High expression of PD-L1 not only inhibits the activity of T cells but also inhibits the activities of APCs such as DCs and macrophages.9,276,277 PD-L1 expression on APCs plays an immunosuppressive role in the TME. APCs with PD-L1 expression play a dominant role in the regulation of T-cell immunity and the response to cancer immunotherapy in the context of cancer.278,279 On the other hand, PD-1 on macrophages inhibits the phagocytosis of tumor cells, and PD-1-positive TAMs are associated with a reduction in 5-year overall survival in the context of cancer.9,235 Therefore, the PD-1-PD-L1 axis interacts with both the innate and adaptive immune systems in the TME.
PD-L1 in other diseases
The PD-1-PD-L1 axis plays important roles in many other diseases, such as autoimmune diabetes, rheumatoid arthritis,280,281 allergic disease,282,283 and neurological disorders. PD-L1 participates in the progression of hypoxia-induced multiple organ injuries, such as injury caused by ischemic stroke, AKI, and obstructive sleep apnea.284 Hypoxia upregulates PD-L1 expression via HIF-1α, and PD-L1 is overexpressed in the spleen and central nervous system (CNS) post-stroke. The overexpression of PD-L1 in microglia reduces acute ischemic brain injury by reducing T-cell infiltration and cytokine release. Previous studies have stressed PD-1-PD-L1 as a T-cell checkpoint; therefore, we mainly focused on the functions of these factors in phagocytosis.
The PD-1-PD-L1 axis also plays a critical role in preventing pathogen infection. Sepsis is an overwhelming reaction to infection, and PD-1 on macrophages/monocytes was obviously upregulated during sepsis, together with macrophage dysfunction. The phagocytic function of macrophages during sepsis relies on their PD-1 expression, indicating the role of the PD-1-PD-L1 axis as a phagocytosis checkpoint in microbial clearance.232 Furthermore, PD-L1 expression is upregulated on synovial fluid myeloid DCs, T cells and macrophages in rheumatoid arthritis due to the high levels of IFN-γ and TNF-α in RA-derived synovial fluid.280,281
MHC-I in diseases and the TME
MHC-I in cancer and the TME
MHC-I on tumor cells interacts with the relevant receptors on almost all immune cells in the TME, thereby affecting tumor immune escape. Downregulation of MHC-I occurs in 40–90% of human tumors and is significantly correlated with poor prognosis.285 In contrast, due to irreversible changes in MHC-I expression in tumors caused by genetic mutations, tumors may temporarily upregulate MHC-I to escape natural immune attacks, such as killing by NK cells286 and phagocytosis by macrophages.10
After anti-CD47 treatment, tumor cells with MHC-I high are more resistant to phagocytosis by macrophages than those with MHC-I low expression. When epithelial cell adhesion molecule or EGFR blockers were used, CD47 and MHC-I double-negative cells were more vulnerable to phagocytosis, whereas the expression of either MHC-I or CD47 alone attenuated macrophage attack, and macrophage resistance was strongest in both double-positive cells. All the above results suggest that MHC-I and CD47 are two independent antiphagocytic signals.10
HLA-G is a nonclassical MHC-I, unlike classical MHC, and it is characterized by a low polymorphism rate and tolerogenic function. HLA-G has three soluble isoforms (HLA-G5, HLA-G6 and HLA-G7) that are secreted into the tumor microenvironment and directly inhibit the activation of immune cells.287 The expression of HLA-G in solid tumors predicts poor prognosis. However, increased plasma levels of soluble HLA-G in B-cell malignancies are not related with poor clinical outcomes. As an inhibitor of B-cell growth, HLA-G probably exerts an inhibitory effect on tumor growth by interacting with LILRB1, suggesting that HLA-G-LILRB1 axis can be applied to the treatment of B-cell malignancies.288
MHC-I in other diseases
MHC-I plays critical roles in transplantation, autoimmune diseases and virus infection.289 MHC are the main antigens that induce the rejection of allogeneic transplants. The higher the similarity of MHC is between the donor and the recipient, the higher the success rate after transplantation. An HLA match test between the donor and the recipient is required before transplantation.290 Regarding autoimmune diseases, more than 50 human diseases have been demonstrated to be related to HLA. For example, high HLA-B27 contributes to the development of ankylosing spondylitis.291 Other diseases associated with specific MHC molecules include multiple sclerosis,292 Crohn’s disease,293 and rheumatoid arthritis.294 In addition, MHC-I-restricted CTLs are important effector cells against viral infection, and during symbiosis of the virus and host, the virus escapes elimination and clearance by the host by interfering with the killing activity of CTLs through different pathways.289 This process inhibits viral peptides expression by MHC and the recognition of MHC-mutant peptide complexes by the TCR. The function of MHC-I in other diseases related to phagocytosis requires further investigation.
CD24 in diseases and the TME
CD24 in cancers and the TME
CD24 is overexpressed in many cancers, including B-cell lymphomas, gliomas, SCLC, HCC, and breast cancer, and appears to be oncogenic.295 CD24 has been demonstrated to be a marker for cancer diagnosis and prognosis. High expression of CD24 on tumor cells not only facilitates tumor progression by affecting the proliferation and migration of tumor cells but also allows tumor cells to escape killing by immune cells via interactions with immune cells around the tumor. When CD24 on tumor cells binds to Siglec-10 on different immune cells, it causes immune cell inhibitory signaling cascades mediated by SHP-1/SHP-2, promoting escape from killing by T and NK cells and engulfment by macrophages.
CD24 in other diseases
In addition to its functions in cancers, CD24 plays critical roles in autoimmune diseases, inflammation, and metabolic disorders.
Regarding autoimmune diseases, CD24 polymorphisms are related to the progression and risk of multiple sclerosis,296 rheumatoid arthritis.297 Mice without CD24 are highly resistant to autoimmune encephalomyelitis experimental.95 The detailed role of CD24 in the regulation of autoimmune disease requires further investigation.
Inflammation is involved in many diseases, such as infection, sepsis, liver injury, and chronic graft-versus-host disease; it is the innate immune response to pathogen infection and tissue damage, and CD24 is able to differentiate between DAMPs and pathogen-associated molecular patterns during inflammation.87 It selectively inhibits the host response to tissue injury via interaction with Siglec G (mouse) or Siglec10 (human). Regarding metabolic diseases, CD24 binding to Siglec-E is a key inhibitor of obesity-related metabolic dysfunction.298
STC-1 in diseases and the TME
STC-1 in cancer and the TME
STC-1 is expressed highly in a variety of cancers, such as colon cancer,299 gastric cancer (GC),206 ovarian cancer,299 breast cancer,114 bladder cancer,300 glioblastoma,301 acute leukemia,302 and hepatocellular carcinoma, and higher expression of STC-1 relates to metastasis, lower survival rate and faster progression. Serum STC-1 serves as a promising tumor marker in GC and ovarian cancer because its expression is higher in such patients than in patients with benign tumors,303,304 and STC-1 is a potentially useful blood marker for predicting tumor progression and invasion in patients with GC.305 STC-1 overexpression increases proliferation, migration, and colony formation in cancer cells. Mechanistically, STC-1 on cancer-associated fibroblasts (CAFs) increases migration and invasion.299,306 STC-1 promotes tumor angiogenesis by upregulating VEGF and promoting gastric tumor growth206 and promotes cancer cell proliferation, migration and invasion during hypoxia via Bcl-2.307 Moreover, STC-1 promotes lipid metabolism and resistance to cisplatin via regulating the FOXC2/ITGB6 pathway in ovarian cancer.308 However, in cervical cancer, STC-1 inhibits cell proliferation and invasion and promotes apoptosis.309 The expression of STC-1 can be induced under hypoxia by HIF-1 in human cancer cells.310,311
STC-1 in other diseases
In contrast to its function in most cancers, high expression of STC-1 in other diseases promotes survival. STC-1 overexpression alleviates oxidative stress-induced injury by inhibiting ROS through the mitochondrial pathway312 and reduces neuroinflammation; STC-1 overexpression also ameliorates cognitive function by inhibiting the ERK1/2 signaling pathway.313 In diabetic nephropathy, STC-1 improves renal injury by inhibiting BNIP3 via the AMPK/SIRT3 signaling, and patients with high levels of STC-1 have a better prognosis.118 Serum STC-1 expression is decreased in asthma patients compared with healthy donors, and STC-1 reduces airway hyperresponsiveness (AHR) and inflammation.314 Furthermore, the STC-1 concentration in the cerebrospinal fluid was reduced in a heterogeneous group of dementias other than Alzheimer’s disease, especially dementia with Lewy bodies and vascular dementia,315 and it also ameliorated cognitive function by inhibiting the ERK1/2 signaling pathway.313 Consistently, low expression of STC-1 results in poor prognosis, and NF-κB upregulates miR-155-5p to inhibit STC-1 expression, leading to hepatic mitochondrial dysfunction in nonalcoholic fatty liver and thereby stimulating the occurrence of nonalcoholic fatty liver disease.316
GD2 in diseases and the TME
GD2 in cancer and the TME
GD2 has limited expression in normal tissues but is overexpressed on tumors, including gliomas,317 melanoma,318 osteosarcoma,319,320 and soft tissue sarcoma.321 The GD2 antibody showed therapeutic effects in all tumors, indicating that GD2 is a promising therapeutic target.
In human neuroblastoma cells, GD2 is the major ganglioside, and progression-free survival was inversely related to circulating GD2 levels, indicating that neuroblastoma tumor gangliosides play a role in accelerating tumor progression.322 However, GD2 levels do not seem to correlate with tumor grade,323 and GD2 is expressed higher in SCLC than in NSCLC or normal lung cells.324,325 In addition, GD2 in melanoma cells is involved in their attachment to extracellular matrix proteins.326,327 Antibodies targeting GD2 cause regression of cutaneous metastatic melanoma, and GD2-specific CAR-T cells have antimelanoma activity.328
GD2 in other diseases
GD2 is most abundant in the central nervous system and modulates the activity of Ca2+ channels and transporters. Mice without complex gangliosides (GM2/GD2 synthase knockout) have impaired Ca2+ regulation after neuronal development.329 Deficiency in neuropathies such as balance, coordination, strength and reflexes develop significantly, indicating the role of GD2 in the maintenance of normal neural physiology.129 GM2/GD2 synthase knockout mice also exhibit morphological changes in synaptic vesicles and the mode of synaptic contact with central terminals and deficits in cognitive function and hippocampal plasticity.330,331 Mutations of B4GALNT1, which encodes GM2/GD2 synthase, are associated with limb spasticity, dysarthria, peripheral neuropathy, and severe intellectual disability.332,333 In addition, the neural ganglioside GD2 is a marker expressed by mesenchymal stem cells (MSCs) isolated from either bone marrow or umbilical cord blood, suggesting that GD2 functions in maintaining stem cell viability.334
The expression and working mechanisms of all the reported phagocytosis checkpoints in various diseases are shown in Table 1).
Targeting phagocytosis checkpoints and clinical applications
Targeting CD47 and its clinical applications
Targeting CD47 in cancer immunotherapy
The role of CD47-SIRPα as an immune checkpoint signaling pathway has been reviewed elsewhere. CD47 is expressed highly on a variety of cancer cells and functions as a key antiphagocytic protein which maintains tumor cells’ resistance to host immune surveillance. The CD47-SIRPα pathway is a phagocytosis checkpoint in macrophages and other innate immune cells, and CD47 has been verified to be a promising therapeutic target due to its antiphagocytic function in tumor cells. Targeting CD47-SIRPα not only disrupts the binding of CD47 and SIRPα and potentiates the phagocytosis ability of cancer cells by stimulating macrophage cytokine secretion and thus stimulating the patients’ immune system7,335 but also kills tumor cells through the NK-cell-mediated ADCC effect336 and even directly induces tumor cell apoptosis.337,338 Moreover, targeting CD47 also enables DCs to phagocytize tumor cells, present the tumor antigen to T cells and activates the adaptive immunity.
Development and clinical applications of CD47 antibodies
CD47 was originally discovered as a missing antigen in Rh-negative RBCs by the antibody 1D8 in 1987,21 and later, it was defined as an antigen recognized by BRIC126, CIKM1 or BRIC125 monoclonal antibodies.339,340 The development of antibodies targeting CD47 has continued since the discovery of CD47. CD47 antibody therapy is mainly divided into three research directions: single drugs, combined therapy with antibody drugs and combined therapy with T-cell checkpoint inhibitors.
Hu5F9-G4 was the 1st humanized antibody targeting CD47, and it was initially used in children with malignant primary brain tumors.261 Hu5F9 has a curative effect against five different childhood brain tumors. It can be used to treat a variety of malignant tumors of the central nervous system. Another antibody, SIRPαD1-Fc, a novel CD47-targeting fusion protein, increases the autophagy of NSCLC cells by inactivating the Akt/mTOR pathway and increasing ROS levels Table 1.
The combination of CD47-SIRPα targeting with other treatments likely achieves better efficacy. Common combination therapies include treatment with other therapeutic antibodies, recruitment of macrophages, combined chemotherapy and radiotherapy and inhibition of tumor metastasis. For example, blocking the CD47-SIRPα pathway together with treatment with sodium stibogluconate (SSG), an antileishmaniasis drug, overcomes the resistance of anti-CD20-opsonized B-cell lymphoma cells to neutrophil killing.341 Hu5F9-G4 (now known as magrolimab) is in a phase II/III clinical study for AML and has shown a favorable safety profile in combination with azathioprine (AZA).342,343 Magrolimab in combination with AZA demonstrated early efficacy in AML patients with mutated TP53.344,345
An increasing number of companies, including overseas companies such as Forty-seven (merged by Gilead in 2020), Celgene, Trillium, Alxoncology and domestic companies such as IMAB and ImmuneOnco, are currently developing drugs targeting CD47, specifically monoclonal antibodies, bispecific antibodies, fusion proteins and small molecules; many of these drugs have entered the clinical research stage, but there are no such drugs on the market yet. In 2019, Forty-seven announced that its CD47 monoclonal antibody magrolimab showed excellent and sustainable clinical efficacy. Since then, a pipeline of research on CD47 has emerged all over the world. The most advanced drugs in development are in phase III clinical trials, and most of the drugs in development are in phase I/II clinical trials; please refer to the detailed list of clinical trials in the USA and China (Table 3).
Challenges of targeting CD47
Red blood cell toxicity
CD47 is universally expressed on normal cells, including RBCs and T lymphocytes. thus, special attention should be given to whether the developed antibodies have adverse effects on normal cells. Targeting CD47 leads to the phagocytosis of RBCs by macrophages and causes the agglutination of the RBCs, ultimately leading to the lysis of RBCs. In addition, NK cells or macrophages may attack RBCs via Fc-mediated effector function, via either ADCC or antibody-dependent cell-mediated phagocytosis (ADCP). Therefore, avoiding binding with RBCs has become a primary concern in CD47 antibody drug development.
T lymphocyte toxicity
CD47 is expressed on T lymphocytes; when a CD47 antibody binds to CD47 on T lymphocytes, it may cause T-cell apoptosis, which may prevent clinical development, as T cells are key immune cells in cancer immunotherapy.
Selection of IgG subclasses
The Fc part of the antibody activates FcR on NK cells, macrophages, or neutrophils, leading to tumor cell lysis via ADCC or ADCP. In addition, antibodies directly activate the complement pathway to enable killing of antibody-coated tumor cells via complement-dependent cytotoxicity (CDC).346,347 The four subtypes of IgG bind different types of FcR with different binding capabilities and different effector functions in ADCC and ADCP, and the IgG subclass must be taken into consideration during antitumor therapeutic antibody selection.348,349,350 Since most CD47 antibodies preferentially bind to RBCs, if IgG1 is selected, immune cells such as NK cells and macrophages will be activated by RBCs. Therefore, to avoid RBC toxicity caused by CD47 antibodies, the IgG4 subtype was selected for all CD47 antibodies in development, but the antitumor activity of these antibodies is reduced.
High blood pressure
The CD47/TSP-1 axis regulates blood pressure,351 and CD47 knockout mice have normal central pulse pressure but elevated peripheral blood pressure. Targeting CD47 achieves vasopressor activity to maintain global hemodynamics under stress.352
Differences in binding affinity between animal models and humans
The selection of an animal model is very important when evaluating CD47 targeting. The binding affinity between CD47 from humans and SIRPα from NSG mice is 10 times higher than that between CD47 and SIRPα from humans, indicating that positive results in mice may not translate to success in human clinical trials. Furthermore, the mice used in the animal model are immuno-comprised animals that lack a complete immune system. Xenotransplantation under ideal conditions warrants further investigation.
Proposed strategies and future perspectives for targeting CD47
Designing a CD47 antibody with weak binding to RBCs
The high expression of CD47 on RBCs means that RBCs bind to CD47 antibodies preferentially,353 but it is still possible to develop antibodies with antitumor activity and without hematological toxicity since the molecular conformation of CD47 on tumor cells is distinct from that of CD47 on RBCs.354,355,356 TJC4 was designed with the above concept and is in a phase II clinical trial currently.357
Designing a CD47 antibody based on the differences between RBCs and tumor cells
Tumor cells are different from RBCs in terms of both morphology and molecular biology, and it is possible to develop antibodies based on these differences.358 AO-176 was designed based on this idea, and it selectively binds to tumor cells rather than RBCs; another antibody, RRX-001, was also designed according to the above concept and does not cause anemia.359
Targeting SIRPα instead of CD47
Targeting SIRPα can also block the CD47-SIRPα pathway. Since SIRPα is not expressed on RBCs, targeting SIRPα will not cause the depletion of RBCs and platelets. The antibodies ADU-185, TTI-621 and ALX148 were designed with high affinity for SIRPα and low affinity for blood cells.360,361,362
Targeting QPCTL-mediated CD47 pyroglutamylation in CD47-SIRPα signaling
The pyroglutamylation of CD47 is essential for the binding between CD47 and SIRPα, and QPCTL is the key enzyme for pyroglytamylation of CD47.338,363 Targeting QPCTL significantly attenuates the binding ability of CD47 to SIRPα and increases phagocytosis of tumor cells by macrophages, thus regulating tumor immunity (Fig. 7a),50,52,364 and targeting QPCTL avoids anemia since QPCTL is not expressed on mature RBCs.

Targeting QPCTL and targeting PD-L1. a QPCTL is the key enzyme for pyroglutamylation of CD47. Targeting QPCTL significantly reduces the binding ability of CD47 to SIRPα and prompts phagocytosis of tumor cells by macrophages thus regulating tumor immunity. b PD-1 is expressed on T cells, the antibody targeting PD-1 can bind to FcR on phagocytes and then be engulfed by the phagocytes, leading to the inhibition of cancer immunotherapy
Targeting PD-1-PD-L1 in phagocytosis and clinical applications
After the discovery of the important role of PD-1-PD-L1 in tumor immune escape, targeting of this pair has been rapidly applied to clinical treatment. PD-1 and PD-L1 blockade have become important clinical treatments. Targeting the PD-1-PD-L1 immune checkpoint has achieved remarkable results in clinical applications, with unprecedented progress; numerous clinical trials are always ongoing. A PD-1 or PD-L1 antibody blocks the immunosuppressive effect of PD-1-PD-L1 and restores the ability of T cells to kill tumors. Since the approval of nivolumab in 2014, many companies have successfully developed and approved PD-1-PD-L1 antibody drugs. There are many such antibody drugs in the clinic for a variety of different indications.
Immune checkpoint inhibitor drugs, such as PD-1 and PD-L1 antibodies, have multiple effects in immunotherapy. They not only suppress the inhibitory interaction between T cells and tumor cells but also block the binding between macrophages and tumor cells. The combination of CD47 and PD-1 blocking antibodies results in a synergistic ability to inhibit tumor growth, providing a new strategy for immunotherapy.9 Recently, bispecific antibodies targeting PD-1-PD-L1 and CD47-SIRPα have also been applied in clinical trials as a new strategy. In addition, TAMs can remove PD-1 blocking antibodies from T cells via FC-FcγR to weaken the immune response (Fig. 7b).365,366 Therefore, Fc-engineered IgG variants that disrupt FCR binding and combination therapies with agents that inhibit FcγR binding are better options for immunotherapy.
TAMs, a type of M2-polarized macrophage, eliminate or suppress T-cell-mediated anti-tumor responses. Carfilzomib is an FDA-approved drug for treating patients with relapsed/refractory multiple myeloma, recent research indicated that Carfilzomib induces M2 macrophages to express cytokines secreted by M1 macrophages, and phagocytizes tumor cells, as well as presents antigens to T cells. Mechanistically, treatment of Carfilzomib elicited unfolded protein response (UPR), activated IRE1α to recruit TRAF2, and enhanced NF-κB activation to transcribe genes encoding M1 markers in M2 macrophages, thus leading to enhanced phagocytosis of macrophages.367 The combination of Carfilzomib with PD-1 antibody exerts the synergistic effect in lung cancers.367
Targeting MHC-I-LILRB1 and clinical applications
MHC-I expression on tumor cells renders their resistance to phagocytosis, which may be because of the inhibitory interaction between the β2M subunit of MHC-I on cancer cells and LILRB1 on phagocytes. Therefore, targeting the MHC-I-LILRB1 axis may enhance the phagocytosis of tumor cells. HLA-G is rarely expressed in normal cells and mainly found in tumor cells, as a ligand, it interacts with LIRB1 receptor with the highest affinity. Monoclonal antibodies against HLA-G have been successfully used against cancer as part of an immune checkpoint suppression strategy.368 Since the most important function of MHC-I is antigen presentation and since MHC-1 is ubiquitously expressed on all types of cells, targeting MHC-I enhances phagocytosis of tumor cells by macrophages but may also lead to the loss of T-cell recognition of antigens and tumor immune evasion. There are currently no MHC-I-targeting drugs on the market.
Although cytotoxic T cells’ antitumor activity is dependent on their interaction with MHC-I, specific blockade of the LILRB1/β2M axis is a potential target for innate immune drugs. LIRB1 is not only present mainly in myeloid cells but also highly expressed in CSCs and probably modulates tumor progression and recurrence directly and determines tumor stem cell activity. Importantly, studies in mice with knockout of the relevant targets showed that LILRB1 does not affect normal development or hematopoiesis. Hence, LILRB1 may be an ideal target for tumor therapy.369 Preclinical data show that BND-22 exerts broad antitumor effects by targeting LILRB1-mediated “don’t eat me” signals in macrophages and activating NK and CD8+ lymphocytes, effectively inhibiting tumor growth in melanoma and colorectal cancer, prolonging the survival of model mice, and inhibiting the spread of cancer cells. Treatment of melanoma and colorectal cancer with BND-22 prolongs the survival of mice and inhibits the spread of cancer cells.370 NGM707 is another novel dual antibody antagonist targeting LIRB1 and LIRB2. Preclinical data suggest that NGM707 stimulates myeloid and lymphocyte activation by blocking LIRB1 and reprograms inhibitory myeloid cells to a stimulatory state by blocking LIRB2.371 The HLA-B57-Fc fusion protein iosH2 binds to LILRB1/2 and KIR3DL1 with high affinity and blocks the binding of HLA-G and ANGPTL to LILRB1/2, promoting the conversion of macrophage in the M2 phenotype to the M1 phenotype and thus enhancing phagocytosis of cancer cells in vitro; iosH2 also increases the cytotoxicity of T cells and NK cells in coculture with cancer cell lines.372
Targeting CD24 and clinical applications
CD24 emerges as a potential therapeutic target due to its important role in cancers. Antibodies targeting CD24 have been widely exploited for cancer treatment. Preclinical studies of CD24 antibody-mediated targeted therapy have been reviewed previously,373 and there has been no clinical study targeting CD24 to date.
Blocking or reducing the interaction between CD24 and Siglec-10 by reducing CD24 expression via monoclonal antibodies or gene editing potently enhances the phagocytosis of tumor cells with high CD24 expression by macrophages.8 Siglec-10 binds to CD24 in a sialic acid-dependent manner. Recent studies have demonstrated that increased tumor sialic acid loss decreases Siglec’s inhibitory effect.374 Selective removal of sialic acid from tumor cells using antibody-sialidase conjugates has been verified to significantly enhance tumor cell susceptibility to ADCC and enable immune cell killing of desialylated cancer cells. For example, in sepsis, treatment of CD24 with sialidase abolishes the interaction between Siglec-10 and CD24.375
Due to its high expression in cancers and its role as a biomarker of some CSCs and an antiphagocytic checkpoint, CD24 may be a promising target for cancer immunotherapy. Although its expression on immune cells leads to harmful adverse effects, investigators have also attempted to explore the efficacy of anti-CD24-based cancer therapy in preclinical models.
Targeting GD2 and clinical applications
Tumor-specific GD2 is the first ganglioside that is demonstrated to be an effective target antigen for cancer immunotherapy with monoclonal antibodies or CAR-T cells, and numerous clinical trials are underway (Table 2). Targeting GD2 with mAbs causes dephosphorylation of focal adhesion kinase (FAK) and inhibits activation of the PI3K/Akt pathways, thus inducing apoptosis and attenuating the migration of cancer cells.376 The murine antibodies m3F and 14.G2a were developed in the 1980s and demonstrated promising effects in vitro and in vivo in neuroblastoma, which provides a strong rationale for clinical trials.377
Later, the human-murine chimeric antibody ch14.18 was used as a variant of 14.G2a was subsequently renamed dinutuximab. It binds to GD2 and induces ADCC and CDC.378 In 2015, the U.S. The Food and Drug Administration (FDA) approved dinutuximab in combination with granulocyte-macrophage colony-stimulating factor, IL-2 and 13-cis retinoic acid (RA) for the treatment of high-grade neuroblastoma in pediatric patients. Dinutuximab is expressed by traditional Sp2/0 cells and contains a Gal-α3Gal glycosylation-modified epitope, which may induce allergy, while the improved version (Ch14.18, named dinutuximab β) expressed in CHO cells had a better glycosylation pattern compared to dinutuximab and avoided virus contamination from mice since it contains almost no Gal-α3Gal; dinutuximab β was approved by the European Medicines Agency (EMA) for high-risk neuroblastoma in 2017.123
Humanized mAbs have been developed since chimeric Abs are less immunogenic than murine mAbs. Hu3F8 (Naxitamab) was approved by the FDA for treating neuroblastoma in 2020.379 Hu14.18K322A was modified from 14G2a to improve its efficacy and is in phase II clinical trials in children with neuroblastoma.123
CD22 in clinical applications
CD22 undergoes constitutive endocytosis and is well suited for the efficient delivery of toxins into cells.380 At present, the drugs targeting CD22 mainly include monoclonal antibody drugs, antibody conjugates (ADC), and CAR-T cells.
Epratuzumab, derived from IgG2 monoclonal antibody (LL2, also called HPB-2), is a humanized IgG1 antibody against CD22 that contributes to BCR signaling by phosphorylating CD22 and induces ADCC.381 Epratuzumab has clinical activity and safety, with a 43% objective response rate in follicular NHL patients.147,381 SM03, another anti-CD22 recombinant IgG1 mAb, is currently being developed to treat rheumatoid arthritis in a phase III clinical trial.382
The FDA has approved two antibody-drug conjugates targeting CD22 to deliver cytotoxic agents to B-cell lymphoma/leukemia cells. Inotuzumab ozogamicin, a calicheamicin-conjugated a monoclonal antibody binding to CD22-expressing tumor cells, can be internalized and release cytotoxic calicheamicin inside the cell, leading to DNA damage and the following cell death.383 Inotuzumab ozogamicin was approved for the treatment of adults with refractory or relapsed leukemia. Moxetumomab pasudotox-tdfk, also called HA22 or CAT-8015, an anti-CD22 monoclonal antibody fused to Pseudomonas exotoxin (PE38), is another FDA-approved antibody-drug conjugate for targeting CD22.384,385 It is approved for application in patients with refractory or relapsed hairy cell leukemia (HCL). Other ADCs targeting CD22 are in clinical trials. DT2219, a bispecific ligand-directed toxin targeting both CD22 and CD19, is conjugated to the catalytic domain of diphtheria toxin and is used for treating refractory or relapsed B-lineage leukemia or lymphoma.386
CD22-targeted and bispecific CARs, such as CD19-CD22 and CD20-CD22 CARs, are the ongoing trials in the treatment of lymphoma and leukemia. Antigen loss is a common cause of resistance to CD19-targeted immunotherapy, but CD22 is also present in most B-ALL cases and is usually retained after CD19 loss.387 Therefore, CD22 is a promising candidate for antigen targeting by CAR T cells in patients with CD19 relapse.388 CD22 is mostly used as a supplement to CD19 or CD20 CAR T-cell therapy.
In summary, phagocytosis can be realized by either targeting phagocytosis checkpoints or interrupting the binding between ligands and receptors. Furthermore, macrophage activation is modulated by various compounds,389 and the compounds library has been built, the related pathway mediating macrophage activation has been elucidated.389
Conclusion and future perspectives
Dual goals of survival improvement and toxicity reduction will be achieved by the promising cancer immunotherapy. Immune checkpoint inhibitors, such as those targeting CTLA-4 and PD-1/PD-L1, have achieved unprecedented clinical applications and ushered in a new phase in the history of cancer treatment. A series of clinical trials have shown that targeting CTLA-4 or PD-1 leads to the proliferation of autoimmune lymphocytes, which increases the risk of adverse autoimmune reactions, such as pneumonia, colitis, hepatitis and vitiligo. Phagocytosis checkpoints have been increasingly recognized, and research on “don’t eat me” signals has also made rapid progress. New phagocytosis checkpoints have been discovered over time, and their evaluations in clinical trials are either in preparation or ongoing.
CD47-SIRPα, as the 1st phagocytosis checkpoint discovered, has already been in clinical trials. Targeting CD47 is less toxic than other approaches; it allows cancer cells to be engulfed by macrophages complelely, with little release of cellular contents after cell death. By enhancing the ADCP of targeted antibodies, disrupting the binding of CD47 to SIRPα has emerged as a promising immunotherapeutic strategy for advanced cancers. Anti-CD47 antibodies are theoretically able to target quiescent tumor stem cells with high expression of CD47.390 In addition to CD47, CD24, PD-L1, MHC-I, STC-1 and CD22 are also phagocytosis checkpoints discovered in recent years. Antibodies targeting these phagocytosis checkpoints are in preclinical or clinical trials. From the perspective of targeting phagocytosis, none of these drugs are on the market yet. Some drugs were developed based on other immune responses, but more research will be performed on phagocytosis checkpoint drugs.
Targeting phagocytosis also faces other potential challenges. The function of phagocytosis checkpoints mainly relies on innate responses that are less specific and may induce tissue damage to normal tissues in addition to tumors, especially when phagocytosis targeting is used combing with other immune-modulating methods, such as STING agonists or cytokine therapies. Regarding the CD47 checkpoint, given that CD47 is expressed highly in circulating blood cells, hematotoxicity has emerged as the most common side effect. To mitigate this toxicity, various methods have been developed during the process of antibody and inhibitor development. Presumably, targeting the phagocytosis checkpoint should complement T-cell responses, such as targeting PD-L1, to maximize antitumor responses. Patients who do not respond to anti-PD-L1 therapy may be sensitive to anti-CD47 treatment. More than half of the ongoing clinical trials are combinational therapies targeting CD47, as listed in Tables 3 and 4. In conclusion, targeting phagocytosis checkpoints has ushered in a new era of immunotherapy but also faces new challenges, and further investigation of the mechanisms underlying tumor-mediated immune evasion might overcome these challenges and promote the development of the first drug targeting phagocytosis checkpoints.
Data availability
The datasets of clinical trials in this study are available at the two below websites:
The clinical trials registered in the US: https://clinicaltrials.gov. The clinical trials registered in China: http://www.chinadrugtrials.org.cn/m_index.html
References
Kennedy, L. B. & Salama, A. K. S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 70, 86–104 (2020).
Mahoney, K. M., Rennert, P. D. & Freeman, G. J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Disco. 14, 561–584 (2015).
Webster, R. The immune checkpoint inhibitors: where are we now? Nat. Rev. Drug Disco. 13, 883–884 (2014).
Zhang, Q. et al. Immune and clinical features of CD96 expression in glioma by in silico analysis. Front Bioeng. Biotechnol. 8, 592 (2020).
Galipeau, J. & Sensebe, L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell 22, 824–833 (2018).
Liotta, F. et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells 26, 279–289 (2008).
Majeti, R. et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem. Cells Cell 138, 286–299 (2009).
Barkal, A. A. et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396 (2019).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Barkal, A. A. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 19, 76–84 (2018).
Lin, H. et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell 39, 480–493.e486 (2021).
Theruvath, J. et al. Anti-GD2 synergizes with CD47 blockade to mediate tumor eradication. Nat. Med. 28, 333–344 (2022).
Feng, M. Y. et al. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586 (2019).
Lian, S. et al. Simultaneous blocking of CD47 and PD-L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine 42, 281–295 (2019).
Hu, J. et al. Glioblastoma Immunotherapy Targeting the Innate Immune Checkpoint CD47-SIRPalpha Axis. Front Immunol. 11, 593219 (2020).
Li, Z. et al. The role of CD47-SIRPalpha immune checkpoint in tumor immune evasion and innate immunotherapy. Life Sci. 273, 119150 (2021).
Yu, W. B., Ye, Z. H., Chen, X., Shi, J. J. & Lu, J. J. The development of small-molecule inhibitors targeting CD47. Drug Disco. Today 26, 561–568 (2021).
Yang, Y., Yang, Z. & Yang, Y. Potential role of CD47-directed bispecific antibodies in cancer immunotherapy. Front. Immunol. 12, 686031 (2021).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Pittet, M. J., Michielin, O. & Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 19, 402–421 (2022).
Miller, Y. E., Daniels, G. L., Jones, C. & Palmer, D. K. Identification of a cell-surface antigen produced by a gene on human chromosome 3 (cen-q22) and not expressed by Rhnull cells. Am. J. Hum. Genet 41, 1061–1070 (1987).
Oldenborg, P. A. et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054 (2000).
Seiffert, M. et al. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood 94, 3633–3643 (1999).
Fenalti, G. et al. Structure of the human marker of self 5-transmembrane receptor CD47. Nat. Commun. 12, 5218 (2021).
Lindberg, F. P., Gresham, H. D., Schwarz, E. & Brown, E. J. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J. Cell Biol. 123, 485–496 (1993).
Reinhold, M. I. et al. In vivo expression of alternatively spliced forms of integrin-associated protein (CD47). J. Cell Sci. 108(Pt 11), 3419–3425 (1995).
Brooke, G., Holbrook, J. D., Brown, M. H. & Barclay, A. N. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 173, 2562–2570 (2004).
Hatherley, D., Harlos, K., Dunlop, D. C., Stuart, D. I. & Barclay, A. N. The structure of the macrophage signal regulatory protein alpha (SIRPalpha) inhibitory receptor reveals a binding face reminiscent of that used by T cell receptors. J. Biol. Chem. 282, 14567–14575 (2007).
Gao, A. G. & Frazier, W. A. Identification of a receptor candidate for the carboxyl-terminal cell binding domain of thrombospondins. J. Biol. Chem. 269, 29650–29657 (1994).
Isenberg, J. S. & Roberts, D. D. The role of CD47 in pathogenesis and treatment of renal ischemia reperfusion injury. Pediatr. Nephrol. 34, 2479–2494 (2019).
Resovi, A., Pinessi, D., Chiorino, G. & Taraboletti, G. Current understanding of the thrombospondin-1 interactome. Matrix Biol. 37, 83–91 (2014).
Ghimire, K. et al. CD47 Promotes age-associated deterioration in angiogenesis, blood flow and glucose homeostasis. Cells. 9, 1695 (2020).
Raugi, G. J., Olerud, J. E. & Gown, A. M. Thrombospondin in early human wound tissue. J. Invest Dermatol. 89, 551–554 (1987).
Kale, A., Rogers, N. M. & Ghimire, K. Thrombospondin-1 CD47 Signalling: From Mechanisms to Medicine. Int. J. Mol. Sci. 22, 4062 (2021).
Lopez-Dee, Z., Pidcock, K. & Gutierrez, L. S. Thrombospondin-1: multiple paths to inflammation. Mediators Inflamm. 2011, 296069 (2011).
Kaur, S. et al. Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci. Rep. 3, 1673 (2013).
Hayat, S. M. G. et al. CD47: role in the immune system and application to cancer therapy. Cell Oncol. (Dordr.) 43, 19–30 (2020).
Barclay, A. N. & van den Berg, T. K. The interaction between signal regulatory protein alpha (SIRP alpha) and CD47: structure, function, and therapeutic target. Annu Rev. Immunol. 32, 25–50 (2014).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016).
Zhang, H. et al. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl Acad. Sci. USA 112, E6215–E6223 (2015).
Virbasius, C. A., Virbasius, J. V. & Scarpulla, R. C. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 7, 2431–2445 (1993).
Lo, J. et al. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology 62, 534–545 (2015).
Zhang, X. et al. Blocking CD47 efficiently potentiated therapeutic effects of anti-angiogenic therapy in non-small cell lung cancer. J. Immunother. Cancer 7, 346 (2019).
Zhao, H. et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci. Rep. 6, 29719 (2016).
Ye, Z. H. et al. Regulation of CD47 expression by interferon-gamma in cancer cells. Transl. Oncol. 14, 101162 (2021).
Chen, J. et al. Macrophages induce CD47 upregulation via IL-6 and correlate with poor survival in hepatocellular carcinoma patients. Oncoimmunology 8, e1652540 (2019).
Liu, F. et al. SRSF10-mediated IL1RAP alternative splicing regulates cervical cancer oncogenesis via mIL1RAP-NF-kappaB-CD47 axis. Oncogene 37, 2394–2409 (2018).
Ye, Z. M. et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 10, 138 (2019).
Hatherley, D. et al. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol. Cell 31, 266–277 (2008).
Wu, Z. Q. et al. Identification of Glutaminyl Cyclase isoenzyme isoQC as a regulator of SIRP alpha-CD47 axis. Cell Res. 29, 502–505 (2019).
Logtenberg, M. E. W. et al. Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRP alpha axis and a target for cancer immunotherapy. Nat. Med. 25, 612 (2019).
Mair, B. et al. High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat. Biomed. Eng. 3, 796–805 (2019).
Sick, E. et al. Activation of CD47 receptors causes proliferation of human astrocytoma but not normal astrocytes via an Akt-dependent pathway. Glia 59, 308–319 (2011).
Hu, T. et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 10, 4056–4072 (2020).
Dong, H., Zhu, G., Tamada, K. & Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 5, 1365–1369 (1999).
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
Dong, H. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 8, 793–800 (2002).
Iwai, Y. et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002).
Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands in tolerance and immunity. Annu Rev. Immunol. 26, 677–704 (2008).
Boussiotis, V. A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 375, 1767–1778 (2016).
Garcia-Diaz, A. et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201 (2017).
Wang, X. et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 184, 7–14 (2017).
Quandt, D., Jasinski-Bergner, S., Muller, U., Schulze, B. & Seliger, B. Synergistic effects of IL-4 and TNFα on the induction of B7-H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J. Transl. Med. 12, 1–12 (2014).
Xu, L. J. et al. Inhibition of IL-6-JAK/Stat3 signaling in castration-resistant prostate cancer cells enhances the NK cell-mediated cytotoxicity via alteration of PD-L1/NKG2D ligand levels. Mol. Oncol. 12, 269–286 (2018).
Yi, M., Niu, M. K., Xu, L. P., Luo, S. X. & Wu, K. M. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 14, 10 (2021).
Yamaguchi, H., Hsu, J. M., Yang, W. H. & Hung, M. C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 19, 287–305 (2022).
Hewitt, E. W. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110, 163–169 (2003).
La Gruta, N. L., Gras, S., Daley, S. R., Thomas, P. G. & Rossjohn, J. Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 18, 467–478 (2018).
Yunis, E. J. 1987 Philip Levine award lecture. MHC haplotypes in biology and medicine. Am. J. Clin. Pathol. 89, 268–280 (1988).
Benacerraf, B. A hypothesis to relate the specificity of T lymphocytes and the activity of I region-specific Ir genes in macrophages and B lymphocytes. J. Immunol. 120, 1809–1812 (1978).
Complete sequence and gene map of a human major histocompatibility complex. The MHC sequencing consortium. Nature 401, 921–923 (1999) https://www.nature.com/articles/44853.
Bjorkman, P. J. et al. Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329, 506–512 (1987).
Bjorkman, P. J. et al. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature 329, 512–518 (1987).
Lentz, R. W., Colton, M. D., Mitra, S. S. & Messersmith, W. A. Innate immune checkpoint inhibitors: the next breakthrough in medical oncology? Mol. Cancer Ther. 20, 961–974 (2021).
Chang, C. C. et al. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 3, 237–243 (2002).
Banchereau, J. et al. Immunoglobulin-like transcript receptors on human dermal CD14+ dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming. Proc. Natl Acad. Sci. USA 109, 18885–18890 (2012).
Dersh, D. et al. Genome-wide Screens Identify Lineage- and Tumor-Specific Genes Modulating MHC-I- and MHC-II-Restricted Immunosurveillance of Human Lymphomas. Immunity 54, 116–131.e110 (2021).
Duong, E. et al. Type I interferon activates MHC class I-dressed CD11b(+) conventional dendritic cells to promote protective anti-tumor CD8(+) T cell immunity. Immunity 55, 308–323.e309 (2022).
Tetruashvily, M. M., Melson, J. W., Park, J. J., Peng, X. & Boulanger, L. M. Expression and alternative splicing of classical and nonclassical MHCI genes in the hippocampus and neuromuscular junction. Mol. Cell Neurosci. 72, 34–45 (2016).
Chapman, D. C. & Williams, D. B. ER quality control in the biogenesis of MHC class I molecules. Semin Cell Dev. Biol. 21, 512–519 (2010).
Apcher, S., Manoury, B. & Fahraeus, R. The role of mRNA translation in direct MHC class I antigen presentation. Curr. Opin. Immunol. 24, 71–76 (2012).
Sijts, A., Zaiss, D. & Kloetzel, P. M. The role of the ubiquitin-proteasome pathway in MHC class I antigen processing: implications for vaccine design. Curr. Mol. Med. 1, 665–676 (2001).
Donaldson, J. G. & Williams, D. B. Intracellular assembly and trafficking of MHC class I molecules. Traffic 10, 1745–1752 (2009).
Le Gall, S., Heard, J. M. & Schwartz, O. Analysis of Nef-induced MHC-I endocytosis. Res Virol. 148, 43–47 (1997).
Choma, M. K., Lumb, J., Kozik, P. & Robinson, M. S. A genome-wide screen for machinery involved in downregulation of MHC class I by HIV-1 Nef. PloS One 10, e0140404 (2015).
Cano, F. et al. The RNA-binding E3 ubiquitin ligase MEX-3C links ubiquitination with MHC-I mRNA degradation. Embo J. 31, 3596–3606 (2012).
Chen, G. Y., Tang, J., Zheng, P. & Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Sci. (N. Y., N. Y.) 323, 1722–1725 (2009).
Elghetany, M. T. & Patel, J. Assessment of CD24 expression on bone marrow neutrophilic granulocytes: CD24 is a marker for the myelocytic stage of development. Am. J. Hematol. 71, 348–349 (2002).
Stefanova, I., Horejsi, V., Ansotegui, I. J., Knapp, W. & Stockinger, H. GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 254, 1016–1019 (1991).
Kay, R., Takei, F. & Humphries, R. K. Expression cloning of a cDNA encoding M1/69-J11d heat-stable antigens. J. Immunol. 145, 1952–1959 (1990).
Fischer, G. F., Majdic, O., Gadd, S. & Knapp, W. Signal transduction in lymphocytic and myeloid cells via CD24, a new member of phosphoinositol-anchored membrane molecules. J. Immunol. 144, 638–641 (1990).
Freile, J. A. et al. CD24 is a potential immunotherapeutic target for mantle cell lymphoma. Biomedicines. 10, 1175 (2022).
O’Neill, L. A. J. When signaling pathways collide: Positive and negative regulation of Toll-like receptor signal transduction. Immunity 29, 12–20 (2008).
Sammar, M. et al. Expression of CD24 and Siglec-10 in first trimester placenta: implications for immune tolerance at the fetal-maternal interface. Histochem Cell Biol. 147, 565–574 (2017).
Bai, X. F. et al. The heat-stable antigen determines pathogenicity of self-reactive T cells in experimental autoimmune encephalomyelitis. J. Clin. Invest 105, 1227–1232 (2000).
Toubai, T. et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood 123, 3512–3523 (2014).
Thomas, S. et al. CD24 is an effector of HIF-1-driven primary tumor growth and metastasis. Cancer Res. 72, 5600–5612 (2012).
Overdevest, J. B. et al. CD24 expression is important in male urothelial tumorigenesis and metastasis in mice and is androgen regulated. P Natl Acad. Sci. USA 109, E3588–E3596 (2012).
Kwon, M. J. et al. CD24 overexpression is associated with poor prognosis in luminal A and triple-negative breast cancer. PloS One 10, e0139112 (2015).
Kaipparettu, B. A. et al. Estrogen-mediated downregulation of CD24 in breast cancer cells. Int J. Cancer 123, 66–72 (2008).
Cao, X., Geradts, J., Dewhirst, M. W. & Lo, H. W. Upregulation of VEGF-A and CD24 gene expression by the tGLI1 transcription factor contributes to the aggressive behavior of breast cancer cells. Oncogene 31, 104–115 (2012).
Vesuna, F., Lisok, A., Kimble, B. & Raman, V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 11, 1318–1328 (2009).
Naumov, I. et al. CD24 knockout prevents colorectal cancer in chemically induced colon carcinogenesis and in APC(Min)/CD24 double knockout transgenic mice. Int J. Cancer 135, 1048–1059 (2014).
Muppala, S. et al. CD24 induces expression of the oncomir miR-21 via Src, and CD24 and Src are both post-transcriptionally downregulated by the tumor suppressor miR-34a. PloS One 8, e59563 (2013).
Ghuwalewala, S. et al. MiRNA-146a/AKT/beta-Catenin activation regulates cancer stem cell phenotype in oral squamous cell carcinoma by targeting CD24. Front Oncol. 11, 651692 (2021).
Altevogt, P., Sammar, M., Huser, L. & Kristiansen, G. Novel insights into the function of CD24: A driving force in cancer. Int J. Cancer 148, 546–559 (2021).
Li, D. et al. CD24-p53 axis suppresses diethylnitrosamine-induced hepatocellular carcinogenesis by sustaining intrahepatic macrophages. Cell Disco. 4, 6 (2018).
Wang, L. et al. Intracellular CD24 disrupts the ARF-NPM interaction and enables mutational and viral oncogene-mediated p53 inactivation. Nat. Commun. 6, 5909 (2015).
Baumann, P. et al. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res 65, 10783–10793 (2005).
Taniuchi, K., Nishimori, I. & Hollingsworth, M. A. Intracellular CD24 inhibits cell invasion by posttranscriptional regulation of BART through interaction with G3BP. Cancer Res. 71, 895–905 (2011).
Wagner, G. F., Hampong, M., Park, C. M. & Copp, D. H. Purification, characterization, and bioassay of teleocalcin, a glycoprotein from salmon corpuscles of Stannius. Gen. Comp. Endocr. 63, 481–491 (1986).
Yeung, B. H. Y., Law, A. Y. S. & Wong, C. K. C. Evolution and roles of stanniocalcin. Mol. Cell Endocrinol. 349, 272–280 (2012).
Varghese, R., Wong, C. K. C., Deol, H., Wagner, G. F. & DiMattia, G. E. Comparative analysis of mammalian stanniocalcin genes. Endocrinology 139, 4714–4725 (1998).
Han, J., Jeon, M., Shin, I. & Kim, S. Elevated STC‑1 augments the invasiveness of triple‑negative breast cancer cells through activation of the JNK/c‑Jun signaling pathway. Oncol. Rep. 36, 1764–1771 (2016).
Chang, A. C., Jellinek, D. A. & Reddel, R. R. Mammalian stanniocalcins and cancer. Endocr. Relat. Cancer 10, 359–373 (2003).
Bishop, A., Cartwright, J. E. & Whitley, G. S. Stanniocalcin-1 in the female reproductive system and pregnancy. Hum. Reprod. Update 27, 1098–1114 (2021).
dos Santos, M. T. et al. Human stanniocalcin-1 interacts with nuclear and cytoplasmic proteins and acts as a SUMO E3 ligase. Mol. Biosyst. 7, 180–193 (2011).
Liu, Z. et al. STC-1 ameliorates renal injury in diabetic nephropathy by inhibiting the expression of BNIP3 through the AMPK/SIRT3 pathway. Lab Invest 99, 684–697 (2019).
Dalvin, L. A. et al. Stanniocalcin-1 is a modifier of oxygen-induced retinopathy severity. Curr. Eye Res. 45, 46–51 (2020).
Schulz, G. et al. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. 44, 5914–5920 (1984).
Yu, A. L. et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 363, 1324–1334 (2010).
Ladenstein, R. et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 19, 1617–1629 (2018).
Nazha, B., Inal, C. & Owonikoko, T. K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. 10, 1000 (2020).
Horwacik, I. et al. Structural basis of GD2 Ganglioside and mimetic peptide recognition by 14G2a antibody. Mol. Cell Proteom. 14, 2577–2590 (2015).
Yuki, N., Yamada, M., Tagaawa, Y. & Takahashi, H. Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J. Neurol. Sci. 149, 127–130 (1997).
Ohmi, Y. et al. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem Res 37, 1185–1191 (2012).
Lopez, P. H. & Schnaar, R. L. Gangliosides in cell recognition and membrane protein regulation. Curr. Opin. Struct. Biol. 19, 549–557 (2009).
Sheikh, K. A. et al. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl Acad. Sci. USA 96, 7532–7537 (1999).
Chiavegatto, S., Sun, J., Nelson, R. J. & Schnaar, R. L. A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp. Neurol. 166, 227–234 (2000).
Muller, J. & Nitschke, L. The role of CD22 and Siglec-G in B-cell tolerance and autoimmune disease. Nat. Rev. Rheumatol. 10, 422–428 (2014).
Tedder, T. F., Tuscano, J., Sato, S. & Kehrl, J. H. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu Rev. Immunol. 15, 481–504 (1997).
Pluvinage, J. V. et al. CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192 (2019).
Nitschke, L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr. Opin. Immunol. 17, 290–297 (2005).
Tuscano, J., Engel, P., Tedder, T. F. & Kehrl, J. H. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22L interactions impairs T-cell proliferation. Blood 87, 4723–4730 (1996).
Santos, L. et al. Dendritic cell-dependent inhibition of B cell proliferation requires CD22. J. Immunol. 180, 4561–4569 (2008).
Sindhava, V. J. et al. Bone marrow dendritic cell-mediated regulation of TLR and B cell receptor signaling in B cells. J. Immunol. 189, 3355–3367 (2012).
Kawasaki, N., Rademacher, C. & Paulson, J. C. CD22 regulates adaptive and innate immune responses of B cells. J. Innate Immun. 3, 411–419 (2011).
Matsubara, N. et al. CD22-binding synthetic sialosides regulate B lymphocyte proliferation through CD22 ligand-dependent and independent pathways, and enhance antibody production in mice. Front. Immunol. 9, 820 (2018).
Ereno-Orbea, J. et al. Molecular basis of human CD22 function and therapeutic targeting. Nat. Commun. 8, 764 (2017).
Schulte, R. J., Campbell, M. A., Fischer, W. H. & Sefton, B. M. Tyrosine phosphorylation of Cd22 during B-cell activation. Science 258, 1001–1004 (1992).
Doody, G. M. et al. A role in B-cell activation for Cd22 and the protein-tyrosine-phosphatase SHP. Science 269, 242–244 (1995).
Nitschke, L., Carsetti, R., Ocker, B., Kohler, G. & Lamers, M. C. CD22 is a negative regulator of B-cell receptor signalling. Curr. Biol. 7, 133–143 (1997).
Muller, J. et al. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. P Natl Acad. Sci. USA 110, 12402–12407 (2013).
O’Keefe, T. L., Williams, G. T., Davies, S. L. & Neuberger, M. S. Hyperresponsive B cells in CD22-deficient mice. Science 274, 798–801 (1996).
Ghosh, S., Bandulet, C. & Nitschke, L. Regulation of B cell development and B cell signalling by CD22 and its ligands alpha 2,6-linked sialic acids. Int Immunol. 18, 603–611 (2006).
Ballet, R. et al. A CD22-Shp1 phosphatase axis controls integrin beta7 display and B cell function in mucosal immunity. Nat. Immunol. 22, 381–390 (2021).
Leonard, J. P. et al. Epratuzumab, a humanized anti-CD22 antibody, in aggressive non-Hodgkin’s lymphoma: Phase I/II clinical trial results. Clin. Cancer Res. 10, 5327–5334 (2004).
Polson, A. G. et al. Anti-CD22-MCC-DM1: an antibody-drug conjugate with a stable linker for the treatment of non-Hodgkin’s lymphoma. Leukemia 24, 1566–1573 (2010).
Li, D. et al. DCDT2980S, an anti-CD22-monomethyl auristatin E antibody-drug conjugate, is a potential treatment for non-Hodgkin lymphoma. Mol. Cancer Ther. 12, 1255–1265 (2013).
Uckun, F. M., Goodman, P., Ma, H., Dibirdik, I. & Qazi, S. CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. P Natl Acad. Sci. USA 107, 16852–16857 (2010).
Ma, D. Y., Suthar, M. S., Kasahara, S., Gale, M. & Clark, E. A. CD22 is required for protection against west nile virus infection. J. Virol. 87, 3361–3375 (2013).
El-Sayed, Z. A., Ragab, S. M., Khalifa, K. A. & El Ashmawy, R. A. Altered CD19/CD22 balance in Egyptian children and adolescents with systemic lupus erythematosus. Egypt J. Immunol. 16, 27–38 (2009).
Suzuki, J. et al. CD19/22 balance relates to improvement of disease activity in systemic lupus erythematosus. Mod. Rheumatol. 16, 235–238 (2006).
Ben Mkaddem, S., Benhamou, M. & Monteiro, R. C. Understanding Fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front. Immunol. 10, 811 (2019).
Bruhns, P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119, 5640–5649 (2012).
Bournazos, S., Gupta, A. & Ravetch, J. V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 20, 633–643 (2020).
Bruhns, P. et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725 (2009).
Nimmerjahn, F. & Ravetch, J. V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Hogarth, P. M. & Pietersz, G. A. Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat. Rev. Drug Disco. 11, 311–331 (2012).
Knorr, D. A. & Ravetch, J. V. Immunotherapy and hyperprogression: unwanted outcomes, unclear mechanism. Clin. Cancer Res. 25, 904–906 (2019).
Tong, B. & Wang, M. Z. CD47 is a novel potent immunotherapy target in human malignancies: current studies and future promises. Future Oncol. 14, 2179–2188 (2018).
Cannons, J. L., Tangye, S. G. & Schwartzberg, P. L. SLAM family receptors and SAP adaptors in immunity. Annu Rev. Immunol. 29, 665–705 (2011).
Li, D. et al. SLAMF3 and SLAMF4 are immune checkpoints that constrain macrophage phagocytosis of hematopoietic tumors. Sci. Immunol. 7, eabj5501 (2022).
Fouquet, G. et al. Rescuing SLAMF3 expression restores sorafenib response in hepatocellular carcinoma cells through the induction of mesenchymal-to-epithelial transition. Cancers (Basel). 14, 910 (2022).
Li, K. & Underhill, D. M. C-type lectin receptors in phagocytosis. Curr. Top. Microbiol. Immunol. 429, 1–18 (2020).
Lopez Robles, M. D. et al. Cell-surface C-type lectin-like receptor CLEC-1 dampens dendritic cell activation and downstream Th17 responses. Blood Adv. 1, 557–568 (2017).
Gauttier, V. et al. Abstract 3259: CLEC-1 is a novel myeloid immune checkpoint for cancer immunotherapy controlling damaged and tumor cells phagocytosis. Cancer Res. 80, 3259–3259 (2020).
Thebault, P. et al. The C-type lectin-like receptor CLEC-1, expressed by myeloid cells and endothelial cells, is up-regulated by immunoregulatory mediators and moderates T cell activation. J. Immunol. 183, 3099–3108 (2009).
Vernon, P. J. & Tang, D. Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox Signal 18, 677–691 (2013).
Cockram, T. O. J., Dundee, J. M., Popescu, A. S. & Brown, G. C. The phagocytic code regulating phagocytosis of mammalian cells. Front Immunol. 12, 629979 (2021).
Krause, K. H. & Michalak, M. Calreticulin. Cell 88, 439–443 (1997).
Chao, M. P. et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci. Transl. Med 2, 63ra94 (2010).
Tai, Y. T. et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 112, 1329–1337 (2008).
Malaer, J. D. & Mathew, P. A. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am. J. Cancer Res. 7, 1637–1641 (2017).
Chen, J. et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 544, 493–497 (2017).
Aderem, A. & Underhill, D. M. Mechanisms of phagocytosis in macrophages. Annu Rev. Immunol. 17, 593–623 (1999).
He, Y. et al. Cancer cell-expressed SLAMF7 is not required for CD47-mediated phagocytosis. Nat. Commun. 10, 533 (2019).
Fujioka, Y. et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol. Cell Biol. 16, 6887–6899 (1996).
Kharitonenkov, A. et al. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386, 181–186 (1997).
Tsai, R. K. & Discher, D. E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 180, 989–1003 (2008).
Timms, J. F. et al. SHPS-1 is a scaffold for assembling distinct adhesion-regulated multi-protein complexes in macrophages. Curr. Biol. 9, 927–930 (1999).
Han, X. et al. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 275, 37984–37992 (2000).
Liu, Y. et al. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J. Biol. Chem. 277, 10028–10036 (2002).
Soto-Pantoja, D. R., Kaur, S. & Roberts, D. D. CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit. Rev. Biochem Mol. Biol. 50, 212–230 (2015).
Chhabra, A. et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci. Transl. Med. 8, 351ra105 (2016).
Briscoe, J. & Therond, P. P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429 (2013).
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of Pd-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell-death. Embo J. 11, 3887–3895 (1992).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest 128, 4654–4668 (2018).
Pathria, P., Louis, T. L. & Varner, J. A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 40, 310–327 (2019).
Izquierdo, E. et al. Extracellular vesicles and PD-L1 suppress macrophages, inducing therapy resistance in TP53-deficient B-cell malignancies. Blood 139, 3617–3629 (2022).
Diskin, B. et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 21, 442–454 (2020).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018).
Cosman, D. et al. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7, 273–282 (1997).
van der Touw, W., Chen, H. M., Pan, P. Y. & Chen, S. H. LILRB receptor-mediated regulation of myeloid cell maturation and function. Cancer Immunol. Immunother. 66, 1079–1087 (2017).
Bruhns, P. et al. Molecular basis of the recruitment of the SH2 domain-containing inositol 5-phosphatases SHIP1 and SHIP2 by fcgamma RIIB. J. Biol. Chem. 275, 37357–37364 (2000).
Daeron, M., Jaeger, S., Du Pasquier, L. & Vivier, E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol. Rev. 224, 11–43 (2008).
Dhatchinamoorthy, K., Colbert, J. D. & Rock, K. L. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 12, 636568 (2021).
Pillai, S., Netravali, I. A., Cariappa, A. & Mattoo, H. Siglecs and immune regulation. Annu Rev. Immunol. 30, 357–392 (2012).
Crocker, P. R., Paulson, J. C. & Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 7, 255–266 (2007).
Zhang, J., Somani, A. K. & Siminovitch, K. A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 12, 361–378 (2000).
Freile, J. & Ustyanovska Avtenyuk, N. CD24 is a potential immunotherapeutic target for mantle cell lymphoma. Biomedicines 10, 1175 (2022).
Bandala-Sanchez, E. et al. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nat. Immunol. 14, 741–748 (2013).
Chen, G. Y., Brown, N. K., Zheng, P. & Liu, Y. Siglec-G/10 in self-nonself discrimination of innate and adaptive immunity. Glycobiology 24, 800–806 (2014).
Zhang, P. et al. Siglec-10 is associated with survival and natural killer cell dysfunction in hepatocellular carcinoma. J. Surg. Res 194, 107–113 (2015).
Abram, C. L. & Lowell, C. A. Shp1 function in myeloid cells. J. Leukoc. Biol. 102, 657–675 (2017).
He, L. F. et al. Stanniocalcin-1 promotes tumor angiogenesis through up-regulation of VEGF in gastric cancer cells. J. Biomed. Sci. 18, 39 (2011).
Liu, G. et al. Stanniocalcin 1 and ovarian tumorigenesis. J. Natl Cancer Inst. 102, 812–827 (2010).
Abe, K. et al. Targeting stanniocalcin-1-expressing tumor cells elicits efficient antitumor effects in a mouse model of human lung cancer. Cancer Med. 10, 3085–3100 (2021).
Eladl, E. et al. Role of CD47 in hematological malignancies. J. Hematol. Oncol. 13, 96 (2020).
Huang, C. Y., Ye, Z. H., Huang, M. Y. & Lu, J. J. Regulation of CD47 expression in cancer cells. Transl. Oncol. 13, 100862 (2020).
de Vries, H. E. et al. Signal-regulatory protein alpha-CD47 interactions are required for the transmigration of monocytes across cerebral endothelium. J. Immunol. 168, 5832–5839 (2002).
Nath, P. R. et al. CD47 expression in natural killer cells regulates homeostasis and modulates immune response to lymphocytic choriomeningitis virus. Front Immunol. 9, 2985 (2018).
Latour, S. et al. Bidirectional negative regulation of human T and dendritic cells by CD47 and its cognate receptor signal-regulator protein-alpha: down-regulation of IL-12 responsiveness and inhibition of dendritic cell activation. J. Immunol. 167, 2547–2554 (2001).
Liu, X. et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 21, 1209–1215 (2015).
Van, V. Q. et al. Expression of the self-marker CD47 on dendritic cells governs their trafficking to secondary lymphoid organs. Embo J. 25, 5560–5568 (2006).
Zen, K. & Parkos, C. A. Leukocyte-epithelial interactions. Curr. Opin. Cell Biol. 15, 557–564 (2003).
Boirivant, M. et al. Stimulated human lamina propria T cells manifest enhanced Fas-mediated apoptosis. J. Clin. Invest 98, 2616–2622 (1996).
Manna, P. P. & Frazier, W. A. The mechanism of CD47-dependent killing of T cells: heterotrimeric Gi-dependent inhibition of protein kinase A. J. Immunol. 170, 3544–3553 (2003).
Pettersen, R. D. CD47 and death signaling in the immune system. Apoptosis 5, 299–306 (2000).
Miller, T. W., Kaur, S., Ivins-O’Keefe, K. & Roberts, D. D. Thrombospondin-1 is a CD47-dependent endogenous inhibitor of hydrogen sulfide signaling in T cell activation. Matrix Biol. 32, 316–324 (2013).
Kaur, S. et al. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J. Biol. Chem. 286, 14991–15002 (2011).
Van, V. Q. et al. Cutting edge: CD47 controls the in vivo proliferation and homeostasis of peripheral CD4+ CD25+ Foxp3+ regulatory T cells that express CD103. J. Immunol. 181, 5204–5208 (2008).
Gallagher, S. et al. CD47 limits antibody dependent phagocytosis against non-malignant B cells. Mol. Immunol. 85, 57–65 (2017).
Park, G. B. et al. Ligation of CD47 Induces G1 Arrest in EBV-transformed B Cells Through ROS Generation, p38 MAPK/JNK Activation, and Tap73 Upregulation. J. Immunol 37, 309–320 (2014).
Lucas, E. D. et al. PD-L1 reverse signaling in dermal dendritic cells promotes dendritic cell migration required for skin immunity. Cell Rep. 33, 108258 (2020).
Peng, Q. et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 11, 4835 (2020).
Hartley, G. P., Chow, L., Ammons, D. T., Wheat, W. H. & Dow, S. W. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol. Res. 6, 1260–1273 (2018).
Cai, H., Zhang, Y. C., Wang, J. & Gu, J. Y. Defects in macrophage reprogramming in cancer therapy: the negative impact of PD-L1/PD-1. Front. Immunol. 12, 690869 (2021).
Zhang, Y. H. et al. The role of the PD-1/PD-L1 axis in macrophage differentiation and function during pregnancy. Hum. Reprod. 34, 25–36 (2019).
Wei, Y. et al. PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp. Cell Res. 402, 112575 (2021).
Chen, W., Wang, J., Jia, L., Liu, J. & Tian, Y. Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis 7, e2115 (2016).
Huang, X. et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl Acad. Sci. USA 106, 6303–6308 (2009).
Shen, L. et al. PD-1/PD-L pathway inhibits M.tb-specific CD4(+) T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci. Rep. 6, 38362 (2016).
Qorraj, M. et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 31, 470–478 (2017).
Jiang, L. R. et al. PD-1-positive tumor-associated macrophages define poor clinical outcomes in patients with muscle invasive bladder cancer through potential CD68/PD-1 complex interactions. Front Oncol. 11, 679928 (2021).
Nishimura, H., Nose, M., Hiai, H., Minato, N. & Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999).
Pulko, V. et al. B7-h1 expressed by activated CD8 T cells is essential for their survival. J. Immunol. 187, 5606–5614 (2011).
Liu, X. et al. B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8(+) T cells. Sci. Rep. 6, 36722 (2016).
Blees, A. et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017).
Satthaporn, S. & Eremin, O. Dendritic cells (I): Biological functions. J. R. Coll. Surg. Edinb. 46, 9–19 (2001).
Arosa, F. A., Esgalhado, A. J., Reste-Ferreira, D. & Cardoso, E. M. Open MHC Class I Conformers: A Look through the Looking Glass. Int. J. Mol. Sci 22, 9738 (2021).
Li, O. et al. Massive and destructive T cell response to homeostatic cue in CD24-deficient lymphopenic hosts. J. Exp. Med. 203, 1713–1720 (2006).
Bai, X. F. et al. CD24 controls expansion and persistence of autoreactive T cells in the central nervous system during experimental autoimmune encephalomyelitis. J. Exp. Med. 200, 447–458 (2004).
Hunte, B. E., Capone, M., Zlotnik, A., Rennick, D. & Moore, T. A. Acquisition of CD24 expression by Lin-CD43+B220(low)ckit(hi) cells coincides with commitment to the B cell lineage. Eur. J. Immunol. 28, 3850–3856 (1998).
Lu, L., Chappel, M. S., Humphries, R. K. & Osmond, D. G. Regulation of cell survival during B lymphopoiesis: increased pre-B cell apoptosis in CD24-transgenic mouse bone marrow. Eur. J. Immunol. 30, 2686–2691 (2000).
Liu, Y. et al. Heat-stable antigen is a costimulatory molecule for CD4 T cell growth. J. Exp. Med. 175, 437–445 (1992).
Hubbe, M. & Altevogt, P. Heat-stable antigen/CD24 on mouse T lymphocytes: evidence for a costimulatory function. Eur. J. Immunol. 24, 731–737 (1994).
Wu, Y., Zhou, Q., Zheng, P. & Liu, Y. CD28-independent induction of T helper cells and immunoglobulin class switches requires costimulation by the heat-stable antigen. J. Exp. Med. 187, 1151–1156 (1998).
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72, 7–33 (2022).
Xie, S.-H., Huang, R.-Q., Liu, Y.-L., Cao, S.-M. & Qian, C.-N. An increase in early cancer detection rates at a single cancer center: Experiences from Sun Yat-sen University Cancer Center. Vis. Cancer Med. 3, 1 (2022).
Wu, C. et al. NNMT-DNMT1 axis is essential for maintaining cancer cell sensitivity to oxidative phosphorylation inhibition. Adv. Sci. (Weinh). 10, e2202642 (2022).
Murata, Y., Saito, Y., Kotani, T. & Matozaki, T. CD47-signal regulatory protein alpha signaling system and its application to cancer immunotherapy. Cancer Sci. 109, 2349–2357 (2018).
Nigro, A. et al. Enhanced expression of CD47 is associated with off-target resistance to tyrosine kinase inhibitor gefitinib in NSCLC. Front. Immunol. 10, 3135 (2020).
Li, F. et al. Blocking the CD47-SIRP alpha axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 7, e1391973 (2017).
Daubon, T. et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat. Commun. 10, 1146 (2019).
Lv, C. et al. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 8, 1036 (2017).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Disco. 17, 887–904 (2018).
Mantovani, A. & Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 212, 435–445 (2015).
Lecoultre, M., Dutoit, V. & Walker, P. R. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J. Immunother. Cancer 8, e001408 (2020).
Gholamin, S. et al. Disrupting the CD47-SIRP alpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 9, eaaf2968 (2017).
Wang, Y. et al. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 21, 1919–1929 (2013).
Ring, N. G. et al. Anti-SIRPalpha antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl Acad. Sci. USA 114, E10578–E10585 (2017).
Iribarren, K. et al. Anticancer effects of anti-CD47 immunotherapy in vivo. Oncoimmunology 8, 1550619 (2019).
Abe, T. et al. Signal regulatory protein alpha blockade potentiates tumoricidal effects of macrophages on gastroenterological neoplastic cells in syngeneic immunocompetent mice. Ann. Gastroenterol. Surg. 2, 451–462 (2018).
Wang, S. et al. Blocking CD47 promotes antitumour immunity through CD103(+) dendritic cell-NK cell axis in murine hepatocellular carcinoma model. J. Hepatol. 77, 467–478 (2022).
Yu, L. et al. Significance of CD47 and its association with tumor immune microenvironment heterogeneity in ovarian cancer. Front Immunol. 12, 768115 (2021).
Labrousse-Arias, D. et al. HIF-2alpha-mediated induction of pulmonary thrombospondin-1 contributes to hypoxia-driven vascular remodelling and vasoconstriction. Cardiovasc Res. 109, 115–130 (2016).
Novelli, E. M. et al. Vascular TSP1-CD47 signaling promotes sickle cell-associated arterial vasculopathy and pulmonary hypertension in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 316, L1150–L1164 (2019).
El-Rashid, M., Ghimire, K., Sanganeria, B., Lu, B. & Rogers, N. M. CD47 limits autophagy to promote acute kidney injury. FASEB J. 33, 12735–12749 (2019).
Maxhimer, J. B., Shih, H. B., Isenberg, J. S., Miller, T. W. & Roberts, D. D. Thrombospondin-1/CD47 blockade following ischemia-reperfusion injury is tissue protective. Plast. Reconstr. Surg. 124, 1880–1889 (2009).
Isenberg, J. S. et al. Thrombospondin-1 limits ischemic tissue survival by inhibiting nitric oxide-mediated vascular smooth muscle relaxation. Blood 109, 1945–1952 (2007).
Cham, L. B., Adomati, T., Li, F. H., Ali, M. & Lang, K. S. CD47 as a potential target to therapy for infectious diseases. Antibodies. 9, 44 (2020).
Ayi, K. et al. CD47-SIRPalpha interactions regulate macrophage uptake of plasmodium falciparum-infected erythrocytes and clearance of malaria in vivo. Infect. Immun. 84, 2002–2011 (2016).
Patel, S. P. & Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 14, 847–856 (2015).
Cai, J. J. et al. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J. Cancer Res. Clin. 145, 1377–1385 (2019).
Karyampudi, L. et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-kappa B. Cancer Res. 76, 239–250 (2016).
Johnson, R. M. G., Wen, T. & Dong, H. Bidirectional signals of PD-L1 in T cells that fraternize with cancer cells. Nat. Immunol. 21, 365–366 (2020).
Tang, H. et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest 128, 580–588 (2018).
Moret, F. M., van der Wurff-Jacobs, K. M., Bijlsma, J. W., Lafeber, F. P. & van Roon, J. A. Synovial T cell hyporesponsiveness to myeloid dendritic cells is reversed by preventing PD-1/PD-L1 interactions. Arthritis Res. Ther. 16, 497 (2014).
Wan, B. et al. Aberrant regulation of synovial T cell activation by soluble costimulatory molecules in rheumatoid arthritis. J. Immunol. 177, 8844–8850 (2006).
Ritprajak, P., Hashiguchi, M., Tsushima, F., Chalermsarp, N. & Azuma, M. Keratinocyte-associated B7-H1 directly regulates cutaneous effector CD8+ T cell responses. J. Immunol. 184, 4918–4925 (2010).
Michaels, A. D. et al. CD47 blockade as an adjuvant immunotherapy for resectable pancreatic cancer. Clin. Cancer Res 24, 1415–1425 (2018).
Sun, Y., Tan, J., Miao, Y. & Zhang, Q. The role of PD-L1 in the immune dysfunction that mediates hypoxia-induced multiple organ injury. Cell Commun. Signal 19, 76 (2021).
Cornel, A. M., Mimpen, I. L. & Nierkens, S. MHC Class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers (Basel) 12, 1760 (2020).
Anfossi, N. et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity 25, 331–342 (2006).
Loustau, M. et al. HLA-G neo-expression on tumors. Front Immunol. 11, 1685 (2020).
Naji, A., Menier, C., Maki, G., Carosella, E. D. & Rouas-Freiss, N. Neoplastic B-cell growth is impaired by HLA-G/ILT2 interaction. Leukemia 26, 1889–1892 (2012).
Koutsakos, M. et al. Downregulation of MHC class I expression by influenza A and B viruses. Front Immunol. 10, 1158 (2019).
Benichou, G., Kant, C. D., Madsen, J. & Tocco, G. Modulation of alloreactivity to MHC-derived peptides and transplantation tolerance. Front Biosci. 12, 4239–4247 (2007).
Chen, B. et al. Role of HLA-B27 in the pathogenesis of ankylosing spondylitis (Review). Mol. Med. Rep. 15, 1943–1951 (2017).
Lorefice, L. et al. Multiple sclerosis and HLA genotypes: A possible influence on brain atrophy. Mult. Scler. (Houndmills, Basingstoke, Engl.) 25, 23–30 (2019).
Oryoji, D. et al. Associations of HLA class I alleles in Japanese patients with Crohn’s disease. Genes Immun. 16, 54–56 (2015).
Mitsunaga, S. et al. Associations between six classical HLA loci and rheumatoid arthritis: a comprehensive analysis. Tissue Antigens 80, 16–25 (2012).
Kristiansen, G., Sammar, M. & Altevogt, P. Tumour biological aspects of CD24, a mucin-like adhesion molecule. J. Mol. Histol. 35, 255–262 (2004).
Zhou, Q. et al. CD24 is a genetic modifier for risk and progression of multiple sclerosis. Proc. Natl Acad. Sci. USA 100, 15041–15046 (2003).
Sanchez, E. et al. Investigating the role of CD24 gene polymorphisms in rheumatoid arthritis. Ann. Rheum. Dis. 67, 1197–1198 (2008).
Wang, X. et al. CD24-Siglec axis is an innate immune checkpoint against metaflammation and metabolic disorder. Cell Metab. 34, 1088–1103.e1086 (2022).
Pena, C. et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 73, 1287–1297 (2013).
Sun, J. L. et al. STC1 is a novel biomarker associated with immune characteristics and prognosis of bladder cancer. INT J. Gen. Med 14, 5505–5516 (2021).
Xiong, Y. & Wang, Q. STC1 regulates glioblastoma migration and invasion via the TGFbeta/SMAD4 signaling pathway. Mol. Med Rep. 20, 3055–3064 (2019).
Abaza, H. M. H., Elmougy, M. I., El Maraghy, H. M. A. & Mahmoud, H. M. Stanniocalcin1 gene expression in patients with acute leukemia: impact on response to therapy and disease outcome. Int J. Lab. Hematol. 38, 81–89 (2016).
Duan, Y. R. et al. Serum autoantibodies against LRDD, STC1, and FOXA1 as biomarkers in the detection of ovarian cancer. Dis. Markers. 2022, 6657820 (2022).
Fang, Z., Tian, Z. Q., Luo, K. L., Song, H. Z. & Yi, J. Clinical significance of stanniocalcin expression in tissue and serum of gastric cancer patients. Chin. J. Cancer Res 26, 602–610 (2014).
Arigami, T. et al. Expression of stanniocalcin 1 as a potential biomarker of gastric cancer. Oncol.-Basel 83, 158–164 (2012).
Zhang, C. F., Wang, B. S., Wang, X. Q., Sheng, X. G. & Cui, Y. C. Sevoflurane inhibits the progression of ovarian cancer through down-regulating stanniocalcin 1 (STC1). Cancer Cell Int. 19, 339 (2019).
Wang, Y. et al. Stanniocalcin1 promotes cell proliferation, chemoresistance and metastasis in hypoxic gastric cancer cells via Bcl2. Oncol. Rep. 41, 1998–2008 (2019).
Lin, F. et al. Stanniocalcin 1 promotes metastasis, lipid metabolism and cisplatin chemoresistance via the FOXC2/ITGB6 signaling axis in ovarian cancer. J. Exp. Clin. Cancer Res. 41, 129 (2022).
Guo, F. et al. Stanniocalcin1 (STC1) inhibits cell proliferation and invasion of cervical cancer cells. PLoS One 8, e53989 (2013).
Yeung, H. Y. et al. Hypoxia-inducible factor-1-mediated activation of stanniocalcin-1 in human cancer cells. Endocrinology 146, 4951–4960 (2005).
Law, A. Y., Ching, L. Y., Lai, K. P. & Wong, C. K. Identification and characterization of the hypoxia-responsive element in human stanniocalcin-1 gene. Mol. Cell Endocrinol. 314, 118–127 (2010).
Zhang, Y., Shan, P. Y., Srivastava, A., Li, Z. Y. & Lee, P. J. Endothelial stanniocalcin 1 maintains mitochondrial bioenergetics and prevents oxidant-induced lung injury via toll-like receptor 4. Antioxid. Redox Signal 30, 1775–1796 (2019).
Wang, P., Li, X. L. & Cao, Z. H. STC1 ameliorates cognitive impairment and neuroinflammation of Alzheimeras disease mice via inhibition of ERK1/2 pathway. Immunobiology 226, 152092 (2021).
Xu, J. Y. et al. Epithelial expression and role of secreted STC1 on asthma airway hyperresponsiveness through calcium channel modulation. Allergy 76, 2475–2487 (2021).
Shahim, P. et al. Cerebrospinal fluid stanniocalcin-1 as a biomarker for Alzheimer’s disease and other neurodegenerative disorders. Neuromol Med 19, 154–160 (2017).
Shen, M., Pan, H., Ke, J. & Zhao, F. NF-kappaB-upregulated miR-155-5p promotes hepatocyte mitochondrial dysfunction to accelerate the development of nonalcoholic fatty liver disease through downregulation of STC1. J. Biochem. Mol. Toxicol. 36, e23025 (2022).
Majzner, R. G. et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 603, 934–941 (2022).
Foon, K. A. et al. Clinical and immune responses in advanced melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. J. Clin. Oncol. 18, 376–384 (2000).
Roth, M. et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer-Am. Cancer Soc. 120, 548–554 (2014).
Heiner, J. P. et al. Localization of Gd2-specific monoclonal-antibody 3f8 in human osteosarcoma. Cancer Res 47, 5377–5381 (1987).
Dobrenkov, K., Ostrovnaya, I., Gu, J., Cheung, I. Y. & Cheung, N. K. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr. Blood Cancer 63, 1780–1785 (2016).
Chan, G. C. & Chan, C. M. Anti-GD2 Directed Immunotherapy for High-Risk and Metastatic Neuroblastoma. Biomolecules 12, 358 (2022).
Wu, Z. L., Schwartz, E., Seeger, R. & Ladisch, S. Expression of Gd2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 46, 440–443 (1986).
Yoshida, S. et al. Ganglioside G(D2) in small cell lung cancer cell lines: enhancement of cell proliferation and mediation of apoptosis. Cancer Res. 61, 4244–4252 (2001).
Cheresh, D. A., Rosenberg, J., Mujoo, K., Hirschowitz, L. & Reisfeld, R. A. Biosynthesis and expression of the disialoganglioside Gd2, a relevant target antigen on small-cell lung-carcinoma for monoclonal antibody-mediated cytolysis. Cancer Res. 46, 5112–5118 (1986).
Cheresh, D. A., Harper, J. R., Schulz, G. & Reisfeld, R. A. Localization of the gangliosides Gd2 and Gd3 in adhesion plaques and on the surface of human-melanoma. Cells P Natl Acad. Sci.-Biol. 81, 5767–5771 (1984).
Cheresh, D. A., Pierschbacher, M. D., Herzig, M. A. & Mujoo, K. Disialogangliosides-Gd2 and Gd3 Are Involved in the Attachment of Human-Melanoma and Neuroblastoma-Cells to Extracellular-Matrix Proteins. J. Cell Biol. 102, 688–696 (1986).
Yvon, E. et al. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin. Cancer Res. 15, 5852–5860 (2009).
Wu, G. S., Xie, X., Lu, Z. H. & Ledeen, R. W. Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. P Natl Acad. Sci. USA 98, 307–312 (2001).
Sha, S. et al. Deficits in cognitive function and hippocampal plasticity in GM2/ GD2 synthase knockout mice. Hippocampus 24, 369–382 (2014).
Sugiura, Y. et al. Sensory nerve-dominant nerve degeneration and remodeling in the mutant mice lacking complex gangliosides. Neuroscience 135, 1167–1178 (2005).
Boukhris, A. et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet 93, 118–123 (2013).
Harlalka, G. V. et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 136, 3618–3624 (2013).
Martinez, C., Hofmann, T. J., Marino, R., Dominici, M. & Horwitz, E. M. Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood 109, 4245–4248 (2007).
Chao, M. P. et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713 (2010).
Kim, M. J. et al. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumour Biol. 29, 28–34 (2008).
Kikuchi, Y. et al. Apoptosis inducing bivalent single-chain antibody fragments against CD47 showed antitumor potency for multiple myeloma. Leuk. Res 29, 445–450 (2005).
Mateo, V. et al. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat. Med. 5, 1277–1284 (1999).
Knapp, W. et al. Towards a better definition of human leucocyte surface molecules. Immunol. Today 10, 253–258 (1989).
Avent, N. et al. Monoclonal antibodies that recognize different membrane proteins that are deficient in Rhnull human erythrocytes. One group of antibodies reacts with a variety of cells and tissues whereas the other group is erythroid-specific. Biochem J. 251, 499–505 (1988).
van Rees, D. J. et al. Sodium stibogluconate and CD47-SIRPalpha blockade overcome resistance of anti-CD20-opsonized B cells to neutrophil killing. Blood Adv. 6, 2156–2166 (2022).
Sallman, D. A. et al. The first-in-class anti-CD47 antibody magrolimab (5F9) in combination with azacitidine is effective in MDS and AML patients: ongoing phase 1b results. Blood 134, 569 (2019).
Sikic, B. I. et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 37, 946–953 (2019).
Saxena, K. & Konopleva, M. An expert overview of emerging therapies for acute myeloid leukemia: novel small molecules targeting apoptosis, p53, transcriptional regulation and metabolism. Expert Opin. Investig. Drugs 29, 973–988 (2020).
Swoboda, D. M. & Sallman, D. A. The promise of macrophage directed checkpoint inhibitors in myeloid malignancies. Best. Pr. Res Clin. Haematol. 33, 101221 (2020).
Jiang, X. R. et al. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Disco. 10, 101–110 (2011).
DiLillo, D. J. & Ravetch, J. V. Fc-receptor interactions regulate both cytotoxic and immunomodulatory therapeutic antibody effector functions. Cancer Immunol. Res 3, 704–713 (2015).
Yu, J. F., Song, Y. P. & Tian, W. Z. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 13, 45 (2020).
Salfeld, J. G. Isotype selection in antibody engineering. Nat. Biotechnol. 25, 1369–1372 (2007).
Jefferis, R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat. Rev. Drug Disco. 8, 226–234 (2009).
Bauer, P. M. et al. Activated CD47 promotes pulmonary arterial hypertension through targeting caveolin-1. Cardiovasc Res. 93, 682–693 (2012).
Isenberg, J. S. et al. Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress. Matrix Biol. 28, 110–119 (2009).
Velliquette, R. W. et al. Monoclonal anti-CD47 interference in red cell and platelet testing. Transfusion 59, 730–737 (2019).
Pan, Y. et al. Studying the mechanism of CD47-SIRPalpha interactions on red blood cells by single molecule force spectroscopy. Nanoscale 6, 9951–9954 (2014).
Sosale, N. G. et al. Cell rigidity and shape override CD47’s “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood 125, 542–552 (2015).
Lv, Z. et al. Loss of cell surface CD47 clustering formation and binding avidity to SIRPalpha facilitate apoptotic cell clearance by macrophages. J. Immunol. 195, 661–671 (2015).
Li, M. et al. Anti-CD47 immunotherapy in combination with BCL-2 inhibitor to enhance anti-tumor activity in B-cell lymphoma. Hematol. Oncol. 40, 596–608 (2022).
Burger, P., Hilarius-Stokman, P., de Korte, D., van den Berg, T. K. & van Bruggen, R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 119, 5512–5521 (2012).
Oronsky, B. et al. RRx-001, a downregulator of the CD47- SIRPα checkpoint pathway, does not cause anemia or thrombocytopenia. Expert Opin. Drug Metab. Toxicol 17, 355–357 (2021).
Petrova, P. S. et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res 23, 1068–1079 (2017).
Kauder, S. E. et al. ALX148 is a high affinity sirp alpha fusion protein that blocks CD47, enhances the activity of anti-cancer antibodies and checkpoint inhibitors, and has a favorable safety profile in preclinical models. Blood 130, 112 (2017).
Weiskopf, K. et al. Engineered SIRP alpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91 (2013).
Liu, Y. E., Shi, Y. & Wang, P. Functions of glutaminyl cyclase and its isoform in diseases. Vis. Cancer Med 4, 1 (2023).
Logtenberg, M. E. W. Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRP alpha axis and target for immunotherapy. Nat Med. 25, 612–619 (2019).
Arlauckas, S. P. et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 9, eaal3604 (2017).
Peranzoni, E. et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. P Natl Acad. Sci. USA 115, E4041–E4050 (2018).
Zhou, Q. et al. Carfilzomib modulates tumor microenvironment to potentiate immune checkpoint therapy for cancer. Embo Mol. Med. 14, e14502 (2022).
Attia, J. V. D. et al. The molecular and functional characteristics of HLA-G and the interaction with its receptors: where to intervene for cancer immunotherapy? Int. J. Mol. Sci. 21, 8678 (2020).
Kang, X. et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle 15, 25–40 (2016).
Mandel, I. et al. BND-22, a first-in-class humanized ILT2-blocking antibody, promotes antitumor immunity and tumor regression. J. Immunother. Cancer 10, e004859 (2022).
Mondal, K. et al. Preclinical evaluation of NGM707, a novel anti-ILT2/anti-ILT4 dual antagonist monoclonal antibody. Cancer Res. 81, LB156 (2021).
Belaunzaran, O. M. et al. Iosh2 exerts potent anti-tumor activity by blocking Lilrb1/2 and Kir3dl1 receptor signaling. J. Immuno Ther. Cancer 9, A906–A906 (2021).
Ni, Y. H., Zhao, X. & Wang, W. CD24, a review of its role in tumor diagnosis, progression and therapy. Curr. Gene Ther. 20, 109–126 (2020).
Munkley, J. & Scott, E. Targeting aberrant sialylation to treat cancer. Medicines (Basel) 6, 10 (2019).
Chen, G. Y. et al. Amelioration of sepsis by inhibiting sialidase-mediated disruption of the CD24-SiglecG interaction. Nat. Biotechnol. 29, 428–435 (2011).
Liu, Y. et al. In situ modulation of dendritic cells by injectable thermosensitive hydrogels for cancer vaccines in mice. Biomacromolecules 15, 3836–3845 (2014).
Sait, S. & Modak, S. Anti-GD2 immunotherapy for neuroblastoma. Expert Rev. Anticancer Ther. 17, 889–904 (2017).
Dhillon, S. Dinutuximab: first global approval. Drugs 75, 923–927 (2015).
Markham, A. Naxitamab: first approval. Drugs 81, 291–296 (2021).
Chen, W. C. et al. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood 115, 4778–4786 (2010).
Leonard, J. P. et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J. Clin. Oncol. 21, 3051–3059 (2003).
Wong, K. L. et al. SM03, an anti-CD22 antibody, converts Cis-to-Trans ligand binding of CD22 against alpha2,6-linked sialic acid glycans and immunomodulates systemic autoimmune diseases. J. Immunol. 208, 2726–2737 (2022).
Lamb, Y. N. Inotuzumab ozogamicin: first global approval. Drugs 77, 1603–1610 (2017).
Kreitman, R. J. & Pastan, I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res 17, 6398–6405 (2011).
Dhillon, S. Moxetumomab pasudotox: first global approval. Drugs 78, 1763–1767 (2018).
Bachanova, V. et al. Phase I study of a bispecific ligand-directed toxin targeting CD22 and CD19 (DT2219) for refractory B-cell malignancies. Clin. Cancer Res. 21, 1267–1272 (2015).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28 (2018).
Pan, J. et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia 33, 2854–2866 (2019).
Hu, G. et al. High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat. Commun. 12, 773 (2021).
Wang, Y., Yin, C., Feng, L., Wang, C. & Sheng, G. Ara-C and anti-CD47 antibody combination therapy eliminates acute monocytic leukemia THP-1 cells in vivo and in vitro. Genet Mol. Res. 14, 5630–5641 (2015).
Kazama, R. et al. Combination of CD47 and signal-regulatory protein-alpha constituting the “don’t eat me signal” is a prognostic factor in diffuse large B-cell lymphoma. Cancer Sci. 111, 2608–2619 (2020).
Mateo, V. et al. Mechanisms of CD47-induced caspase-independent cell death in normal and leukemic cells: link between phosphatidylserine exposure and cytoskeleton organization. Blood 100, 2882–2890 (2002).
Chao, M. P. et al. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood 118, 4890–4901 (2011).
Goto, H. et al. Efficacy of anti-CD47 antibody-mediated phagocytosis with macrophages against primary effusion lymphoma. Eur. J. Cancer 50, 1836–1846 (2014).
Kitai, Y. et al. CD47 promotes T-cell lymphoma metastasis by up-regulating AKAP13-mediated RhoA activation. Int Immunol. 33, 273–280 (2021).
Jain, S. et al. Targeted inhibition of CD47-SIRPalpha requires Fc-FcgammaR interactions to maximize activity in T-cell lymphomas. Blood 134, 1430–1440 (2019).
Herrmann, H. et al. Delineation of target expression profiles in CD34+/CD38- and CD34+/CD38+ stem and progenitor cells in AML and CML. Blood Adv. 4, 5118–5132 (2020).
Jiang, H. J. et al. CD47 is expressed abnormally on hematopoietic cells in myelodysplastic syndrome. Leuk. Res 37, 907–910 (2013).
Sun, J. et al. Targeting CD47 as a novel immunotherapy for multiple myeloma. Cancers (Basel) 12, 305 (2020).
Rastgoo, N. et al. Targeting CD47/TNFAIP8 by miR-155 overcomes drug resistance and inhibits tumor growth through induction of phagocytosis and apoptosis in multiple myeloma. Haematologica 105, 2813–2823 (2020).
Orozco-Morales, M. et al. Clinicopathological and prognostic significance of CD47 expression in lung neuroendocrine tumors. J. Immunol. Res. 2021, 6632249 (2021).
Liu, X. J. et al. CD47 promotes human glioblastoma invasion through activation of the PI3K/Akt pathway. Oncol. Res 27, 415–422 (2019).
Kosaka, A. et al. CD47 blockade enhances the efficacy of intratumoral STING-targeting therapy by activating phagocytes. J. Exp. Med. 218, e20200792 (2021).
Liu, R. et al. CD47 promotes ovarian cancer progression by inhibiting macrophage phagocytosis. Oncotarget 8, 39021–39032 (2017).
Vaeteewoottacharn, K. et al. Attenuation of CD47-SIRPalpha signal in cholangiocarcinoma potentiates tumor-associated macrophage-mediated phagocytosis and suppresses intrahepatic metastasis. Transl. Oncol. 12, 217–225 (2019).
Shi, L. et al. CD47 deficiency ameliorates autoimmune nephritis in Fas(lpr) mice by suppressing IgG autoantibody production. J. Pathol. 237, 285–295 (2015).
Park, J. K., Lee, Y. J., Park, J. S., Lee, E. B. & Song, Y. W. CD47 potentiates inflammatory response in systemic lupus erythematosus. Cells 10, 1151 (2021).
Ghimire, K. et al. Deficiency in SIRP-alpha cytoplasmic recruitment confers protection from acute kidney injury. FASEB J. 33, 11528–11540 (2019).
Torrez Dulgeroff, L. B. et al. CD47 blockade reduces the pathologic features of experimental cerebral malaria and promotes survival of hosts with Plasmodium infection. Proc. Natl Acad. Sci. USA 118, e1907653118 (2021).
Gwag, T., Ma, E., Zhou, C. & Wang, S. Anti-CD47 antibody treatment attenuates liver inflammation and fibrosis in experimental non-alcoholic steatohepatitis models. Liver Int. 42, 829–841 (2022).
McLaughlin, K. M. et al. A potential role of the CD47/SIRPalpha axis in COVID-19 pathogenesis. Curr. Issues Mol. Biol. 43, 1212–1225 (2021).
Tal, M. C. et al. Upregulation of CD47 is a host checkpoint response to pathogen recognition. mBio 11, e0129320 (2020).
Wang, F., Wu, X., Lu, Z., Tang, C. & Cao, X. Overexpression of CD47 inhibits apoptosis of SW480 human colon cancer cells by blocking Fas/FasL pathway. Xi Bao Yu Fen. Zi Mian Yi Xue Za Zhi 37, 904–909 (2021).
Zhang, Y., Sime, W., Juhas, M. & Sjolander, A. Crosstalk between colon cancer cells and macrophages via inflammatory mediators and CD47 promotes tumour cell migration. Eur. J. Cancer 49, 3320–3334 (2013).
Okunuki, Y., Tabor, S. J., Lee, M. Y. & Connor, K. M. CD47 deficiency ameliorates ocular autoimmune inflammation. Front Immunol. 12, 680568 (2021).
Yao, D. Z., Xia, J. Y., Li, J. H. & Xu, J. CD47 is associated with the up-regulation of the PD-1 oncogenic signaling pathway. Int J. Clin. Exp. Pathol 11, 5612–5621 (2018).
Sharp, R. C., Brown, M. E., Shapiro, M. R., Posgai, A. L. & Brusko, T. M. The immunoregulatory role of the signal regulatory protein family and CD47 signaling pathway in type 1 diabetes. Front Immunol. 12, 739048 (2021).
Beguier, F. et al. The 10q26 risk haplotype of age-related macular degeneration aggravates subretinal inflammation by impairing monocyte elimination. Immunity 53, 429–441.e428 (2020).
Li, Y. et al. CD47 antibody suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Am. J. Transl. Res. 12, 5908–5923 (2020).
Zuo, Z. et al. CD47 deficiency attenuates isoproterenol-induced cardiac remodeling in mice. Oxid. Med Cell Longev. 2019, 7121763 (2019).
Kojima, Y. et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Xi, Q. et al. Restoration of miR-340 controls pancreatic cancer cell CD47 expression to promote macrophage phagocytosis and enhance antitumor immunity. J. Immunother. Cancer 8, e000253 (2020).
Wang, C. J. et al. Protective role of programmed death 1 ligand 1 (PD-L1)in nonobese diabetic mice: the paradox in transgenic models. Diabetes 57, 1861–1869 (2008).
Nguyen, J. et al. Overexpression of programmed death ligand 1 in refractory inflammatory bowel disease. Hum. Pathol. 126, 19–27 (2022).
Carter, L. L. et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 182, 124–134 (2007).
Salama, A. D. et al. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J. Exp. Med 198, 71–78 (2003).
Kim, J. Y. et al. Tonsil-derived mesenchymal stem cells (T-MSCs) prevent Th17-mediated autoimmune response via regulation of the programmed death-1/programmed death ligand-1 (PD-1/PD-L1) pathway. J. Tissue Eng. Regen. M 12, E1022–E1033 (2018).
Gotsman, I. et al. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Invest 117, 2974–2982 (2007).
Bu, D. X. et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. 31, 1100–U1411 (2011).
Cochain, C. et al. Programmed cell death-1 deficiency exacerbates T cell activation and atherogenesis despite expansion of regulatory T Cells in atherosclerosis-prone mice. Plos One 9, e93280 (2014).
Thompson, R. H. et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. P Natl Acad. Sci. USA 101, 17174–17179 (2004).
Thompson, R. H. et al. Tumor B7-H1 is associated with poor prognosis in renal cell carcinoma patients with long-term follow-up. Cancer Res. 66, 3381–3385 (2006).
Lu, L. G. et al. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut 71, 2551–2560 (2022).
Zhang, J. et al. Circulating PD-L1 in NSCLC patients and the correlation between the level of PD-L1 expression and the clinical characteristics. Thorac. Cancer 6, 534–538 (2015).
Azuma, K. et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall- cell lung cancer. Ann. Oncol. 25, 1935–1940 (2014).
Hino, R. et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer-Am. Cancer Soc. 116, 1757–1766 (2010).
Massi, D. et al. PD-L1 marks a subset of melanomas with a shorter overall survival and distinct genetic and morphological characteristics. Ann. Oncol. 25, 2433–2442 (2014).
Li, C. W. et al. Eradication of triple-negative breast cancer cells by targeting Glycosylated PD-L1. Cancer Cell 33, 187–201 e110 (2018).
Sun, C., Mezzadra, R. & Schumacher, T. N. Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 (2018).
Zhang, X. et al. NEK2 inhibition triggers anti-pancreatic cancer immunity by targeting PD-L1. Nat. Commun. 12, 4536 (2021).
Gu, X. B. et al. Elevated PD-L1 expression predicts poor survival outcomes in patients with cervical cancer. Cancer Cell Int. 19, 146 (2019).
Li, Y. et al. The prognostic and clinicopathological roles of PD-L1 expression in colorectal cancer: a systematic review and meta-analysis. Front. Pharmacol. 10, 139 (2019).
Shan, T. et al. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int J. Clin. Exp. Pathol 12, 1764–1769 (2019).
Ito, S. et al. Expression of PD-L1 and HLA class I in esophageal squamous cell carcinoma: prognostic factors for patient outcome. Ann. Surg. Oncol. 23, 508–515 (2016).
Kiyasu, J. et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 126, 2193–2201 (2015).
Lamano, J. B. et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin. Cancer Res. 25, 3643–3657 (2019).
Li, H. et al. The immune checkpoint regulator PDL1 is an independent prognostic biomarker for biochemical recurrence in prostate cancer patients following adjuvant hormonal therapy. J. Cancer 10, 3102–3111 (2019).
Li, Y. et al. The clinicopathologic and prognostic significance of programmed cell death ligand 1 (PD-L1) expression in patients with prostate cancer: a systematic review and meta-analysis. Front. Pharmacol. 9, 1494 (2019).
Gu, L. H. et al. PD-L1 and gastric cancer prognosis: A systematic review and meta-analysis. Plos One 12, e0182692 (2017).
Valentino, L. et al. Shed tumor gangliosides and progression of human neuroblastoma. Blood 75, 1564–1567 (1990).
Kailayangiri, S. et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 106, 1123–1133 (2012).
Chang, H. R., Cordoncardo, C., Houghton, A. N., Cheung, N. K. V. & Brennan, M. F. Expression of disialogangliosides Gd2 and Gd3 on human soft-tissue sarcomas. Cancer-Am. Cancer Soc. 70, 633–638 (1992).
Mount, C. W. et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 24, 572–579 (2018).
Hu, Y. et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 27, 2764–2772 (2021).
Tamura, S. et al. Clinical significance of STC1 gene expression in patients with colorectal cancer. Anticancer Res. 31, 325–329 (2011).
Ding, H., Wei, M., Bao, Y., Xiong, X. & Yi, W. J. I. J. C. E. M. Prognostic value of STC 1 expression in ovarian cancer. Int. J. Clin. Exp. Med. 12, 5433–5439 (2019).
Han, J., Jeon, M., Shin, I. & Kim, S. Elevated STC1 augments the invasiveness of triplenegative breast cancer cells through activation of the JNK/cJun signaling pathway. Oncol. Rep. 36, 1764–1771 (2016).
Cai, S. et al. Stanniocalcin-1 relates to tumor recurrence and unfavorable prognosis of urothelial bladder cancer. Int J. Clin. Exp. Pathol. 9, 5429–5436 (2016).
Yeung, B. H., Shek, F. H., Lee, N. P. & Wong, C. K. Stanniocalcin-1 reduces tumor size in human hepatocellular carcinoma. PLoS One 10, e0139977 (2015).
Kasper, M. et al. Intraocular dendritic cells characterize HLA-B27-associated acute anterior uveitis. Elife 10, e67396 (2021).
Friis, J. et al. HLA-B27 in juvenile chronic arthritis. J. Rheumatol. 12, 119–122 (1985).
Balamtekin, N. et al. The HLA groups and their relationship with clinical features in Turkish children and adolescents with celiac disease. Turk. J. Pediatr. 63, 118–125 (2021).
Tandon, N., Mehra, N. K., Taneja, V., Vaidya, M. C. & Kochupillai, N. HLA antigens in Asian Indian patients with Graves’ disease. Clin. Endocrinol. (Oxf.) 33, 21–26 (1990).
Guirguis, F. K. et al. HLA-A, -B and -C specificities in insulin dependent diabetes mellitus in the Egyptian population. Ann. Biol. Clin. (Paris) 43, 233–237 (1985).
Khong, H. T., Wang, Q. J. & Rosenberg, S. A. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J. Immunother. 27, 184–190 (2004).
Maleno, I., Lopez-Nevot, M. A., Cabrera, T., Salinero, J. & Garrido, F. Multiple mechanisms generate HLA class I altered phenotypes in laryngeal carcinomas: high frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Cancer Immunol. Immunother. 51, 389–396 (2002).
Meissner, M. et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin. Cancer Res. 11, 2552–2560 (2005).
Garrido, M. A. et al. HLA class I alterations in breast carcinoma are associated with a high frequency of the loss of heterozygosity at chromosomes 6 and 15. Immunogenetics 70, 647–659 (2018).
Squire, R., Fowler, C. L., Brooks, S. P., Rich, G. A. & Cooney, D. R. The relationship of class I MHC antigen expression to stage IV-S disease and survival in neuroblastoma. J. Pediatr. Surg. 25, 381–386 (1990).
Turcotte, S. et al. Tumor MHC class I expression improves the prognostic value of T-cell density in resected colorectal liver metastases. Cancer Immunol. Res. 2, 530–537 (2014).
Andersson, E. et al. Correlation of HLA-A02* genotype and HLA class I antigen down-regulation with the prognosis of epithelial ovarian cancer. Cancer Immunol. Immunother. 61, 1243–1253 (2012).
Hanagiri, T. et al. Prognostic implications of human leukocyte antigen class I expression in patients who underwent surgical resection for non-small-cell lung cancer. J. Surg. Res. 181, E57–E63 (2013).
Li, O., Zheng, P. & Liu, Y. CD24 expression on T cells is required for optimal T cell proliferation in lymphopenic host. J. Exp. Med. 200, 1083–1089 (2004).
Tsioulos, G. et al. Insights into CD24 and exosome physiology and potential role in view of recent advances in COVID-19 therapeutics: a narrative review. Life (Basel) 12, 1472 (2022).
Ronaghi, M., Vallian, S. & Etemadifar, M. CD24 gene polymorphism is associated with the disease progression and susceptibility to multiple sclerosis in the Iranian population. Psychiatry Res. 170, 271–272 (2009).
Rostoker, R. et al. CD24(+) cells fuel rapid tumor growth and display high metastatic capacity. Breast Cancer Res. 17, 78 (2015).
Wang, W. F. et al. CD24-dependent MAPK pathway activation is required for colorectal cancer cell proliferation. Cancer Sci. 101, 112–119 (2010).
Bretz, N. et al. CD24 promotes tumor cell invasion by suppressing tissue factor pathway inhibitor-2 (TFPI-2) in a c-Src-dependent fashion. Clin. Exp. Metastasis 29, 27–38 (2012).
Zhou, Z. et al. The CD24(+) cell subset promotes invasion and metastasis in human osteosarcoma. EBioMedicine 51, 102598 (2020).
Pei, Z. et al. CD24 promotes the proliferation and inhibits the apoptosis of cervical cancer cells in vitro. Oncol. Rep. 35, 1593–1601 (2016).
Yang, X. R. et al. CD24 is a novel predictor for poor prognosis of hepatocellular carcinoma after surgery. Clin. Cancer Res. 15, 5518–5527 (2009).
Schostak, M. et al. Quantitative real-time RT-PCR of CD24 mRNA in the detection of prostate cancer. BMC Urol. 6, 7 (2006).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos. 31830053, 31920103007, 8207112072, 82122056), the National Key Research and Development Program of China (2020YFA0803201), and the Science Technology Commission of Shanghai Municipality (No. 20S11900700).
Author information
Authors and Affiliations
Contributions
P.W. designed the project, reviewed and revised the manuscript. Y.L., Y.W., Y.Y. summarized the literature and wrote part of the manuscript. Y.L. drew figures of the article and revised the article. L.W., Q.W., and P.Z. sourced the literatures, Q.W., J.Z. polished the language. L.F. and Y.S. gave suggestions for revision and helped organize and revise the manuscript. All authors contributed to the article, read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
Y.S. holds a share of Nanjing Shijiang Medicine Technology Co. LTD, which develops mitochondrial targeting reagents for human health. All the other authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Wang, Y., Yang, Y. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Sig Transduct Target Ther 8, 104 (2023). https://doi.org/10.1038/s41392-023-01365-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01365-z
This article is cited by
-
Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy
Journal of Hematology & Oncology (2024)
-
Targeting the innate immune system in pediatric and adult AML
Leukemia (2024)
-
Survival mechanisms of circulating tumor cells and their implications for cancer treatment
Cancer and Metastasis Reviews (2024)
-
Targeting CD24/Siglec-10 signal pathway for cancer immunotherapy: recent advances and future directions
Cancer Immunology, Immunotherapy (2024)
-
Targeting CD24 as a novel immunotherapy for solid cancers
Cell Communication and Signaling (2023)
