
Abstract
Drug resistance is mainly responsible for cancer recurrence and poor prognosis. Epigenetic regulation is a heritable change in gene expressions independent of nucleotide sequence changes. As the common epigenetic regulation mechanisms, DNA methylation, histone modification, and non-coding RNA regulation have been well studied. Increasing evidence has shown that aberrant epigenetic regulations contribute to tumor resistance. Therefore, targeting epigenetic regulators represents an effective strategy to reverse drug resistance. In this review, we mainly summarize the roles of epigenetic regulation in tumor resistance. In addition, as the essential factors for epigenetic modifications, histone demethylases mediate the histone or genomic DNA modifications. Herein, we comprehensively describe the functions of the histone demethylase family including the lysine-specific demethylase family, the Jumonji C-domain-containing demethylase family, and the histone arginine demethylase family, and fully discuss their regulatory mechanisms related to cancer drug resistance. In addition, therapeutic strategies, including small-molecule inhibitors and small interfering RNA targeting histone demethylases to overcome drug resistance, are also described.
Similar content being viewed by others
Introduction
Increasing evidence has suggested that the incidence of multiple cancers has been rising year by year in the world.1 Although great progress has been made in the treatment of cancer, it is still the main cause of death. Chemotherapy, radiotherapy, immunotherapy, surgical resection, and targeted cancer therapies are usually used in cancer treatment.2,3,4,5 Generally, good therapeutic effects can be achieved in the early stage of cancer, but after long-term treatment with chemotherapeutic drugs, tumor cells could develop drug resistance. Drug resistance is closely related to poor prognosis and cancer recurrence, which is one of the main reasons for cancer treatment failure. Therefore, it is desirable to elucidate the resistance mechanisms in tumor cells during treatment.
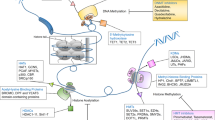
To date, there are many identified drug resistance mechanisms (Fig. 1). As a member of ATP-binding cassette (ABC) transporters, P-glycoprotein (P-gp) can promote the efflux of drugs and make the drugs lose their therapeutic effect, inhibition of P-gp can significantly reverse drug resistance.6,7,8,9,10 Additionally, cytoprotective autophagic response often counteracts apoptosis triggered by anticancer drugs, potentially contributing to acquired drug resistance.11,12 Notably, it is generally believed that eliminating cancer stem cells (CSCs) and reversing epithelial-mesenchymal transition (EMT) are also effective means to overcome drug resistance.13,14,15,16 Additionally, gene mutations also contribute to drug resistance, especially the resistance to tyrosine kinase inhibitors. EGFR mutation can usually lead to resistance to tyrosine kinase inhibitors, and Bruton’s tyrosine kinase mutation can also result in resistance to ibrutinib.17,18,19

Common drug resistance mechanisms. There are seven main reasons that can lead to cancer drug resistance, including overexpressed ABC transporters, CSCs, autophagy, apoptosis, gene mutations, EMT, and epigenetic regulation
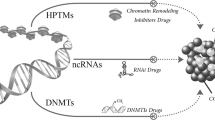
In addition to these factors mentioned above, epigenetic regulation is also important in mediating drug resistance (Fig. 2). Epigenetic modifications refer to the heritable changes in gene expression without changes in DNA sequence, including DNA methylation, histone modification, X-chromatin remodeling, non-coding RNA, nucleosome localization and genomic imprinting.20,21 Of note, modifications of DNA and histone not only affect the function of transcription factors, but also tightly associate with other epigenetic modifications such as chromatin remodeling and non-coding RNAs to co-regulate neoplastic processes.22,23 Generally, DNA methylation often affects gene expression, transcription, and activity. Under the action of DNA methyltransferase, a methyl group is covalently added to the C-5 position of the DNA cytosine ring, and hypermethylation of gene promoters usually leads to transcriptional inhibition, resulting in decreased gene expression.24,25,26

Epigenetic regulation mechanisms of drug resistance. Non-coding RNAs, DNA methylation, and histone modifications including histone acetylation and methylation play important roles in tumor resistance. DNA methylation is maintained by DNA methyltransferases and DNA demethylases, histone acetylation is regulated by histone acetyltransferases and histone deacetylases, and histone methylation is maintained by histone demethylases and histone methyltransferases
Similarly, covalent histone modification is also an important epigenetic model, which includes acetylation, phosphorylation, methylation, ADP ribosylation, ubiquitination, and citrullination, etc.27,28,29,30,31Among these modifications, most of the studies have focused on the acetylation, methylation, and phosphorylation. Numerous studies have shown that covalent post-translational modification of histone tails is critical to the occurrence and development of cancer, including histone demethylases, histone methyltransferases, histone deacetylases, histone acetyltransferases (HATs), and ADP ribosyltransferases.32,33,34,35 For example, it has been shown that histone acetylation is closely associated with gene transcription and the alterations of histone acetylation are tightly associated with cancer phenotypes in multiple cancers.36,37 Furthermore, cancer cells can produce significant resistance to chemotherapeutic drugs through epigenetic changes, especially abnormal modification of histone or genomic DNA.38,39 Their dysregulation usually results in the activation of oncogenes or the inactivation of tumor suppressor genes as well as the dysfunction of many signaling pathways. Therefore, targeting epigenetic modifications could be a promising strategy to overcome drug resistance. Herein, in this review, we first summarize the roles of different epigenetic regulators in tumor resistance. We further summarize the classification and functions of histone demethylases and describe the relationship between histone demethylases and cancer drug resistance in detail.
DNA methylation and drug resistance
In eukaryotes, DNA methylation is an enzymatic reaction catalyzed by a series of DNA methyltransferases, in which a methyl group is covalently added to the 5-carbon of the cytosine ring within a CpG dinucleotide.23 Studies have shown that the activity of some tumor suppressor genes is suppressed due to DNA methylation, which is the basic pathogenesis of multiple cancers. The retinoblastoma tumor suppressor (RB1) was the first identified tumor suppressor gene that is hypermethylated in tumor tissues.40 In breast and ovarian cancers, the DNA repair gene BRCA1 is also silenced because of hypermethylation.41
There are five members in the DNA methyltransferase family, namely DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L. Among them, only three have catalytic methyltransferase activity.24,42 Considered a maintenance methyltransferase, DNMT1 preferentially methylates hemimethylated DNA and is responsible for replicating parental DNA methylation patterns to newly synthesized DNA daughter strands.43 On the contrary, regarded as de novo methyltransferases, DNMT3A and DNMT3B are more biased towards methylating unmethylated CpG dinucleotides.44 The remaining two DNA methyltransferases are generally thought to lack cytosine methyltransferase activity, although DNMT3L can increase the binding capacity of DNMT3A and DNMT3B to the methyl donor S-adenosyl-L-methionine (SAM) to increase their activity.45
Studies have shown that DNA methyltransferase is closely related to tumor resistance (Fig. 3). CSCs are key factors in cancer resistance, and under the regulation of DNMT1, brain-expressed X-linked protein 1 (BEX1) can activate Wnt/β-catenin signaling to maintain the self-renewal capacity of liver stem cells.46 In glioma stem cells (GSCs), the interaction between CD133, a marker of CSCs, and DNMT1 inhibited the nuclear translocation of DNMT1 and maintained the self-renewal and tumorigenesis of GSCs, thus promoting GSCs resistant to the chemotherapeutic agent temozolomide.47 In addition, DNMT1-induced hypermethylation of the miR-34a promoter region mediated its silence and aberrant activation of the Notch pathway, while treatment with decitabine, a small-molecule inhibitor of DNMT1, can increase the sensitivity of pancreatic cancer cells to sorafenib.48 DNA methyltransferases DNMT3A and DNMT3B are overexpressed in Rhabdomyosarcoma and tamoxifen-resistant breast cancers, which indicated that targeting DNA methyltransferases could serve as a potential strategy for radiosensitization and chemosensitization.49,50 Moreover, DNMT3B was also increased in sorafenib-resistant cells, and DNMT3B inhibition by nanaomycin A significantly increased the sensitivity of HCC cells to sorafenib.51 Similarly, DNMT3B mRNA expression levels are also negatively correlated with decitabine sensitivity in pancreatic cancers.52 However, Jia Yu et al. found that triple-negative breast cancers with high expression of DNMTs were more sensitive to decitabine treatment.53 And in ovarian cancers, decitabine sensitivity also correlated more closely with high expression of DNMT1.54 Taken together, this evidence indicates that DNA methyltransferases play a dual role in tumor resistance.

The roles of different epigenetic modifications in tumor resistance. DNA methylation is co-regulated by DNA methyltransferases and DNA demethylases, mainly influencing apoptosis, stemness, EMT, and cell proliferation through Notch and Wnt/β-catenin signaling pathways to modulate drug resistance. Non-coding RNAs mainly include four small nucleotides and two large nucleotides, which promote or inhibit tumor resistance. Similarly, histone methylation and acetylation are histone modifications. They keep gene expressions in balance through histone demethylases and methyltransferases, histone acetyltransferases and histone deacetylases, respectively, and function as a double-edged sword in cancer resistance
In addition to DNA methyltransferases, DNA demethylases also play an important role in drug resistance (Fig. 3). Ten-eleven translocations (TETs) are a family of DNA demethylases that work with DNMTs to maintain the balance of DNA methylation, which consist of TET1, TET2, and TET3.55 As a founding member of the TET family, TET1 catalyzes the oxidation of methyl cytosine to hydroxymethyl cytosine, and then lead to further hydroxylation and oxidation to remove the methyl group from methylated cytosine.55,56 In 5-fluorouracil-resistant colon cancer cells, TET1 binds to the NADPH oxidase DUOX2 promoter and induces its transcription by regulating its methylation, thereby promoting EMT and increasing ROS level.57 Besides, enriched in the promoter region of CRABP2, TET1 can increase its expression, thereby inhibiting Bax-dependent apoptosis to induce oxaliplatin resistance in gastric cancer cells.58 Interestingly, TET1 knockdown can cause lung cancer cells resistant to gefitinib.59 Similarly, the knockdown of TET2 triggered resistance in BRCA2 deficient cells to multiple poly (ADP-ribose) polymerase (PARP) inhibitors and cisplatin.60 And loss of TET2 also reduced ERα expression, thereby causing endocrine resistance in breast cancer cells.61 As a tumor suppressor, inactivation of the Retinoic acid receptor-β (RARβ) gene by methylation of its promoter contributes to tumorigenesis and drug resistance in various cancers,62 whereas TET2 overexpression can reverse its reduction and increase the sensitivity of squamous cell lines to All-trans-retinoic acid (ATRA).63
Compared with the above two DNA demethylases, TET3 has been less studied in tumor resistance. Negatively correlated with TLX and positively correlated with laminin-integrin α6, upregulation of TET3 is able to increase the levels of tumor suppressor genes, thereby inhibiting the growth and self-renewal ability in glioblastoma stem cells.64,65 Regulation of stemness is perhaps responsible for the reduction of glioblastoma resistance induced by TET3, however, the underlying mechanisms of TET3-regulated drug resistance in other cancers require further investigation. More importantly, in addition to the roles in regulating tumor development by influencing gene expression, DNA methylation can interact with other epigenetic modifications to synergistically regulate chromatin formation, including histone modification, nucleosome localization, etc. Related contents have been described in detail in previously published article.24
Up to now, many small-molecule inhibitors targeting DNA methylation have been developed. Among these, DNMT inhibitors azacytidine, guadecitabine (SGI-110), and decitabine have already entered clinical trials.66,67 In 2018, a phase III clinical trial of azacitidine has been completed to evaluate its safety profile and to determine whether azacitidine can cure patients with acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML) or myelodysplastic syndromes (MDS) following allogeneic (donor) stem cell transplantation (ClinicalTrials.gov Identifier: NCT00887068). In the same year, a phase II clinical trial of azacitidine for injectable suspension has been completed to explore its effect on patients with prostate cancer (ClinicalTrials.gov Identifier: NCT00384839). In this study, a total of 35 patients with hormone-refractory prostate cancer received azacitidine for 5 consecutive days of each 28-day cycle and response was assessed after a minimum of 2 cycles. In addition, its roles in head and neck squamous cell carcinoma have also been studied in a phase I trial (ClinicalTrials.gov Identifier: NCT02178072). Decitabine also inhibited DNMT1 activity, and its effect on multiple cancers has also been studied in patients with AML (ClinicalTrials.gov Identifier: NCT00416598), refractory diffuse-large B-cell lymphoma (ClinicalTrials.gov Identifier: NCT03579082), follicular thyroid cancer (ClinicalTrials.gov Identifier: NCT00085293), etc. However, although several clinical trials have been carried out for these two drugs, their impact on drug resistance is rarely involved. Of note, although most of the clinical trials of SGI-110 has been focused on AML, a phase I clinical trial of SGI-110 was initiated to evaluate the efficacy for restoring cisplatin sensitivity in refractory germ cell tumor (ClinicalTrials.gov Identifier: NCT02429466), indicating that targeting DNA methylation is an effective therapeutic measure to overcome tumor resistance. A total of 15 subjects were enrolled in this study and SGI-110 was subcutaneously given daily, 30 mg/m2 on days (1–5) followed by cisplatin 100 mg/m2 on day 8 every 4 weeks.
Non-coding RNA and drug resistance
As another important part of epigenetics, non-coding RNAs (ncRNAs) have been recognized as key regulators of virtually every cellular process which is closely associated with gene expression. In addition, they act as oncogenes or tumor suppressors in various cancers as well.68 Generally, non-coding RNAs can be divided into small nucleotides and large nucleotides (Fig. 3), and small ncRNAs are further divided into small nucleolar RNAs (snoRNAs), P-element induced WImpy testis (PIWI)-interacting RNAs (piRNAs), miRNAs, and transfer RNA (tRNA)-derived small RNAs (tsRNAs).69,70
MiRNAs can bind with complementary sequences in the 3’ untranslated region (3’UTR) of target mRNAs, leading to translational repression or degradation of the target mRNA.71 So far, a large number of miRNAs have been found, and different miRNAs have different roles in tumors. For instance, miR-155,72 miR-2173 and miR-10b74 can act as oncogenes and promote tumor initiation and progression, whereas miRlet-7,75 miR-15a, miR-16-176 and miR-34a77 can function as tumor suppressors. tsRNAs are derived from mature transfer RNAs (tRNAs) and have similar functions to miRNAs.68 TRF/miR-1280 has been reported to inhibit epithelial-mesenchymal transformation and CSCs by inhibiting the Notch pathway in colorectal cancer.78 Moreover, aberrant expression of piRNAs is strongly associated with the development of human malignancies by loading onto members of the PIWI subfamily of argonaute proteins to silence transposons.79,80,81 PiRNAs can also act as promoters or suppressors of tumor development by regulating DNMT expression and DNA methylation.82 Mainly found in the nucleolus, small nuclear RNAs (snoRNAs) not only direct posttranscriptional modifications of ribosomal RNAs and some spliceosomal RNAs, but also participate in the nucleolytic processing of original rRNA transcripts.83,84 SnoRNAs can be divided into two main families: C/D box snoRNAs (SNORDs) and H/ACA box snoRNAs (SNORAs), and jointly regulate the development of cancer.85,86 SNORA42, an H/ACA box snoRNA, is overexpressed in non-small-cell lung cancer and its expression is negatively correlated with clinical survival.87 In contrast, the expression of C/D box snoRNAs U50 has been reported to be downregulated in breast and prostate cancers.88,89
Large ncRNAs include circular RNAs (circRNAs) and long non-coding RNAs. CircRNAs are single-stranded RNAs with covalently closed circular structure, which are produced by back-splicing process of linear precursor RNAs.90,91 Acting as oncogenic factors or tumor suppressors, circRNAs are able to directly bind to proteins and interact with miRNAs to regulate cancer initiation.92,93 Likewise, long non-coding RNAs are able to bind to both proteins and DNA to exert either tumor-promoting or tumor-suppressive effect. For example, MEG3 can upregulate p53 and promote the binding of p53 to the promoter of growth differentiation factor 15 (GDF15), thereby inhibiting tumor growth.94 On the contrary, as an oncogene, lncRNA HOX transcript antisense RNA overexpression can increase the metastatic and invasive abilities in various tumors.95,96
The relationship between non-coding RNAs and tumor resistance has been reported (Fig. 3). Recent studies have described the effects of non-coding RNAs on drug resistance in colorectal and lung cancer, including lncRNA, miRNA, and circRNAs, indicating that ncRNAs can be used as biomarkers to predict drug resistance.97,98,99 Fatemeh Najafi et al. also focused on the involvement of miR-424 and miR-631 in the regulation of tumor resistance and sensitivity.100 EMT is an important mechanism by which tumors develop drug resistance, and HaShem khanbabaei et al. summarized the relationship between non-coding RNAs and EMT in cancers, suggesting that ncRNAs might serve as important regulators in tumor resistance.70 In addition to regulating EMT, non-coding RNAs also participate in the development of drug resistance by regulating CSCs, apoptosis, and autophagy.101 More interestingly, evidences have indicated that piRNAs may act as a double-edged sword in tumor resistance. Piwi-interacting RNA 1037 could enhance cisplatin resistance in oral squamous cell carcinoma (OSCC) cells by inhibiting cell apoptosis.102 Conversely, Piwi-interacting RNA piR-39980 induced intracellular doxorubicin accumulation, DNA damage, and cell apoptosis, thereby increasing fibrosarcoma sensitivity to doxorubicin.103
Additionally, the roles of lncRNA in drug resistance have also been studied extensively. For example, lncRNA Miat could promote the resistance of stem-like medulloblastoma (MB) cells to radiotherapy by downregulating p53 signaling pathway and reducing radiation-induced cell death.104 lncRNA MCF2L-AS1 activated the IGF2/MEK/ERK pathway by interacting with insulin-like growth factor-2 mRNA binding protein 1 (IGF2BP1), and knockdown of MCF2L-AS1 increased the sensitivity of ovarian cancer cells to cisplatin.105 Combined with miR-4496, AC116025.2 promoted the resistance of esophageal cancer cells to 5-FU by reducing cell apoptosis.106 Importantly, LncRNA can interact with LSD1 to regulate drug resistance. Overall, non-coding RNAs are not represented as unimportant, although these non-coding RNAs cannot be translated into proteins, they can interact with DNA, RNA, and proteins to participate in various of cellular activities and modulate tumor resistance.
Based on the characteristics of non-coding RNAs, using ncRNA or directly targeting ncRNA may become an effective strategy for the precise treatment of cancer patients. Up to now, many studies about non-coding RNA have entered clinical trials.107 And most clinical trials aim to validate the roles of non-coding RNAs as cancer biomarkers, such as long non-coding RNA HOTAIR as a biomarker in thyroid cancer (ClinicalTrials.gov Identifier: NCT03469544) and long non-coding RNAs WRAP53 and UCA-1 as biomarkers in hepatocellular carcinoma (ClinicalTrials.gov Identifier: NCT05088811). However, the role of non-coding RNAs in drug resistance has not been clinically studied.
Histone modifications and drug resistance
Histone acetyltransferases (HATs)
Many lysine residues on histone tails are capable of acetylation, and studies have shown that histone acetylation contributes to cancer development by regulating intracellular pH and affecting the gene transcriptional activity and chromatin structure.108 Histone acetylation is a dynamic process, which is regulated by two enzymes with opposite functions: HATs and histone deacetylases (HDACs), and the imbalance often leads to the occurrence of tumors.109 HATs are able to catalyze the transfer of an acetyl group from the donor acetyl coenzyme A to histone lysine side chains, eliminating the positive charge of lysine and thus unfolding the local structure of chromatin.110 HAT is mainly composed of three families located in the nucleus: the MYST family (Moz-Ybf2/Sas3-Sas2-Tip60), the p300/CREB-binding protein family (CBP/CREBBP), and the GCN5-related N-acetyltransferases family (GNAT).111,112,113 In addition, there is also a histone acetyltransferase Hat1 in the both nucleus and cytoplasm, and it is mainly responsible for acetylating newly synthesized histone H3 and H4.114
As a member of the MYST family, MYST1, also named hMOF, can acetylate H4K16 and is abnormally expressed in many cancers, including gastric cancer, breast cancer, non-small cell lung cancer, etc.115,116,117 TIP60, another member of MYST family, can also acetylate histone H2A, H3, and H4 and a variety of non-histones, such as p53, STAT3, NF-κB, etc.118,119 CBP/p300 can inhibit H3K27 acetylation to block estrogen receptor α in breast cancer, and treatment with the CBP/p300 inhibitor A-485 specifically reduced CBP/p300 mediated histone acetylation and led to growth arrest of cells by activating the autophagy pathway in NSCLC.120,121 Moreover, the upregulation of GCN5 is usually closely related to the poor prognosis of tumors.122 In prostate cancer, GCN5-mediated acetylation of LIFR promoted its homodimerization, subsequently promoted LIFR-S1044 phosphorylation, thus activating AKT signaling.123 Interestingly, these HATs can also affect the activity of histone methyltransferases in vitro.124
Many studies have reported the roles of HATs in drug resistance (Fig. 3). High expression of histone acetyltransferase 1 (HAT1) can enhance the resistance of castration-resistant prostate cancer and pancreatic cancer to enzalutamide and gemcitabine.125,126 Mechanistically, HAT1 can promote PVT1 transcription and improve the stability of EZH2 protein to increase drug resistance.126 Besides, inhibition of lysine acetyltransferase 6 A (KAT6A), a member of MYST family, can induce apoptosis and enhance the sensitivity of ovarian cancer cells to cisplatin.127 KAT2A can also contribute to tamoxifen resistance in breast cancer by reducing p53 levels, and KAT2A knockdown sensitizes prostate cancer cells to abiraterone.128,129 Interestingly, in addition to these HATs, P300 has been reported to be able to regulate drug resistance in tumors as a double-edged sword. In pancreatic cancer, loss of P300 could mediate Wnt/β-catenin independent tumor growth, thus leading to resistance to PORCN inhibition.130 P300 inhibition also rendered bladder cancer cells resistant to doxorubicin.131 On the contrary, P300 encoding gene EP300 was able to combine with the COL1A2 promoter to activate its expression, thus promoting apatinib resistance in GC cells.132 And upregulation of P300 promoted pERK1/2 rebound and subsequent resistance of melanoma cells to BRAF inhibitors.133 In conclusion, most HATs can promote drug resistance and act as therapeutic targets to prevent or overcome drug resistance in tumors.
Therefore, targeting HATs to develop small-molecule inhibitors holds great promise as a promising measure to reverse drug resistance in the treatment of cancer. Many previously developed HAT inhibitors have been summarized in other articles.134,135 However, few inhibitors have entered clinical trials and the existed inhibitors mainly target CBP/P300. Phase I/II clinical studies of CBP/P300 inhibitor CCS1477 were initiated for the treatment of hematologic malignancies, prostate cancer, breast cancer, and NSCLC (ClinicalTrials.gov Identifier: NCT03568656; NCT04068597). In 2022, a phase I clinical trial was started to evaluate the safety and maximum tolerated dose of EP31670, a dual inhibitor of BET and CBP/P300, in advanced solid tumors (ClinicalTrials.gov Identifier: NCT05488548).
Histone deacetylases (HDACs)
The balance between histone deacetylases and acetyltransferases is a key regulatory mechanism of gene expression and is involved in various activities and disease occurrence. Contrary to the HATs, histone deacetylases can remove acetyl groups from histone or non-histone lysine residues, thus concentrating chromatin and weakening gene transcription activity.136,137 According to the structure and sequence similarities, histone deacetylases can be grouped into four families comprising a total of 18 isozymes: Class I, II, III, and IV. Class I RPD3-like proteins consist of HDAC1, 2, 3, 8, which mainly located in the nucleus. Class II HDACs consist of class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10) which can shuttle between cytoplasm and nucleus to regulate the cytoplasmic substrate. As nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, Class III HDACs mainly include Sirtuin (SIRT)1-7, which are homologous to the Sir2 protein in yeast. And located in the nucleus, the single member of the class IV HDACs, HDAC11, has gained prominence in epigenetics.107,111 Targeting histones or non-histone proteins, HDACs are able to act as both tumor promotors and suppressors, and dually regulate cancer progression by influencing cellular activities such as stemness, proliferation, apoptosis, differentiation, angiogenesis, migration, and invasion.111,138,139
Generally, high expression of HDAC will promote tumor resistance and amongst of these HDACs, histone deacetylase HDAC6 is widely concerned in regulating drug resistance.140 In NSCLC and melanoma, HDAC6 can enhance the stability of EGFR and tubulin β3 and reduce apoptosis, subsequently resulting in tumor resistance.141,142 HDAC1/2/6 induced deacetylation of specificity protein 1 (Sp1) promoted CSC-like cell proliferation, thus enhancing the resistance of gliobalastoma to temozolomide.143 Notably, ubiquitin-specific peptidase 10 (USP10) was also able to interact with HDAC6 and increase its stabilization, further increasing cisplatin resistance in NSCLC.144 In addition to HDAC6, SIRT7 can deacetylate p53 and reduce the sensitivity of hepatocellular carcinoma to adriamycin.145 SIRT1 increased β-catenin expression and nuclear translocation, thus enhancing the resistance of colorectal cancer (CRC) cells to radiotherapy.146 Surprisingly, SIRT3 and SIRT1 have also been reported to be tightly associated with insulin resistance.147,148 Significantly overexpressed in glioblastoma, the class I HDACs HDAC1/3/8 also contributed to temozolomide resistance.149 In summary, aberrant expression of histone deacetylases promotes the development of tumor resistance, and targeting histone deacetylases can limit the generation of drug resistance (Fig. 3).
Up to now, HDAC inhibitors have been extensively developed and become the focus of cancer treatment. Four HDAC inhibitors have been approved by the food and Drug Administration for clinical treatment, including romidepsin, belinostat, panobinostat, and vorinostat.150 In addition, many other HDAC is are undergoing clinical evaluation now, and the combination of HDAC is with mTOR inhibitors, EGFR inhibitors, PI3K inhibitors, and immune checkpoint inhibitors has also become an important part of clinical and preclinical studies.151,152
Histone methyltransferases
Histone methyltransferases (HMTs), also known as protein methyltransferases (PMTs), can be divided into histone lysine methyltransferases (HKMTs or PKMTs) and histone arginine methyltransferases (HRMTs or PRMTs). Depending on the products generated, histone arginine methyltransferases can be further classified into three classes: class I includes PRMT1, PRMT2, PRMT3, PRMT4 (CARM1), PRMT6, and PRMT8, which primarily catalyze the formation of monomethyl arginine and asymmetric dimethylarginine; class II consists of PRMT5 and PRMT9, which mainly catalyze the formation of monomethyl arginine and symmetric dimethylarginine; class III only includes PRMT7 which is responsible for catalyzing the formation of monomethyl arginine.153,154 Arginine methylation usually occurs in histones H3R2/R17/R26 and H4R3, which exerts an activating effect on gene expression. And the abnormal expression of PRMTs is conducive to the occurrence, development, and invasion of various tumors, including lung cancer, breast cancer, colorectal cancer, bladder cancer, and leukemia.155,156 However, HKMTs can perform opposite functions when acting on different substrates. Methylation of H3K4, H3K26, H3K36, H3K79, and H4K12 is mainly involved in gene activation, while methylation of H3K9, H3K27, H3K56, H4K5, and H4K20 is related to gene silencing.157 Highly expressed in tumors, SUV39H1, SETDB2 and G9a mainly target H3K9 for methylation, and H3K4 is the methylated target of KMT2A-E and KMT7. KMT3A-G can methylate H3K36 and is overexpressed in multiple cancers. In addition, DOT1L is the only histone lysine methyltransferase responsible for H3K79 methylation which can promote the occurrence of leukemia. Notably, much attention has been paid to the fact that EZH2 is mainly involved in tumorigenesis by regulating histone H3 lysine 27 (H3K27) methylation, whose overexpression is associated with poor prognosis.157,158,159,160
Histone methyltransferases are closely related to drug resistance (Fig. 3). High expression of EZH2 can activate cell survival pathways to promote ovarian cancer resistance to cisplatin.161 EZH2 can also activate PI3K/AKT pathway, thereby leading to acquired resistance to gefitinib in NSCLC.162 On the contrary, EZH2 inhibitor GSK126 can increase the expression of MEIS1 and make CRC cells sensitive to oxaliplatin.163 Additionally, SETDB1 can interact with PELP1 and activate AKT, thereby promoting the resistance of breast cancer to tamoxifen.164 Leukemic stem cells have been identified as an important cause of TKI resistance, and inhibition of G9A can increase the expression of tumor suppressor gene SOX6, thereby significantly inhibiting the survival and self-renewal ability of leukemia stem cells.165 In addition to lysine methyltransferases, elevated histone arginine methyltransferase PRMT5 level is also related to the malignant progression of tumor, and PRMT5 inhibition by GSK3186000A sensitized human AML cell lines to PARP inhibition.166 Similarly, class I PRMT inhibitor MS023 can decrease the resistance of ovarian cancer cells to PARP inhibitor BMN-673.167 Taken together, histone methyltransferases hold great promise as targets for cancer therapy to overcome tumor resistance. Up to now, many inhibitors have been developed for these histone methyltransferases, and many phase I/II clinical trials have been carried out for different cancers.168,169,170 Among these inhibitors, most of them target EZH2. Notably, in these clinical trials, combination of histone methyltransferase inhibitors with other therapeutics has also received widespread attention, suggesting an important role for drug combinations in cancer treatment.
Histone demethylases (HDMTs)
As a reversible dynamic regulatory process, abnormal histone methylation can directly or indirectly affect physiological and pathological processes. In this process, histone demethylases have gained much attention due to their important roles in tumor regulation. Many studies have revealed that histone demethylases (HDMTs) are tightly related to drug resistance and regulated by inhibitors and siRNA, histone demethylases can modulate drug resistance by affecting multiple activities including autophagy, EMT and metabolism (Fig. 4). Therefore, in this review, we summarize the classification and functions of histone demethylases, inhibitors in clinical trials, the roles of histone demethylases in tumor resistance and effective strategies targeting histone demethylases for reversing resistance.

Regulation of histone demethylases in tumor resistance. Histone demethylases can contribute to the development of drug resistance by regulating gene transcription, promoting autophagy, reducing apoptosis, affecting cellular metabolic processes, and promoting epithelial-mesenchymal transition. Conversely, targeting histone demethylases by inhibitors or small interfering RNAs can reverse drug resistance
Classification, function of histone demethylases
Histone demethylases include histone lysine demethylases and histone arginine demethylases. A large number of histone lysine demethylases have been reported before, while histone arginine demethylases are relatively less reported (Fig. 5). There are three main families of histone demethylases: the lysine-specific demethylase (LSD) family, the Jumonji C(JmjC)-domain-containing demethylase (JMJD) family and the histone arginine demethylases.

Classification of histone demethylases and their demethylation sites. There are three families of histone lysine demethylases (shown in different color): lysine-specific demethylase (LSD) family, JMJD family consisting of six families with 21 jmjC domain-containing proteins, and histone arginine demethylases. And different demethylases can demethylate different methylation sites of histones
The lysine-specific demethylase (LSD) family
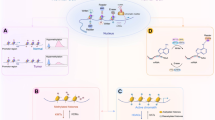
The lysine-specific demethylase (LSD) family consists of LSD1 and LSD2. Lysine-specific demethylase 1 (LSD1) is the first histone lysine demethylase which was found in 2004.171 LSD1, also known as KDM1A, BHC110, and AOF2, belongs to the flavin adenine dinucleotide (FAD) dependent ammonia oxidase superfamily.172,173,174 LSD1 is composed of three main domains: (1) the SWIRM domain, which is located at the N-terminal and responsible for the interaction with other proteins.175 (2) the Tower domain, which consists of two oppositely parallel α Helix composition and ensure the normal function of LSD1 demethylation.176 (3) the AOL domain, which is an amine oxidase-like domain locating at the C-terminal of LSD1 and is divided into two regions by the Tower domain, one is used for substrate binding and recognition, and the other is defined as the binding site of non-covalent FAD (Fig. 6).177

Structure domain of LSD family and catalytic mechanism. a Structure domain difference between LSD1 and LSD2. b Structure of histone and the demethylase catalytic process, every two molecules of histone H2A, H2B, H3, and H4 constitute a core protein octamer, and then about 146 bp of DNA is bound around the octamer to form a nucleosome, demethylases can function by removing methyl groups from histones
The combination of LSD1 with different co-factors or substrates can produce different functions. When interacted with CoREST,178 CtBP,179 NuRD,180 LSD1 can demethylate H3K4me1/me2, thus enhancing the expression of DNMTs and redemethylating DNA. In this way, it negatively regulates gene expression and inhibits gene transcription.181 However, in the presence of estrogen or androgen, LSD1 can demethylate H3K9 and activate gene expression.182 In addition, LSD1 can demethylate H3K20me2 and activate neural regulatory genes.177 At last, LSD1 can also interact with other proteins such as p53, p62, E2F1, and ERR non-histones and regulate their functions by demethylation.183,184,185,186,187
LSD2, also known as KDM1B and AOF1, a homolog of LSD1, is another FAD-dependent ammonia oxidase (Fig. 6).188 Both LSD2 and LSD1 have AOL and SWIRM domains, but the difference is that LSD2 contains N-terminal zinc finger domain, which is very important for its demethylation activity.189 LSD2 is an H3K4me1/me2 demethylase, it can inhibit the expression of p53 through H3K4me1/2 demethylation, promote the proliferation of colorectal cancer and inhibit its apoptosis.190 Besides, LSD2 can demethylate H3K4me1 and regulate the expression of TFPI-2, which plays a survival-promoting role in small cell lung cancer (SCLC).191 Besides, LSD2 also has E3 ubiquitin ligase activity, which can promote the proteasome degradation of O-GlcNAc transferase and inhibit the growth of lung cancer A549 cells.192 Compared with LSD1, the functions of LSD2 have been less studied, and more mechanisms need to be further explored.
The JmjC domain-containing proteins families
There are six families with 21 jmjC domain-containing proteins that can demethylate histone lysine and arginine.
The FBXL (KDM2) family: The FBXL family includes two proteins: FBXL11 and FBXL10.193 FBXL11, also known as JHDM1A or KDM2A, is the first discovered JmjC domain-containing demethylase, which can demethylate H3K36me1/me2 in a Fe (II) and α- ketoglutarate dependent manner.194 The high expression of KDM2A is influenced by multiple factors, including microRNA, inflammation, and hypoxia.195,196 Moreover, it plays an essential role in regulating the occurrence and development of gastric cancer and breast cancer.197,198,199 FBXL10, also named JHDM1B or KDM2B, has a specific H3K4me3 demethylase activity, which is able to inhibit ribosomal RNA and negatively regulate cell proliferation (Fig. 5).200,201 Furthermore, KDM2B can inhibit tumor growth in a p53-dependent manner.202 Besides, it can regulate and decrease the expression of p15INK4B by demethylating H3K4me2, so as to play a key role in the development and maintenance of leukemia.203
The JMJD1 (KDM3) family: There are three proteins in the JMJD1 family, including JMJD1A (JDHM2A, KDM3A), JMJD1B (JDHM2B, KDM3B) as well as JMJD1C (JDHM2C), all of which can demethylate H3K9me1/me2 (Fig. 5).204,205 It has been reported that JMJD1A can upregulate DCLK1 and CDK6 in a demethylation-dependent manner and maintain the occurrence and development of pancreatic cancer.206,207
The JMJD2 (KDM4) family: The JMJD2 (KDM4) family consists of six proteins (JMJD2A-F),208,209 all the proteins have the ability to demethylate H3K9me2/me3 and H3K36me2/me3, which are conductive to breast, colorectal, prostate and other cancers formation (Fig. 5).210 Studies have shown that overexpression of KDM4 proteins can change the transcription and chromatin remodeling to induce cell proliferation.211 However, knockout of KDM4 demethylases can reduce the expression of Taf1b and Nom1 genes by increasing the accumulation of H3K9me3 on the initiation site, so as to downregulate the maintenance of hematopoietic stem cells.212
The JARID1 (KDM5) family: There are four proteins named JARID1A, JARID1B, JARID1C, and JARID1D in the JARID1 (KDM5) family, which can demethylate methyl marks of H3K4.213,214 The structure of KDM5A contains three PHD domains, especially, the PHD1 domain in KDM5A can preferentially recognize unmethylated H3K4 histone tails and stimulate its activity (Fig. 5).215 It is worth noting that KDM5B is up-regulated in breast cancer and prostate cancer. Knocking down KDM5B can activate AMPK signaling pathway, through this manner, KDM5B reverses epithelial-mesenchymal transition (EMT), induces lipid reprogramming, and inhibits cell proliferation and migration, while its overexpression can enhance PI3K/AKT signal pathway.216,217 In clear cell renal cell carcinoma (ccRCC), JARID1C can significantly inhibit tumor growth by reducing H3K4me3 level.218 But JARID1C can regulate BRMS1 and its silence inhibits invasion and metastasis of breast cancer.219 Interestingly, as a transcription factor, KDM5D can demethylate Z2F1 to inhibit its expression, then inhibit its binding to FKBP4 and decrease the transcription of FKBP4.220
The UTX/JMJD3 (KDM6) family: In this family, UTX, also known as KDM6A and JMJD3 (KDM6B) discovered in 2007 can demethylate H3K27me2/me3,221 while UTY (KDM6C) has no enzymatic activity (Fig. 5).222 An increasing number of studies have indicated that JMJD3 plays an important role in maintaining the function of stem cells. JMJD3 can enhance neural commitment by regulating Pax6, Nestin, and Sox1 in order to influence the differentiation of Embryonic Stem Cell (ESC).223 In multiple myeloma, NF-κB pathway can upregulate KDM6B, and overexpression of KDM6B increases the expression of PRKCB and FOS genes related to MAPK pathway, thereby promoting the growth and survival of multiple myeloma cells.224 Notably, UTX is closely related to bladder cancer by affecting p53 and FGFR3 expressions.225,226
The KDM7 family: In KDM7 family, it is generally believed that KIAA1718 (KDM7A) and PHF8 (KDM7B) can participate in demethylating H3K27me1/2, H3K9me1/2 and H4K20me1 and promoting cancer progress (Fig. 5), while PHF2 is considered to inhibit tumor growth.227,228 Specifically, PHF8 is generally elevated in hepatocellular carcinoma (HCC), which is directly related to the occurrence and migration.229 Besides, PHF2 can modulate the expression of cell cycle-related genes and regulate DNA replication.230
In addition, there are many other proteins containing jumonji domain, for instance, similar to JARID1 family, although JARID2 does not have demethylase activity, its expression inhibits the formation of differentiation markers, and regulates keratinocyte differentiation genes.231 Besides, Hspbap1 containing jumonji C domain (jmjd) can interact with heat shock protein HSPb1, but its enzymatic activity needs further study and it only has the possibility of becoming a histone demethylase.232
Histone arginine demethylases
Like lysine methylation, histone arginine methylation is also a reversible process, but compared with demethylases for histone lysine, only a few histone arginine demethylases have been reported. So far, only two histone arginine demethylases have been reported.233 Peptidylarginine deiminase 4 (PAD4) can regulate arginine methylation and gene expression by removing methyl groups from H3R2, H3R8, H3R17, and H3R26, and then converting arginine to citrulline.233,234 When PAD4 activity is decreased, the expression of mesenchymal markers is increased, causing damages to cancer growth and metastasis.235 At the same time, knocking down PAD4 could promote cell autophagy and induce cell apoptosis, thereby inhibiting cell proliferation.236 JMJD6, another JmjC-containing iron and 2-oxoglutarate-dependent dioxygenase, can demethylate histone H3 arginine 2 (H3R2me2) and histone H4 arginine 3 (H4R3me2) as an arginine demethylase by removing methyl groups.233,237,238 Furthermore, JMJD1B can regulate the demethylation of H3K9me2, and then regulate the demethylation of H4R3me2/me1, which is closely related to the growth of hematopoietic stem cells (Fig. 5).239
Histone demethylase inhibitors in clinical development
Given the importance of demethylases in tumorigenesis, the development of small-molecule inhibitors has become a central theme for cancer treatment. At present, many histone demethylase inhibitors have been developed, and some of them have been approved for use in clinical trials to evaluate their efficacy and safety in patients (Fig. 7, Table 1).240,241

Representative inhibitors against histone demethylases in clinical trials
Originally developed as a MAO inhibitor to treat mood and anxiety disorders, tranylcypromine (TCP) can inhibit the activity of MAO-A and MAO-B.242,243 In addition, as an irreversible LSD1 inhibitor, TCP also inhibits cancer proliferation and invasion.244 A clinical phase I/II study investigating whether TCP could sensitize ATRA in patients with non-M3 AML or MDS in 2015 was initiated by Michael Luebbert and the University Hospital Freiburg (ClinicalTrials.gov Identifier: NCT02717884). In phase I trial, four dose levels of TCP (20 mg, 40 mg, 60 mg, 80 mg on days 1–28) were examined in combination with fixed dose of ATRA (45 mg/m2 on days 10–28) and fixed-dose of AraC (40 mg on days 1–10) to determine the dose used in phase II clinical trial, and then phase II trial was designed to evaluate the efficacy of TCP at recommended dose in patients with AML or MDS. In addition, two other clinical trials have been carried out for the combination of TCP and ATRA (ClinicalTrials.gov Identifier: NCT02261779; NCT02273102). Phase I/II clinical trials initiated by Martin-Luther-Universität Halle-Wittenberg was designed to explore the feasibility, safety, and efficacy of combining TCP with all-trans retinoic acid (ATRA) in relapsed or refractory AML.245 And another phase I clinical trial sponsored by University of Miami has been completed in 2020, it indicated that these two drugs combination was safe and effective, and LSD1 inhibition can sensitize AML cells to ATRA.
ORY-1001 is a highly potent and selective covalent LSD1 inhibitor.246 A phase I study on the safety, pharmacodynamics, and pharmacokinetics of ORY-1001 in relapsed or refractory AML sponsored by Oryzon Genomics has been successfully completed in 2016 (EudraCT 2013-002447-29). A phase I clinical trial to determine the maximum tolerated dose in participants with relapsed, extensive-stage disease small cell lung cancer has also been completed (ClinicalTrials.gov Identifier: NCT02913443), Moreover, the combination of ORY-1001 with driamycin, an inhibitor of DNA methylation, also entered phase II clinical trials (EudraCT 2018-000482-36). Very recently, a phase I study was sponsored by Oryzon Genomics S.A. to investigate the safety of the combination of ORY-1001 with gilteritinib in FLT3-mutated R/R AML (ClinicalTrials.gov Identifier: NCT05546580). ORY-2001 (vafidemstat) is another LSD1/MAO-B dual inhibitor developed by Oryzon Genomics.239 However, its clinical studies were mainly designed for treating Alzheimer’s disease (EudraCT 2019-001436-54; ClinicalTrials.gov Identifier: NCT03867253), acute respiratory distress syndrome (ARDS) (EudraCT 2020-001618-39), multiple sclerosis (MS) (EudraCT 2017-002838-23), borderline personality disorder (ClinicalTrials.gov Identifier: NCT04932291), etc, rather than tumors. GSK2879552 is a selective irreversible inhibitor developed by GlaxoSmithKline.247 Clinical trials evaluating its safety, pharmacokinetics and pharmacodynamics in recurrent or refractory SCLC (ClinicalTrials.gov Identifier: NCT02034123), AML (ClinicalTrials.gov Identifier: NCT02177812) and MDS (ClinicalTrials.gov Identifier: NCT02929498) have been terminated because of their poor risk benefit.
INCB059872 is a novel LSD1 inhibitor,239 and a phase I/II clinical study was started to evaluate its safety, tolerability and efficacy in patients with advanced malignant tumors in 2016, which was conducted in 4 parts (ClinicalTrials.gov Identifier: NCT02712905). Part 1 determined the recommended dose of INCB059872 based on maximum tolerated dose. Part 2 further determined the safety, tolerability, efficacy, PK, and PD of the selected monotherapy dose in various malignant tumors, including AML/MDS, SCLC, myelofibrosis, ewing sarcoma, and poorly differentiated neuroendocrine tumors. Part 3 determined the recommended dose of INCB059872 in combination with azacitadine and ATRA in AML and in combination with nivolumab in SCLC. And part 4 further determined the safety, tolerability, efficacy, PK, and PD of the selected combination dose in Part 3. However, this study has been terminated according to strategic business decision. In addition, clinical trials evaluating the safety and activity of INCB059872 in subjects with sickle cell disease (ClinicalTrials.gov Identifier: NCT03132324) and relapsed or refractory Ewing sarcoma (ClinicalTrials.gov Identifier: NCT03514407) have also been discontinued according to business decisions. A phase I/II study in subjects with advanced or metastatic solid tumors to evaluate the combination of INCB059872 with programmed death receptor-1 (PD-1) inhibitor pembrolizumab and the indoleamine 2,3-dioxygenase (IDO-1) inhibitor epacadostat has also been terminated by sponsor (ClinicalTrials.gov Identifier: NCT02959437).
As a selective LSD1 inhibitor developed by Imago BioSciences, there are ten studies that have been registered in clinicaltrials.gov website under IMG-7289. Very recently, a phase I clinical trial was started to evaluate the safety and tolerability of IMG-7289 and Bomedemstat combination therapy in relapsed or refractory acute myeloid leukemia (ClinicalTrials.gov Identifier: NCT05597306). A phase I clinical trial sponsored by Imago BioSciences to evaluate the safety, steady-state pharmacokinetics, and pharmacodynamics of IMG-7289 alone or in combination with ATRA in the treatment of AML and MDS patients has been completed (ClinicalTrials.gov Identifier: NCT02842827). In addition, a phase I/II study initiated by the University of Washington to explore the combination of IMG-7289 with the PD-L1 inhibitor atezolizumab in patients with small cell lung cancer is also recruiting volunteers (ClinicalTrials.gov Identifier: NCT05191797). Subjects involved in the study received atezolizumab intravenously on day 1 and IMG-7289 once daily on days 1-21, and cycles were repeated every 21 days in the absence of disease progression or unacceptable toxicity. Then these patients were followed up at 30 days and every 12 weeks thereafter after treatment completion. Moreover, Imago BioSciences also launched a phase II clinical study in 2021 to evaluate the safety and effectiveness of IMG-7289 in patients with myopathic neuroplasms (ClinicalTrials.gov Identifier: NCT05223920). And the remaining clinical studies were to evaluate the effect of IMG-7289 on the essential thrombocythemia (ClinicalTrials.gov Identifier: NCT04254978; NCT04262141; NCT04081220), Polycythemia Vera (ClinicalTrials.gov Identifier: NCT05558696) and Myelofibrosis (ClinicalTrials.gov Identifier: NCT03136185; NCT05569538). Another LSD1 small-molecule inhibitor TAK-418,239 a phase I study initiated by Takeda to evaluate its safety and tolerability when administered as a single oral dose in healthy participants has been completed (ClinicalTrials.gov Identifier: NCT03228433). The participants were divided into five groups, six of them received TAK-418 and two received matching placebo. The results showed that TAK-418 was well tolerated and exhibited a nearly linear pharmacokinetic profile with a t1/2 of 4.35–5.36 h.248 Moreover, other two phase I studies in healthy volunteers after oral dose of TAK-418 administration have been terminated because of administrative reasons and business decision, respectively (ClinicalTrials.gov Identifier: NCT04202497, NCT03501069).
In addition to covalent irreversible LSD1 inhibitors, two LSD1 reversible inhibitors have also entered clinical trials.249,250 Most clinical trials of the first reversible LSD1 inhibitor CC-90011 are related to drug combinations. For example, a phase I/II study was supported by Celgene to assess the safety, tolerability, and preliminary efficacy of CC-90011 given concurrently with venetoclax and azacytidine in AML, this clinical trial was completed in 2022 (ClinicalTrials.gov Identifier: NCT04748848). It included three parts: a dose escalation part in R/R AML, a dose escalation part in newly diagnosed AML (ndAML), and a randomized dose expansion part in ndAML of Venetoclax and Azacitidine with or without CC-90011. Another phase I study aimed to evaluate the safety and efficacy of CC-90011 in combination with itraconazole and rifampicin in patients with relapsed and refractory solid tumors and non-Hodgkin’s lymphomas was initiated in 2016 (ClinicalTrials.gov Identifier: NCT02875223). The study comprised two parts, the dose escalation part (part A) aimed to estimate the maximum tolerated dose (MTD) of CC-90011, and the expansion part (part B) further evaluated the safety and efficacy of CC-90011 administered at or below the MTD in 3 selected expansion cohorts in order to define the recommended phase 2 dose (RP2D). In addition, a phase I trial of CC-90011 in combination with cisplatin and etoposide (ClinicalTrials.gov Identifier: NCT03850067) and a phase II trial in combination with nivolumab (ClinicalTrials.gov Identifier: NCT04350463) in SCLC has also been initiated. In addition to the drug combination, a phase I study aimed to assess whether CC-90011 can induce AR expression to re-sensitize metastatic castration-resistant prostate cancer to anti-hormonal therapy is recruiting (ClinicalTrials.gov Identifier: NCT04628988). SP-2577 is also a reversible LSD1 inhibitor developed by Salarius Pharmaceuticals. A phase I clinical study of SP-2577 in patients with advanced solid tumors has been completed (clinicalTrials.gov Identifier: NCT03895684). In this study, SP-2577 was given as oral tablets in patients with advanced solid tumors with a 28-day cycle. And clinical trials of SP-2577 in combination with topotecan and cyclophosphamide (ClinicalTrials.gov Identifier: NCT03600649), as well as azacytidine (ClinicalTrials.gov Identifier: NCT04734990), have also been initiated.
Compared with LSD1 inhibitors, very few inhibitors targeting other demethylases have entered clinical trials. Coffeic acid inhibits KDM4C expression.251 Two phase III studies were initiated by the First Affiliated Hospital of Henan University of Science and Technology to investigate the efficacy and safety of coffeic acid in Chinese advanced esophageal squamous cell cancer (ClinicalTrials.gov Identifier: NCT04648917, NCT03070262). In addition, a phase III clinical trial, a multicenter randomized study of caffeic acid tablets as second-line therapy for the treatment of immune thrombocytopenia (ITP), was initiated by Shandong University in 2012 (ClinicalTrials.gov Identifier: NCT02351622). Then a phase IV trial was also initiated by Shandong University to investigate the efficacy and safety of caffeic acid tablets combined with dexamethasone in ITP (ClinicalTrials.gov Identifier: NCT02556814).
As an emerging target for cancer therapy, LSD1 has important biological roles in multiple biological processes and diseases. Until now, many LSD1 inhibitors have been reported, including reversible and irreversible inhibitors. TCP has been recognized as a privileged scaffold for new irreversible LSD1 inhibitors and six TCP-based irreversible inhibitors alone or in combination with other therapeutic agents have been in clinical trials for disease treatment (Table 1). However, these LSD1 irreversible inhibitors can be covalently conjugated to FAD, resulting in a long-term inhibitory effect on the FAD-dependent targets, therefore, it may increase the off-target effect and potential toxicity.241 For example, the phase I clinical trial initiated by University of Miami showed that common adverse effects of TCP are febrile neutropenia and increased creatinine (ClinicalTrials.gov Identifier: NCT02273102). And all three clinical trials of GSK2879552 were terminated due to its potential toxic effects, poor disease control, and poor risk benefit. In contrast, reversible inhibitors have safer profiles, so developing highly active and selective reversible LSD1 inhibitors is a central focus currently. The discovery of CC-90011 and SP-2577 proves the therapeutic potential of reversible inhibitors (Table 1). Notably, LSD1 has interactions with other proteins, and combination therapy of LSD1 inhibitors and other drugs may have synergistic effects. Currently, all the combinations of TCP with ATRA, ORY-1001 with gilteritinib, INCB059872 with azacitadine and ATRA, IMG-7289 with bomedemstat, and atezolizumab, CC-90011 with venetoclax and azacytidine, SP-2577 with topotecan and cyclophosphamide, as well as azacytidine have initiated clinical trials, demonstrating the importance of drug combinations. Unfortunately, compared to LSD1, few other demethylases inhibitors have entered clinical trials.
Histone demethylases and cancer drug resistance
Epigenetic changes, especially histone methylation, and demethylation, play an important role in drug resistance.252 As regulatory factors, histone demethylases are closely related to drug resistance, and targeting the demethylases in various tumors represents a promising strategy to overcome drug resistance (Fig. 3). In this section, we mainly discussed the functions and effects of different histone demethylases on drug resistance.
The lysine-specific demethylase (LSD) family and drug resistance
LSD1 is an important histone demethylation enzyme and can regulate the drug resistance of various cancer cells by changing the methylation levels of H3K4 and H3K9.181,182 Satoi Nagasawa et al. found that the increased LSD1 mRNA level is a potential prognostic factor of poor prognosis in basal-like breast cancer, and the increased expression of LSD1 protein is related to the poor prognosis of triple-negative breast cancer.253
It is generally acknowledged that cancer stem cells have a strong ability for self-renewal, differentiation, and proliferation.254,255,256 Therefore, conventional chemotherapy cannot completely eliminate CSCs in cancer cells, and there remain a lot of residues of tumor stem cells after chemotherapy, which is an important reason for drug resistance.257,258 As a critical epigenetic enzyme, LSD1 can further regulate cell resistance by modulating the function of stem cells (Fig. 8). As reported, John Verigos et al. found that overexpression of LSD1 increased the ability of mammary gland formation and the stem cell potential, thereby increasing the resistance of breast cancer cells to adriamycin, while knocking down LSD1 led to the opposite effect.259 In this sense, the high level of LSD1 is associated with poor prognosis in breast cancer patients (Table 2). In addition to breast cancer, LSD1 can also regulate drug resistance in many other cancers by changing cell viability, such as colorectal cancer, gastric cancer, and hepatocellular carcinoma. Tumor stem cells have many surface markers, such as CD13, CD133 and CD44.260,261 Hu et al. have demonstrated that CD13 can prevent LSD1 from protein ubiquitination and degradation to stabilize LSD1 via deacetylating LSD1 by HDAC5. Next, LSD1 enhanced the demethylation level of p65 protein to improve the stability of p65 and activate NF-κB consequently, eventually producing sorafenib resistance.262 Based on this, knocking down LSD1 can be used as an important target for the treatment of colorectal cancer by attenuating CD133+ cell stemness characteristics (Table 2).263 In addition, gastric cancer cells can release small extracellular vesicles containing LSD1, which can increase the expression of CD44, SOX2, and OCT4, thus promoting chemoresistance to oxaliplatin.264 Significantly, it also indicated that LSD1 plays an important role in cancer stem cells after long-term sorafenib therapy in HCC (Table 2).265 Decreasing LSD1 can inhibit Wnt/β-catenin signaling pathway to enhance the sensitivity of drug-resistant cells to sorafenib.265 And a recent study showed that LSD1 increased cancer stemness by activating Wnt/β-catenin signaling pathway, inducing thyroid cancer resistance to doxorubicin.266 At the same time, LSD1 can inhibit the expression of some suppressors, especially spirgle1 and APC in Lgr5+ cancer-initiating cells by regulating H3K4me1/2 methylation, resulting in high viability of Lgr5+ cells and occurrence of cancer cell drug resistance.267

The roles of lysine-specific demethylase (LSD) family on drug resistance. LSD1 can maintain the function of stem cells by activating NF-κB and Wnt/β-catenin signaling pathway, promote EMT and interact with long-chain non-encoding RNA, thus promoting drug resistance. LSD2 can enhance stem cell characteristics, induce cell apoptosis, and regulate other enzyme expressions to promote drug resistance
Long chain non-encoding RNA is a transcription factor consisting of >200 nucleotides, and it lacks protein encoding potential. LncRNAs can participate in regulating drug resistance and multiple cellular processes through changing epigenetic and gene expressions in translation level (Table 2).268 Many studies have shown that LncRNAs can interact with and recruit LSD1 in order to regulate cell resistance through demethylation modification (Table 2, Fig. 8).269,270,271,272 Zheng et al. found that the poor prognosis in NSCLC patients was closely related to overexpression of LncRNA FTH1P3, which can inhibit TIMP3 protein by recruiting LSD1 and increased cell resistance to gefitinib.269 Moreover, HAS2-AS1 also inhibited EphB3 by recruiting LSD1, promoting tumorigenesis and gefitinib-resistance in NSCLC.270 Li et al. have found that in gefitinib-resistance cell PC-9, the expression of Hox transcript antisense intergenic RNA (HOTAIR) was up-regulated, while the level of LSD1 decreased significantly, which increased the recruitment of H3K27me3 to p16 and p21 promoters.272 In oxaliplatin resistant HepG2 cells, the high expressions of LINC01134, p62 and LSD1 were positively correlated. LSD1 can activate the expression of LINC01134 and increase the resistance of HCC to oxaliplatin through the LINC01134-SP1-p62 axis.271 In summary, LncRNAs can interact with LSD1 and regulate the generation of drug resistance collectively.
In addition to the above mechanisms, LSD1 has also been reported to promote drug resistance in breast cancer and NSCLC by regulating EMT (Fig. 8).273,274 LSD1 is essential for hypoxia-induced gefitinib-resistance, and knockout LSD1 can reverse resistance and enhance drug sensitivity by blocking the associated EMT.273 Boulding et al. have proved for the first time that LSD1 can interact with PKC-θ during the EMT process in breast cancer and promote gene induction.274 Importantly, silencing LSD1 can also increase the BRCA1 mRNA level in breast cancer cells (Table 2).275 Taken together, LSD1 can induce cell resistance through various mechanisms, such as maintaining the function of cancer stem cells, regulating EMT process and interacting with small interfering RNA. Interestingly, although it has been proved that multidrug resistance genes and ABC binding proteins are important factors in the development of drug resistance, the clear relationship between LSD1 and them, especially the P-glycoprotein, has not yet been elucidated.6,276 Therefore, more attention might need to be devoted to the effect of LSD1 on P-gp expression for overcoming drug-induced resistance.
Up to now, compared with LSD1, the functions of LSD2 on drug resistance are less studied. A recent study has indicated that LSD2 is highly expressed in PANC-1, and knocking down LSD2 can inhibit cell proliferation by increasing the apoptosis rate and the related apoptotic proteins expressions.277 Similarly, Tiffany et al. found that knocking down LSD2 can inhibit cell clonogenic ability, induce cell apoptosis, and block cells in S phase, thereby making breast cancer cell sensitive to 5-aza-deoxycytidine (DAC), a DNMT inhibitor (Table 2).278 Chen et al. proved for the first time that the overexpression of LSD2 increased NANOG, SOX2, and mRNA levels of LSD1, KDM4B and KDM5B at the same time, and promoted the characteristics of CSCs, suggesting that LSD2 might interact with other enzymes in tumor development.279 Moreover, LSD2 gene may be the key to the resistance to cisplatin in ovarian cancer. When LSD2 was inhibited, the expression of DNA damage repair gene DCLRE1B decreased, and the sensitivity of cells to cisplatin increased significantly, thus inhibiting cell growth.280
The JmjC domain-containing protein family and drug resistance
KDM2 family and drug resistance: There are two members in KDM2 family. Recent studies have shown that the KDM2 family can promote the emergence of cell resistance. As the target of Zinc-fingers and homeoboxes (ZHX2), a tumor suppressor, KDM2A is negatively correlated with ZHX2 in hepatoma cells. KDM2A knockdown can lessen its downstream SOX2, NANOG, and OCT4 to reduce the formation of tumor stem cells and decrease the resistance to sorafenib (Table 3).281 Another member of the KDM2 family, KDM2B, also plays a significant role in the regulation of stem cells, thereby regulating cell sensitivity to drugs. Staberg et al. reported that targeting KDM2B can reduce the expression of EZH2, CD133, and SOX2 and impair the differentiation ability of stem cells, which is necessary for glioblastoma (GBM). Moreover, the deletion of KDM2B also increased the apoptotic cells by inducing the expression of p21CIP1/WAF1 and cleaved PARP, thus making cells more sensitive to chemotherapeutic agents (Table 3).282 Besides, inhibition of KDM2B in GBM led to a higher expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and reduced tumor growth and angiogenesis in mice (Fig. 9).283

The roles of KDM2 and KDM3 families on drug resistance. KDM2A can promote cell stemness by upregulating the expression of stemness markers Sox2 and Oct4. KDM2B can enhance drug resistance by increasing stem cells and apoptosis at the same time. In addition, KDM3A is able to decrease p53 expression at the transcriptional level and KDM3C can inhibit EMT through the ERK/MAPK signal pathway
KDM3 family and drug resistance: Several evidences have shown that KDM3A is closely related to radiation and drug resistance. Under hypoxia, KDM3A was increased and co-localized with hypoxia-inducible factor HIF-1α in esophageal squamous cell carcinoma (ESCC). When KDM3A was knocked down, the sensitivity of cells to radiation therapy can be increased, and the survival rate of cells under hypoxia conditions was significantly reduced.284 Notably, p53 is an important target of KDM3A, and KDM3A can inhibit the transcriptional activity of p53 by demethylating p53. On the contrary, when KDM3A was inhibited, the binding of p53 to PUMA and NOXA promoter was strengthened, and the expression of related apoptosis proteins was increased, thus restoring cell sensitivity.285,286 Moreover, Ramadoss et al. also found that in cisplatin-resistant ovarian cancer cells, KDM3A can activate SOX2 and inhibit p53 activity through demethylating p53, thereby downregulating the expression of apoptosis-related protein BCL-2 and p21, while inhibiting KDM3A can block the drug-resistant cells in G2/M phase (Fig. 9, Table 3).287 In addition to affecting p53, Wade et al. demonstrated that KDM3A is essential for the expression of ER-targeted genes in tamoxifen-resistant cells, and its deletion can inhibit cell proliferation by blocking cells in G1 phase.288 Overall, KDM3A plays a crucial role in drug resistance. It mainly affects the function of p53 by methylation modification, and then regulates downstream genes of p53 to promote drug resistance and cell proliferation.285,286,287 Therefore, targeting KDM3A could be a feasible strategy to overcome resistance.
Although studies on KDM3B and KDM3C are relatively rare, these homologs play a crucial role in promoting tumor growth and drug resistance. The function of JMJD1A and JMJD1B to demethylate H3K9 is necessary to maintain the function of embryonic stem cells (ESC). Their depletion increased the rate of apoptotic cells and weakened the versatile nature of ESC.289 Moreover, apart from H3K9me2, KDM3B can also demethylate H4R3me2, which affects the development and function of hematopoietic stem cells.239 Notably, the expression of JMJD1C increased significantly in paclitaxel-resistant ESCC. When JMJD1C was knocked down, the cell metastasis ability was reduced, and cell apoptosis rate was increased, so that it could reduce resistance (Table 3).290 Similarly, Schimek et al. also found that the sensitivity of cells to gemcitabine and carboplatin was increased after knocking down JMJD1C.291 Moreover, ESC self-renewing needs JMJD1C, which is likely to inhibit ERK/MAPK signal transduction and EMT process activation by regulating the miR-200 family and miR-290/295 cluster (Fig. 9).292
KDM4 family and drug resistance: There are six members of the KDM4 family. In addition to JMJD2E and JMJD2F, other four members are frequently overexpressed in cancer and closely involved in the development of drug resistance. Metzger et al. first demonstrated that KDM4A controlled the proliferation and self-renewal of breast cancer stem cells (BCSCs) which have been suggested to be responsible for therapy resistance.293 Besides, JMJD2A is tightly related to the sensitivity of gastric cancer cells to anticancer drugs, which can be regulated by the interaction between JMJD2A and its substrate CCDC8.294 In castrated resistant prostate cancer (CRPC), the expression of KDM4A-AS1 was increased, which could regulate the stability of USP14-AR and prevent AR degradation, and this was why cells were resistant to enzalutamide (Table 2).295 Apart from KDM4A, KDM4B also plays an important role in prostate cancer. Mechanically, HIF-1α induced expression of KDM4B to promote cell proliferation by activating the Wnt/β-catenin pathway and activating autophagy.296 It has been found that KDM4B can interact with C-Myc and promote metabolic genes LDHA, ENO1, and PFK levels to regulate cellular metabolism (Fig. 10).297 Furthermore, upon interacting with AR, it can also increase the transcription of C-Myc through its demethylation of H3K9, and its upregulation promotes cell resistance to enzyluamine (Table 3).298 Taken together, these results suggest that targeting KDM4B could be a potential strategy for treating CRPC.

The role of KDM4 family in drug resistance. Small-molecule inhibitors NSC636819 and JIB-04 inhibit KDM4A activity and increase cell sensitivity by affecting CCDC8 expression and apoptosis. Under hypoxia, KDM4B can induce autophagy and increase metabolism through activating the Wnt/β-catenin pathway and transcribing C-Myc respectively. At the same time, KDM4C is able to promote cancer progress by influencing cell cycle and transcription of CDC6, and KDM4D can increase the CSC property through the Wnt/β-catenin and Notch signaling pathways
Like KDM4A and KDM4B, high expression of KDM4C was also closely related to drug resistance. KDM4C can increase the expression of MALAT1 and MALAT1 inhibits its downstream miR-328-3p expression, and then activates cyclin CCND2, making the acute myeloid leukemia (AML) cells resistant to cytarabine (Fig. 10, Table 3).299 In addition, UHRF1 can recruit KDM4C and further regulate the transcription of CDC6 through the demethylation of H3K9 by KDM4C, which is crucial for cell resistance.300 Similar to the function of LSD1, KDM4C, and KDM4D are also necessary for the maintenance of cancer stem cell. Overexpression of KDM4C could activate aldehyde dehydrogenase ALDH1A3 transcription, promoting stemness and chemoresistance of gastric cancer stem cells.301 Chen et al. reported that KDM4C could bind to OCT4 promoter and affect its expression. When the expression of KDM4C was decreased, the migration and spheroid formation ability of cells was reduced, and the CSC property was inhibited.302 And it has been proved that JMJD2D can enhance the transcription of EpCAM and Sox9, which are the target genes of Wnt/β-catenin signaling pathway and Notch signaling pathway, thereby promoting the self-renewal ability of liver cancer stem-like cells (LSCS) (Fig. 10).303 Although the role of KDM4D on drug resistance is clear in hepatocellular carcinoma, the drug resistance mechanisms of KDM4D in other cancers are less studied. The roles of JMJD2E and JMJD2F on drug resistance are currently still unclear.
KDM5 family and drug resistance: The KDM5 family include JARID1A, JARID1B, JARID1C, and JARID1D, all of which have a significant impact on drug resistance. KDM5A is highly expressed in various cancers.304,305,306 In 2015, Romani et al. reported that in temozolomide-resistant glioblastoma cells, the expression of KDM5A was significantly elevated and its knockdown could promote cell apoptosis.304 In tamoxifen-resistant ER (+) breast tumors, KDM5A can activate IGF1R and ErbB signaling, causing the activation of PI3K/AKT/mTOR pathway and leading to the occurrence of tamoxifen resistance (Fig. 11, Table 3).305 What is more, inhibition of KDM5A can enhance the anti-proliferative ability of WEE1 inhibitor AZD1775 in drug-resistant acute myeloid leukemia cells.306 KDM5A also plays a key role in regulating P-gp and EMT. In detail, KDM5A can increase the expression of P-gp, inhibit cell apoptosis and promote cell proliferation (Table 3), at the same time, overexpression of KDM5A can also promote EMT) and metastasis of paclitaxel-resistant lung adenocarcinoma cells (Fig. 11).307

The roles of KDM5-7 on drug resistance. In KDM5 family, KDM5A can promote PI3K/AKT/mTOR pathway through ErbB and induce EMT in combination with KDM5D inhibition, thereby leading to drug resistance. However, KDM5C exerts opposite effects by regulating PTEN and p53, PBIT can inhibit the demethylase activity of KDM5B, which in turn affects the MAPK pathway and cell stemness. Besides, KDM6A and KDM6B can enhance drug resistance by promoting cell stemness and regulating the cell cycle, respectively. Importantly, PHF8 can also upregulate the expression of IL6 and FOXA2 to promote drug resistance
Like KDM5A, KDM5B is also highly expressed in many cancers and can promote drug resistance. In NSCLC, KDM5B can promote stem cell phenotypes by activating c-Met signaling pathway. While knockdown of KDM5B inhibits cell proliferation, migration, and colony-forming ability.308 Xu et al. demonstrated that KDM5B was highly expressed in cisplatin-resistant gastric cancer cells. It recruited XRCC1 through demethylation of H3K4, thus inhibiting cell apoptosis and increasing cisplatin resistance (Table 3).309 KDM5B is also highly expressed in paclitaxel-resistant endometrial cancer cells, which is regulated by microRNA-29c-3p (a tumor suppressor), thereby promoting drug resistance (Fig. 11).310 Importantly, KDM5B is also closely related to the drug resistance of melanoma, and the expression of KDM5B increased significantly after radiotherapy.311 KDM5B can induce the transformation of CD34 into CD34+ and decrease the sensitivity to BRAF inhibitors in mouse melanoma cells.312 What is more, it can be co-expressed with Ki67 and positively correlated with the proliferative ability of cells in canine tissues (Fig. 11).313
KDM5C is also a drug resistance regulator, but it has been recently reported to have two opposite effects. KDM5C can promote cell proliferation, but can restrain drug resistance. Hong et al. found that in CRPC, KDM5C can promote cell proliferation by inhibiting tumor suppressor PTEN regulated by BRD4 in vivo and in vitro (Fig. 11).314 And overexpression of KDM5C can promote nasopharyngeal carcinoma (NPC) cell proliferation and inhibit its apoptosis, while KDM5C knockdown exhibited the opposite results.315 KDM5C is highly expressed in gastric cancer cells and can promote cell proliferation and migration by inhibiting the expression of p53 and its downstream p21 and p27.316 And the latest study indicates that Homebox D3 can increase the expression of KDM5C and then inhibit p53, thereby promoting the proliferation of diffuse-large B-cell lymphoma (DLBCL).317 But interestingly, Lin et al. have demonstrated that KDM5C overexpression can reduce the expression of ABCC1 through its demethylation, thus inhibiting the resistance of colorectal cancer cells to drugs, such as oxaliplatin and irinotecan (Fig. 11).318 At the same time, knockdown of KDM5C can promote tumor growth and the resistance of ccRCC to ferroptosis (Table 3).319 Collectively, this evidence indicated that KDM5C might play different roles in different cancer cells. Compared with other KDM5s, the roles of KDM5D on drug resistance are less investigated. However, contrary to other members in KDM5 family, the decreased expression of KDM5D is beneficial to the drug resistance of cancer cells. It has been reported that low expression of KDM5D can promote the metastasis of gastric cancer cells by inducing EMT and demethylating H3K4 on CUL4A promoter (Fig. 11).320 Besides, Komura et al. indicated that the deletion or decrease of KDM5D is the major cause of docetaxel resistance. KDM5D can directly regulate the transcriptional activity of androgen, thereby affecting the sensitivity of CRPC to docetaxel.321 Additionally, silencing KDM5D could elevate MYBL2, thereby inducing docetaxel resistance in prostate cancer cells.322 As reported, miR-4661-5p is the upstream regulator of KDM5D. When it is inhibited, the expression of KDM5D is increased, and the expression of Mars2 is decreased, leading to a decrease in malignant behavior of cells (Fig. 11).323 Collectively, the KDM5 family are critical in the drug resistance, and targeting the KDM5 family is an important way to improve the curative effect.
KDM6 family and drug resistance: The KDM6 family plays an important role in drug resistance and cancer recurrence by demethylation of H3K27. Amongst them, KDM6A is a novel relapse-associated gene in AML.324 The reduction of KDM6A expression may decrease ENT1 level and is associated with a decreased cytarabine sensitivity.325 Besides, KDM6A can upregulate TRKA expression through YY1 independent of its demethylation activity, thereby promoting cell resistance to imatinib (Table 3).326 In addition to leukemia, KDM6A is also a key regulator in osteosarcoma, colorectal cancer, and prostate cancer. One study suggested that KDM6A was up-regulated in prostate cancer. USP7 can increase KDM6A expression by deubiquitinating it, while targeting KDM6A can inhibit tumor growth (Fig. 11).327 Furthermore, the low level of H3K27me3 is closely related to cell stemness and oxaliplatin resistance in colorectal cancer. PCGF1 can increase the expression of KDM6A, further reducing H3K27me3 level to activate the transcription of stem cell markers and promoting the proliferation of stem cells.328,329 What is more, the transcription factor GATA3, which is closely related to the poor prognosis of ovarian cancer, can recruit UTX and enhance the stemness of ovarian HGSC cells.330
Like KDM6A, KDM6B is highly expressed in various cancers and is conducive to tumor development and drug resistance. In DLBCL, inhibition of KDM6B can significantly increase the sensitivity of cells to chemotherapeutic drugs.331 Besides, KDM6B increased the expression of IGFBP5, and its inhibition can induce apoptosis and increase the sensitivity of breast cancer cells to GDC-0941.332 Notably, KDM6B can facilitate the expression of C-Myc and its target gene CyclinD1, thereby promoting the proliferation of prostate cancer cells and tumor growth.333,334 Interestingly, KDM6B is overexpressed in neuroblastoma and can activate the CDK4/6-pRB-E2F pathway, which is closely related to palbociclib resistance by demethylation of H3K27 (Fig. 11, Table 3).335 These data suggest that inhibition of KDM6B could become an effective therapeutic strategy to overcome drug resistance.
KDM7 family and drug resistance: Until now, the roles of the KDM7 family on drug resistance have less been studied. KDM7A can upregulate AR transcription activity via demethylating H3K27me2, and KDM7A inhibitor TC-E 5002 can significantly reduce cell viability in cisplatin-resistant bladder cancer cell lines.336 PHF8, also named KDM7B, can enhance the expression of FOXA2, which is essential for the development of neuroendocrine prostate cancer (NEPC) through demethylation modification (Table 3).337 Besides, PHF8 and HER2 can interact with each other in breast cancer, thus promoting the resistance to trastuzumab and other anti-HER2 drugs by regulating the expression of IL6 (Fig. 11).338
Other jumonji containing proteins and drug resistance: In addition to the above six families, some other jumonji containing proteins, particularly JARID2, play an important role on drug resistance and tumor recurrence. Although JARID2 does not have demethylase activity, it can affect the cell stemness by increasing the expression of Notch1 and promoting resistance of non-small lung cell to cisplatin (Table 3).339 Interestingly, JARID2 was downregulated in glioblastoma, and JARID2 overexpression could reduce CCND1 expression and promote apoptosis after treated with temozolomide.340
Histone arginine demethylases and drug resistance
PAD4, which can transform arginine into citrulline, is also closely related to drug resistance. In gefitinib-resistant NSCLC, the expression of PAD4 is significantly downregulated. On the contrary, PAD4 overexpression can inhibit EMT by decreasing Elk1 expression, thereby inhibiting drug resistance.341 Zhou et al. demonstrated that PAD4 was decreased in adriamycin-resistant breast cancer. Overexpression of PAD4 promoted apoptosis and increased the expression of p53 and GSK3β so as to increase the sensitivity of cells to doxorubicin (Fig. 12).342 Interestingly, PAD4 can also induce autophagy and increase LC3B expression in hepatocellular carcinoma, thus promoting cell chemoresistance (Fig. 12).343 Although PAD4 has different influence on drug resistance in different cancers, this evidence suggest that targeting PAD4 might be a feasible strategy to decrease drug resistance.

The opposite roles of histone arginine demethylase PAD4 in drug resistance. PAD4 can not only inhibit the production of drug resistance through regulating EMT and apoptosis, but also promote autophagy and enhance drug resistance
Targeting histone demethylases for overcoming drug resistance
Studies have shown that drug resistance is an important cause of poor prognosis and relapse in cancer patients, and the relationship between histone demethylation and cancer resistance has attracted extensive attention.344 Evidences have suggested that histone demethylase inhibitors or other molecules targeting histone demethylation can effectively control drug resistance. In this section, we will discuss the effects of histone demethylase inhibitors and small interfering RNA on drug resistance.
Targeting LSD family for overcoming drug resistance
LSD1 is closely related to poor prognosis and can promote drug resistance in many cancer cells. Many studies have shown that LSD1 inhibitors can increase the sensitivity of cells and reverse drug resistance.241,345,346,347 Arborinine, a natural product isolated from G. parva, is a reversible LSD1 inhibitor, which can inhibit the proliferation of adriamycin-resistant gastric cancer cells by increasing H3K4me1 and H3K9me1/2 levels and inhibiting EMT (Table 4).345 The LSD1 inhibitor HCI-2509 significantly inhibited colony formation, decreased the expression of C-MYC protein, and blocked the cell cycle to inhibit cell proliferation of PC3 and DU145 cells and docetaxel-resistant prostate cancers (Table 4).346 Additionally, GSK2879552 and pargyline can decrease the number of Lgr5+ CSCs significantly to enhance the sensitivity of drug-resistant cells to sorafenib.267 GSK2879552 can significantly reduce the level of LINC01134 in HepG2 and MHCC97H cells and increase drug sensitivity (Table 4).271 Similarly, Etani et al. first reported that NCL1, a highly selective LSD1 inhibitor, can inhibit cell proliferation and autophagy in a dose-dependent manner both in vivo and in vitro (Table 4).347 More interestingly, targeting the PELP1-KDM1 axis with NCL1 can inhibit the transcription activation of LSD1 mediated ERα target genes and reverse drug resistance to tamoxifen or letrozole.348 Furthermore, 2-PCPA89 and SP2059100 can reverse the drug resistance of breast cancer and NSCLC. To conclude, targeting LSD1 or in combination with LSD1 inhibitors is an effective method to reverse drug resistance. In contrast, the development of LSD2 inhibitors and the capability of reversing drug resistance are poorly studied.
Targeting the JmjC domain-containing proteins for overcoming drug resistance
Increasing evidences have demonstrated that regulating the expression of KDM family can affect the emergence of cell drug resistance, some small-molecule inhibitors and small interfering RNAs (siRNA) capable of overcoming drug resistance have been reported. NSC636819, a KDM4A/B inhibitor, can increase the expression of DR5 and TRAIL and activate exogenous apoptosis by TRAIL-DR5, leading to tumor growth inhibition in vivo and in vitro.349 The KDM4B inhibitor ML324 inhibited the growth of enzalutamide-resistant tumors (Table 4).298 Mar et al. showed that the KDM4A inhibitor JIB-04 can increase H3K36me3 levels and restore the sensitivity of leukemia cells to cytosine arabinoside (Fig. 9).350 Besides, JIB-04 can inhibit KDM6B and play a synergistic role with GSK-J4 in inhibiting the proliferation of TMZ-resistant cells (Table 4).351 Recently, Yang et al. reported that the KDM5A inhibitor ZINC33576 can effectively inhibit the demethylation activity of KDM5A, block the cell in G1 phase, and induce cell senescence, thereby playing an anti-proliferative effect (Table 4).352 The KDM5A inhibitor YUKA1 significantly inhibited the proliferation of gefitinib-resistant EGFR-mutant lung cancer cells.353 In NSCLC, PBIT, an inhibitor of KDM5B, can reduce KDM5B expression and inhibit lung cancer stem cells (LSCS) like phenotype to increase cell sensitivity to cisplatin and doxorubicin.308 When PBIT was used, the cell survival rate of cisplatin-resistant canine melanoma cell line decreased, indicating that inhibiting KDM5B can reduce the resistance of cells to cisplatin (Fig. 10).313 Interestingly, CPI-455 was identified as a specific inhibitor of the KDM5 family.354,355 CPI-455 can decrease the survival of erlotinib-tolerant cells after lethal drug exposures in multiple cell culture models and significantly inhibited the proliferation of temozolomide-resistant glioblastoma cells.356,357 In addition, CPI-455 can inhibit KDM5A to sensitize NB4 cells to all-trans retinoic acid which induced cell differentiation in acute promyelocytic leukemia (APL).358
The KDM6 inhibitor GSK-J4 is effective in reversing resistance of various cancers, such as colorectal cancer, chondrosarcoma, and lymphoma (Table 4). GSK-J4 can inhibit the expression of Notch2, thereby increasing the sensitivity of colorectal cancer to oxaliplatin.329 Significantly, GSK-J4 can cooperate with 5-FU to inhibit the proliferation of colorectal cancer and reduce cell stemness, thus increasing sensitivity.359 In addition, Lhuissier et al. found that chondrosarcoma is resistant to both radiotherapy and chemotherapy. And the combination of GSK-J4 and cisplatin can significantly inhibit the proliferation of chondrosarcoma cells.360 Moreover, when GSK-J4 is combined with vincristine and doxorubicin, it can significantly promote the apoptosis of DLBCL.332 Interestingly, in addition to KDM6A and KDM6B, GSK-J4 also inhibited KDM2B, reduced the expression of KDM2B, and increased the H3K36 methylation levels, thereby reducing the self-renewal ability of stem cells and increased the sensitivity of cells to drugs.282
Besides, siRNA can also regulate the expression of KDM family proteins. Circ0006168, a cyclic RNA having a complementary miR-194-5p binding sequence with KDM3C, can positively regulate the expression of KDM3C, and its downregulation can inhibit the growth of tumors and enhance the sensitivity of cells to paclitaxel by decreasing the expression of KDM3C (Table 4).290 In conclusion, the KDM family is an important regulator of tumor resistance. Except for the inhibitory role of KDM5C in cancer resistance, the other KDM family members often promote tumor resistance. Therefore, targeting the KDM family proteins could be an effective strategy to overcome drug resistance. Of note, although there exist many PAD4 inhibitors, their association with drug resistance has not been described.
Conclusions and perspectives
Malignant tumors seriously threaten human health, and the inevitable emergence of drug resistance in tumors has posed continuous challenges to chemotherapeutics. The mechanisms for tumor resistance are complex, and epigenetic regulation could mediate cancer resistance. In this review, we mainly discuss the association of DNA methylation, non-coding RNAs, histone acetylation, and histone methylation with tumor resistance. Evidence has suggested that epigenetic regulators may act as promoters or inhibitors in different tumors. Many epigenetic regulators are overexpressed in tumors and contribute to drug resistance mainly by inhibiting apoptosis, inducing autophagy and EMT, enhancing stemness, and activating multiple signaling pathways including the PI3K/Akt, Notchand NF-κB pathways, etc. In addition, p53 repression is also an important mechanism for epigenetic regulators to promote tumor resistance. As a tumor suppressor, p53 expression can be suppressed by lncRNA Miat, KAT2A, LSD2, KDM3A, and KDM5C, which in turn promote drug resistance. For these regulators that promote tumor progression and drug resistance, targeted inhibition can effectively increase tumor sensitivity to chemotherapy. Interestingly, there are also some regulators that can act as a double-edged sword, such as DNA methyltransferase DNMT1, DNA demethylases TET1 and TET2, HAT p300, and histone demethylases KDM5C. Therefore, further elucidation of their specific roles in different cancers is urgently required to achieve the goal of targeting epigenetics to overcome drug resistance precisely.
Clearly, histone demethylases can regulate tumorigenesis in a manner dependent or independent of their demethylase activity. Currently, more than 20 histone demethylases have been discovered, and these demethylases are involved in the development of drug resistance through multiple mechanisms. As mentioned above, KDM5C showed the opposite effects in different cancers. For example, in colon cancer and clear cell renal cell carcinoma, overexpression of KDM5C can inhibit drug resistance, while in other tumors, such as gastric cancer and CRPC, KDM5C promotes tumor growth and drug resistance. However, other demethylases, which are always highly expressed in various cancers, often promote the development of drug resistance. Until now, many histone demethylase inhibitors have been developed, and several have already entered clinical trials, providing a direction for the development of targeted inhibitors. However, in addition to LSD1 inhibitors, almost all other demethylase inhibitors are in preclinical studies. Unfortunately, although many histone demethylase inhibitors and siRNA have been reported to be able to overcome drug resistance, these inhibitors mentioned above have not entered the clinical stage. Additionally, the issue of how to improve the selectivity and specificity of epigenetic inhibitors to avoid disturbing the overall epigenetic status should also be considered.
Notably, when used alone or in combination with other drugs, these inhibitors can often increase the efficacy of chemotherapeutics. Considering the interaction between some epigenetic regulators to co-regulate tumor development, the combination of two or more inhibitors with different targets may exert better effects to reverse drug resistance. Therefore, the development of multiple target inhibitors, proteolysis targeting chimeric (PROTAC) molecules, allosteric inhibitors, and those inhibitors regulating non-enzymatic functions may also provide new strategies for reversing tumor resistance. Overall, the regulatory mechanisms of epigenetics in cancer resistance provide a molecular basis for personalized cancer therapy, and targeting epigenetics holds great promise in overcoming tumor resistance.
References
Ugai, T. et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat. Rev. Clin. Oncol. 19, 656–673 (2022).
Nagasaka, M. & Gadgeel, S. M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert Rev. Anticancer Ther. 18, 63–70 (2018).
Sato, H., Demaria, S. & Ohno, T. The role of radiotherapy in the age of immunotherapy. Jpn. J. Clin. Oncol. 51, 513–522 (2021).
Orcutt, S. T. & Anaya, D. A. Liver resection and surgical strategies for management of primary liver cancer. Cancer Control 25, 1073274817744621 (2018).
Chow, A., Perica, K., Klebanoff, C. A. & Wolchok, J. D. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat. Rev. Clin. Oncol. 19, 775–790 (2022).
Robey, R. W. et al. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 18, 452–464 (2018).
Wang, S. Q. et al. Preclinical studies of the triazolo[1,5-a]pyrimidine derivative WS-716 as a highly potent, specific and orally active P-glycoprotein (P-gp) inhibitor. Acta Pharm. Sin. B 12, 3263–3280 (2022).
Yuan, S. et al. Discovery of new 4-indolyl quinazoline derivatives as highly potent and orally bioavailable p-glycoprotein inhibitors. J. Med. Chem. 64, 14895–14911 (2021).
Fletcher, J. I., Williams, R. T., Henderson, M. J., Norris, M. D. & Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 26, 1–9 (2016).
Wang, S. et al. Discovery of the triazolo[1,5-a]pyrimidine-based derivative WS-898 as a highly efficacious and orally bioavailable ABCB1 inhibitor capable of overcoming multidrug resistance. J. Med. Chem. 64, 16187–16204 (2021).
Das, S., Shukla, N., Singh, S. S., Kushwaha, S. & Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 26, 512–533 (2021).
Hussein, N. A. et al. The role of endolysosomal trafficking in anticancer drug resistance. Drug Resist. Updat. 57, 100769 (2021).
Zhou, H. M., Zhang, J. G., Zhang, X. & Li, Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct. Target Ther. 6, 62 (2021).
Takebe, N. et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat. Rev. Clin. Oncol. 12, 445–464 (2015).
Pan, G., Liu, Y., Shang, L., Zhou, F. & Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 41, 199–217 (2021).
De Las Rivas, J. et al. Cancer drug resistance induced by EMT: novel therapeutic strategies. Arch. Toxicol. 95, 2279–2297 (2021).
Sun, Y. et al. Allosteric SHP2 inhibitor, IACS-13909, overcomes EGFR-dependent and EGFR-independent resistance mechanisms toward osimertinib. Cancer Res. 80, 4840–4853 (2020).
Zhang, X. et al. Alterations in the global proteome and phosphoproteome in third generation EGFR TKI resistance reveal drug targets to circumvent resistance. Cancer Res. 81, 3051–3066 (2021).
Woyach, J. A. et al. BTK(C481S)-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J. Clin. Oncol. 35, 1437–1443 (2017).
Hake, S. B., Xiao, A. & Allis, C. D. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br. J. Cancer 90, 761–769 (2004).
Zhang, L., Lu, Q. & Chang, C. Epigenetics in health and disease. Adv. Exp. Med. Biol. 1253, 3–55 (2020).
Wajapeyee, N. & Gupta, R. Epigenetic alterations and mechanisms that drive resistance to targeted cancer therapies. Cancer Res. 81, 5589–5595 (2021).
Pasculli, B., Barbano, R. & Parrella, P. Epigenetics of breast cancer: biology and clinical implication in the era of precision medicine. Semin. Cancer Biol. 51, 22–35 (2018).
Jin, B., Li, Y. & Robertson, K. D. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer 2, 607–617 (2011).
Robertson, K. D. DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 (2005).
Yang, X. et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 26, 577–590 (2014).
Smith, M. M. Histone structure and function. Curr. Opin. Cell Biol. 3, 429–437 (1991).
Tessarz, P. & Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 15, 703–708 (2014).
Jarome, T. J. et al. Ubiquitination of histone H2B by proteasome subunit RPT6 controls histone methylation chromatin dynamics during memory formation. Biol. Psychiatry 89, 1176–1187 (2021).
Messner, S. & Hottiger, M. O. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 21, 534–542 (2011).
Zhu, D., Zhang, Y. & Wang, S. Histone citrullination: a new target for tumors. Mol. Cancer 20, 90 (2021).
Messner, S. et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 38, 6350–6362 (2010).
Højfeldt, J. W., Agger, K. & Helin, K. Histone lysine demethylases as targets for anticancer therapy. Nat. Rev. Drug Discov. 12, 917–930 (2013).
Chen, Y. et al. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Signal Transduct. Target Ther. 5, 143 (2020).
Wiesel-Motiuk, N. & Assaraf, Y. G. The key roles of the lysine acetyltransferases KAT6A and KAT6B in physiology and pathology. Drug Resist. Updat. 53, 100729 (2020).
Seligson, D. B. et al. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 174, 1619–1628 (2009).
Chen, Y. C., Koutelou, E. & Dent, S. Y. R. Now open: evolving insights to the roles of lysine acetylation in chromatin organization and function. Mol. Cell 82, 716–727 (2022).
Garcia-Martinez, L., Zhang, Y., Nakata, Y., Chan, H. L. & Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nat. Commun. 12, 1786 (2021).
Asano, T. Drug resistance in cancer therapy and the role of epigenetics. J. Nippon Med. Sch. 87, 244–251 (2020).
Greger, V., Passarge, E., Höpping, W., Messmer, E. & Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 83, 155–158 (1989).
Esteller, M. et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl Cancer Inst. 92, 564–569 (2000).
Al-Yozbaki, M., Jabre, I., Syed, N. H. & Wilson, C. M. Targeting DNA methyltransferases in non-small-cell lung cancer. Semin. Cancer Biol. 83, 77–87 (2022).
Probst, A. V., Dunleavy, E. & Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol. 10, 192–206 (2009).
Okano, M., Bell, D. W., Haber, D. A. & Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999).
Kareta, M. S., Botello, Z. M., Ennis, J. J., Chou, C. & Chédin, F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 281, 25893–25902 (2006).
Wang, Q. et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 75, 1142–1153 (2021).
Wei, Y. et al. The interaction between DNMT1 and high-mannose CD133 maintains the slow-cycling state and tumorigenic potential of glioma stem cell. Adv. Sci. 9, e2202216 (2022).
Ma, Y. et al. DNA methyltransferase mediates the hypermethylation of the microRNA 34a promoter and enhances the resistance of patient-derived pancreatic cancer cells to molecular targeting agents. Pharmacol. Res. 160, 105071 (2020).
Jahangiri, R., Mosaffa, F., Emami Razavi, A., Teimoori-Toolabi, L. & Jamialahmadi, K. Altered DNA methyltransferases promoter methylation and mRNA expression are associated with tamoxifen response in breast tumors. J. Cell. Physiol. 233, 7305–7319 (2018).
Camero, S. et al. DNMT3A and DNMT3B targeting as an effective radiosensitizing strategy in embryonal rhabdomyosarcoma. Cells 10, 2956 (2021).
Lai, S. C. et al. DNMT3b/OCT4 expression confers sorafenib resistance and poor prognosis of hepatocellular carcinoma through IL-6/STAT3 regulation. J. Exp. Clin. Cancer Res. 38, 474 (2019).
Simó-Riudalbas, L., Melo, S. A. & Esteller, M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes Chromosomes Cancer 50, 527–534 (2011).
Yu, J. et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J. Clin. Invest. 128, 2376–2388 (2018).
Stewart, M. L. et al. KRAS genomic status predicts the sensitivity of ovarian cancer cells to decitabine. Cancer Res. 75, 2897–2906 (2015).
Liu, H. et al. Regulation of T cell differentiation and function by epigenetic modification enzymes. Semin. Immunopathol. 41, 315–326 (2019).
Branco, M. R., Ficz, G. & Reik, W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat. Rev. Genet. 13, 7–13 (2011).
Kang, K. A. et al. DUOX2-mediated production of reactive oxygen species induces epithelial mesenchymal transition in 5-fluorouracil resistant human colon cancer cells. Redox Biol. 17, 224–235 (2018).
Tang, X. et al. Upregulation of CRABP2 by TET1-mediated DNA hydroxymethylation attenuates mitochondrial apoptosis and promotes oxaliplatin resistance in gastric cancer. Cell Death Dis. 13, 848 (2022).
Forloni, M. et al. Oncogenic EGFR represses the TET1 DNA demethylase to induce silencing of tumor suppressors in cancer cells. Cell Rep. 16, 457–471 (2016).
Kharat, S. S. et al. Degradation of 5hmC-marked stalled replication forks by APE1 causes genomic instability. Sci. Signal. 13, eaba8091 (2020).
Kim, M. R., Wu, M. J., Zhang, Y., Yang, J. Y. & Chang, C. J. TET2 directs mammary luminal cell differentiation and endocrine response. Nat. Commun. 11, 4642 (2020).
Li, Y., Lu, D. G., Ma, Y. M. & Liu, H. Association between Retinoic acid receptor-β hypermethylation and NSCLC risk: a meta-analysis and literature review. Oncotarget 8, 5814–5822 (2017).
Zhang, X. et al. Aberrant epigenetic regulation of RARβ by TET2 is involved in cutaneous squamous cell carcinoma resistance to retinoic acid. Int. J. Biochem. Cell Biol. 145, 106190 (2022).
Cui, Q. et al. Downregulation of TLX induces TET3 expression and inhibits glioblastoma stem cell self-renewal and tumorigenesis. Nat. Commun. 7, 10637 (2016).
Herrmann, A. et al. Integrin α6 signaling induces STAT3-TET3-mediated hydroxymethylation of genes critical for maintenance of glioma stem cells. Oncogene 39, 2156–2169 (2020).
Quagliano, A., Gopalakrishnapillai, A. & Barwe, S. P. Understanding the Mechanisms by Which Epigenetic Modifiers Avert Therapy Resistance in Cancer. Front. Oncol. 10, 992 (2020).
Singh, M. et al. Current paradigms in epigenetic anticancer therapeutics and future challenges. Semin. Cancer Biol. 83, 422–440 (2022).
Slack, F. J. & Chinnaiyan, A. M. The Role of Non-coding RNAs in Oncology. Cell 179, 1033–1055 (2019).
Hussain, S. et al. Role of epigenetics in carcinogenesis: Recent advancements in anticancer therapy. Semin. Cancer Biol. 83, 441–451 (2022).
Khanbabaei, H. et al. Non-coding RNAs and epithelial mesenchymal transition in cancer: molecular mechanisms and clinical implications. J. Exp. Clin. Cancer Res. 41, 278 (2022).
Ha, M. & Kim, V. N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524 (2014).
Babar, I. A. et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl Acad. Sci. USA 109, E1695–E1704 (2012).
Medina, P. P., Nolde, M. & Slack, F. J. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467, 86–90 (2010).
El Fatimy, R., Subramanian, S., Uhlmann, E. J. & Krichevsky, A. M. Genome editing reveals glioblastoma addiction to MicroRNA-10b. Mol. Ther. 25, 368–378 (2017).
Trang, P. et al. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol. Ther. 19, 1116–1122 (2011).
Klein, U. et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 17, 28–40 (2010).
Concepcion, C. P. et al. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 8, e1002797 (2012).
Huang, B. et al. tRF/miR-1280 suppresses stem cell-like cells and metastasis in colorectal cancer. Cancer Res. 77, 3194–3206 (2017).
Ozata, D. M., Gainetdinov, I., Zoch, A., O’Carroll, D. & Zamore, P. D. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20, 89–108 (2019).
Yan, H. et al. piRNA-823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia 29, 196–206 (2015).
Chattopadhyay, T., Biswal, P., Lalruatfela, A. & Mallick, B. Emerging roles of PIWI-interacting RNAs (piRNAs) and PIWI proteins in head and neck cancer and their potential clinical implications. Biochim. Biophys. Acta Rev. Cancer 1877, 188772 (2022).
Jia, D. D. et al. The regulatory function of piRNA/PIWI complex in cancer and other human diseases: the role of DNA methylation. Int. J. Biol. Sci. 18, 3358–3373 (2022).
Tollervey, D. & Kiss, T. Function and synthesis of small nucleolar RNAs. Curr. Opin. Cell Biol. 9, 337–342 (1997).
Weinstein, L. B. & Steitz, J. A. Guided tours: from precursor snoRNA to functional snoRNP. Curr. Opin. Cell. Biol. 11, 378–384 (1999).
Williams, G. T. & Farzaneh, F. Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer 12, 84–88 (2012).
Wong, C. M., Tsang, F. H. & Ng, I. O. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 15, 137–151 (2018).
Mei, Y. P. et al. Small nucleolar RNA 42 acts as an oncogene in lung tumorigenesis. Oncogene 31, 2794–2804 (2012).
Dong, X. Y. et al. Implication of snoRNA U50 in human breast cancer. J. Genet. Genomics 36, 447–454 (2009).
Dong, X. Y. et al. SnoRNA U50 is a candidate tumor-suppressor gene at 6q14.3 with a mutation associated with clinically significant prostate cancer. Hum. Mol. Genet. 17, 1031–1042 (2008).
Chen, L. L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 21, 475–490 (2020).
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019).
Yang, F. et al. Cis-acting circ-CTNNB1 promotes β-catenin signaling and cancer progression via DDX3-mediated transactivation of YY1. Cancer Res. 79, 557–571 (2019).
Chen, S. et al. Widespread and functional RNA circularization in localized prostate cancer. Cell 176, 831–843.e822 (2019).
Zhou, Y. et al. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 282, 24731–24742 (2007).
Loewen, G., Jayawickramarajah, J., Zhuo, Y. & Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 7, 90 (2014).
Dhamija, S. & Diederichs, S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int. J. Cancer 139, 269–280 (2016).
Lampropoulou, D. I., Pliakou, E., Aravantinos, G., Filippou, D. & Gazouli, M. The role of exosomal non-coding RNAs in colorectal cancer drug resistance. Int. J. Mol. Sci. 23, 1473 (2022).
Wei, S., Hu, W., Feng, J. & Geng, Y. Promotion or remission: a role of noncoding RNAs in colorectal cancer resistance to anti-EGFR therapy. Cell Commun. Signal. 20, 150 (2022).
Li, J. et al. Emerging role of noncoding RNAs in EGFR TKI-resistant lung cancer. Cancers 14, 4423 (2022).
Najafi, F. et al. The role of miRNA-424 and miR-631 in various cancers: focusing on drug resistance and sensitivity. Pathol. Res. Pract. 239, 154130 (2022).
Zhou, X. et al. Non-coding RNA in cancer drug resistance: underlying mechanisms and clinical applications. Front. Oncol. 12, 951864 (2022).
Li, G. et al. Piwi-interacting RNA1037 enhances chemoresistance and motility in human oral squamous cell carcinoma cells. Onco Targets Ther. 12, 10615–10627 (2019).
Das, B., Jain, N. & Mallick, B. piR-39980 mediates doxorubicin resistance in fibrosarcoma by regulating drug accumulation and DNA repair. Commun. Biol. 4, 1312 (2021).
Peng, K. L. et al. Miat and interacting protein Metadherin maintain a stem-like niche to promote medulloblastoma tumorigenesis and treatment resistance. Proc. Natl Acad. Sci. USA 119, e2203738119 (2022).
Zhu, Y., Yang, L., Wang, J., Li, Y. & Chen, Y. SP1-induced lncRNA MCF2L-AS1 promotes cisplatin resistance in ovarian cancer by regulating IGF2BP1/IGF2/MEK/ERK axis. J. Gynecol. Oncol. 33, e75 (2022).
Zhang, S. et al. Tumor-derived extracellular vesicles confer 5-fluorouracil resistance in esophageal cancer via long noncoding RNA AC116025.2 delivery. Mol. Carcinog. 61, 1177–1190 (2022).
Shi, Y. et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Signal Transduct. Target Ther. 7, 200 (2022).
McBrian, M. A. et al. Histone acetylation regulates intracellular pH. Mol. Cell 49, 310–321 (2013).
Huang, R. et al. The role of HDAC2 in chromatin remodelling and response to chemotherapy in ovarian cancer. Oncotarget 7, 4695–4711 (2016).
Shahbazian, M. D. & Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100 (2007).
Yang, Q. et al. Epigenetics in ovarian cancer: premise, properties, and perspectives. Mol. Cancer 17, 109 (2018).
Roth, S. Y., Denu, J. M. & Allis, C. D. Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 (2001).
Ding, P. et al. Lysine acetylation/deacetylation modification of immune-related molecules in cancer immunotherapy. Front. Immunol. 13, 865975 (2022).
Parthun, M. R. Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 26, 5319–5328 (2007).
Cao, L. et al. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. Int. J. Oncol. 44, 1207–1214 (2014).
Zhao, L., Wang, D. L., Liu, Y., Chen, S. & Sun, F. L. Histone acetyltransferase hMOF promotes S phase entry and tumorigenesis in lung cancer. Cell. Signal. 25, 1689–1698 (2013).
Pfister, S. et al. The histone acetyltransferase hMOF is frequently downregulated in primary breast carcinoma and medulloblastoma and constitutes a biomarker for clinical outcome in medulloblastoma. Int. J. Cancer 122, 1207–1213 (2008).
Jaiswal, B. & Gupta, A. Modulation of nuclear receptor function by chromatin modifying factor TIP60. Endocrinology 159, 2199–2215 (2018).
Jaiswal, B., Agarwal, A. & Gupta, A. Lysine acetyltransferases and their role in AR signaling and prostate cancer. Front. Endocrinol. 13, 886594 (2022).
Waddell, A., Mahmud, I., Ding, H., Huo, Z. & Liao, D. Pharmacological inhibition of CBP/p300 blocks estrogen receptor alpha (ERα) function through suppressing enhancer H3K27 acetylation in luminal breast cancer. Cancers (Basel) 13, 2799 (2021).
Ansari, M. S. Z. et al. Pharmacological targeting of CBP/p300 drives a redox/autophagy axis leading to senescence-induced growth arrest in non-small cell lung cancer cells. Cancer Gene Ther. 30, 124–136 (2023).
Haque, M. E. et al. The GCN5: its biological functions and therapeutic potentials. Clin. Sci. 135, 231–257 (2021).
Ding, Y. et al. Leukemia inhibitory factor receptor homodimerization mediated by acetylation of extracellular lysine promotes prostate cancer progression through the PDPK1/AKT/GCN5 axis. Clin. Transl. Med. 12, e676 (2022).
Trush, V. V. et al. Enzymatic nucleosome acetylation selectively affects activity of histone methyltransferases in vitro. Biochim. Biophys. Acta Gene Regul. Mech. 1865, 194845 (2022).
Hong, Z. et al. Histone acetyltransferase 1 upregulates androgen receptor expression to modulate CRPC cell resistance to enzalutamide. Clin. Transl. Med. 11, e495 (2021).
Sun, Y. et al. Histone acetyltransferase 1 promotes gemcitabine resistance by regulating the PVT1/EZH2 complex in pancreatic cancer. Cell Death Dis. 12, 878 (2021).
Liu, W. et al. KAT6A, a novel regulator of β-catenin, promotes tumorigenicity and chemoresistance in ovarian cancer by acetylating COP1. Theranostics 11, 6278–6292 (2021).
Lu, D. et al. KAT2A-mediated AR translocation into nucleus promotes abiraterone-resistance in castration-resistant prostate cancer. Cell Death Dis. 12, 787 (2021).
Oh, J. H. et al. Elevated GCN5 expression confers tamoxifen resistance by upregulating AIB1 expression in ER-positive breast cancer. Cancer Lett. 495, 145–155 (2020).
Zhong, Z., Harmston, N., Wood, K. C., Madan, B. & Virshup, D. M. A p300/GATA6 axis determines differentiation and Wnt dependency in pancreatic cancer models. J. Clin. Invest. 132, e156305 (2022).
Takeuchi, A. et al. p300 mediates cellular resistance to doxorubicin in bladder cancer. Mol. Med. Rep. 5, 173–176 (2012).
Yu, G. et al. TWIST1-EP300 expedites gastric cancer cell resistance to apatinib by activating the expression of COL1A2. Anal. Cell. Pathol. 2022, 5374262 (2022).
Zhang, F. et al. Targeting the p300/NONO axis sensitizes melanoma cells to BRAF inhibitors. Oncogene 40, 4137–4150 (2021).
Huang, M., Huang, J., Zheng, Y. & Sun, Q. Histone acetyltransferase inhibitors: an overview in synthesis, structure-activity relationship and molecular mechanism. Eur. J. Med. Chem. 178, 259–286 (2019).
He, Z. X. et al. Current development of CBP/p300 inhibitors in the last decade. Eur. J. Med. Chem. 209, 112861 (2021).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009).
Shvedunova, M. & Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 23, 329–349 (2022).
Alseksek, R. K., Ramadan, W. S., Saleh, E. & El-Awady, R. The role of HDACs in the response of cancer cells to cellular stress and the potential for therapeutic intervention. Int. J. Mol. Sci. 23, 8141 (2022).
Brancolini, C., Gagliano, T. & Minisini, M. HDACs and the epigenetic plasticity of cancer cells: target the complexity. Pharmacol. Ther. 238, 108190 (2022).
Yu, S. et al. Targeting HSP90-HDAC6 regulating network implicates precision treatment of breast cancer. Int. J. Biol. Sci. 13, 505–517 (2017).
Wang, Z., Hu, P., Tang, F. & Xie, C. HDAC6-mediated EGFR stabilization and activation restrict cell response to sorafenib in non-small cell lung cancer cells. Med. Oncol. 33, 50 (2016).
Kim, Y., Kim, H. & Jeoung, D. Tubulin Beta3 SERVES AS A TARGET of HDAC3 and mediates resistance to microtubule-targeting drugs. Mol. Cells 38, 705–714 (2015).
Yang, W. B. et al. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro Oncol. 22, 1439–1451 (2020).
Hu, C. et al. The USP10-HDAC6 axis confers cisplatin resistance in non-small cell lung cancer lacking wild-type p53. Cell Death Dis. 11, 328 (2020).
Zhao, J. et al. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 38, 252 (2019).
Yang, M. et al. FOXQ1-mediated SIRT1 upregulation enhances stemness and radio-resistance of colorectal cancer cells and restores intestinal microbiota function by promoting β-catenin nuclear translocation. J. Exp. Clin. Cancer Res. 41, 70 (2022).
Brunetta, H. S. et al. Nitrate consumption preserves HFD-induced skeletal muscle mitochondrial ADP sensitivity and lysine acetylation: a potential role for SIRT1. Redox Biol. 52, 102307 (2022).
Martino, E. et al. SIRT3 modulates endothelial mitochondrial redox state during insulin resistance. Antioxid. (Basel.) 11, 1611 (2022).
Hanisch, D. et al. Class I HDAC overexpression promotes temozolomide resistance in glioma cells by regulating RAD18 expression. Cell Death Dis. 13, 293 (2022).
Singh, A. K., Bishayee, A. & Pandey, A. K. Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients 10, 731 (2018).
Jenke, R., Reßing, N., Hansen, F. K., Aigner, A. & Büch, T. Anticancer therapy with HDAC inhibitors: mechanism-based combination strategies and future perspectives. Cancers 13, 634 (2021).
Zhou, M. et al. Combining histone deacetylase inhibitors (HDACis) with other therapies for cancer therapy. Eur. J. Med. Chem. 226, 113825 (2021).
Baldwin, R. M., Morettin, A. & Côté, J. Role of PRMTs in cancer: could minor isoforms be leaving a mark? World J. Biol. Chem. 5, 115–129 (2014).
Karkhanis, V., Hu, Y. J., Baiocchi, R. A., Imbalzano, A. N. & Sif, S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 36, 633–641 (2011).
Hwang, J. W., Cho, Y., Bae, G. U., Kim, S. N. & Kim, Y. K. Protein arginine methyltransferases: promising targets for cancer therapy. Exp. Mol. Med. 53, 788–808 (2021).
Jarrold, J. & Davies, C. C. PRMTs and arginine methylation: cancer’s best-kept secret? Trends Mol. Med. 25, 993–1009 (2019).
Cao, Y. C. et al. Histone lysine methylation modification and its role in vascular calcification. Front. Endocrinol. 13, 863708 (2022).
Audia, J. E. & Campbell, R. M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 8, a019521 (2016).
Luo, M. Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 118, 6656–6705 (2018).
Singh, P. K. Histone methyl transferases: a class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur. J. Med. Chem. 166, 351–368 (2019).
Sun, J. et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene 38, 564–580 (2019).
Liu, X. et al. LINC00665 induces acquired resistance to gefitinib through recruiting EZH2 and activating PI3K/AKT pathway in NSCLC. Mol. Ther. Nucleic Acids 16, 155–161 (2019).
Li, Y. et al. Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct. Target Ther. 7, 87 (2022).
Liu, Z. et al. SETDB1 interactions with PELP1 contributes to breast cancer endocrine therapy resistance. Breast Cancer Res. 24, 26 (2022).
Zhou, M. et al. Targeting protein lysine methyltransferase G9A impairs self-renewal of chronic myelogenous leukemia stem cells via upregulation of SOX6. Oncogene 40, 3564–3577 (2021).
Hamard, P. J. et al. PRMT5 regulates DNA repair by controlling the alternative splicing of histone-modifying enzymes. Cell Rep. 24, 2643–2657 (2018).
Dominici, C. et al. Synergistic effects of type I PRMT and PARP inhibitors against non-small cell lung cancer cells. Clin. Epigenetics 13, 54 (2021).
Duan, R., Du, W. & Guo, W. EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104 (2020).
Kaniskan, H., Martini, M. L. & Jin, J. Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 118, 989–1068 (2018).
Song, Y., Wu, F. & Wu, J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 9, 49 (2016).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Maiques-Diaz, A. & Somervaille, T. C. LSD1: biologic roles and therapeutic targeting. Epigenomics 8, 1103–1116 (2016).
Yang, M. et al. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 14, 535–539 (2007).
Fu, D. J., Li, J. & Yu, B. Annual review of LSD1/KDM1A inhibitors in 2020. Eur. J. Med. Chem. 214, 113254 (2021).
Aravind, L. & Iyer, L. M. The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin-modifying activities. Genome Biol. 3, Research0039 (2002).
Stavropoulos, P., Blobel, G. & Hoelz, A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 13, 626–632 (2006).
Gu, F. et al. Biological roles of LSD1 beyond its demethylase activity. Cell. Mol. Life Sci. 77, 3341–3350 (2020).
Song, Y. et al. Mechanism of crosstalk between the LSD1 demethylase and HDAC1 deacetylase in the CoREST complex. Cell Rep. 30, 2699–2711.e8 (2020).
Shi, Y. et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 (2003).
Wang, Y. et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 (2009).
Wang, J. et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 41, 125–129 (2009).
Metzger, E. et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439 (2005).
Huang, J. et al. p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108 (2007).
Carnesecchi, J., Cerutti, C., Vanacker, J. M. & Forcet, C. ERRα protein is stabilized by LSD1 in a demethylation-independent manner. PLoS One 12, e0188871 (2017).
Chao, A. et al. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 8, 74434–74450 (2017).
He, Y. et al. LSD1 promotes S-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation. Oncogene 37, 534–543 (2018).
Zheng, Y. C. et al. A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev. 35, 1032–1071 (2015).
Fang, R. et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 39, 222–233 (2010).
Zhang, Q. et al. Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Res. 23, 225–241 (2013).
Cai, S. et al. Lysine-specific histone demethylase 1B (LSD2/KDM1B) represses p53 expression to promote proliferation and inhibit apoptosis in colorectal cancer through LSD2-mediated H3K4me2 demethylation. Aging 12, 14990–15001 (2020).
Mino, K. et al. Regulation of tissue factor pathway inhibitor-2 (TFPI-2) expression by lysine-specific demethylase 1 and 2 (LSD1 and LSD2). Biosci. Biotechnol. Biochem. 78, 1010–1017 (2014).
Yang, Y., Yin, X., Yang, H. & Xu, Y. Histone demethylase LSD2 acts as an E3 ubiquitin ligase and inhibits cancer cell growth through promoting proteasomal degradation of OGT. Mol. Cell 58, 47–59 (2015).
Vacík, T., Lađinović, D. & Raška, I. KDM2A/B lysine demethylases and their alternative isoforms in development and disease. Nucleus 9, 431–441 (2018).
Tsukada, Y. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 (2006).
Shou, T., Yang, H., Lv, J., Liu, D. & Sun, X. MicroRNA‑3666 suppresses the growth and migration of glioblastoma cells by targeting KDM2A. Mol. Med. Rep. 19, 1049–1055 (2019).
Yang, H. et al. Secreted frizzled-related protein 2 promotes the osteo/odontogenic differentiation and paracrine potentials of stem cells from apical papilla under inflammation and hypoxia conditions. Cell Prolif. 53, e12694 (2020).
Wang, F. et al. LINC00460 modulates KDM2A to promote cell proliferation and migration by targeting miR-342-3p in gastric cancer. Onco Targets Ther. 11, 6383–6394 (2018).
Liu, H., Liu, L., Holowatyj, A., Jiang, Y. & Yang, Z. Q. Integrated genomic and functional analyses of histone demethylases identify oncogenic KDM2A isoform in breast cancer. Mol. Carcinog. 55, 977–990 (2016).
Rizwani, W., Schaal, C., Kunigal, S., Coppola, D. & Chellappan, S. Mammalian lysine histone demethylase KDM2A regulates E2F1-mediated gene transcription in breast cancer cells. PLoS One 9, e100888 (2014).
De Nicola, I. et al. Combined expression levels of KDM2A and KDM2B correlate with nucleolar size and prognosis in primary breast carcinomas. Histol. Histopathol. 35, 1181–1187 (2020).
Frescas, D., Guardavaccaro, D., Bassermann, F., Koyama-Nasu, R. & Pagano, M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature 450, 309–313 (2007).
Penzo, M. et al. JHDM1B expression regulates ribosome biogenesis and cancer cell growth in a p53 dependent manner. Int. J. Cancer 136, E272–E281 (2015).
He, J., Nguyen, A. T. & Zhang, Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood 117, 3869–3880 (2011).
Sui, Y., Gu, R. & Janknecht, R. Crucial functions of the JMJD1/KDM3 epigenetic regulators in cancer. Mol. Cancer Res. 19, 3–13 (2021).
Jeon, H. Y., Ryu, H., Pornour, M. & Qi, J. Histone demethylase JMJD1A in cancer progression and therapeutic resistance. Mol. Carcinog. 61, 392–396 (2022).
Dandawate, P. et al. The histone demethylase KDM3A, increased in human pancreatic tumors, regulates expression of DCLK1 and promotes tumorigenesis in mice. Gastroenterology 157, 1646–1659.e11 (2019).
Hou, X. et al. KDM1A and KDM3A promote tumor growth by upregulating cell cycle-associated genes in pancreatic cancer. Exp. Biol. Med. 246, 1869–1883 (2021).
Katoh, M. & Katoh, M. Identification and characterization of JMJD2 family genes in silico. Int. J. Oncol. 24, 1623–1628 (2004).
Manni, W., Jianxin, X., Weiqi, H., Siyuan, C. & Huashan, S. JMJD family proteins in cancer and inflammation. Signal Transduct. Target Ther. 7, 304 (2022).
Berry, W. L. & Janknecht, R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 73, 2936–2942 (2013).
Young, L. C. & Hendzel, M. J. The oncogenic potential of Jumonji D2 (JMJD2/KDM4) histone demethylase overexpression. Biochem. Cell Biol. 91, 369–377 (2013).
Agger, K. et al. The KDM4/JMJD2 histone demethylases are required for hematopoietic stem cell maintenance. Blood 134, 1154–1158 (2019).
Harmeyer, K. M., Facompre, N. D., Herlyn, M. & Basu, D. JARID1 Histone Demethylases: Emerging Targets in Cancer. Trends Cancer 3, 713–725 (2017).
Fu, X., Yang, C. & Yu, B. Targeting KDM5 demethylases: inhibition and degradation. Curr. Top. Med. Chem. 20, 261–263 (2020).
Torres, I. O. et al. Histone demethylase KDM5A is regulated by its reader domain through a positive-feedback mechanism. Nat. Commun. 6, 6204 (2015).
Li, G. et al. KDM5B is essential for the hyperactivation of PI3K/AKT signaling in prostate tumorigenesis. Cancer Res. 80, 4633–4643 (2020).
Zhang, Z. G. et al. KDM5B promotes breast cancer cell proliferation and migration via AMPK-mediated lipid metabolism reprogramming. Exp. Cell Res. 379, 182–190 (2019).
Niu, X. et al. The von Hippel-Lindau tumor suppressor protein regulates gene expression and tumor growth through histone demethylase JARID1C. Oncogene 31, 776–786 (2012).
Wang, Q., Wei, J., Su, P. & Gao, P. Histone demethylase JARID1C promotes breast cancer metastasis cells via down regulating BRMS1 expression. Biochem. Biophys. Res. Commun. 464, 659–666 (2015).
Liu, M. & Gao, N. KDM5D inhibits the transcriptional activation of FKBP4 by suppressing the expression of E2F1 in colorectal cancer in males. Biochem. Pharmacol. 194, 114814 (2021).
Hong, S. et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl Acad. Sci. USA 104, 18439–18444 (2007).
Zhang, X., Liu, L., Yuan, X., Wei, Y. & Wei, X. JMJD3 in the regulation of human diseases. Protein Cell 10, 864–882 (2019).
Burgold, T. et al. The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One 3, e3034 (2008).
Ohguchi, H. et al. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 31, 2661–2669 (2017).
Kobatake, K. et al. Kdm6a deficiency activates inflammatory pathways, promotes M2 macrophage polarization, and causes bladder cancer in cooperation with p53 dysfunction. Clin. Cancer Res. 26, 2065–2079 (2020).
Barrows, D., Feng, L., Carroll, T. S. & Allis, C. D. Loss of UTX/KDM6A and the activation of FGFR3 converge to regulate differentiation gene-expression programs in bladder cancer. Proc. Natl Acad. Sci. USA 117, 25732–25741 (2020).
Chaturvedi, S. S., Ramanan, R., Waheed, S. O., Karabencheva-Christova, T. G. & Christov, C. Z. Structure-function relationships in KDM7 histone demethylases. Adv. Protein Chem. Struct. Biol. 117, 113–125 (2019).
Chaturvedi, S. S. et al. Conformational dynamics underlies different functions of human KDM7 histone demethylases. Chem. Eur. J. 25, 5422–5426 (2019).
Cheng, Y. et al. MicroRNA-383 inhibits proliferation, migration, and invasion in hepatocellular carcinoma cells by targeting PHF8. Mol. Genet. Genom. Med. 8, e1272 (2020).
Pappa, S. et al. PHF2 histone demethylase prevents DNA damage and genome instability by controlling cell cycle progression of neural progenitors. Proc. Natl Acad. Sci. USA 116, 19464–19473 (2019).
Al-Raawi, D. et al. A novel form of JARID2 is required for differentiation in lineage-committed cells. EMBO J. 38, e98449 (2019).
Oh, S., Shin, S. & Janknecht, R. The small members of the JMJD protein family: enzymatic jewels or jinxes? Biochim. Biophys. Acta Rev. Cancer 1871, 406–418 (2019).
Zhang, J., Jing, L., Li, M., He, L. & Guo, Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol. Med. Rep. 19, 3963–3971 (2019).
Mondal, S. & Thompson, P. R. Protein arginine deiminases (PADs): biochemistry and chemical biology of protein citrullination. Acc. Chem. Res. 52, 818–832 (2019).
Yuzhalin, A. E. et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 9, 4783 (2018).
Chen, H. et al. LINC00324 suppresses apoptosis and autophagy in nasopharyngeal carcinoma through upregulation of PAD4 and activation of the PI3K/AKT signaling pathway. Cell Biol. Toxicol. 38, 995–1011 (2021).
Yang, J. et al. Jumonji domain-containing protein 6 protein and its role in cancer. Cell Prolif. 53, e12747 (2020).
Wang, K. et al. Role of the epigenetic modifier JMJD6 in tumor development and regulation of immune response. Front. Immunol. 13, 859893 (2022).
Li, S. et al. JMJD1B demethylates H4R3me2s and H3K9me2 to facilitate gene expression for development of hematopoietic stem and progenitor cells. Cell Rep. 23, 389–403 (2018).
Fang, Y. et al. Natural products as LSD1 inhibitors for cancer therapy. Acta Pharm. Sin. B 11, 621–631 (2020).
Fang, Y., Liao, G. & Yu, B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J. Hematol. Oncol. 12, 129 (2019).
Yang, M. et al. Structural basis for the inhibition of the LSD1 histone demethylase by the antidepressant trans-2-phenylcyclopropylamine. Biochemistry 46, 8058–8065 (2007).
Binda, C. et al. Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833 (2010).
Song, Y., Yang, X. & Yu, B. Repurposing antidepressants for anticancer drug discovery. Drug Discov. Today 27, 1924–1935 (2022).
Wass, M. et al. A proof of concept phase I/II pilot trial of LSD1 inhibition by tranylcypromine combined with ATRA in refractory/relapsed AML patients not eligible for intensive therapy. Leukemia 35, 701–711 (2021).
Maes, T. et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33, 495–511.e412 (2018).
Mohammad, H. P. et al. A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69 (2015).
Yin, W. et al. Safety, pharmacokinetics and pharmacodynamics of TAK-418, a novel inhibitor of the epigenetic modulator lysine-specific demethylase 1A. Br. J. Clin. Pharmacol. 87, 4756–4768 (2021).
Kanouni, T. et al. Discovery of CC-90011: a potent and selective reversible inhibitor of lysine specific demethylase 1 (LSD1). J. Med. Chem. 63, 14522–14529 (2020).
Sorna, V. et al. High-throughput virtual screening identifies novel N’-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 56, 9496–9508 (2013).
Xiao, Z. et al. GASC1 promotes glioma progression by enhancing NOTCH1 signaling. Mol. Med. Rep. 23, 310 (2021).
Shen, D. W., Pouliot, L. M., Hall, M. D. & Gottesman, M. M. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 64, 706–721 (2012).
Nagasawa, S. et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS One 10, e0118002 (2015).
Eun, K., Ham, S. W. & Kim, H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 50, 117–125 (2017).
Walcher, L. et al. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front. Immunol. 11, 1280 (2020).
Makena, M. R., Ranjan, A., Thirumala, V. & Reddy, A. P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165339 (2020).
Qin, K. et al. Study on the proliferation and drug-resistance of human brain tumor stem-like cells. Cell. Mol. Neurobiol. 30, 955–960 (2010).
Huang, T. et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 10, 8721–8743 (2020).
Verigos, J. et al. The histone demethylase LSD1/ΚDM1A mediates chemoresistance in breast cancer via regulation of a stem cell program. Cancers 11, 1585 (2019).
Liu, Y. C., Yeh, C. T. & Lin, K. H. Cancer stem cell functions in hepatocellular carcinoma and comprehensive therapeutic strategies. Cells 9, 1331 (2020).
Castelli, G., Pelosi, E. & Testa, U. Liver cancer: molecular characterization, clonal evolution and cancer stem cells. Cancers 9, 127 (2017).
Hu, B. et al. CD13 promotes hepatocellular carcinogenesis and sorafenib resistance by activating HDAC5-LSD1-NF-κB oncogenic signaling. Clin. Transl. Med. 10, e233 (2020).
Chen, J. et al. Knocking down LSD1 inhibits the stemness features of colorectal cancer stem cells. Braz. J. Med. Biol. Res. 53, e9230 (2020).
Zhao, L. J. et al. Lysine demethylase LSD1 delivered via small extracellular vesicles promotes gastric cancer cell stemness. EMBO Rep. 22, e50922 (2021).
Huang, M. et al. Targeting KDM1A attenuates Wnt/β-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett. 398, 12–21 (2017).
Zhang, W. et al. KDM1A promotes thyroid cancer progression and maintains stemness through the Wnt/β-catenin signaling pathway. Theranostics 12, 1500–1517 (2022).
Lei, Z. J. et al. Lysine-specific demethylase 1 promotes the stemness and chemoresistance of Lgr5(+) liver cancer initiating cells by suppressing negative regulators of β-catenin signaling. Oncogene 34, 3188–3198 (2015).
Han, T. S., Hur, K., Cho, H. S. & Ban, H. S. Epigenetic associations between lncRNA/circRNA and miRNA in hepatocellular carcinoma. Cancers (Basel) 12, 2622 (2020).
Zheng, G. et al. E2F1-induced ferritin heavy chain 1 pseudogene 3 (FTH1P3) accelerates non-small cell lung cancer gefitinib resistance. Biochem. Biophys. Res. Commun. 530, 624–631 (2020).
Sun, P., Sun, L., Cui, J., Liu, L. & He, Q. Long noncoding RNA HAS2-AS1 accelerates non-small cell lung cancer chemotherapy resistance by targeting LSD1/EphB3 pathway. Am. J. Transl. Res. 12, 950–958 (2020).
Ma, L. et al. LSD1-demethylated LINC01134 confers oxaliplatin resistance through SP1-induced p62 transcription in HCC. Hepatology 74, 3213–3234 (2021).
Li, W. et al. HOTAIR promotes gefitinib resistance through modification of EZH2 and silencing p16 and p21 in non-small cell lung cancer. J. Cancer 12, 5562–5572 (2021).
Lu, Y., Liu, Y., Oeck, S. & Glazer, P. M. Hypoxia promotes resistance to EGFR inhibition in NSCLC cells via the histone demethylases, LSD1 and PLU-1. Mol. Cancer Res. 16, 1458–1469 (2018).
Boulding, T. et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 8, 73 (2018).
Sengodan, S. K. et al. β-hCG-induced mutant BRCA1 ignites drug resistance in susceptible breast tissue. Carcinogenesis 40, 1415–1426 (2019).
Li, W. et al. Overcoming ABC transporter-mediated multidrug resistance: molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 27, 14–29 (2016).
Wang, Y., Sun, L., Luo, Y. & He, S. Knockdown of KDM1B inhibits cell proliferation and induces apoptosis of pancreatic cancer cells. Pathol. Res. Pract. 215, 1054–1060 (2019).
Katz, T. A. et al. Inhibition of histone demethylase, LSD2 (KDM1B), attenuates DNA methylation and increases sensitivity to DNMT inhibitor-induced apoptosis in breast cancer cells. Breast Cancer Res. Treat. 146, 99–108 (2014).
Chen, L. et al. Functional characterization of lysine-specific demethylase 2 (LSD2/KDM1B) in breast cancer progression. Oncotarget 8, 81737–81753 (2017).
Lee, Y. K. et al. Promotion of cell death in cisplatin-resistant ovarian cancer cells through KDM1B-DCLRE1B modulation. Int. J. Mol. Sci. 20, 2443 (2019).
Lin, Q. et al. ZHX2 restricts hepatocellular carcinoma by suppressing stem cell-like traits through KDM2A-mediated H3K36 demethylation. EBioMedicine 53, 102676 (2020).
Staberg, M. et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 12, 406–420 (2018).
Kurt, I. C. et al. KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis. 8, e2897 (2017).
Macedo-Silva, C. et al. JmjC-KDMs KDM3A and KDM6B modulate radioresistance under hypoxic conditions in esophageal squamous cell carcinoma. Cell Death Dis. 11, 1068 (2020).
He, C. et al. Lysine demethylase KDM3A regulates nanophotonic hyperthermia resistance generated by 2D silicene in breast cancer. Biomaterials 255, 120181 (2020).
Ramadoss, S., Guo, G. & Wang, C. Y. Lysine demethylase KDM3A regulates breast cancer cell invasion and apoptosis by targeting histone and the non-histone protein p53. Oncogene 36, 47–59 (2017).
Ramadoss, S. et al. Lysine-specific demethylase KDM3A regulates ovarian cancer stemness and chemoresistance. Oncogene 36, 1537–1545 (2017).
Wade, M. A. et al. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 43, 196–207 (2015).
Kuroki, S. et al. Combined loss of JMJD1A and JMJD1B reveals critical roles for H3K9 demethylation in the maintenance of embryonic stem cells and early embryogenesis. Stem Cell Rep. 10, 1340–1354 (2018).
Qu, F. et al. Circular RNA circ_0006168 enhances Taxol resistance in esophageal squamous cell carcinoma by regulating miR-194-5p/JMJD1C axis. Cancer Cell Int. 21, 273 (2021).
Schimek, V., Björn, N., Pellé, L., Svedberg, A. & Gréen, H. JMJD1C knockdown affects myeloid cell lines proliferation, viability, and gemcitabine/carboplatin-sensitivity. Pharmacogenet. Genomics 31, 60–67 (2021).
Xiao, F. et al. JMJD1C ensures mouse embryonic stem cell self-renewal and somatic cell reprogramming through controlling MicroRNA expression. Stem Cell Rep. 9, 927–942 (2017).
Metzger, E. et al. KDM4 inhibition targets breast cancer stem-like cells. Cancer Res. 77, 5900–5912 (2017).
Nakagawa, T. et al. JMJD2A sensitizes gastric cancer to chemotherapy by cooperating with CCDC8. Gastric Cancer 23, 426–436 (2020).
Zhang, B. et al. Targeting KDM4A-AS1 represses AR/AR-Vs deubiquitination and enhances enzalutamide response in CRPC. Oncogene 41, 387–399 (2022).
Sha, J. et al. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J. Cell. Physiol. 235, 2129–2138 (2020).
Wu, M. J. et al. Targeting KDM4B that coactivates c-Myc-regulated metabolism to suppress tumor growth in castration-resistant prostate cancer. Theranostics 11, 7779–7796 (2021).
Tang, D. E. et al. Targeting the KDM4B-AR-c-Myc axis promotes sensitivity to androgen receptor-targeted therapy in advanced prostate cancer. J. Pathol. 252, 101–113 (2020).
Xue, L., Li, C., Ren, J. & Wang, Y. KDM4C contributes to cytarabine resistance in acute myeloid leukemia via regulating the miR-328-3p/CCND2 axis through MALAT1. Ther. Adv. Chronic Dis. 12, 2040622321997259 (2021).
Gao, Y. et al. UHRF1 promotes androgen receptor-regulated CDC6 transcription and anti-androgen receptor drug resistance in prostate cancer through KDM4C-Mediated chromatin modifications. Cancer Lett. 520, 172–183 (2021).
Lang, T. et al. Disruption of KDM4C-ALDH1A3 feed-forward loop inhibits stemness, tumorigenesis and chemoresistance of gastric cancer stem cells. Signal Transduct. Target Ther. 6, 336 (2021).
Chen, G. Q. et al. Histone demethylase KDM4C is required for ovarian cancer stem cell maintenance. Stem Cells Int. 2020, 8860185 (2020).
Deng, Y. et al. Histone demethylase JMJD2D promotes the self-renewal of liver cancer stem-like cells by enhancing EpCAM and Sox9 expression. J. Biol. Chem. 296, 100121 (2021).
Banelli, B. et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 14, 3418–3429 (2015).
Choi, H. J. et al. Role of RBP2-induced ER and IGF1R-ErbB signaling in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 110, https://doi.org/10.1093/jnci/djx207 (2018).
Garcia, T. B. et al. Increased HDAC activity and c-MYC expression mediate acquired resistance to WEE1 inhibition in acute leukemia. Front. Oncol. 10, 296 (2020).
Xu, L., Wu, H. & Hu, X. Histone demethylase KDM5A enhances cell proliferation, induces EMT in lung adenocarcinoma cells, and have a strong causal association with paclitaxel resistance. Acta Biochim. Pol. 68, 593–602 (2021).
Kuo, K. T. et al. Histone demethylase JARID1B/KDM5B promotes aggressiveness of non-small cell lung cancer and serves as a good prognostic predictor. Clin. Epigenetics 10, 107 (2018).
Xu, W. et al. KDM5B demethylates H3K4 to recruit XRCC1 and promote chemoresistance. Int. J. Biol. Sci. 14, 1122–1132 (2018).
Li, L., Shou, H., Wang, Q. & Liu, S. Investigation of the potential theranostic role of KDM5B/miR-29c signaling axis in paclitaxel resistant endometrial carcinoma. Gene 694, 76–82 (2019).
Shannan, B. et al. Sequence-dependent cross-resistance of combined radiotherapy plus BRAF(V600E) inhibition in melanoma. Eur. J. Cancer 109, 137–153 (2019).
Liu, X. et al. KDM5B promotes drug resistance by regulating melanoma-propagating cell subpopulations. Mol. Cancer Ther. 18, 706–717 (2019).
Tobin, S. J., Chang, H., Kent, M. S. & Davies, A. E. JARID1-targeted histone H3 demethylase inhibitors exhibit anti-proliferative activity and overcome cisplatin resistance in canine oral melanoma cell lines. Vet. Comp. Oncol. 19, 518–528 (2021).
Hong, Z. et al. KDM5C is transcriptionally regulated by BRD4 and promotes castration-resistance prostate cancer cell proliferation by repressing PTEN. Biomed. Pharmacother. 114, 108793 (2019).
Liu, J., Zhu, M. & Tang, Q. Human umbilical cord mesenchymal stem cells-derived exosomal microRNA-181a retards nasopharyngeal carcinoma development by mediating KDM5C. J. Cancer Res. Clin. Oncol. 147, 2867–2877 (2021).
Xu, L. et al. Enhancement of proliferation and invasion of gastric cancer cell by KDM5C via decrease in p53 expression. Technol. Cancer Res. Treat. 16, 141–149 (2017).
Cui, Y. et al. HOXD3 up-regulating KDM5C promotes malignant progression of diffuse large B-cell lymphoma by decreasing p53 expression. Balk. Med. J. 39, 30–38 (2022).
Lin, H. et al. KDM5c inhibits multidrug resistance of colon cancer cell line by down-regulating ABCC1. Biomed. Pharmacother. 107, 1205–1209 (2018).
Zheng, Q. et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics 11, 8674–8691 (2021).
Shen, X. et al. KDM5D inhibit epithelial-mesenchymal transition of gastric cancer through demethylation in the promoter of Cul4A in male. J. Cell. Biochem. 120, 12247–12258 (2019).
Komura, K. et al. Resistance to docetaxel in prostate cancer is associated with androgen receptor activation and loss of KDM5D expression. Proc. Natl Acad. Sci. USA 113, 6259–6264 (2016).
Yoshikawa, Y. et al. Increased MYBL2 expression in aggressive hormone-sensitive prostate cancer. Mol. Oncol. 16, 3994–4010 (2022).
Gu, J. & Chu, K. Increased Mars2 expression upon microRNA-4661-5p-mediated KDM5D downregulation is correlated with malignant degree of gastric cancer cells. Cell Biol. Int. 45, 2118–2128 (2021).
Ribera, J. et al. Copy number profiling of adult relapsed B-cell precursor acute lymphoblastic leukemia reveals potential leukemia progression mechanisms. Genes Chromosomes Cancer 56, 810–820 (2017).
Stief, S. M. et al. Loss of KDM6A confers drug resistance in acute myeloid leukemia. Leukemia 34, 50–62 (2020).
Zhang, C. et al. KDM6A promotes imatinib resistance through YY1-mediated transcriptional upregulation of TRKA independently of its demethylase activity in chronic myelogenous leukemia. Theranostics 11, 2691–2705 (2021).
Tang, D. et al. Targeting KDM6A suppresses SREBP1c-dependent lipid metabolism and prostate tumorigenesis. Cancer Res. https://doi.org/10.1158/0008-5472.CAN-22-1465 (2021).
Ji, G. et al. PCGF1 promotes epigenetic activation of stemness markers and colorectal cancer stem cell enrichment. Cell Death Dis. 12, 633 (2021).
Wang, Q. et al. Elevating H3K27me3 level sensitizes colorectal cancer to oxaliplatin. J. Mol. Cell Biol. 12, 125–137 (2020).
Chen, H. J. et al. GATA3 as a master regulator and therapeutic target in ovarian high-grade serous carcinoma stem cells. Int. J. Cancer 143, 3106–3119 (2018).
Mathur, R. et al. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 102, 373–380 (2017).
Wang, W. et al. KDM6B counteracts EZH2-mediated suppression of IGFBP5 to confer resistance to PI3K/AKT cer. Mol. Cancer Ther. 17, 1973–1983 (2018).
Yıldırım-Buharalıoğlu, G. Lysine demethylase 6B regulates prostate cancer cell proliferation by controlling c-MYC expression. Mol. Pharmacol. 101, 106–119 (2022).
Cao, Z. et al. KDM6B is an androgen regulated gene and plays oncogenic roles by demethylating H3K27me3 at cyclin D1 promoter in prostate cancer. Cell Death Dis. 12, 2 (2021).
D’Oto, A. et al. KDM6B promotes activation of the oncogenic CDK4/6-pRB-E2F pathway by maintaining enhancer activity in MYCN-amplified neuroblastoma. Nat. Commun. 12, 7204 (2021).
Lee, K. H. et al. Histone demethylase KDM7A regulates androgen receptor activity, and its chemical inhibitor TC-E 5002 overcomes cisplatin-resistance in bladder cancer cells. Int. J. Mol. Sci. 21, 5658 (2020).
Liu, Q. et al. Histone demethylase PHF8 drives neuroendocrine prostate cancer progression by epigenetically upregulating FOXA2. J. Pathol. 253, 106–118 (2021).
Liu, Q. et al. Contribution of synergism between PHF8 and HER2 signalling to breast cancer development and drug resistance. EBioMedicine 51, 102612 (2020).
Wang, Q. et al. JARID2 promotes stemness and cisplatin resistance in non-small cell lung cancer via upregulation of Notch1. Int. J. Biochem. Cell Biol. 138, 106040 (2021).
Kuang, W., Jiang, W., Chen, Y., Tian, Y. & Liu, Z. The function and mechanism of the JARID2/CCND1 axis in modulating glioma cell growth and sensitivity to temozolomide (TMZ). Cancer Biol. Ther. 22, 392–403 (2021).
Duan, Q., Pang, C., Chang, N., Zhang, J. & Liu, W. Overexpression of PAD4 suppresses drug resistance of NSCLC cell lines to gefitinib through inhibiting Elk1-mediated epithelial-mesenchymal transition. Oncol. Rep. 36, 551–558 (2016).
Zhou, Q. et al. Peptidylarginine deiminase 4 overexpression resensitizes MCF-7/ADR breast cancer cells to adriamycin via GSK3β/p53 activation. Cancer Manag. Res. 11, 625–636 (2019).
Fan, T. et al. Peptidylarginine deiminase IV promotes the development of chemoresistance through inducing autophagy in hepatocellular carcinoma. Cell Biosci. 4, 49 (2014).
Sterling, J., Menezes, S. V., Abbassi, R. H. & Munoz, L. Histone lysine demethylases and their functions in cancer. Int. J. Cancer https://doi.org/10.1002/ijc.33375 (2020).
Chu, Y. et al. Arborinine, a potential LSD1 inhibitor, inhibits epithelial-mesenchymal transition of SGC-7901 cells and adriamycin-resistant gastric cancer SGC-7901/ADR cells. Invest. N. Drugs 39, 627–635 (2021).
Gupta, S. et al. Reversible lysine-specific demethylase 1 antagonist HCI-2509 inhibits growth and decreases c-MYC in castration- and docetaxel-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 19, 349–357 (2016).
Etani, T. et al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses castration-resistant prostate cancer growth via regulation of apoptosis and autophagy. J. Clin. Med. 8, 442 (2019).
Cortez, V. et al. Targeting the PELP1-KDM1 axis as a potential therapeutic strategy for breast cancer. Breast Cancer Res. 14, R108 (2012).
Wang, J. et al. Silencing the epigenetic silencer KDM4A for TRAIL and DR5 simultaneous induction and antitumor therapy. Cell Death Differ. 23, 1886–1896 (2016).
Mar, B. G. et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 130, 2631–2641 (2017).
Romani, M., Daga, A., Forlani, A., Pistillo, M. P. & Banelli, B. Targeting of histone demethylases KDM5A and KDM6B inhibits the proliferation of temozolomide-resistant glioblastoma cells. Cancers 11, 878 (2019).
Yang, G. J., Ko, C. N., Zhong, H. J., Leung, C. H. & Ma, D. L. Structure-based discovery of a selective KDM5A inhibitor that exhibits anti-cancer activity via inducing cell cycle arrest and senescence in breast cancer cell lines. Cancers 11, 92 (2019).
Gale, M. et al. Screen-identified selective inhibitor of lysine demethylase 5A blocks cancer cell growth and drug resistance. Oncotarget 7, 39931–39944 (2016).
Gehling, V. S. et al. Identification of potent, selective KDM5 inhibitors. Bioorg. Med. Chem. Lett. 26, 4350–4354 (2016).
Fu, Y. D. et al. Targeting histone demethylase KDM5B for cancer treatment. Eur. J. Med. Chem. 208, 112760 (2020).
Vinogradova, M. et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol. 12, 531–538 (2016).
Banelli, B. et al. Small molecules targeting histone demethylase genes (KDMs) inhibit growth of temozolomide-resistant glioblastoma cells. Oncotarget 8, 34896–34910 (2017).
Xu, S. et al. KDM5A suppresses PML-RARα target gene expression and APL differentiation through repressing H3K4me2. Blood Adv. 5, 3241–3253 (2021).
Zhang, J. et al. Targeted inhibition of KDM6 histone demethylases eradicates tumor-initiating cells via enhancer reprogramming in colorectal cancer. Theranostics 10, 10016–10030 (2020).
Lhuissier, E. et al. Antiproliferative effect of the histone demethylase inhibitor GSK-J4 in chondrosarcomas. IUBMB Life 71, 1711–1719 (2019).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (nos. 22277110, 81973177, and 81903770), Natural Science Foundation of Henan Province (no. 222300420069), Program for Science & Technology Innovation Talents in Universities of Henan Province (no. 21HASTIT045), Key scientific and technological projects of Henan Province (no. 222102310125) and China Postdoctoral Science Foundation (No. 2019M662556).
Author information
Authors and Affiliations
Contributions
B.Y. conceived this project, revised the draft, and submitted this review on behalf of other authors. T.M. revised the draft. N.W. wrote the draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, N., Ma, T. & Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Sig Transduct Target Ther 8, 69 (2023). https://doi.org/10.1038/s41392-023-01341-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01341-7
This article is cited by
-
Tubulin alpha-1b chain was identified as a prognosis and immune biomarker in pan-cancer combing with experimental validation in breast cancer
Scientific Reports (2024)
-
Exploring epigenetic strategies for the treatment of osteoporosis
Molecular Biology Reports (2024)
-
TMEM120A-mediated regulation of chemotherapy sensitivity in colorectal cancer cells
Cancer Chemotherapy and Pharmacology (2024)
-
The emerging roles of histone demethylases in cancers
Cancer and Metastasis Reviews (2024)
-
Laser-activatable oxygen self-supplying nanoplatform for efficiently overcoming colorectal cancer resistance by enhanced ferroptosis and alleviated hypoxic microenvironment
Biomaterials Research (2023)
