
Abstract
In recent years, immunotherapy represented by immune checkpoint inhibitors (ICIs) has led to unprecedented breakthroughs in cancer treatment. However, the fact that many tumors respond poorly or even not to ICIs, partly caused by the absence of tumor-infiltrating lymphocytes (TILs), significantly limits the application of ICIs. Converting these immune “cold” tumors into “hot” tumors that may respond to ICIs is an unsolved question in cancer immunotherapy. Since it is a general characteristic of cancers to resist apoptosis, induction of non-apoptotic regulated cell death (RCD) is emerging as a new cancer treatment strategy. Recently, several studies have revealed the interaction between non-apoptotic RCD and antitumor immunity. Specifically, autophagy, ferroptosis, pyroptosis, and necroptosis exhibit synergistic antitumor immune responses while possibly exerting inhibitory effects on antitumor immune responses. Thus, targeted therapies (inducers or inhibitors) against autophagy, ferroptosis, pyroptosis, and necroptosis in combination with immunotherapy may exert potent antitumor activity, even in tumors resistant to ICIs. This review summarizes the multilevel relationship between antitumor immunity and non-apoptotic RCD, including autophagy, ferroptosis, pyroptosis, and necroptosis, and the potential targeting application of non-apoptotic RCD to improve the efficacy of immunotherapy in malignancy.
Similar content being viewed by others
Background
Cell death is classified into two categories based on the rate at which it occurs and whether drugs or genes may influence it: accidental cell death and regulated cell death (RCD).1 Accidental cell death results from the biological process, while RCD is mediated by signal transduction pathways and well-defined mechanisms of action.1 RCD plays a vital role in homeostasis maintenance and diseases development. Based on different morphological, biochemical, immunological, and genetic characteristics, RCD is subdivided into apoptotic and non-apoptotic categories.2,3 Non-apoptotic RCD can be subdivided into autophagy, ferroptosis, pyroptosis, and necroptosis (Table 1 and Fig. 1). Immunogenic cell deaths (ICD) mentioned in Table 1. will be described in detail below. Resistance to apoptosis is a general characteristic of cancer.4 Research on apoptosis has been conducted for more than 30 years. Nevertheless, therapeutic agents targeting apoptosis regulators such as apoptosis-related caspases or B-cell lymphoma-2 (BCL-2) family proteins have poor effects in antitumor therapy.5 On the contrary, non-apoptotic RCD affects the development of cancer and its response to therapy.1,2,3 For example, in genetic engineering mice, enhanced sensitivity of tumors to ferroptosis significantly inhibited the formation and progression of pancreatic cancer.6 The KRAS mutation-driven lung cancer model, however, suggests that autophagy is necessary for maintaining mitochondrial function and providing energy for cells to survive and grow.7 Inflammasome, a key component of pyroptosis, plays a critical role in chemoresistance in oral squamous cell carcinoma and insensitivity to radiotherapy in glioblastoma.8,9 Key mediators in the process of necroptosis are thought to promote head and neck squamous cell carcinoma metastasis and progression as well as negatively affect the prognosis in glioblastoma; however, necroptosis has also been reported to act as a defense mechanism, playing a tumor-suppressive role when tumor cell apoptosis is impaired in leukemia and colorectal cancer.10,11,12,13 Therefore, targeting non-apoptotic RCD has attracted much attention in the field of antitumor therapy.

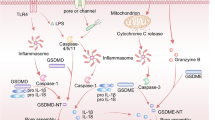
Core molecular mechanisms of autophagy, pyroptosis, ferroptosis, and necroptosis. a The ULK complex initiates autophagy by responding to nutrient stress signals from mTOR and energy stress signals from AMPK, which eventually activate VPS34. VPS34 complex generates PI3P at membrane to recruit and assemble ubiquitin-like coupling systems. In LC3 lipidation, ATG7, ATG3, and ATG5-ATG12-ATG16L complexes are ubiquitin enzymes that recruit loads to cargo receptors such as SQSTM1/P62 and NBR. In the presence of ATG9, the phagosome expands and eventually closes to form autophagosomes, which are subsequently fused with lysosomes to form autolysosomes mediated by SNAREs and the HOPS complex. Finally, cargoes are degraded by lysosomal hydrolases and nutrients are recycled. b Cytoplasmic sensor proteins such as NOD-like receptor family members (NLRP1, NLRP3, and NLRP4), AIM2 and Pyrin proteins are stimulated by PAMPs or DAMPs, recruiting and activating CASP1 via ASC. CASP4/5/11 are activated in the cytoplasm binding to LPS. Finally, activated CASP1 and CASP4/5/11 cause the cleavage and production of GSDMD-N, which leads to pyroptosis by activating typical and atypical inflammasomes. Pyroptosis regulated by potassium efflux triggers the release of HMGB1 and K+. c During ferroptosis, two fundamental processes trigger oxidative membrane damage: iron accumulation and lipid peroxidation. The transferrin–transferrin receptor (TF–TFRC) complex, iron export transporter, and ferritinophagy contribute to ferroptosis by increasing iron accumulation through increased iron uptake, restricted iron efflux, and decreased iron storage, respectively. The ACSL4–LPCAT3–ALOXs pathway plays a critical role in promoting ferroptosis by activating lipid peroxidation to produce PLOOH from PUFA with the involvement of RAB7A-dependent lipophagy. Several antioxidant systems such as Xc-system–GSH–GPX4, AIFM2-CoQ10, GTP-GCH1-BH4, or ESCRT-III membrane repair system inhibit lipid peroxidation. d After TNFα binds to the receptor, the intracellular tails of TNFR1 recruit multiple proteins to form Complex I. Lys63-linked polyubiquitination (Lys63-Ub) of RIP1 mediated by cIAP is essential for the survival pathway. Deubiquitination of RIP1 by CYLD promotes the conversion of Complex I to Complex II. When CASP8 is activated in complex II, apoptosis is initiated. When CASP8 is inhibited, MLKL, RIPK1, and RIPK3 are recruited to assemble the necrosome through phosphorylation. The phosphorylation-mediated activation of MLKL and subsequent MLKL-mediated membrane pore formation results in necroptosis
The immune system contributes to preventing the occurrence, progression, and metastasis of tumor and regulating tumor response to therapy. Immune surveillance provides a way to identify, control and kill tumor cells.14,15,16 However, tumor cells evade immune surveillance by reducing immunogenicity and forming an immunosuppressive network.14,15,16 Immunotherapy harnesses the immune system against tumors by stimulating antitumor immune responses, including immune checkpoint inhibitors (ICIs),17,18,19 chimeric antigen receptor T cells (CAR-T cells),20 dendritic cell vaccines,21 and cytokine therapies.22 For the past few years, ICIs have made significant breakthroughs in the field of antitumor therapy.23,24,25 Mechanically, ICIs inhibit cancer development by restoring the function of effector T cells.17,19 Traditionally, it was believed that immunotherapy-activated CD8+ T cells induce tumor cell death mainly through the perforin-granzyme pathway and the Fas-Fas ligand (FASL) pathway.26,27 However, many studies have surprisingly revealed that CD8+ T cells can suppress tumors by inducing ferroptosis and pyroptosis.28,29,30,31 Similarly, recent studies have shown that non-apoptotic RCD participates in the survival, differentiation, activation, and translocation of immune cells and its function performance (both antitumor and tumor-promoting effect cells).32,33 Meanwhile, tumor-autonomous non-apoptotic RCD can affect tumor growth by modulating immune responses.34,35 It is worth stating that the process of tumor cell deaths that stimulates an adaptive immune response is called ICD.36 During ICD, damage-associated molecular patterns (DAMPs) including a variety of biomolecules including high mobility group box 1 (HMGB1), mitochondrial DNA, and ATP, and pathogen-associated molecular patterns (PAMPs) including various microbial pathogen components such as lipopolysaccharide (LPS) can be identifiable by pattern recognition receptors (PRRs) and play a dual role in tumor immunity.37 Besides the antitumor immune responses, the release of cytokines and chemokines facilitates the inflammatory responses that promote tumor growth.38,39 It is possible for ICD to occur in the context of autophagy, ferroptosis, pyroptosis, and necroptosis.40,41,42,43 We, therefore, hypothesize that these non-apoptotic RCD processes may be a double-edged sword for tumor immune responses.
In this context, we review autophagy, ferroptosis, pyroptosis, and necroptosis on tumor development, the multilevel relationships with tumor immune responses, and the critical roles in immunotherapy. In addition, we discuss the potential application of targeting non-apoptotic RCD to enhance the efficacy of immunotherapy in malignancy.
Overview of autophagy
Eukaryotic cells utilize autophagy for maintaining homeostasis and managing lipid metabolism.44,45 Activated by various stress states, autophagic membrane structures are formed to engulf and degrade intracellular structures, including damaged organelles, unfolded proteins, and pathogens.42,46 Autophagy was initially thought to be a “bulk degradation” process. Still, new findings suggest that specific cargoes such as organelles and proteins can be recognized by selective autophagic receptors (SARs) and be degraded.47,48
The autophagy initiation is mediated by the unc-51-like kinase (ULK) complex,49 which shifts to an active state when mTOR complex 1 (mTORC1) is inhibited, or 5′-AMP-activated protein kinase (AMPK) is activated stimulated by stress signals then activating vacuolar protein sorting 34 (VPS34).50,51 The VPS34 complex acts as a phosphatidylinositol 3-phosphate kinase (PI3K) to generate phosphatidylinositol 3-phosphate (PI3P) which acts as a scaffold to recruit PI3P-binding molecules, forming an isolated pre-autophagosomal structure called phagosome.52,53 Specifically, PI3P recruits and assembles two ubiquitin-like coupling systems that are involved in LC3 lipidation and autophagosome formation.53,54,55 During LC3 lipidation, LC3 is sheared to the soluble form LC3I as the precursor of LC3II which is a docking site covalently attaching to the membrane of phagosomes for cargo receptors.48,56,57 The receptor binds to specific cargoes through ubiquitin labeling, which is central to the selective recruitment of loads during autophagy.48,57 Subsequently, the phagosome extends and eventually closes to form a separate compartment called autophagosome.58,59 Autophagosomes are transported to the perinuclear region, where they fuse with proximal lysosomes to form autolysosomes.60,61 In the presence of lysosomal hydrolases, cargoes are degraded, and nutrients are recycled.62
The dysregulation of autophagy contributes to tumor growth and progression. Studies in 1999 showed that a single allele of Beclin1 was absent in 40–75% of disseminated human breast and ovarian cancers, which was the first time that autophagy was reported to be associated with human cancer.63,64 Similarly, a heterozygous deletion of ATG5 at chromosome 6q21 is a prominent feature of advanced melanoma in humans and affects KRAS-driven pancreatic tumor development and metastasis.65,66 In addition to effects on tumor cells, autophagy defects can indirectly promote tumorigenesis through inflammation.67 The Thr300Ala mutation in ATG16L1 may lead to chronic inflammatory Crohn’s disease, thereby predisposing patients to colorectal cancer.68 Interestingly, a KRAS mutation-driven pancreatic cancer model revealed that tumor growth is facilitated by autophagy in the cancer mesenchymal region. Pancreatic stellate cells secrete alanine through autophagy by pancreatic tumor cells to promote growth-friendly mitochondrial metabolism.69
Overview of ferroptosis
The term ferroptosis introduced in 2012 refers to iron-dependent RCD caused by the excessive amount of lipid peroxidation, resulting in the ruptured plasma membrane.70 When ferroptosis occurs, iron accumulation and lipid peroxidation both contribute to oxidative membrane damage.71,72 Increased iron accumulation is a key trigger of ferroptosis in animal models.73 Specifically, transferrin promotes ferroptosis by mediating iron uptake through the transferrin receptor (TFRC).74,75 Degrading intracellular iron storage proteins or iron export transporter solute carrier family 40 member 1 (SLC40A1) by the autophagy increases iron accumulation, thereby initiating or enhancing ferroptosis.76,77,78
Excess intracellular iron can contribute to subsequent lipid peroxidation through the production of reactive oxygen species (ROS) and the activation of iron-containing enzymes such as arachidonic acid lipoxygenases (ALOXs).79,80,81 In the presence of long-chain fatty acid–CoA ligase 4 (ACSL4) and lysophospholipid acyltransferase 5 (LPCAT3), polyunsaturated fatty acid (PUFA) is catalyzed to develop phospholipids-polyunsaturated fatty acid (PL-PUFA).82,83 Finally, PL-PUFA is mediated by ALOXs to produce phospholipid hydroperoxides (PL-PUFA-OOH), which can promote ferroptosis.84 Ferroptosis is primarily a process of balancing oxidative and antioxidant damage.85 Glutathione (GSH)-glutathione peroxidase 4 (GPX4) antioxidant system plays an essential role in protecting cells from ferroptosis. Xc-system is responsible for the import of cyst(e)ine as a rate-limiting substrate for GSH synthesis in exchange for intracellular glutamate (Glu).71 GPX4 uses GSH as a reducing cofactor that reduces PLOOH to fatty alcohol, thereby inhibiting ferroptosis in tumor cells.86,87,88 Other antioxidant systems, such as the coenzyme apoptosis-inducing factor mitochondrial 2-coenzyme Q10 (AIFM2-Q10),89 tetrahydrobiopterin (BH),90 as well as sorting complexes in the endosomes as a requirement for transport III (ESCRT-III) membrane repair system,91 all contribute to antagonize ferroptosis in solid tumors.92 Ferroptosis was initially regarded as the cell death process that did not depend on autophagy.70 However, recent studies have revealed that iron accumulation and lipid peroxidation are promoted by excessive activation of selective autophagy, resulting in ferroptosis.93,94 Selective autophagy mainly includes nuclear receptor coactivator 4 (NCOA4)-induced ferritinophagy,95,96 heat shock protein 90 (HSP90)-regulated chaperone protein-dependent autophagy,97 RAS oncogene family member RAB7A-mediated lipophagy,98 and clockophagy associated with SQSTM1,99 respectively, to selectively degrade ferritin, GPX4, lipid droplets, thereby increasing intracellular iron and free fatty acid levels and accelerating the peroxidation of lipids to promote ferroptosis.
It has been progressively recognized that several oncogenic pathways are closely associated with ferroptosis.100 For example, most KRAS mutation-driven pancreatic cancers are sensitive to ferroptosis activators, Erastin.101,102 Furthermore, new evidence suggests that as cancer suppressor gene, p53 inhibits cyst(e)ine uptake and sensitizes cells to ferroptosis by suppressing the expression of Xc-system.81,103,104 However, it has also been shown that p53 can limit Erastin-induced ferroptosis in a transcription-independent manner by blocking dipeptidyl peptidase-4 (DPP-4) activity.105
Overview of pyroptosis
Pyroptosis is an ICD caused by caspases found in immune cells during microbial infections.106 Inflammasome-associated caspases, such as CASP1, CASP4, CASP5, and CASP11 (mouse), are mainly responsible for regulating pyroptosis,107,108 whereas some caspases associated with apoptosis such as CASP3109 and CASP8110 also play a role in pyroptosis. And the cleavage of gasdermin (GSDM) family members such as GSDMD110 and GSDME109 mediated by caspase is crucial to trigger pyroptosis. In typical and atypical pyroptosis pathways, CASP1/4/5/11 has been reported for GSDMD cleavage.107,108 Under particular circumstances, apoptosis-dependent CASP8 can directly cleave GSDMD, which triggers pyroptosis.110 CASP8-dependent cleavage of GSDMD promotes host defense against infection while also enhances tumor necrosis factor (TNF) lethality.111 In addition, GSDME can be cleaved by CASP3/8, thereby converting non-inflammatory apoptosis to pyroptosis. Granzyme B (GZMB) acts at the same site to cleave GSDME, activating caspase-independent pyroptosis in target cells.31,112 Similarly, GSDMB is cleaved by CASP1113 or granzyme A (GZMA).114 Here is not detailed explanation of GSDMA/C-mediated pyroptosis.115
In the typical inflammasome activation pathway, PAMPs or DAMPs are detected by cytoplasmic sensor proteins such as NOD-like receptor family members (NLRP), absent in melanoma 2 (AIM2), and Pyrin proteins.116,117,118,119 For example, AIM2 are activated by detecting and then binding precisely to cytoplasmic double-stranded DNA.116 NLRP3 responds to components such as ATP, crystals, and viruses, causing potassium efflux to trigger NLRP3 activity.120,121 Activated sensor proteins recruit and activate CASP1 via apoptosis-associated speck-like protein containing a CARD (ASC) which together constitute the inflammasome.117 In the atypical inflammasome activation pathway, CASP11 in mice or CASP4/5 in humans is activated in cytoplasm binding directly to LPS.108 Finally, CASP1 or CASP4/5/11 causes the release of active GSDMD N-terminal fragment (GSDMD-N) which binds to acidic phospholipids on the plasma membrane and forms oligomeric death-inducing pores, increasing intracellular osmolality thus leading to cytolysis to mediate pyroptosis.122,123,124
Pyroptosis appears to play a dual role in tumor development, either promoting tumor or causing tumor regression which depends on the context in which tumor cells are located. For example, in pancreatic cancer cells, macrophage-stimulating factor 1 (MST1) promotes CASP1-dependent pyroptosis by inducing the production of ROS.125 Gao et al. have shown that the levels of GSDMD protein were extremely increased in NSCLC. High GSDMD expression was associated with aggressiveness of NSCLC, including larger tumor volume and higher TNM stage.126 Nevertheless, activation of pyroptosis can also induce potent antitumor activity.127 For example, in hepatocellular carcinoma (HCC) cells, pyroptosis induced by NLRP3 inflammasomes significantly impedes tumor growth characteristics and metastatic potential.128 Aside from the digestive system,129,130 pyroptosis acts an equally important part in the development of cancers in respiratory,131 reproductive,132 and hematopoietic systems.133
Overview of necroptosis
Necroptosis, introduced in 2005 by Degterev et al. is another form of ICD in which specific death receptors (DRs) including FAS and tumor necrosis factor receptor 1 (TNFR1), etc. or PRRs such as toll-like receptor3 (TLR3) recognize unfavorable signals from the intra- and extra-cellular microenvironment to initiate necroptosis.134,135,136 Necroptosis, triggered by the same stimuli as apoptosis, is similar to necrosis in its morphology (e.g., organelle swelling and ruptured plasma membrane).137,138 Necroptosis appears to be a backup mechanism of apoptosis, in which a key component of necroptosis, necrosome, assembles in TNFR1 stimulation in response to viral infection when CASP8 involved in apoptosis is inhibited.139,140 Moreover, the reduction of intracellular ATP occurs during the transition from apoptosis to necroptosis or necrosis.141
In response to TNFα, the intracellular tails of TNFR1 recruit a variety of proteins that together form a signaling complex called “Complex I” in which the ubiquitination of RIPK1 is regulated by cellular inhibitor of apoptosis protein (cIAP), which is indispensable for nuclear factor kappa-B (NF-κB) and MAPK activation involved in the survival pathway.142,143,144,145 The conversion of Complex I into Complex II is facilitated by deubiquitination of RIPK1 by cylindromatosis (CYLD).142 When CASP8 is activated in complex II, apoptosis is initiated.143,146 However, in RIPK3-rich cells, when CASP8 is inhibited, intracellular junctional molecules sequentially recruit RIPK1, RIPK3, and mixed lineage kinase domain-like (MLKL) to complete necrosome assembly after phosphorylation events.147,148,149 RIPK3 can also be activated when TLR3 is sensed by double-stranded RNA (dsRNA) in the endosome or ZBP1 is sensed by cytosolic DNA.135,150 When MLKL is activated by RIPK3, oligomerization and subsequent translocation occurs. As a result, plasma membrane permeability increases, causing membrane rupture and the release of DAMPs.151
There has been evidence to suggest that necroptosis acts as a tumor suppressor in most cases.152,153 Two-thirds of samples in a study of more than 60 cancer cell lines showed decreased levels of RIPK3, which indicates that the cancer cells prefer to escape necroptosis and survive. Furthermore, necroptosis is strongly associated with cancer prognosis. The Cox proportional risk model showed that the expression of RIPK3 is an independent prognostic factor in colorectal cancer patients with regards to overall survival and disease-free survival.154 Recently, a study has shown that the expression of RIPK1, RIPK3, and MLKL was linked to better overall survival in HCC.155 Furthermore, methylation near the transcription start site silences RIPK3 expression in cancer cells. Therefore, hypomethylation drug treatment can improve prognosis by restoring RIPK3 expression and increasing sensitivity to chemotherapeutic agents.156
Autophagy, ferroptosis, pyroptosis, and necroptosis synergize antitumor immune response
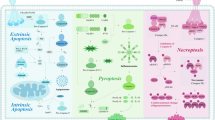
The organism can initiate autophagy, ferroptosis, pyroptosis, and necroptosis as defense in the face of various intra- and extra-cellular stress stimuli, acting to inhibit the proliferation of cancer cells. This defense is achieved in large part through a synergistic antitumor immune response. Specifically, non-apoptotic RCD is involved in the survival, differentiation, activation, transport, and functional performance of immune cells (both antitumor and tumor-promoting effect cells). Meanwhile, tumor-autonomous non-apoptotic RCD can alter tumor growth by modulating immune responses (Figs. 2, 3, and 4).

Crosstalk between T cells and dying cancer cells in the tumor microenvironment. a In dead cancer cells, autophagy increases the production of autophagosomes with TAA, which promotes DC-mediated cross-presentation. When TCR is stimulated, activated T cells have enhanced levels of autophagy, which is linked to rapidly increased calcium levels. By reprogramming metabolic pathways, autophagy is vital for mitochondrial integrity, which maintains T cells’ homeostasis. High levels of lactate in tumors inhibit autophagy and induce apoptosis in naive T cells. Furthermore, NBR1-mediated MHCI degradation through autophagy reduces MHCI expression on the surface of cancer cells and impairs CD8+ T cells recognition of antigens. b On the one hand, tumor cells via pyroptosis pathway facilitate the recruitment of CD8+ T cells by releasing danger signals. On the other hand, CD8+ T cells induce cancer cell pyroptosis by secreting GZMA and GZMB, which can cleave GSDMB/D/E. NLRP3 inflammasomes promote IL-18 and IL-1β secretion, which have tumor-promoting or antitumor effect dependent on the context of TME. c Significant lipid peroxidation activity can occur in CD36-positive CD8+ T cells, which results in ferroptosis induced by GPX4 inhibitors, leading to reduced release of IFN-γ. IFN-γ released by CD8+ T cells induces tumor cells ferroptosis through the activation of JAK1-STAT1 signaling, which transcriptionally regulates the expression of Xc- component, SLC7A11and SLC3A2. d Two strategies have been reported to trigger antitumor immunity through necroptosis. (1) DAMPs released from tumor cells through necroptosis promote cross-priming of DCs, and subsequent cytotoxic effects of CD8+ T cells. (2) Fibroblasts in the TME through necroptosis induce the robust immune response via NF-κB signaling. Besides, the necroptosis-induced release of regulatory cytokines such as IL-1α by CD8+ T cells triggers inflammation and promotes tumor growth by facilitating proliferation and migration of cancer cells

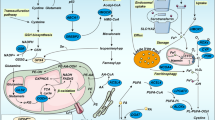
Crosstalk between Macrophages I/II cells and dying cancer cells in the tumor microenvironment. Tumor cells affect the function of macrophages by releasing DAMPs such as KRAS-G12D, HMGB1, 8-OHG, and PGE2 through ferroptosis. Pancreatic cancer cells can release KRAS-G12D during ferroptosis, whose exocytosis is largely dependent on autophagosome-derived amphisomes. KRAS-G12D triggers M2 cells polarization by binding to AGER which might induce adaptive immunosuppression. In addition, iron-addicted cancer cells activate STING-dependent DNA sensor pathways in M1 cells through the release of 8-OHG to create an inflammatory microenvironment for tumor growth. Similarly, PGE2, induced by ferroptotic cancer cells can act on DNMT3A, causing DNA methylation thus suppressing immunogenic genes. M1 cells are more resistant to ferroptosis than M2 cells, even in the absence of GPX4. Mechanically, iNOS which is highly expressed in M1 cells but inhibited in M2 cells produces more NO•, replacing GPX4 as a negative regulator of ferroptosis. Furthermore, excessive lipid accumulation in macrophages can prevent autophagy in obese mice, thus promoting the conversion of macrophages into pro-inflammatory M1 cells. In the TME, IL-6, and CCL2 trigger autophagy by binding to IL-6R and CCR2, respectively, which is essential for macrophage polarization to M2 phenotype. Furthermore, in M2 cells, TLR2 signaling inhibits the NF-κB signaling pathway through selective autophagy. TLR2 signaling also promotes sustained phosphorylation of MAPK1 and MAPK3, which stimulates autophagy-dependent NF-κB regulation. Autophagy can be inhibited in M2 cells regulated by TLR4-mTOR pathway. In addition, triggered by NLRP3 inflammasome, tumor cell-derived IL-1β and IL-18 recruit M1/2 cells to inhibit or promote tumor progression

The role of autophagy, ferroptosis, pyroptosis, and necroptosis in immunogenic cell death. During ICD, cancer cells can release specific DAMPs such as HMGB1, ATP, lipid mediator, etc. and cytokines such as IL-18, IL-1β, etc. through specific cell death such as autophagy, pyroptosis, ferroptosis, and necroptosis to act on tumor-promoting immune cells including M2, MDSCs and Treg cells or antitumor immune cells including M1, NK, and CD8+ T cells by binding to receptor specifically. It’s worth noting that mitophagy, a selective form of autophagy removes damaged mitochondria, suppresses type I IFN production and inhibits inflammasome activation thus reducing IL1β and IL18 production. The occurrence of glycolysis in MDSCs reduces autophagy, which reduces the expression of G-CSF and GM-CSF and prevents MDSCs proliferation. And MDSCs selectively accumulate AA-tags and PGE2 but not PL-PUFA and LPOs, thus forming ferroptosis resistance. Moreover, MDSCs with high Xc-system expression consume cyst(e)ine which is not transported to the microenvironment due to the absence of ASC transporter proteins, thereby depriving of the cyst(e)ine required for T cells activation. Tregs express high levels of ARG2, leading to activation of autophagy. When TCR/CD28 is co-stimulated, ferroptosis is reduced by the expression of GPX4 in Tregs. In addition, hypoxia induces HIF-2 to transport to the nucleus and activates the autophagy sensor ITPR1 to degrade NK cell-derived GZMB. Furthermore, lipid peroxidation in ferroptosis inhibits glucose metabolism in NK cells leading to NK cells dysfunction
Autophagy synergizes antitumor immune response
Recent research has shown that autophagy contributes to antitumor immunity such as innate immunity, antigen presentation, immune cell development, and inhibition of immune evasion.157 Autophagy substrates, DAMPs and PAMPs trigger innate immunity whose elimination through autophagy is necessary for immune homeostasis to protect the cells from exposed membranes and other organelles.158 For example, mitophagy is a selective form of autophagy in response to diverse stimuli that removes damaged mitochondria, suppresses type I IFN production, and inhibits inflammasome activation thus reducing the production of IL1β and IL18 as a result of preventing the accumulation of mitochondrial-derived DAMPs, such as ROS and mitochondrial DNA (mtDNA).159 Autophagy inactivation increases the production and secretion of inflammatory cytokines such as type I, II IFNs and TNFα. As well, mice with essential autophagy genes produce fewer type I and II IFNs, TNFα, and C-C Motif Chemokine Ligand 2 (CCL2).160,161 Therefore, we may conclude that even though autophagy-deficient primary tumors infiltrated with the pro-inflammatory TME may be suppressed, induction of inflammation-related cancer probably results from autophagy deficiency and inability to remove bacteria, organelles, and damaged proteins.162 In autophagy-deficient mice, unchecked innate immune activation and damage in normal tissues leads to human diseases related to defective autophagy genes, including Crohn’s disease, the risk factor for colon cancer.159
Autophagy serves as an ICD that promotes ATP secretion by facilitating the migration of ATP-containing lysosomes toward the plasma membrane.163 Due to ATP’s crucial role as a chemotactic signal, chemotherapeutic agents are less likely to trigger a robust antitumor immune response when autophagy is lost in tumor cells.164 Besides ATP, other signals from the intra- and extra-cellular can be presented to antigen-presenting cells (APCs) through autophagy. Pathogens are engulfed by autophagosomes and degradation products are delivered to the major histocompatibility complex II (MHCII) of APCs, thereby activating CD4+ T cells.165 A study has concluded that when ATG5 is defective, the fusion of lysosomes with phagosomes is delayed, thereby inhibiting antigen presentation by dendritic cells (DCs) via the MHCII and activation of CD4+ T cells.166,167 Autophagy may also promote the presentation of extracellular antigens to MHCII through ATG8/ LC3-related phagocytosis (LAP), one of the atypical autophagy pathways. LAP participates in the uptake and degradation of dying cells by macrophages, which subsequently present antigens to immune effector cells. In the absence of LAP, inflammation is caused by an imbalance of pro-inflammatory and anti-inflammatory cytokines.168,169,170 Furthermore, autophagy contributes to MHCI-mediated cross-antigen presentation. α-Tocopherylacetic acid (α-TEA) induces autophagy and produces autophagosome-rich supernatant fraction, α-TAGS, which acts as a carrier of antigen for cross-presentation to specific CD8+ T cells via MHCI.171,172
Studies have demonstrated that autophagy is closely related to T cells survival, activation, proliferation, differentiation, and functional performance.173 The survival of peripheral naive T cells is dependent on T cell receptor (TCR) interaction with stromal cells and the process of IL-7 signaling, which is involved with ATG3-dependent autophagy proteins.174 On the contrary, in tumor-bearing mice, tumor-infiltrating T cells (especially naive T cells) often exhibit impaired autophagy and undergo apoptosis, thus supporting tumor immune escape which is caused by tumor-derived lactate suppressing FIP200 expression in T cells by disrupting the balance between pro-and anti-apoptotic BCL2 family members.175 Once TCR is stimulated, activated T cells have enhanced levels of autophagy, which is associated with rapid elevation of calcium levels that activate ULK1 complex phosphorylated by AMPK to promote autophagy.176,177 When ATG3, 5, and 7 genes are defective in activated T cells, cyclin-dependent kinase inhibitor1B (CDKN1B) cannot be degraded, resulting in the inability of T cells to proliferate efficiently.178 Autophagy regulates T cells differentiation by affecting different metabolic programs.179,180 When activated T cells are more likely to induce mTOR, T cells differentiate into T helper cells (Th cells) due to enhanced glycolysis. When the activated level of AMPK is higher, primitive T cells undergo lipid peroxidation and preferentially differentiate into T regulatory cells (Treg cells).181 In addition, autophagy maintains T cells homeostasis by regulating the mitochondria content during T cells development. Defective autophagy leads to inadequate degradation of mitochondria components and increases ROS production thereby disrupting T cells development and function.182 Interestingly, lipophagy may be involved in the fatty acid β-oxidation process and promote the formation of memory T cells.183 The effector CD8+ T cell that lacks autophagy is incapable of establishing a lasting memory for providing antitumor immunity.179,184
In addition to contributing to the antitumor immune effect of T cells, autophagy is involved in B cells development, differentiation, and antibody production. It was demonstrated that ATG5 was required for effector B cells development and maintenance of B1 CD5+ (B1a) cell numbers. Knockdown of ATG5 resulted in impaired development of B cells in the bone marrow and reduced numbers of B1a cells in the peripheral blood.185 On the contrary, another study showed that autophagy was not necessary for the transition of progenitor B cells to pre-B cells and B cells activation, but was required for plasmocyte differentiation and specific production of IgM and IgG in response to LPS stimulation.186 The tumor-derived autophagosomes (DRibbles) stimulate the activation of B cells, which secrete antibodies and cytokines.187 Mitochondrial autophagy is required to maintain the survival and function of reactive B memory cells. Mice lacking mitochondrial autophagy genes accumulate mitochondria and experience oxidative phosphorylation and fatty acid synthesis, leading to the loss of B memory cells.188 Similarly, autophagy facilitates the differentiation of monocytes to macrophages stimulated by colony-stimulating factor 1 (CSF1) and CSF2. Mechanically, CSF1 promotes autophagy by increasing the expression and phosphorylation of ULK1.189 CSF2 helps Beclin1 release from Bcl-2 protein and thus stimulates autophagy by activating c-Jun N-terminal kinase (JNK) and blocking ATG5 cleavage.190 Furthermore, defective autophagy promotes inflammation by promoting M1 polarization. Recent evidence suggests that in obese mice, excessive lipid accumulation in macrophages can promote the conversion of macrophages to pro-inflammatory M1 cells via inhibited autophagy pathway, leading to the progression of liver inflammation and liver injury.191
As we know, cancer immunotherapy can be improved by blocking PD-1/PD-L1 immune checkpoints by binding PD-L1 on cancer cells to PD-1 on T cells resulting in T cells inactivation, and consequently cancer immune invasion.192,193 A growing body of evidence suggests autophagy may affect cancer cells’ immune escape through the degradation of immune checkpoint protein. A recent study has shown that as autophagy receptor for PD-L1 binding, Huntingtin-interacting protein 1-related (HIP1R) induces PD-L1 degradation in lysosomes, subsequently suppressing the tumor growth via activation of T cells.194 However, cancer cells inhibit the degradation of PD-L1 by autophagy via transcriptional modification. For example, in a breast tumor model, epidermal growth factor receptor (EGFR)/β1,3-N-acetylglucosaminyltransferase-3 (B3GNT3) pathway-mediated PD-L1 glycosylation inhibits autophagic degradation of PD-L1, leading to tumor immune escape.195 Likewise, a colon tumor model shows that palmitoylation of acyl transferase DHHC3-induced PD-L1 decreases its autophagic degradation, causing the immune suppression and tumor growth.196 In addition to the modification of PD-L1, the cell membrane chemokine-like factor super family 6 (CMTM6) binds to PD-L1, inhibits endocytosed degradation of PD-L1, leading to tumor immunity evasion.197 Nevertheless, another study demonstrates that the activation of autophagy increases the expression of PD-L1 by 5-hydroxytryptamine receptor 1A (5-HT1AR)/autophagy/STAT3 phosphorylation pathway in lung cancer patients suffering from depression that results in immune escape which remains to be determined.198 As another immune tolerance checkpoint, cytolytic T lymphocyte-associated antigen-4 (CTLA-4) is an effectively therapeutic target for cancer patients. In the presence of CTLA-4, PI3K/AKT/mTOR pathways are activated significantly and translocation of forkhead box protein O1 (FOXO1) to the nucleus is induced, which constrains LC3β transcription and autophagosomes formation, consequently inducing autophagy deficiency.199 Nevertheless, the activation of autophagy can increase CTLA-4 expression, restore CTLA-4 suppressor activity and expand Tregs which can inhibit inflammation and suppress inflammatory cancer.200
As another immunologic tolerance molecule, indoleamine 2,3 dioxygenase (IDO) induced by tumor cells, tumor-associated myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs) have been shown to suppress the CTLs responses and inflammatory DCs maturation, augment tolerogenic APCs, and stimulate Tregs differentiation, thereby alleviating effective antitumor immunity, facilitating immunological tolerance, and promoting the tumor growth.174 Inflammation-mediated IDO production can be inhibited by suppressing inflammation via autophagy.201 In turn, IDO can inhibit expression of mTOR, leading to autophagy via LC3 production. General control nonderepressible 2 (GCN2), recognized as a key effector of the IDO pathway inhibits the translation of initiation factor 2α (eIF2α), reduces protein synthesis, and blocks cell growth which are important in inflammatory carcinogenesis.202 There is a possibility that the IDO1/GCN2 autophagy pathway may play a significant role in human inflammatory conditions, since autophagy induced by IDO or GCN2 can protect organisms from death-causing inflammatory disorders.203
As well as PD-1/PD-L1, CTLA4, and IDO, SIRPα/CD47 immune checkpoints act as “don’t eat me” signals to prevent macrophage phagocytosis of cancer cells.204,205 CD47, which is highly expressed on cancer cells, binds to SIRPα on macrophages, inhibiting phagocytosis.204 Exosomal CD47 can inhibit pancreatic cancer cells from being cleared by phagocytes.206 It’s worth noting that PD-L1 or CD47 can be released by exosomes, cellular secreted vesicles (30–150 nm) with double-layer membrane not degraded by lysosomes, which is key to regulate crosstalk between cells.207 However, the relationship between exosomes and autophagy is still unclear, which requires further investigation.
Pyroptosis synergizes antitumor immune response
Two concurrently published studies have found that tumor cells released danger signals that recruited antitumor immune cells through pyroptosis while immune cells induced pyroptosis in tumor cells, thereby establishing a positive feedback loop.29,31 A bioorthogonal system was built to release GSDMA3 into tumor cells and Wang et al. found that only 15% of tumor cells required pyroptosis to eliminate the whole tumor. Further studies concluded that the number of CD4+ T, CD8+ T, natural killer cells (NK cells), and M1 macrophages increased in tumors that underwent pyroptosis, while the number of monocytes, neutrophils, MDSCs, and M2 macrophages decreased.29 Along with increased levels of IL-1β, IL-18, and HMGB1, many effector genes for immunostimulatory and antitumor effects were upregulated, whereas various effector genes for immunosuppressive and tumor-promoting effects were downregulated.29
Zhang et al. reported that CD8+ T cells and NK cells induce pyroptosis of tumor cells independent of caspases through the GSDME-GZMB axis in their study.31 Recent studies have shown CD8+ T cells and NK cells can evoke tumor pyroptosis through the GSDMB-GZMA axis, which is induced by interferon-γ(IFNγ). GZMA may be delivered by immune cells to GSDMB-expressing cancer cells to promote antitumor immunity.114 In addition, a previous study showed that GSDMD plays a key role in antitumor function of CD8+ T cells.131 GSDMD and GZMB coexist near immune synapse and GSDMD deficiency has been shown to reduce the cell-killing capacity of CD8+ T cells. Considering that release of cytotoxic molecules into immune synapse is a key pathway for CTLs killing capacity, we hypothesize that GSDMD-GZMB axis may be a potential mechanism for CTLs to exert cytotoxicity. In the past, perforin has been thought to be the only protein responsible for pore formation on CD8+ T cells,208 but it was suggested by the authors that GSDMD could be a new pore-forming protein utilized by effector T cells to form pores in tumor cells.40 Nevertheless, the mechanism of GSDMD transportation from CD8+ T cells to tumor cells remains to be further explored.
Moreover, nuclear PD-L1 is able to modulate the non-canonical pyroptosis pathway mediated by GSDMC/CASP8 to induce tumor necrosis in cancer cells in hypoxic conditions. The nuclear PD-L1 family can switch TNFα-induced apoptosis into pyroptosis by upregulating GSDMC expression, leading to tumor necrosis and promoting tumor growth.209 Under hypoxic stress, the phosphorylated form of STAT3 interacts with PD-L1 to promote nuclear translocation of PD-L1, which in turn activates mRNA transcription of the GSDMC gene. In addition, TNFα treatment cleaves GSDMC via CASP8, releasing its N-terminal domain from the cell membrane and causing pyroptosis to occur.209
Together with the important role of GSDM family proteins in antitumor immune responses, inflammasomes are also key players. The antitumor role of inflammasomes in colitis-associated cancers has been extensively studied, and NLRP3 inflammasomes promote IL-18 secretion by bone marrow-derived cells and intestinal epithelial cells, thereby enhancing the activity of NK cells and CD4+ T cells to protect enterocytes from drug-induced damage in early colitis.210,211 Similarly, IL-18 induced through NLRP3 inflammasomes promote hepatic NK cell maturation, expression of the death ligand FasL, and lethality in tumors sensitive to FasL, thus inhibiting liver metastasis of colorectal cancer.212 Consistent with this, monocytes can be differentiated into DCs and maturation of DCs occurs through IL-1β induced by pyroptosis. What’s more, IL-1β can hyperactive DCs to facilitate tumor lysates as immunogens and bind to the surface of lymphocytes to drive antigen-specific cytotoxic CD8+ T cells responses.213,214 IL-1 has been shown to be effective in regressing different types of transplanted syngeneic tumors, as shown in the above research.215,216,217 In addition to its therapeutic effects, Allen and colleagues demonstrated that IL-1β protected against chemically induced colitis and colon cancer in animal models.218 Regardless, despite the fact that recombinant IL-1 has been shown to exert antitumor effects in various mouse studies, its systemic application has only produced limited benefits and significant toxicity on hematologic and solid tumors in a number of clinical trials.219 To prevent intense cytotoxicity, IL-1 is encapsulated into microspheres which is preferentially internalized by macrophages, thus promoting APCs activation.220 A system that delivers IL-1 intratumorally into fibrosarcoma-burdened mice can effectively cause tumor cell necrosis as well as strong leukocyte infiltration, which delays tumor growth.220 Furthermore, the release of IL-6 from pyroptotic cells contributes to the adaptive response by increasing cell trafficking, differentiation, and antibody production of CD8+ T cells, inhibiting Tregs differentiation and macrophages death.221,222
Interestingly, it is increasingly recognized that members of the intracellular sensor protein NLR family act independently of inflammasomes.223,224 Janowski et al. found that NLRC4 in TAMs inhibits melanoma progression by enhancing T cells function. When NLRC4 is defective in mice, macrophages are less able to produce cytokines and chemokines, and subsequently less able to recruit T cells near the tumor, which promotes tumor growth, independent of the inflammasome components ASC and CASP1.225
Along with inflammatory cytokines, the DNA binding protein, HMGB1, is also released during pyroptosis as well as necroptosis. Once released, HMGB1 binds to the RAGE receptors on tumor or immune cells, or TLR2/4 on the surface of immune cells.226,227 It’s worth noting that differential effects of these receptors on tumor growth are evident. HMGB1 through pyroptosis mediated by GSDME in epithelial cells, binds to RAGE and activates the extracellular regulated protein kinases (ERK1/2) signaling increasing cell migration by activating Rac1 and Cdc42.228 It has been reported that an elevated HMGB1 level is associated with invasion and metastasis in many cancer types. The inhibition of tumor growth by blocking HMGB1 and RAGE signaling was observed in a murine lung cancer model.229 In addition to RAGE receptors, HMGB1 signaling acts on neutrophils, monocytes, macrophages, etc. through TLR2 and TLR4 receptors activating transcription factors NF-κB and AP-1, triggering inflammation and cytokines’ production such as IL-6, TNFα, IL-8 needed for CD8+ T cells activation.226 Through HMGB1 signaling, chemotherapy and radiotherapy-induced cell death leads to an increase in antigen processing and cross-presentation on DCs.230 Thus, we may conclude HMGB1 can play a dual role by signaling through RAGE or TLRs in tumor growth.
As cells undergo pyroptosis and necroptosis, another DAMP, ATP released from cells, binds mainly to the purinergic P2X and P2Y receptors, exerting different antitumor effects depending upon each receptor.231 As a result of P2YR signaling, IL-8 is secreted,232 increasing neutrophil recruitment and phagocytosis.233 ATP binds to its purinergic receptor P2RX7, activates NLRP3 inflammasomes on myeloid APCs, and stimulates IL-1 signaling.234 A recent study found that mice lacking the ability to activate NLRP3 inflammasomes and signal IL-1 and IL-17 did not respond to ICD inducers, such as anthracyclines, a chemotherapy agent.235 However, even though together with dying tumor cells that released ATP, an inflammasome inducer, DCs lacking inflammasome activity could not effectively activate CTLs which warrants further investigation.234 Other representative DAMPs released by ICD and molecular mechanisms for DAMP-mediated activation of the immune system in TME have been elaborated in detail by Hernández et al., etc.236
Ferroptosis synergizes the antitumor immune response
Similar to other ICDs, ferroptotic cells may release lipid mediators as ‘find me’ signals, which recruit APCs and other immune cells to ferroptotic tumor cells microenvironment.100 As well as contributing to the oxygenation of esterified PUFAs as ferroptotic signals, the oxidation products released by ferroptotic cells may also be immunomodulatory.100 In response to inducible GPX4 depletion, eicosanoids can be released by cancer cells through ferroptosis.237 Nevertheless, with the stimulation of TNF or IL-1β, enhancing GPX4 activity reduces the activation of pro-inflammatory lipid mediators such as LTB4 mediated by NF-κB signals.238 It’s noticed that LTB4 is one of the most established pro-inflammatory leukotrienes which plays a key role in carcinogenesis.239 With a deeper understanding of free eicosanoids as signaling molecules in modulating immune responses, the interest in esterified eicosanoids biological roles is increasingly growing, which are derived from phospholipids by lipoxygenase (LOXs) activity or from eicosanoids by re-esterification into phospholipids.240 In addition, the biological effects of extracellular oxidized PEs or their degradation by oxidation or hydrolysis have been much less uncovered, but it has been demonstrated that lyso-phospholipids can promote APCs to induce apoptosis.241 It has also been shown that oxidative state of externalized phospholipids can increase macrophage activity in engulfing and clearing apoptotic cells and macrophages are more likely to phagocytize apoptotic cells whose outer plasma membranes carry peroxidized phosphatidylserine (PS) than cells lacking PS.242 Ferroptotic cells secrete an oxidized PL, 1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH), which is an important “eat-me” signal activating macrophages to phagocytose.243 In principle, oxidized PL from ferroptotic cancer cells may modulate immune cells’ activity and response, but this claim has not been experimentally tested.
Some immunosuppressive cells, such as M2-type macrophages, Treg cells, and MDSCs antagonize ferroptosis by high expression of GPX4 or other components to maintain cells activation. Induction of ferroptosis in these cells may cause cell death and reversal of their tumor-promoting function. M1 cells are highly resistant to ferroptosis compared to M2 cells even in the absence of GPX4.244 Mechanically, M1 cells express high levels of nitric oxide synthase (iNOS) and produce more NO radicals (NO•) which are inhibited in M2 cells. NO• could react with lipid radicals or lipid peroxidation reactions intermediates, thus replacing GPX4 as a negative regulator of ferroptosis. Thus, in the presence of ferroptosis inducers, M2 cells can undergo ferroptosis or repolarize to the M1 phenotype and subsequently exert antitumor effects.244 Similarly, in Tregs activated by TCR/CD28 co-stimulation, GPX4 expression is promoted thereby reducing the occurrence of ferroptosis.245 The deletion of the GPX4 gene can lead to excessive accumulation of lipid peroxides (LPOs) and ferroptosis which promote IL-1β production to enhance T helper 17 (Th17) cell antitumor immune response.246 Likewise, the function of MDSCs is closely related to lipid transport and metabolism. Polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) can depend on myeloperoxidase to undergo lipid peroxidation and transfer lipids to DC cells, blocking the cross-presentation of DC cells and thus exerting tumor-promoting activity.247 In addition, MDSCs selectively accumulate arachidonic acid esterified triglycerides (AA-tags), oxidized AA-tags, and prostaglandin E2 (PGE2) but not PL-PUFA and associated lipid peroxides(LPOs) which lead to ferroptosis, thus forming ferroptosis resistance.248 What’s more, MDSCs with high expression of the Xc- system consume extracellular cyst(e)ine, but do not transport cysteine to the microenvironment due to the lack of ASC transporter proteins, thereby depriving of the cyst(e)ine required for T cells activation.249 Notably, the process of ferroptosis occurring in MDSCs is regulated by the p53 pathway. When p53 protein stability is increased, the production of ROS is inhibited, thereby suppressing ferroptosis in MDSCs.250
Immune cells exert antitumor immune functions by releasing cytokines that promote ferroptosis activity in tumor cells. For example, IFNγ released by CTLs activates the Janus tyrosine kinase (JAK) signal and signal transducer and activator of transcription 1 (STAT1) pathway, which downregulates the Xc-system expression and increases intracellular stored iron content thereby inducing ferroptosis.30 Similarly, transforming growth factor-β (TGF-β1) released by macrophages can inhibit the Xc-system transcription via SMAD signaling thereby promoting ferroptosis.251
Necroptosis synergizes the antitumor immune response
As we know, necroptosis is a form of ICD due to the release of DAMPs. However, effectors in necroptosis such as RIPK1 and RIPK3 can directly regulate the function of immune cells independently of cell death.252,253 In support of this, RIPK3-mediated phosphatase phosphoglycerate mutase 5 (PGAM5) activation promotes natural killer T cells-mediated antitumor immune responses by nuclear translocation of nuclear factor of activated T-cells (NFAT) and dephosphorylation of dynamin-related protein 1 (Drp1) in a process independent of the necroptotic pathway.254 Necroptotic tumor cells activated by RIPK3 were injected into pre-existing tumors to enhance antitumor immunity in syngeneic melanoma and lung adenocarcinoma models.255 The RIPK3 knockout model of lung carcinoma and lymphoma reduced the efficacy of chemotherapy in vivo, which was linked to decreased CD8+ T cell infiltration.256 Similarly, RIPK3(−/−) mice exhibit NF-κB inactivation and impaired secretion of cytokines IL-1β, IL-23, and IL-22, which in turn lead to DC cell dysfunction in damaging inflammation and tissue repair.257 It is revealed RIPK3 plays an important role in NF-κB activation, expression of innate inflammatory cytokines, and involvement in tissue repair of DC cells.257 Likewise, another study demonstrated the presence of inflammatory gene expression indepentent of plasma membrane rupture caused by necroptosis.258 Forced dimerization of MLKL induced-necroptosis promotes inflammatory cytokine release at much lower levels than that of necroptosis induced with TNFα-RIPK-MLKL-NF-κB pathway, suggesting that cell-autonomous inflammatory cytokine expression synergizes with DAMPs release to mount an immune response.259,260
Combined with the above, two strategies are currently available to trigger antitumor immunity through necroptosis.40 In 2016, Aes et al. first demonstrated that necroptosis of tumor cells is one of the ICD, through which necroptotic cells can release DAMPs to DC cells to trigger antigen presentation and thus activate cytotoxic CD8+ T lymphocytes.151,261,262 Furthermore, unlike the study by Aes et al., another study found that fibroblasts in the tumor microenvironment(TME) through necroptosis induced the robust immune response via NF-κB signaling rather than MLKL-mediated cytolytic-dependent DAMPs release.255 In mice tumor model in which DAMPs receptor expression is absent, fibroblasts undergoing necroptosis still inhibited tumor growth.255 Similarly, Yatim et al. also emphasized the necessity of NF-κB for initiating the immune response and the interaction between necroptosis and TME. During necroptosis, inflammatory mediators released from dead cells are not sufficient to initiate CD8+ T cells, whereas separating NF-κB signaling from necroptosis decreases the efficiency of immune response initiation.263
Interestingly, in contrast to the above, high expression levels of RIPK1 and RIPK3 in human pancreatic cancer cells predicted enhanced migration and invasion of tumor cells,41 whereas low expression of MLKL was linked to reduced overall survival (OS) in patients with early resectable pancreatic cancer and reduced recurrence-free survival and OS in pancreatic cancer patients receiving adjuvant chemotherapy,264 which suggests necroptosis effectors differentially influence tumor pathogenesis in different contexts, and this heterogeneity has not been explained so far. Furthermore, in some cases, RIPK1 is not essential for tumor development. For example, researchers have found that RIPK1 inhibitors do not suppress tumor growth in genetically engineered mice models of pancreatic cancer.265 And it has been shown that in mouse mammary tumors, knockout of ZBP1 and MLKL, but not RIPK1, reduces lung metastasis.266 We may therefore conclude that RIPK1 regulates tumor growth through its scaffolding function rather than its kinase activity.260
The role of cytokines and DAMPs such as HMGB1, ATP, etc. has been described in detail in the pyroptosis section of this paper. In addition to the factors secreted directly by cells undergoing ICD, the immune response is further amplified by the activity of APCs. The role of APCs in the clearance of dying cells is dependent on the mode of death, leading to either anti-inflammatory responses (when apoptotic cells are cleared) or pro-inflammatory responses (when pyroptotic or necroptotic cells are cleared). To be specific, when apoptotic cells are engulfed, phagocytes induce secretion of anti-inflammatory factors such as IL-10 and TGF-β and inhibit the release of pro-inflammatory cytokines and chemokines such as IL-6, IL-1β, CCL2, CCL3.267,268 In contrast, as a result of phagocytosis of necroptotic colon carcinoma cells, DCs mature and cross-present to CD8+ T cells, promoting the activity of CD8+ T cells and the production of IFNγ.261 Thus, the release of inflammatory mediators directly from the cells through ICD along with DCs maturation and CD8+ T cells activation causes a strong immune response.269 We can easily draw a conclusion that pyroptosis and necroptosis are able to induce inflammation through the release of DAMPs and cytokines as well as the change of APCs responsible for phagocytosing dying cells.
Autophagy, pyroptosis, ferroptosis, and necroptosis antagonize the antitumor immune response
Although autophagy, pyroptosis, ferroptosis, or necroptosis are generally considered to contribute to the immune response against tumors, studies have concluded that the survival, proliferation, differentiation, and activation of immunosuppressive cells including Treg cells, M2 macrophages, MDSCs, etc. are also dependent on these RCDs under certain circumstances. Moreover, immune-promoting cells are negatively regulated by several RCDs. In addition, the release of DAMPs during ICD promotes the development of inflammatory responses favoring tumor growth in addition to stimulating antitumor immune responses (Figs. 2, 3, and 4).
Autophagy antagonizes the antitumor immune response
Autophagy may enhance or inhibit the growth, development, and functional performance of immune cells, depending on whether they have tumor-promoting or antitumor function.159,270,271 In addition, autophagy is also involved in antigen-presentation component of the adaptive immune response.42 Overall, autophagy facilitates tumor cell evasion from immune surveillance, leading to intrinsic resistance to antitumor immunotherapy.
Autophagy is required by Treg cells to suppress antitumor immune responses. For example, human melanoma-infiltrating Treg cells express high levels of arginase 2 (ARG2), leading to intracellular arginine degradation and inhibiting activation of arginine-mediated mTOR, which may activate autophagy.272 In autophagy-deficient Treg cells, enhanced glycolytic activity and loss of the characteristic forkhead box protein P3 (FOXP3) expression are induced through the mTOR-MYC pathway thereby increasing apoptosis.273,274 Consistently, silencing of key molecules involved in autophagy including Beclin1, ATG5, and PI3 kinase class III (PI3K3) may lead to impaired function of Treg cells.275,276 In the TME, IL-6 and CCL2 trigger autophagy by binding to interleukin 6 receptor (IL-6R) and CC chemokine receptor 2 (CCR2), respectively, which is essential for macrophage polarization to the immunosuppressive M2 phenotype.277,278 Furthermore, it was recently shown that HCC-acquired TLR2 signaling inhibited the NF-κB signaling pathway and thus derived macrophage polarization toward M2 phenotype, which was achieved by the selective autophagy-mediated degradation of NF-κB p65.279 Inhibition of autophagy can restore NF-κB activity and induce high levels of M1-like cytokine production by M2-polarized macrophages. In addition, TLR2 signaling can promote sustained phosphorylation of MAPK1 and MAPK3, which stimulates autophagy-dependent NF-κB regulation.279 However, autophagy may be inhibited in M2 macrophages through LPS or bacterial infection, which is regulated by mTOR pathway activated by TLR4.280 MDSCs are also supported by autophagy for survival and development. Glycolysis reduces partial hepatic enrichment activating protein expression in triple-negative breast cancer by blocking AMPK-ULK1 signaling and autophagy formation, which reduces granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression, thus preventing MDSC developments.281 MDSCs, in turn, activate AMPK, stimulate autophagy, and promote the expression of anti-apoptotic factors MCL-1 and BCL-2, thereby promoting multiple myeloma development.282
Autophagy helps tumors evade surveillance of CTLs, thereby developing immune tolerance. For example, autophagy induced by 5- hydroxytryptamine/5-hydroxytryptamine 1a receptor (5HT/5-HT1aR) signaling pathway facilities an immunosuppressive NSCLC environment and tumor cell resistance to CTLs-mediated lysis through STAT3 phosphorylation.198 Further, autophagy-deficient host mice have tumor-rejecting T cells that are more active than those with adequate autophagy.256 Compared with autophagy-proficient tumor models, the augmented infiltration of immune cells and gene expression signatures of activated type I/II IFN pathway can be found in autophagy-deficient tumor models which can be explained by STING activation. In addition, inactivating both Sting and ATG7 gene led to the restoration of tumor growth in mice, showing that tumors are inhibited by innate immunity activation via STING by autophagy impairment.256 In addition to type I /II IFN, interestingly, gene expression profiling of tumor models showed that CTLs produced higher levels of IFNγ specifically in autophagy-deficient tumor models. IFNγ gene (IFNG) is also involved in antigen presentation and tumor suppression on autophagy-deficient hosts.256 Besides, the IFNG and ATG7 gene defect restored defective growth of tumors, showing the killing effects on tumor of IFNG induced by loss of host autophagy. For instance, the immunosuppressive TME of the liver and immune evasion is attributed to autophagy activation which suppresses innate immune response and thereby antitumor activity of T cells. T cells and IFNG are both required to induce tumor rejection by specific deletion of autophagy in liver hepatocytes.256 In order to block selectively autophagy to overcome hepatic autophagy immune tolerance, we need to uncover the exact mechanism by which autophagy loss activates STING-type I/II IFN pathway and IFNG/IFNγ activation in the hepatocytes.
In B16-F10 and 4T1 mouse tumor models, autophagy of tumor cells induced by hypoxia degrades NK cell-derived GZMB thereby impairing the tumor lysis function of NK cells.283 Mechanically, hypoxia-inducible factor-2α (HIF-2α) transports into the nucleus and activates the autophagy sensor inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) to degrade GZMB. Similarly, hypoxia negatively affects DC cells function which is associated with HIF-1α accumulation and DC cells’ autophagy/apoptosis regulated by the PI3K/AKT pathway.284
Besides, tumor cells can evade immune surveillance through autophagy to degrade MHCI complexes.198,285 For example, in pancreatic cancer cells resistant to ICIs, the MHCI complex is re-transported to the lysosome for selective autophagy by ubiquitin-binding receptor NBR1 and degraded, thereby preventing T cells recognition.286 By contrast, inhibition of autophagy restores the level of MHCI complex and improves antigen presentation, enhancing antitumor T cells responses and therefore reducing tumor growth. Similarly, in the presence of ATG5 and ATG7, as a result of endocytosis and autophagic degradation of the MHCI complex in DCs, antigen presentation and CD8+ T cells priming are inhibited, which is reversed in DCs with absence of autophagy.287 Recently, one study shows that radiotherapy-induced autophagy increases CD8+ T cells infiltration by modulating MHCI expression in NSCLC, but the direct relationship of MCHI expression with autophagy is still unclear.288 Likewise, E3 ubiquitin ligase leads to MHCII complex degradation in MDSCs, causing tumor immunity evasion. Conversely, ATG5 deficiency restores MHCII expression on MDSC surfaces.289 Results from these studies suggest that tumor immune escape might be facilitated by autophagic degradation of MHCI/II complex in both cancer cells and immune cells.
Pyroptosis antagonizes the antitumor immune response
It is noticed that as effector molecules during pyroptosis, whether cytokines play a synergistic or antagonistic role in antitumor immunity depends on the tumor microenvironment. During pyroptosis, activation of inflammasomes can promote the maturation and release of inflammatory factors such as IL-18, IL-1β, IL-10, etc. which may inhibit antitumor immune effects or cause an inflammatory cascade response, thereby promoting tumor development under particular circumstances. Expression or secretion level of IL-18 is detected in different cancer cells in comparison with normal tissue.290 For example, in lymphoma, NLRP3 inflammasome-induced IL-18 contributes to promoting proliferation, inhibiting apoptosis in cancer cells and reducing drug resistance by interfering with the balance of c-Myc/TP53 protein and Bcl-2/Bcl-2 associated with Bax protein.291 Similarly, the inflammatory adapter ASC/IL-18 signaling pathway has a tumor-promoting effect in gastric cancer. Further analysis revealed that the specific effect of IL-18 was associated with high expression of IL-18 gene in gastric cancer epithelial cells, whereas IL-1β was preferentially expressed in immune cells whose knockdown did not inhibit gastric carcinogenesis.292 And IL-18 induces migration of breast cancer cells through downregulation of claudin-12 as well as activation of the p38-MAPK pathway.191 Besides, IL-18 is able to induce angiogenesis which leads to increased migration/invasion in tumors and immune escape.293,294
Aside from IL-18, in cancer patients and experimental tumor models, raised levels of IL-1β are also associated with a worse prognosis, carcinogenesis, and cancer invasion.295,296 For instance, IL-1β mediates the proliferation and invasion of colon epithelial cancer cells through the stromal cyclooxygenase-2 (COX-2) signaling pathway.297 In prostate cancer, IL-1β promotes cancer cells proliferation and metastasis by activating MAPK-mediated IL-8 production.298 By maintaining a microenvironment for cancer stem cells and promoting angiogenesis, IL-1β contributes to tumor growth and progression. For example, dependently expressing vascular endothelial growth factor (VEGF) by IL-1β can pave the way for metastasis and modulate adaptive immune response.299,300 Notably, in a pancreatic cancer model, triggered by NLRP3 inflammasomes, tumor cell-derived IL-1β is involved in the construction of an immunosuppressive TME in which infiltrating CD8+ cytotoxic T cells are reduced while M2 macrophages, MDSCs, CD1HICD5+ regulatory B cells, and Th17 cells are increased, thus promoting pancreatic tumorigenesis in a xenograft mouse model.301 Specifically, macrophages infiltrated in tumors are stimulated by tumor cells and T cell-derived cytokines such as IL-1 to induce a polarized M2 phenotype characterized by IL-4 and IL-10, etc.302,303 IL-1β production induces CCL2 expression via inflammasome activation in TAMs and tumor cells as well, thereby governing the recruitment of myeloid cells into tumors, providing an inflammatory microenvironment and promoting breast cancer progression.304,305 In addition to TAMs, IL-1β also plays a role in the proliferation and migration of MDSCs regulated by the IL-1RI/NF-κB pathway.306 An experiment involving cells that were transfected with an IL-1β expression vector and injected into mice has demonstrated that mice that received transfected 4T1 tumor cells infiltrated with MDSCs at increased levels.307 In addition, the correlation between increased levels of serum IL-1β and a greater number of MDSCs and Tregs in peripheral blood reflects the importance of IL-1β as a proliferating factor for MDSCs.308 Furthermore, IL-1β upregulated COX-2, which encodes prostaglandins that are responsible for MDSCs expansion.309 Tissue-resident endothelial cells are activated by MDSCs to produce VEGF and other angiogenic factors with the stimulation of IL-1β and other pro-inflammatory agents.310 Aside from direct effects of IL-1β and its target genes on MDSCs, elevated CCL2 induced by IL-1β within TME promoted the recruitment of MDSCs.304
IL-1β has already been implicated in the transition from chronic inflammation to tumor development. It has been established that various tumors, including colorectal cancer, gastric cancer, liver cancer, lung cancer, and bladder cancer, can be triggered by persistent inflammation.311 Hepatitis C virus infection, for instance, induces hepatic inflammation that triggers the evolution from fatty liver disorder to fibrogenesis, and finally HCC induced by IL-1β.312 Helicobacter pylori (HP) infection, the most typical bacterial infection which is closely related to gastric cancer can also induce the amount of IL-1β production.313 The development of gastritis and gastric tumor have been linked to genetic polymorphisms in the IL-1β gene.314 An additional mechanism underlying the increased risk of gastric cancer associated with IL-1β is its ability to induce aberrant DNA methylation.315 Besides, it seems that IL-1β can influence chronic obstructive pulmonary disease (COPD) airway inflammation by upregulation in small airway epithelial cells of COPD patients.316 Although as a known risk factor, specific pathways of lung cancer developed by COPD is poorly known and IL-1β seems to play a profound role in this regard. In addition, 3-methylcholanthrene (3MCA), a chemical carcinogen, induces the development and invasiveness of tumors if it is exposed to IL-1β.317 Furthemore, chronic inflammation results in the induction of immune-suppressive MDSCs, TAMs, and NK cells mediated by IL-1β.318 To sum up, the key mechanisms of IL-1β-mediated tumor development include infiltration of immunosuppressive cells,319 tumor angiogenesis320, and driving chronic inflammation.312,313
IL-10-induced adaptive immunosuppression is also involved in the development of pancreatic cancer.321 IL-33 has also been regarded as a new type of danger signal released from pyroptotic cell that targets various immune cells and boasts anti- or pro-inflammatory properties depending upon the disorder.322 In reference to the above mentioned, IL-1β and IL-18, etc. as immunomodulatory cytokines have been attributed to either initiate adaptive antitumor responses or inhibit antitumor immune effects depending on the makeup of cytokine milieu.269,323 Furthermore, Liu’s study found that CAR-T cells could promote GSDME-mediated tumor cell pyroptosis by releasing perforin and GZMB in B-lymphocytic leukemia and solid tumor cells. Nevertheless, pyroptosis can be triggered again in macrophages by pyroptosis releasing factors from tumor cells, leading to the release of cytokines such as IL-6 and IL-1β, which in turn triggers cytokine release syndrome (CRS).324
Ferroptosis antagonizes the antitumor immune response
It is intriguing to speculate that a small part of cells undergoing ferroptosis in the TME may inhibit the immune system, which is mediated by the DAMPs such as KRAS-G12D, HMGB1, 8-hydroxyguanosine (8-OHG) through ferroptosis. During ferroptosis, KRAS-G12D may be released by pancreatic cancer cells, whose exocytosis is dependent largely on the ability to form amphisomes by fusing autophagosomes with multivesicular bodies.325 KRAS-G12D promotes M1 phenotype polarization to M2 phenotype by binding to advanced glycosylation end product-specific receptor (AGER) and induces adaptive immunosuppression by releasing arginine (ARG), IL-10, and TGF-β thereby stimulating tumor growth.325 Likewise, HMGB1 released by iron-addicted cancer cells promotes inflammatory responses in macrophages by binding to AGER.326 Furthermore, iron-addicted cancer cells activate STING-dependent DNA sensor pathways in macrophages by releasing 8-OHG in the presence of GPX4 depletion, promoting the release of cytokines such as IL-6 and nitric oxide synthase 2 (NOS2) to form an inflammatory tumor microenvironment that supports pancreatic cancer.327
Similarly, it has been demonstrated that ferroptosis in cancer cells is linked to increased expression of post-transcriptional gene silencing (PTGS2) and the release of PGE2.100 We may infer, if the sufficient levels are achieved, antitumor immune response will be converted to immunosuppressive responses,328 leading to progressive tumor growth, although more related experiments requires to be validated.329 It is interesting to note that PGE2 is released far prior to all cell deaths, suggesting that suppressed GPX4 activity may indeed be sufficient to sustain the PTGS2-active state.100 PGE2, induced by cancer cells through ferroptosis promotes the recruitment and activation of MDSCs and M2 macrophages and inhibits the antitumor function of NK cells, DCs, and cytotoxic T cells. Mechanically, in myeloid cells, PGE2 can activate DNA methyltransferase 3A (DNMT3A), leading to DNA methylation and suppressing immunogenic gene expression.330 A study has shown that PGE2 can exert immunosuppressive effect in cell lines based on a melanoma mouse model engineered to express BrafV600E mutation, the most prevalent mutation in patients. It was found that PGE2 production is sufficient to inhibit DC-dependent antitumor immunity mediated by CD8+ T cells in this model.331 Besides, considered as a major immunosuppressive mediator, PGE2 directly suppressed cytotoxic T cells activity, consequently interfering with multiple aspects of anticancer immunity.332 PGE2 also compromise DCs directly by reducing the amount of chemokine receptors that induce the recruitment into tumors.100 Further research found that PGE2 reduced the amount of DCs infiltrated into TME by suppressing chemokines CCL5 and XCL1.329 Although the action of PGE2 and its downstream signaling has not been elaborated more detailedly, current study has provided strong evidence that PGE2 has immunosuppressive effect towards NK cells.333
As the stimulation of ferroptosis, ROS acts a vital role in the modulation of immunity in human malignancies in addition to oxidative stress.334 The presence of high ROS inhibits T cells activation and proliferation, while low ROS can restore the cytotoxic effects of T cells.335 ROS suppress the formation of TCR and MHC antigen complexes in T cells, thus inhibiting immune responses.335 What’s more, ROS scavengers is able to improve the CTLs activation by activating superoxide dismutase 2 (SOD2).336 In addition, the ability of CAR-T cells to kill cancer cells has been linked to lower levels of intracellular oxidative stress.337 Thus, CAR-T cells in combination with catalase (ROS inhibitors) demonstrated better antitumor response even when exposed to oxidative stress on the extracellular surface.337 Besides, researchers also observed that CTLs extracted from murine with the treatment of PD-1/L1 antibody might have high mitochondria ROS and elevated O2− microenvironment, which results in compromised CTLs action and inhibited immune response. Thus, Metformin (a ROS inhibitor) combined with PD-1 inhibition enhanced intratumor T cell activation and proliferation, resulting in tumor clearance and alleviating tumor inflammation through the decreased level of tumor hypoxia.338 In addition to CTLs, multiple studies have shown that oxidative stress or ROS caused Tregs to suppress the immune system within a tumor niche. The mitochondrial complex III appears to be required for the inhibition of Tregs function.339 Kunisada and colleagues also found that metformin reduced the amount of tumor-infiltrating Tregs by suppressing the differentiation of naive CD4+ T cells.340 The MDSCs induced by tumors also inhibited T cells proliferation and increased colorectal carcinoma cell growth by producing ROS341 while the negative effect of MDSCs can be suppressed by catalase, thus restoring T cells action.342 ROS are also involved in the activation of macrophage signaling. It has been demonstrated by Lin X et al. that ROS may stimulate an invasive phenotype in TAMs derived from melanoma through the secretion of TNFα.343 Researchers have found several mitochondrial genes highly expressed in TAMs derived from melanomas, indicating ROS is the major cause of oxidative stress within TAMs.334 To sum up, the key mechanisms of ROS involved in TME in modulating tumor immunity remain to be unknown, which need more research.
In addition, many immune cells are sensitive to ferroptosis, including CD8+ T cells, NK cells, and DC cells. Stimulation of ferroptosis by inhibition of GPX4 activity can reduce the specific killing function of these immune cells. CD36 expression on the cell surface has been reported to be crucial for fatty acid or oxidized lipid-induced ferroptosis. Significant lipid peroxidation activity can occur in CD36-positive CD8+ T cells, which results in ferroptosis induced by GPX4 inhibitors, leading to reduced release of IFNγ and thus inducing immunosuppression.344,345 Although there are no relevant studies directly with ferroptosis in NK and DC cells, Poznanski et al. demonstrated that protein expression associated with ferroptosis, lipid peroxidation, and oxidative damage was increased and had a similar cell morphology to that of cells undergoing ferroptosis in NK cells. Furthermore, oxidative stress associated with lipid peroxidation inhibited glucose metabolism in NK cells leading to their dysfunction.346 Similarly, tumor-associated DC cells usually exhibit reduced antigen-presentation capacity due to elevated lipid levels, which is associated with ferroptosis susceptibility.98,347 For example, the 12/15-lipoxygenase(12/15-LO) inhibits DCs maturation and activation as well as dampens the differentiation of T helper 17 cells by generating phospholipid oxidation products that induce antioxidant responses dependent on nuclear factor erythroid 2-related factor 2 (NRF2).348 Similarly, Ramakrishnan et al. showed that various oxidized lipids in DCs, blocked the cross-presentation of exogenous antigens by reducing the expression of MHCI complexes on cell surface.349
Necroptosis antagonizes the antitumor immune response
Induction of necroptosis can generate an immunosuppressive TME in which tumor growth is allowed. A mouse model of pancreatic cancer showed that knocking out RIPK3 or RIPK1 inhibited oncogenic progression. RIPK3-dependent necroptosis of pancreatic cancer cells results in increased expression of sin3A-associated protein 130 (SAP130) and release of chemokines such as C-x-c motif chemokine ligand 1 (CXCL1) and CXCL5, leading to the recruitment of immunosuppressive cells such as MDSCs to form immunosuppressive TME mediating the migration and invasion of cancer cells.350,351 Similarly, RIPK3 signaling in MDSCs increases tumor size by expanding IL17-producing T cells in tumor models.352 In addition, RIPK1 expression was found to be upregulated and then STAT1 was inhibited in TAMs which were differentiated into immunosuppressive M2 cells.353 Inhibition of RIPK1 results in cytotoxic T cell activation as well as T helper cell differentiation to a mixed Th1/Th17 cell phenotype, stimulating antitumor immunity to suppress ductal adenocarcinoma of pancreas (PDA) growth in mice.353 Notably, the tumor-promoting effect of RIPK1 occurring here is independent of the synergistic effect of RIPK3 on it. However, the inhibition of RIPK1 in a mouse model of pancreatic ductal adenocarcinoma, however, did not result in an improvement in overall survival or tumor growth.265 Noteworthy, there may be differences in immunogenicity between the two groups based on the differences in pancreatic mouse models. And phase I/II trials (NCT03681951) about GSK3145095, one RIPK1 inhibitor, have been aborted in pancreatic cancer and headed back to the company’s research and development. In conjunction with this withdrawal, Patel et al. published results showing that RIPK1 inhibition did not inhibit cancer growth or metastasis. Moreover, the TNFα-induced systemic inflammatory response syndrome is effectively blocked in vivo by RIPK1 inhibitor PK68. Both melanoma cells and lung cancer cells are repressed by pretreatment with PK68 achieved by reprogramming of intra-tumoral macrophages. Also, conflicting results have been generated in the study on the impact of RIPK1 inhibitor on metastasis, showing GNE684 had no effect as compared to PK68.265,354 Therefore, different RIPK1 inhibitors have shown varying results and reinforced a nebulous role of RIPK1 in cancer, thus, require further study. In addition, deletion of MLKL in breast cancer cells reduced lung metastasis.355 In some cancer types, tumor cells have also shown a tendency to induce necroptosis in endothelial cells, causing the transendothelial migration and metastasis of tumor cells via expression of amyloid precursor protein.356 There is some uncertainty whether the effects on metastasis result from necroptosis itself or disruption of endothelial barriers. However, we can infer that the activation of necrosome enhances cancer progression. A more complex interaction has been observed between different cell types induced necroptosis and early disease outcomes.
Multiple of evidence suggests that DAMPs released from cells through necroptosis may also recruit inflammatory cells and release regulatory cytokines such as IL-1α, IL-18, etc. to trigger inflammation and promote tumor development by promoting angiogenesis, cancer cell proliferation, and thus metastasis.4,41,357,358,359 The role of cytokines and DAMPs such as HMGB1, ATP, etc. has been described in detail in the pyroptosis section of this paper. Interestingly, MLKL signaling activates NLRP3 inflammasomes, thereby activating CASP1 and triggering the release of the pro-inflammatory cytokine IL-1β which suggests that pyroptosis is involved in the pro-inflammatory process of necroptosis.360,361 Recently, Gutierrez et al. found that GSDMD required for cell lysis as a substrate for CASP1 during pyroptosis was not necessary for MLKL-dependent necroptosis and IL-1β secretion.360,361 We therefore infer that the activation of cytokine release by MLKL may occur before cell lysis, suggesting that MLKL is an endogenous activator of NLRP3 inflammasomes in a GSDMD-independent manner. Activated inflammatory cells may also release reactive nitro intermediates (RNI) and ROS, which promote tumorigenesis by damaging deoxyribonucleic acid and causing genomic instability.357 We have detailedly discussed the effects of ROS on immune modulation in the ferroptosis section of this paper.
New advances in targeting autophagy, pyroptosis, ferroptosis, and necroptosis in immunotherapy
Anticancer drugs targeting autophagy, pyroptosis, ferroptosis, and necroptosis
Targeting autophagy, pyroptosis, ferroptosis, and necroptosis to develop new anticancer drugs for clinical application is a long process, whereas current studies have shown that many drugs approved for clinical application exert potent antitumor activity by inducing (or inhibiting) these types of non-apoptotic RCD40 (Table 2). Currently, the only clinically approved autophagy inhibitors are chloroquine derivatives, which have a long history in the treatment of malaria and rheumatic diseases. To be specific, chloroquine (CQ) and hydroxychloroquine (HCQ) inhibit autophagosome degradation by inhibiting the lysosomal acidification.362 These drugs, especially HCQ, have been re-used in many clinical trials for the treatment of various cancers.363,364,365 Metformin inhibits cancer cell proliferation by inducing indirect pyroptosis activation through CASP3.366 In detail, metformin causes mitochondrial dysfunction and activates the AMPK/sirtuin1/NF-κB pathway, promoting the accumulation of Bax which results in CASP3 activation and GSDME cleavage. In esophageal squamous cell carcinoma (ESCC), metformin can induce GSDMD-mediated pyroptosis by targeting the miR-497-PELP1 axis.367 Sorafenib, an FDA-approved anticancer drug for the treatment of HCC, renal cell carcinoma, and thyroid cancer, as the Xc- system inhibitor, prompts ferroptosis by depleting the antioxidant GSH making the GPX4 system inactive.368,369 Nevertheless, some cancers are inevitably resistant to sorafenib, which may be caused by the upregulation of the non-GSH-dependent thioredoxin antioxidant pathway.370,371 Conversely, octreotide, which is FDA-approved for ovarian cancer treatment, can directly target and inhibit GPX4 to induce ferroptosis, which greatly overcomes the limitations of conventional drugs.372 In addition, the combination of lapatinib, a tyrosine kinase inhibitor used for breast cancer treatment, and siramesine, a lysosomal-disrupting hemolysin drug, can synergistically cause ferroptosis by disrupting iron transport and inducing lipid peroxidation in cancer cells.373 Shikonin, a natural naphthoquinone, is the first reported small-molecule drug to prompt necroptosis. It has been found to exert antitumor effects via the RIPK1/RIPK3-dependent necroptosis pathway in various cancers such as pancreatic cancer, and triple-negative breast cancer.374,375
The development of new compounds targeting autophagy, pyroptosis, ferroptosis, and necroptosis is ongoing. Lysosomal drugs based on the CQ structure are in development including Lys05,376 DQ661,377 and DC661.378 It has been shown that these compounds bind then inhibit the lysosomal enzyme palmitoyl protein thioesterase 1 (PPT1) to inhibit autophagy.379 In addition, studies on inhibitors of different targets in autophagy including ULK1 inhibitors such as SBI-0206965380 and ULK101,381 VPS34 inhibitors such as SAR405,382 compound 13,383 SB02024,384 and ATG4B inhibitors including S130385 and FMK-9a,386 and NSC185058,387 all of which have been shown to have excellent antitumor activity. Besides, the autophagy activator adiponectin ADIPOQ and 2-aminonicotinonitrile compound (w09) significantly inhibit breast cancer growth by stimulating autophagy through the serine/threonine kinase 11 (STK11)-AMPK-ULK1 and EGFR-mediated RAS-RAF1-MAP2K-MAPK1/3 pathway, respectively.388,389
α-NETA induces pyroptosis in epithelial ovarian cancer cells through the GSDMD/CASP4 pathway.390 In colorectal cancer cell lines expressing high levels of GSDME, the combination of TNFα and CHX activates members of the BCL2 family, BAK/BAX, which leads to mitochondrial outer membrane permeability (MOMP) and mediates pyroptosis.391 Polyphyllin VI (PPVI) induces pyroptosis in NSCLC via the ROS/NF-κB/NLRP3/GSDMD signaling, thereby inhibiting the proliferation of NSCLC.392 Bionanoparticles (BNP) loaded with chemotherapeutic agents have antitumor activity in breast cancer, showing that mitochondrial damage and activation of CASP3 is induced upon photoactivation, which subsequently leads to GSDME-mediated pyroptosis.393 Similarly, aurora kinase A (AURKA) inhibitors exert an inhibitory effect on pancreatic cancer growth by inhibiting the activity of AURKA to initiate the assembly and activation of necrosome thereby inducing necroptosis.394 The death receptor ligand TNF-related apoptosis-inducing ligand (TRAIL) acts on TNFR1, mediated by RIPK1/RIPK3 to lead to necroptosis, which is notably regulated by ROS as well as related caspases.395 CD95 ligand (CD95L) binds to its receptor CD95 and induces necroptosis by downregulating cIAPs.396 In addition, selenium nanoparticles induce ROS-mediated necroptosis in prostate cancer cell lines, whereas this cell line only relies on RIPK1 and does not require the activation of RIPK3 and MLKL.397 Ag/Au bimetallic nanoparticles trigger mixed programmed cell death including necroptosis and pyroptosis in p53 deficient cells while they also trigger the release of IL-1β and HMGB1.398
Besides, zero-valent-iron nanoparticle (ZVI-NP) in preclinical models could lead to mitochondrial dysfunctions, oxidative stress as well as lipid peroxidation, inducing ferroptosis in lung carcinoma cell lines.399 Ferroptosis inducers, BSO affects GSH synthesis, rendering GPX4 inactive, which in turn reduces tumor burden in mice371 and increases the sensitivity of melanoma and neuroblastoma cells to chemotherapy.400,401 Similarly, cyst(e)inase, a compound that increases the efficiency of GSH consumption and thus inhibits GPX4 activity, suppresses the growth of prostate and breast cancer xenografts and increases the survival of mice with chronic lymphocytic leukemia models.402 Likewise, drugs loaded GSH-bioimprinted nanocomposites are engineered to enhance anti-leukemogenesis by depleting GSH and disrupting intracellular redox status, thus inducing ferroptosis.403 In addition to inhibiting GPX4 by affecting GSH synthesis, consumption and depletion, withaferin A increases lipid peroxidation through heme oxygenase 1-mediated heme degradation, thereby inducing ferroptosis in neuroblastoma.404
Autophagy, pyroptosis, ferroptosis, and necroptosis in immunotherapy
Nowadays, ICIs, especially those targeting PD-1 and PD-L1, have been approved for the treatment of various cancers.23,24,25,405 Great success has been achieved in some solid tumors such as lung cancer and melanoma.405,406 Nevertheless, ICIs are limited by the fact that only about one-third of patients respond to ICIs. One of the main factors contributing to primary resistance to ICIs is the lack of tumor-infiltrating T cells, which is one of the characteristics of “cold” tumors whose TME is infiltrated with various immunosuppressive cells such as stromal cells, M2 macrophages, MDSCs, and Treg cells.407,408 ICIs may be most effective when in combination with therapies that increase the amount of CD8+ T cells for the treatment of cold tumors. Therefore, we hypothesize that there may be strong potential for the combination of ICIs with ICD induction or inhibitors. In the following, we will address the interaction of targeting autophagy, pyroptosis, ferroptosis, and necroptosis therapy with ICIs (Fig. 5).

Targeting autophagy, ferroptosis, pyroptosis, and necroptosis for tumor immunotherapy in cancer immunity cycle. Five key processes of cancer immunity cycle are noted in the figure. Agents that target autophagy, ferroptosis, pyroptosis, and necroptosis act synergistically with antitumor immunotherapy on different step of cancer immunity cycle (Autophagy inhibitor: CQ, SB02024, SAR405, 3MA; Autophagy inducer: ADIPOQ, W09, Mitoxantrone, Oxaliplatin, Sunitinib, SA-49, 2-BP, IPAG, H1A; Ferroptosis inducer: Lapatinib, Stains, Trigonelline, Sorafenib, Sulfasalazine, Cyst(e)inase, ZVI-NP; Pyroptosis inducer: Radiotherapy, Methotrexate, PD1-NKG2D-41BB NK92 cell, CA-Re; Necroptosis inducer: SMAC mimetic, Birinapant)
As mentioned previously, autophagy is involved in the survival, activation, and functional performance of immune cells.157,175 However, in preclinical models of melanoma and breast cancer, the autophagy inhibitor, CQ, did not impair T cells function, suggesting that the immune system may be tolerant to autophagy inhibitors in a certain intensity.409 Given the fact that autophagy may antagonize antitumor immune response, autophagy inhibitors may enhance the effectiveness of immunotherapy and overcome resistance to immunotherapy.410 For example, CQ blocks autophagy-mediated degradation of MHCI complexes, which together with dual ICIs treatment (anti-PD1 and anti-CTLA4 antibodies) has a synergistic effect and leads to an enhanced antitumor immune response in a mouse model of pancreatic cancer.286 Vps34 inhibitor SB02024 or SAR405 results in elevated levels of CCL5, IFNγ, and other chemokines in TME, causing high levels of NK cells and T cells infiltration in myeloma and colorectal cancer models.411 Furthermore, in an osteosarcoma model, tumor immunogenicity promoted by photodynamic therapy can be counteracted by ROS-induced autophagy, which can be enhanced by the autophagy inhibitor 3MA by suppressing PD-L1 expression, thereby enhancing the efficacy of photodynamic therapy.412 In addition to autophagy inhibitors, induction of autophagy may also enhance the efficacy of ICIs. It has been shown that in human melanoma, expression of the key autophagosomal component LC3-β and other autophagy activators reduces melanoma antigen-A (MAGE-A) protein levels and suppresses the MAGE-Tripartite Motif Containing 28 (TRIM28) complex which predicts resistance uniquely to blockade of CTLA-4, suggesting exploitation of autophagy inducer such as ADIPOQ, w09 for potential therapeutic synergy with CTLA-4 inhibitors.388,389,413
Ferroptosis can limit the function of immunosuppressive cells such as TAMs and Treg cells in cold tumors, transforming immunosuppressive TME into an inflammatory TME rich in antitumor immune cells.244,246,414,415 Therefore, ferroptosis inducer including Lapatinib, Stains, Trigonelline, etc. may help reverse primary resistance to immunotherapy and enhance the efficacy of ICIs.373,416,417 Nevertheless, in tumors with high levels of MDSCs infiltration, ferroptosis inducers may not be a good choice, stemming from the resistance of MDSCs to ferroptosis.249,250 MDSCs compete with immune cells for cyst(e)ine, thus depriving T cells of cyst(e)ine and inhibiting T cells activation due to the high expression level of system Xc- but the absence of ASC transporter. From this perspective, ferroptosis inducers targeting system Xc- such as Sorafenib and Sulfasalazine may alleviate MDSC-mediated deprivation of cyst(e)ine, thereby promoting T cells survival and restoring antitumor immune response.368,369,418 In preclinical models, in combination GPX4 inhibitor, cyst(e)inase with immunotherapy can synergistically improve T-cell-induced antitumor immunity and triggers cancer cells undergoing ferroptosis.419 Moreover, ferroptosis inducer, ZVI-NP also has a key role in augmenting antitumor immune response through polarizing M2 macrophages to M1, reducing the amount of Tregs, lowering the expression of PD-L1 on tumor cells as well as PD-1/CTLA4 on CD8+ T cells, thus maximizing the antitumor effects.399 In addition, ferroptosis inhibitor ferrostatin-1 prevents ferroptosis of CD8+ T cells by targeting fatty acids mediated by CD36 and inhibiting lipid peroxidation, exhibits elevated cytokine production and enhances tumor eradication.344 More importantly, combined with anti-PD-1 antibodies, ferrostatin-1 has greater antitumor effectiveness.
As pyroptosis enhances the tumor-killing activity of immune cells, it may improve the efficacy of ICIs as the killing mechanism of cytotoxic lymphocytes.114 Wang’s study found that ICIs were effective in killing cold tumor cells only in the presence of pyroptosis. Similarly, pyroptosis induction alone did not trigger effective tumor suppression, emphasizing the importance of pyroptosis inducers in combination with ICIs for the treatment of cold tumors.29 Lu et al. designed NK92 cells containing a chimeric co-stimulatory transforming receptor (CCCR) that converted inhibitory PD-1 signals into activation signals, effectively enhancing its activity against H1299 lung cancer cells and significantly inhibiting tumor growth in vivo.420 Further analysis concluded that it was achieved through GSDME-mediated pyroptosis. A carbonic anhydrase IX (CAIX)-anchored rhenium(I) photosensitizer (CA-Re) shows favorable efficacy in photodynamic treatment for effectively stimulating tumor immunogenicity under hypoxic conditions through GSDMD-independent pyroptosis.421 Moreover, the maturation and antigen-presentation capacity of DC cells as well as the activation of CTLs is enhanced by CA-Re, thus killing tumors. Likewise, Zhang et al. designed the engineering covalent organic frameworks (COFs) that could induce durable antitumor immunity through robust induction of GSDME-mediated pyroptosis and the remodeling of TME, thus improving immunotherapy response and restraining tumor metastasis and relapse.421 However, induction of pyroptosis may not benefit all immunotherapies. Recent studies have shown that CAR-T cells can quickly causes pyrolysis in targeted tumor cells via GZMB/GSDME/CASP3 pathway. Then, CASP1 is activated via pyroptosis releasing factor, which cleaves GSDMD in macrophages, causing the release of cytokine and cytokine release syndrome (CRS), a serious side event characterized by fever, hypotension, and respiratory failure.422 Thus, the development of more efficient drugs causing tumor-specific pyrolysis and decreasing pyrolysis in normal tissues is in urgent need.
Necroptosis inducer can synergize with ICIs in an antitumor context. SMAC mimetics bind and degrade cIAPs, inducing necroptosis to promote antitumor immune responses. In melanoma, the SMAC mimetic, Birinapant sensitizes tumor cells to TNFα-mediated T cells killing and directly regulates immune cell function including B cells, myeloid-derived cells, and cytotoxic lymphocytes by modulating the NF-κB signaling pathway, thus improving the response to ICIs.423,424 Similarly, in a mice tumor model, RIPK1-dependent cell death through SMAC mimetics improves the survival benefit of immune checkpoint blockade by activating CD8+ T cells and NK cells.425 These data are further supported by studies in glioblastoma, where SMAC mimetics can exert synergistic effects when combined with ICIs or innate immune stimulants to produce durable cures in which single-agent therapy is ineffective.426 Moreover, SMAC mimetics has also been suggested to enhance the efficacy of CAR-T cells in the treatment of acute lymphoblastic leukemia in term of mechanism that death receptor signaling is a key mediator of CAR-T cells cytotoxicity.427 Vaccinia virus (VACV) is a novel type of immuno-oncolytic therapy based on the mechanism that it can selectively replicate in cancer cells and trigger danger signaling thus augmenting antitumor immunity.428 Especially, in syngeneic mouse models, delivering MLKL into tumor cells through VACV causes necroptosis and boosts antitumor immune response directly against neo-epitopes.428 Moreover, RIPK3-mediated necroptosis can downregulate the expression of immunosuppressive BACH2/GATA-3 by suppressing KRAS-loaded exosomes, which can potentiate the cytolytic effects targeting tumor cells.429
RCD in chemotherapy, radiotherapy, and targeted therapy
In addition to cold tumors infiltrated by immunosuppressive cells, immune desert tumors are also less responsive to immunotherapy due to their low immunogenicity and lack of immune cell infiltration, therefore, radiotherapy, chemotherapy, and targeted agents may be more effective.430 Some targeted therapy (e.g., sorafenib), chemotherapy (e.g., paclitaxel), and radiotherapy may increase the immunogenicity of tumor cells and promote immune cell infiltrated within TME through autophagy, pyroptosis, ferroptosis, and necroptosis, thus enhancing the efficacy of ICIs. In clinical practice, targeted therapy, radiotherapy, and chemotherapy combined with immunotherapy have been applied successfully in various tumors.431,432,433 Therefore, we summarized clinical trials of some novel autophagy, pyroptosis, ferroptosis, and necroptosis inducers/inhibitors, chemotherapy, radiotherapy, and targeted therapy combined with immunotherapy (Table 3).
Standard chemotherapy has been considered to inhibit tumor growth mainly through apoptosis.5 However, studies have further revealed that chemotherapy can trigger non-apoptotic RCD through calreticulin exposure, autophagic ATP release, and HMGB1 upregulation, leading to increased immune cell infiltration.434,435,436 Gao et al. found that in cholangiocarcinoma cells, methotrexate-induced GSDME-mediated pyroptosis led to activation of tumor-derived macrophages and recruitment of neutrophils at the tumor site to exert antitumor effects.437 Decitabine in combination with chemotherapy nanodrugs in cancer treatment can elicit pyroptosis through epigenetics, thereby enhancing the immunological effects of chemotherapy.438 In the presence of mitoxantrone and oxaliplatin, autophagy can be activated in mice with CT26 colon tumors, inducing infiltration of DC cells and T cells in TME.164 In addition, autophagy in CT26 cells facilitates the secretion of ATP during mitoxantrone treatment.164 Extracellular ATP can lead to NLRP3-mediated inflammasome activation, subsequently, recruit APCs into TME to produce IL-1β and activate the antitumor adaptive immune response.234 Notably, some chemotherapy agents lead to unfolded protein responses by inducing endoplasmic reticulum stress, which promotes the release of DAMPs to cause ICD.38 Compared with oxaliplatin, cisplatin is not effective in inducing autophagy in prostate cancer cells which may be related to invalid unfolded protein response, thus weakens a robust immune response.439
The direct effect of radiotherapy on cancer cells is DNA damage, leading to cell cycle arrest or apoptosis.440 However, it was found that in some solid tumors, DNA fragments generated by ionizing radiation can be recognized by intracellular DNA sensors such as AIM2 or ZBP1 thereby activating inflammatory signaling and inducing pyroptosis.441,442 Cytosolic DNA can also activate another DNA sensor, the STING pathway, providing additional immune stimulation through the production of type I interferon.443 Together with inducing pyroptosis, in tumor models, radiotherapy-activated DNA damage response associated kinases such as ataxia-telangiectasia mutated proteins (ATM) inhibit Xc- system leading to reduced cyst(e)ine uptake which enhances tumor lipid oxidation and induce ferroptosis.28 This acts synergistically with interferons released by immunotherapy-activated CD8+ T cells, further demonstrating the robust antitumor effects of immunotherapy combined with radiotherapy. Besides, the release of calreticulin and ATP from colon tumors cells through autophagy activated by radiotherapy promotes phagocytosis of tumor cells by DC cells.444,445 Stereotactic body radiation therapy (SBRT) combined with oncolytic virus enhances antitumor immunity by altering the M1/M2 ratio of macrophages through necroptosis.446 Targeted therapies can also promote ICD and produce immunogenic effects while inhibiting tumor proliferation. Inhibition of cell cycle protein-dependent kinases in a mouse model promotes the release of DAMPs thereby improving antitumor immune response and increasing response to ICIs in mice.447 In addition, it is worth noting that many of the clinical kinase inhibitors such as Bcr-Abl inhibitor also target RIPK1 and RIPK3 to improve the efficacy of current cancer-targeted therapies.448 MEK inhibitor in combination with autophagy inhibitor can activate TAMs toward an immunogenic M1-like phenotype through STING/type I interferon pathway, which is an attractive therapeutic approach for PDA immunotherapy development.449 During a phase III trial of patients with metastatic breast cancer, the receptor protein tyrosine kinase inhibitor sunitinib enhanced the efficacy of ICIs by inducing p62-mediated selective autophagy to downregulate tumor PD-L1 expression.450 Likewise, SA-49 treatment facilitates PKCα/GSK3β/MITF-mediated PD-L1 autophagic degradation,451 and DHHC3 inhibitor 2-bromopalmitate (2-BP) induces PD-L1 autophagic degradation by abolishing PD-L1 palmitoylation,196 thereby improving the efficacy of cancer immunotherapy in a colon tumor model. Due to the interaction of SIGMA I with glycosylated PD-L1, IPAG, the SIGMA I inhibitor restored T cells activity in breast/prostate cancer cells by preventing PD-L1 autophagy.452 Furthermore, PD-L1 antibody H1A inhibits PD-L1 interaction with CMTM6, which results in PD-L1 autophagy.453
Conclusions and perspectives
The study of non-apoptotic RCD is an extensive and rapidly developed field. An emerging view is that targeting autophagy, pyroptosis, ferroptosis, and necroptosis in localized tumors profoundly affects the immune cells infiltrated in TME and the response to immunotherapy. Despite the growing importance of ICIs in cancer therapy, their application is greatly limited by the fact that only about one-third of patients respond to ICIs in most cancer types. In order to break the limitations of immunotherapy, in this review, we explore the broad interaction between non-apoptotic cell death mechanisms and antitumor immunity based on the available evidence from laboratory and clinical studies. The roles of autophagy, pyroptosis, ferroptosis, and necroptosis in tumor immunity are still ambiguous as they synergize the antitumor immune response while playing an antagonistic role. In addition, the effects of effectors such as RIPK1/3 and inflammasomes as well as released cytokines and DAMPs through ICD on immune cells and immune response are still controversial. There is a more complex interaction revealed by these findings between non-apoptotic RCD and immunity in different tumor types and contexts. It will also be crucial to understand how distinct TME cell types such as immune cells, tumor cells, and stromal cells interact with each other to inhibit or promote tumor progression by immunity or metabolism reprogramming.
In this context, targeting non-apoptotic cell death seems to be an increasingly promising strategy to improve the efficacy of immunotherapy in the field of cancer therapy. However, it is not certain that non-apoptotic RCD induced by tumoricidal drugs is beneficial for tumor patients in the long perspective because other normal cells could also die when stimulated by DAMPs released from tumor cells through non-apoptotic RCD. Thus, the development of more specific cell death-inducing drugs that act on tumor cells with minimal side effects on normal tissues are extremely urgent. In the meantime, preclinical testing of the order/timing of these drugs in combination with ICIs as well as chemotherapy, radiation, and targeted therapies will be likely crucial to balance therapeutic goals and likely adverse effects. Soon, clinical trials of combination therapies should be actively encouraged to be conducted to assess their efficacy and safety, providing more references for subsequent in-depth studies in order to benefit more cancer patients.
References
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Tang, D. et al. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Koren, E. & Fuchs, Y. Modes of regulated cell death in cancer. Cancer Discov. 11, 245–265 (2021).
Hanahan, D. & Weinberg, R. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Carneiro, B. & El-Deiry, W. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Badgley, M. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Guo, J. et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011).
Li, L. & Liu, Y. Aging-related gene signature regulated by Nlrp3 predicts glioma progression. Am. J. Cancer Res. 5, 442–449 (2015).
Feng, X. et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 36, 81 (2017).
McCormick, K. et al. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis 37, 522–529 (2016).
Park, S. et al. The receptor interacting protein 1 inhibits p53 induction through NF-kappaB activation and confers a worse prognosis in glioblastoma. Cancer Res. 69, 2809–2816 (2009).
Höckendorf, U. et al. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell 30, 75–91 (2016).
Feng, X. et al. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 62, 592–601 (2015).
Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 5, 263–274 (2005).
Mohme, M., Riethdorf, S. & Pantel, K. Circulating and disseminated tumour cells—mechanisms of immune surveillance and escape. Nat. Rev. Clin. Oncol. 14, 155–167 (2017).
Dersh, D., Hollý, J. & Yewdell, J. A few good peptides: MHC class I-based cancer immunosurveillance and immunoevasion. Nat. Rev. Immunol. 21, 116–128 (2021).
Wei, S., Duffy, C. & Allison, J. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8, 1069–1086 (2018).
Goodman, A., Patel, S. & Kurzrock, R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol. 14, 203–220 (2017).
Carlino, M., Larkin, J. & Long, G. Immune checkpoint inhibitors in melanoma. Lancet 398, 1002–1014 (2021).
Wagner, D. et al. Immunogenicity of CAR T cells in cancer therapy. Nat. Rev. Clin. Oncol. 18, 379–393 (2021).
Harari, A., Graciotti, M., Bassani-Sternberg, M. & Kandalaft, L. Antitumour dendritic cell vaccination in a priming and boosting approach. Nat. Rev. Drug Discov. 19, 635–652 (2020).
Hernandez, R., Põder, J., LaPorte, K. & Malek, T. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Rev. Immunol. https://doi.org/10.1038/s41577-022-00680-w (2022).
Cella, D. et al. Patient-reported outcomes with first-line nivolumab plus cabozantinib versus sunitinib in patients with advanced renal cell carcinoma treated in CheckMate 9ER: an open-label, randomised, phase 3 trial. Lancet Oncol. 23, 292–303 (2022).
Doki, Y. et al. Nivolumab combination therapy in advanced esophageal squamous-cell carcinoma. N. Engl. J. Med. 386, 449–462 (2022).
Kang, Y. et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): a randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 23, 234–247 (2022).
Nguyen, L. & Ohashi, P. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat. Rev. Immunol. 15, 45–56 (2015).
Sun, C., Mezzadra, R. & Schumacher, T. Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 (2018).
Lang, X. et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 9, 1673–1685 (2019).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020).
Wang, W. et al. CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Zhang, Z. et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579, 415–420 (2020).
Rothlin, C., Hille, T. & Ghosh, S. Determining the effector response to cell death. Nat. Rev. Immunol. 21, 292–304 (2021).
Wallach, D. & Kang, T. Programmed cell death in immune defense: knowledge and presumptions. Immunity 49, 19–32 (2018).
Lévy, J. et al. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol. 17, 1062–1073 (2015).
Yang, A. et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 8, 276–287 (2018).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Kroemer, G., Galassi, C., Zitvogel, L. & Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022).
Galluzzi, L. et al. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 17, 97–111 (2017).
Tang, D. et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249, 158–175 (2012).
Tang, R. et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 13, 110 (2020).
Chen, X. et al. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat. Rev. Gastroenterol. Hepatol. 18, 804–823 (2021).
Xia, H., Green, D. & Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 21, 281–297 (2021).
Cadwell, K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 16, 661–675 (2016).
Nixon, R. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Dikic, I. & Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364 (2018).
Morishita, H. & Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 35, 453–475 (2019).
Kirkin, V. & Rogov, V. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol. Cell 76, 268–285 (2019).
Gatica, D., Lahiri, V. & Klionsky, D. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242 (2018).
Hara, T. et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510 (2008).
Backer, J. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 473, 2251–2271 (2016).
Ohashi, Y., Tremel, S. & Williams, R. VPS34 complexes from a structural perspective. J. Lipid Res. 60, 229–241 (2019).
Lahiri, V., Hawkins, W. & Klionsky, D. Watch what you (self-) eat: autophagic mechanisms that modulate metabolism. Cell Metab. 29, 803–826 (2019).
Ktistakis, N. T. & Tooze, S. A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 26, 624–635 (2016).
Mizushima, N. et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679–1688 (2003).
Romanov, J. et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 31, 4304–4317 (2012).
Tanida, I., Ueno, T. & Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 (2004).
Lamark, T., Kirkin, V., Dikic, I. & Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell cycle 8, 1986–1990 (2009).
Shibutani, S. T. & Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 24, 58–68 (2014).
Orsi, A. et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell 23, 1860–1873 (2012).
Wang, Y. et al. SNARE-mediated membrane fusion in autophagy. Semin. Cell Dev. Biol. 60, 97–104 (2016).
Zhao, Y., Codogno, P. & Zhang, H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 22, 733–750 (2021).
Kimmelman, A. & White, E. Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043 (2017).
Aita, V. et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59, 59–65 (1999).
Liang, X. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 (1999).
García-Fernández, M. et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 12, 1776–1790 (2016).
Görgülü, K. et al. Levels of the autophagy-related 5 protein affect progression and metastasis of pancreatic tumors in mice. Gastroenterology 156, 203–217.e220 (2019).
Degenhardt, K. et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10, 51–64 (2006).
Lassen, K. et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl Acad. Sci. USA 111, 7741–7746 (2014).
Sousa, C. et al. Erratum: Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 540, 150 (2016).
Dixon, S. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Tang, D. & Kroemer, G. Ferroptosis. Curr. Biol.: CB 30, R1292–R1297 (2020).
Chen, X. et al. Ferroptosis: machinery and regulation. Autophagy 17, 2054–2081 (2020).
Chen, X., Yu, C., Kang, R. & Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 8, 590226 (2020).
Wang, Y. et al. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem. Biophys. Res. Commun. 531, 581–587 (2020).
Yang, W. & Stockwell, B. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Geng, N. et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 22, 3826–3836 (2018).
Li, J. et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 17, 3361–3374 (2021).
Brown, C. et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell 51, 575–586.e574 (2019).
Yang, W. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. U.S.A. 113, E4966–E4975 (2016).
Stockwell, B. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Kagan, V. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Dixon, S. et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Kuang, F., Liu, J., Tang, D. & Kang, R. Oxidative damage and antioxidant defense in ferroptosis. Front. Cell Dev. Biol. 8, 586578 (2020).
Yang, W. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Ursini, F. & Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 152, 175–185 (2020).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422.e421 (2018).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Kraft, V. et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent. Sci. 6, 41–53 (2020).
Dai, E. et al. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem. Biophys. Res. Commun. 522, 415–421 (2020).
Dai, E. et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem. Biophys. Res. Commun. 523, 966–971 (2020).
Liu, J. et al. Autophagy-Dependent Ferroptosis: machinery and Regulation. Cell Chem. Biol. 27, 420–435 (2020).
Gao, H. et al. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556 (2018).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Wu, Z. et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl Acad. Sci. USA 116, 2996–3005 (2019).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019).
Yang, M. et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019).
Friedmann Angeli, J., Krysko, D. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019).
Eling, N. et al. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2, 517–532 (2015).
Zhu, S. et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 77, 2064–2077 (2017).
Hafner, A., Bulyk, M., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 20, 199–210 (2019).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Cookson, B. & Brennan, M. Pro-inflammatory programmed cell death. Trends Microbiol. 9, 113–114 (2001).
Nyström, S. et al. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 32, 86–99 (2013).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128 (2017).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Demarco, B. et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 6, eabc3465 (2020).
Aizawa, E. et al. GSDME-dependent incomplete pyroptosis permits selective IL-1α release under caspase-1 inhibition. iScience 23, 101070 (2020).
Li, L., Li, Y. & Bai, Y. Role of GSDMB in pyroptosis and cancer. Cancer Manag. Res. 12, 3033–3043 (2020).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, eaaz7548 (2020).
Tang, L., Lu, C., Zheng, G. & Burgering, B. Emerging insights on the role of gasdermins in infection and inflammatory diseases. Clin. Transl. Immunol. 9, e1186 (2020).
Broz, P. & Dixit, V. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Dunn, J., Ellis, L. & Fujita, M. Inflammasomes as molecular mediators of inflammation and cancer: potential role in melanoma. Cancer Lett. 314, 24–33 (2012).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl Acad. Sci. USA 107, 21635–21640 (2010).
Janowski, A., Kolb, R., Zhang, W. & Sutterwala, F. Beneficial and detrimental roles of NLRs in carcinogenesis. Front. Immunol. 4, 370 (2013).
He, Y. et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357 (2016).
Muñoz-Planillo, R. et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Sborgi, L. et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778 (2016).
Aglietti, R. et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl Acad. Sci. USA 113, 7858–7863 (2016).
Cui, J. et al. MST1 suppresses pancreatic cancer progression via ROS-induced pyroptosis. Mol. Cancer Res. 17, 1316–1325 (2019).
Gao, J. et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in non-small cell lung cancer. Oncol. Rep. 40, 1971–1984 (2018).
Xia, X. et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 10, 650 (2019).
Zhang, Y. et al. Alpinumisoflavone suppresses hepatocellular carcinoma cell growth and metastasis via NLRP3 inflammasome-mediated pyroptosis. Pharmacol. Rep. 72, 1370–1382 (2020).
Williams, T. et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunother. 194, 3369–3380 (2015).
Wei, Q. et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Invest. 94, 52–62 (2014).
Xi, G. et al. GSDMD is required for effector CD8 T cell responses to lung cancer cells. Int. Immunopharmacol. 74, 105713 (2019).
So, D. et al. Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 37, 5191–5204 (2018).
Johnson, D. et al. DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid leukemia. Nat. Med. 24, 1151–1156 (2018).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Kaiser, W. et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279 (2013).
Upton, J., Kaiser, W. & Mocarski, E. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 26, 564 (2019).
Galluzzi, L., Kepp, O., Chan, F. & Kroemer, G. Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130 (2017).
Weinlich, R., Oberst, A., Beere, H. & Green, D. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Kaiser, W. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Temkin, V. et al. Inhibition of ADP/ATP exchange in receptor-interacting protein-mediated necrosis. Mol. Cell. Biol. 26, 2215–2225 (2006).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Bertrand, M. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Häcker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. Signal. 2006, re13 (2006).
Van Antwerp, D. et al. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 274, 787–789 (1996).
Wang, L., Du, F. & Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133, 693–703 (2008).
Cho, Y. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
He, S. et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009).
Zhang, D. et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009).
Maelfait, J. et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017).
Kaczmarek, A., Vandenabeele, P. & Krysko, D. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 (2013).
Newton, K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 25, 347–353 (2015).
Lawlor, K. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Feng, X. et al. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 62, 592–601 (2015).
Nicolè, L. et al. RIPK1, RIPK3Necroptosis-driving genes and are associated with intratumoral CD3 and CD8 T cell density and predict prognosis in hepatocellular carcinoma. J. Immunother. Cancer 10, e004031 (2022).
Koo, G. et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 25, 707–725 (2015).
Clarke, A. & Simon, A. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 19, 170–183 (2019).
Deretic, V. & Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 14, 243–251 (2018).
White, E., Lattime, E. C. & Guo, J. Y. Autophagy regulates stress responses, metabolism, and anticancer immunity. Trends Cancer 7, 778–789 (2021).
Karsli-Uzunbas, G. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Disco. 4, 914–927 (2014).
Poillet-Perez, L. et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 1, 923–934 (2020).
Mathew, R. et al. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 55, 916–930 (2014).
Martins, I. et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 21, 79–91 (2014).
Michaud, M. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577 (2011).
Zhong, Z., Sanchez-Lopez, E. & Karin, M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell 166, 288–298 (2016).
Lee, H. et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 32, 227–239 (2010).
Seto, S., Tsujimura, K., Horii, T. & Koide, Y. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS ONE 8, e86017 (2013).
Florey, O. et al. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 13, 1335–1343 (2011).
Martinez, J. et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl Acad. Sci. USA 108, 17396–17401 (2011).
Münz, C. Non-canonical roles of autophagy proteins in endocytosis and exocytosis. Biochem. Soc. Trans. 49, 2841–2851 (2021).
Hahn, T. & Akporiaye, E. α-TEA as a stimulator of tumor autophagy and enhancer of antigen cross-presentation. Autophagy 9, 429–431 (2013).
Li, Y. et al. The vitamin E analogue α-TEA stimulates tumor autophagy and enhances antigen cross-presentation. Cancer Res. 72, 3535–3545 (2012).
Pan, H. et al. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget 7, 21235–21246 (2016).
Jiang, G. et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 18, 17 (2019).
Xia, H. et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naïve T cell apoptosis and affects tumor immunity. Sci. Immunol. 2, eaan4631 (2017).
Hubbard, V. et al. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunother. 185, 7349–7357 (2010).
Botbol, Y., Patel, B. & Macian, F. Common γ-chain cytokine signaling is required for macroautophagy induction during CD4+ T-cell activation. Autophagy 11, 1864–1877 (2015).
Jia, W. et al. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 11, 2335–2345 (2015).
Xu, X. et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 15, 1152–1161 (2014).
Bronietzki, A., Schuster, M. & Schmitz, I. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol. 93, 25–34 (2015).
Michalek, R. et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunother. 186, 3299–3303 (2011).
Pua, H., Guo, J., Komatsu, M. & He, Y. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunother. 182, 4046–4055 (2009).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Carleton, G. & Lum, J. Autophagy metabolically suppresses CD8 T cell antitumor immunity. Autophagy 15, 1648–1649 (2019).
Miller, B. et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 4, 309–314 (2008).
Arnold, J. et al. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. 23, 853–864 (2016).
Zhou, M. et al. Macrophages enhance tumor-derived autophagosomes (DRibbles)-induced B cells activation by TLR4/MyD88 and CD40/CD40L. Exp. Cell Res. 331, 320–330 (2015).
Kodali, S. et al. Protection of quiescence and longevity of igg memory B cells by mitochondrial autophagy. J. Immunother. 208, 1085–1098 (2022).
Jacquel, A., Obba, S., Solary, E. & Auberger, P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy 8, 1141–1143 (2012).
Zhang, Y. et al. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 119, 2895–2905 (2012).
Liu, K. et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 11, 271–284 (2015).
Iwai, Y., Hamanishi, J., Chamoto, K. & Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 24, 26 (2017).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 24, 207–212 (2012).
Wang, H. et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat. Chem. Biol. 15, 42–50 (2019).
Li, C. W. et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187–201.e110 (2018).
Yao, H. et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 3, 306–317 (2019).
Burr, M. L. et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017).
Liu, Y. et al. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur. J. Cancer 114, 8–24 (2019).
Alissafi, T. et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Invest. 127, 2789–2804 (2017).
Kato, H. & Perl, A. Blockade of treg cell differentiation and function by the interleukin-21-mechanistic target of rapamycin axis via suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol. 70, 427–438 (2018).
Folgiero, V. et al. IDO1 involvement in mTOR pathway: a molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget 7, 52900–52911 (2016).
Metz, R. et al. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: a novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 1, 1460–1468 (2012).
McGaha, T. L. IDO-GCN2 and autophagy in inflammation. Oncotarget 6, 21771–21772 (2015).
Jaiswal, S. et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 138, 271–285 (2009).
Casey, S. C. et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231 (2016).
Kamerkar, S. et al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 546, 498–503 (2017).
Colombo, M., Raposo, G. & Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 (2014).
Young, J., Hengartner, H., Podack, E. & Cohn, Z. Purification and characterization of a cytolytic pore-forming protein from granules of cloned lymphocytes with natural killer activity. Cell 44, 849–859 (1986).
Hou, J. et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 22, 1264–1275 (2020).
Du, Q. et al. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 105, 42–54 (2016).
Nowarski, R. et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 163, 1444–1456 (2015).
Dupaul-Chicoine, J. et al. The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763 (2015).
Ben-Sasson, S. et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J. Exp. Med. 210, 491–502 (2013).
Zhivaki, D. & Kagan, J. NLRP3 inflammasomes that induce antitumor immunity. Trends Immunol. 42, 575–589 (2021).
Nakamura, S. et al. Antitumor effect of recombinant human interleukin 1 alpha against murine syngeneic tumors. Jpn. J. Cancer Res. 77, 767–773 (1986).
North, R. J., Neubauer, R. H., Huang, J. J., Newton, R. C. & Loveless, S. E. Interleukin 1-induced, T cell-mediated regression of immunogenic murine tumors. Requirement for an adequate level of already acquired host concomitant immunity. J. Exp. Med. 168, 2031–2043 (1988).
Andina, N., Bonadies, N. & Allam, R. Inflammasome ACtivation in Myeloid Malignancies-friend Or Foe? Front. Cell Dev. Biol. 9, 825611 (2021).
Allen, I. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Veltri, S. & Smith, J. W. Interleukin 1 trials in cancer patients: a review of the toxicity, antitumor and hematopoietic Effects. Oncologist 1, 190–200 (1996).
Mullerad, J., Cohen, S., Benharroch, D. & Apte, R. N. Local delivery of IL-1 alpha polymeric microspheres for the immunotherapy of an experimental fibrosarcoma. Cancer Invest. 21, 720–728 (2003).
Tanaka, T., Narazaki, M. & Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 6, a016295 (2014).
Gou, X. et al. IL-6 Prevents lung macrophage death and lung inflammation injury by inhibiting GSDME- and GSDMD-mediated pyroptosis during pneumococcal pneumosepsis. Microbiol. Spectr. 10, e0204921 (2022).
Zheng, J. et al. A novel function of NLRP3 independent of inflammasome as a key transcription factor of IL-33 in epithelial cells of atopic dermatitis. Cell Death Dis. 12, 871 (2021).
Ahn, H. et al. NLRP3 triggers attenuate lipocalin-2 expression independent with inflammasome activation. Cells 10, 1660 (2021).
Janowski, A. et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J. Clin. Invest. 126, 3917–3928 (2016).
Yu, M. et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26, 174–179 (2006).
Tian, J. et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 (2007).
Tan, G., Huang, C., Chen, J. & Zhi, F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J. Hematol. Oncol. 13, 149 (2020).
Sims, G. P. et al. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 28, 367–388 (2010).
Apetoh, L. et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59 (2007).
Gombault, A., Baron, L. & Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. 3, 414 (2012).
Säve, S. & Persson, K. Extracellular ATP and P2Y receptor activation induce a proinflammatory host response in the human urinary tract. Infect. Immun. 78, 3609–3615 (2010).
Beste, M. T., Lomakina, E. B., Hammer, D. A. & Waugh, R. E. Immobilized IL-8 triggers phagocytosis and dynamic changes in membrane microtopology in human neutrophils. Ann. Biomed. Eng. 43, 2207–2219 (2015).
Ghiringhelli, F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 15, 1170–1178 (2009).
Mattarollo, S. R. et al. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 71, 4809–4820 (2011).
Hernández, Á. P. et al. Restoring the immunity in the tumor microenvironment: insights into immunogenic cell death in onco-therapies. Cancers 13, 2821 (2021).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Li, C. et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J. Med. Chem. 62, 266–275 (2019).
Wang, D. & Dubois, R. N. Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 (2010).
Morgan, A. H. et al. Phosphatidylethanolamine-esterified eicosanoids in the mouse: tissue localization and inflammation-dependent formation in Th-2 disease. J. Biol. Chem. 284, 21185–21191 (2009).
Lauber, K. et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730 (2003).
Tyurin, V. A. et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ. 21, 825–835 (2014).
Luo, X. et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 28, 1971–1989 (2021).
Kapralov, A. et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290 (2020).
Drijvers, J. et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8 T cells. Cancer Immunol. Res. 9, 184–199 (2021).
Xu, C. et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 35, 109235 (2021).
Ugolini, A. et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI insight 5, e138581 (2020).
Veglia, F. et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569, 73–78 (2019).
Srivastava, M. et al. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 70, 68–77 (2010).
Zhu, H. et al. Asah2 represses the p53-Hmox1 axis to protect myeloid-derived suppressor cells from ferroptosis. J. Immunother. 206, 1395–1404 (2021).
Kim, D. et al. TGF-β1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 11, 406 (2020).
Liu, Z. & Chan, F. Regulatory mechanisms of RIPK1 in cell death and inflammation. Semin. Cell Dev. Biol. 109, 70–75 (2021).
Moriwaki, K. et al. Distinct kinase-independent role of RIPK3 in CD11c mononuclear phagocytes in cytokine-induced tissue repair. Cell Rep. 18, 2441–2451 (2017).
Kang, Y. et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat. Commun. 6, 8371 (2015).
Snyder, A. G. et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci. Immunol. 4, eaaw2004 (2019).
Yang, H. et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 5, e1149673 (2016).
Moriwaki, K. et al. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 41, 567–578 (2014).
Orozco, S. et al. RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Rep. 28, 2275–2287.e2275 (2019).
Zhu, K. et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 9, 500 (2018).
Rucker, A. & Chan, F. Tumor-intrinsic and immune modulatory roles of receptor-interacting protein kinases. Trends Biochem. Sci. 47, 342–351 (2022).
Aaes, T. et al. Vaccination with necroptotic cancer cells induces efficient anti-tumor immunity. Cell Rep. 15, 274–287 (2016).
Pasparakis, M. & Vandenabeele, P. Necroptosis and its role in inflammation. Nature 517, 311–320 (2015).
Yatim, N. et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 350, 328–334 (2015).
Colbert, L. et al. Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 119, 3148–3155 (2013).
Patel, S. et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 27, 161–175 (2020).
Baik, J. Y. et al. ZBP1 not RIPK1 mediates tumor necroptosis in breast cancer. Nat. Commun. 12, 2666 (2021).
Heckmann, B. L. et al. LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429, 3561–3576 (2017).
Kim, S., Elkon, K. B. & Ma, X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity 21, 643–653 (2004).
Rosenbaum, S. R., Wilski, N. A. & Aplin, A. E. Fueling the fire: inflammatory forms of cell death and implications for cancer immunotherapy. Cancer Discov. 11, 266–281 (2021).
Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 54, 437–453 (2021).
Yamazaki, T. et al. Autophagy in the cancer-immunity dialogue. Adv. Drug Deliv. Rev. 169, 40–50 (2021).
Lowe, M. et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI insight 4, e129756 (2019).
Wei, J. et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 17, 277–285 (2016).
Zeng, H. et al. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490 (2013).
Chen, L. et al. Foxp3-dependent transformation of human primary CD4+ T lymphocytes by the retroviral protein tax. Biochem. Biophys. Res. Commun. 466, 523–529 (2015).
Parekh, V. et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J. Immunother. 190, 5086–5101 (2013).
Mantovani, A. et al. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229, 176–185 (2013).
Li, N. et al. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol. Ther. 16, 297–306 (2015).
Chang, C., Su, Y., Hu, C. & Lei, H. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 20, 515–523 (2013).
Chen, W. et al. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 72, 1363–1372 (2012).
Li, W. et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 28, 87–103.e106 (2018).
De Veirman, K. et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. 442, 233–241 (2019).
Baginska, J. et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl Acad. Sci. USA 110, 17450–17455 (2013).
Monaci, S. et al. Hypoxia enhances the expression of RNASET2 in human monocyte-derived dendritic cells: role of PI3K/AKT pathway. Int. J. Mol. Sci. 22, 7564 (2021).
Zhan, L., Zhang, J., Wei, B. & Cao, Y. Selective autophagy of NLRC5 promotes immune evasion of endometrial cancer. Autophagy 18, 942–943 (2022).
Yamamoto, K. et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105 (2020).
Loi, M. et al. Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8(+) T cell responses. Cell Rep. 15, 1076–1087 (2016).
Zeng, H., Zhang, W., Gong, Y. & Xie, C. Radiotherapy activates autophagy to increase CD8(+) T cell infiltration by modulating major histocompatibility complex class-I expression in non-small cell lung cancer. J. Int. Med. Res. 47, 3818–3830 (2019).
Alissafi, T. et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Invest. 128, 3840–3852 (2018).
Park, S., Cheon, S. & Cho, D. The dual effects of interleukin-18 in tumor progression. Cell. Mol. Immunol. 4, 329–335 (2007).
Zhao, X. et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 8, 108571–108583 (2017).
Deswaerte, V. et al. Inflammasome adaptor ASC suppresses apoptosis of gastric cancer cells by an IL18-mediated inflammation-independent mechanism. Cancer Res. 78, 1293–1307 (2018).
Jae, L. W., Kang, J. S., Youngil, H. & Hoon, S. D. Endogenous IL-18 modulates immune escape and metastasis of stomach cancer via the suppression of CD70 expression and the sustenance of the high expression of CD44 and VEGF. Mol. Cancer Ther. 6, C24 (2007).
Cai, D. et al. LRG1 in pancreatic cancer cells promotes inflammatory factor synthesis and the angiogenesis of HUVECs by activating VEGFR signaling. J. Gastrointest. Oncol. 13, 400–412 (2022).
Apte, R. et al. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 42, 751–759 (2006).
Maker, A. et al. Cyst fluid interleukin-1beta (IL1beta) levels predict the risk of carcinoma in intraductal papillary mucinous neoplasms of the pancreas. Clin. Cancer Res. 17, 1502–1508 (2011).
Zhu, Y., Zhu, M. & Lance, P. IL1β-mediated Stromal COX-2 signaling mediates proliferation and invasiveness of colonic epithelial cancer cells. Exp. Cell Res. 318, 2520–2530 (2012).
Tsai, C., Lee, T., Kou, Y. & Wu, Y. Glucosamine inhibits IL-1beta-mediated IL-8 production in prostate cancer cells by MAPK attenuation. J. Cell. Biochem. 108, 489–498 (2009).
Mantovani, A., Barajon, I. & Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 281, 57–61 (2018).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Das, S. et al. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 80, 1088–1101 (2020).
Gabrilovich, D. I., Ostrand-Rosenberg, S. & Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012).
Mantovani, A. et al. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Guo, B. et al. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 6, 36107 (2016).
Lim, S. Y., Yuzhalin, A. E., Gordon-Weeks, A. N. & Muschel, R. J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710 (2016).
Tu, S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
Bunt, S. K. et al. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 176, 284–290 (2006).
Jiang, H. et al. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 136, 2352–2360 (2015).
Sinha, P., Clements, V. K., Fulton, A. M. & Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 67, 4507–4513 (2007).
Carmi, Y. et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J. Immunol. 190, 3500–3509 (2013).
Nickoloff, B. J., Ben-Neriah, Y. & Pikarsky, E. Inflammation and cancer: is the link as simple as we think? J. Invest. Dermatol. 124, x-xiv (2005).
Del Campo, J. A., Gallego, P. & Grande, L. Role of inflammatory response in liver diseases: therapeutic strategies. World J. Hepatol. 10, 1–7 (2018).
Hong, J. B., Zuo, W., Wang, A. J. & Lu, N. H. Helicobacter pylori Infection synergistic with IL-1β gene polymorphisms potentially contributes to the carcinogenesis of gastric cancer. Int. J. Med. Sci. 13, 298–303 (2016).
El-Omar, E. M. et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404, 398–402 (2000).
Huang, F. Y. et al. Interleukin-1β increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol. Lett. 11, 2919–2924 (2016).
Yi, G. et al. A large lung gene expression study identifying IL1B as a novel player in airway inflammation in COPD airway epithelial cells. Inflamm. Res. 67, 539–551 (2018).
Krelin, Y. et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67, 1062–1071 (2007).
Kanterman, J., Sade-Feldman, M. & Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 22, 307–318 (2012).
Chen, L. et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. life Sci.: CMLS 75, 2045–2058 (2018).
Steel, J. et al. Prospective analyses of cytokine mediation of sleep and survival in the context of advanced cancer. Psychosom. Med. 80, 483–491 (2018).
Daley, D. et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 214, 1711–1724 (2017).
Neumann, K., Schiller, B. & Tiegs, G. NLRP3 inflammasome and IL-33: novel players in sterile liver inflammation. Int. J. Mol. Sci. 19, 2732 (2018).
Lüthi, A. et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98 (2009).
Liu, Y. et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 5, eaax7969 (2020).
Dai, E. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083 (2020).
Wen, Q. et al. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283 (2019).
Dai, E. et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 11, 6339 (2020).
Wang, D. & DuBois, R. N. The role of prostaglandin E(2) in tumor-associated immunosuppression. Trends Mol. Med. 22, 1–3 (2016).
Böttcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037.e1014 (2018).
Johnson, A., Kleczko, E. & Nemenoff, R. Eicosanoids in cancer: new roles in immunoregulation. Front. Pharmacol. 11, 595498 (2020).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Wang, D. & DuBois, R. N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36, 1085–1093 (2015).
Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 (2012).
Kirtonia, A., Sethi, G. & Garg, M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell. Mol. Life Sci. 77, 4459–4483 (2020).
Weinberg, S. E., Sena, L. A. & Chandel, N. S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417 (2015).
Guerin, A. et al. Acute deafness and desferrioxamine. Lancet 2, 39–40 (1985).
Ligtenberg, M. A. et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J. Immunol. 196, 759–766 (2016).
Scharping, N. E. et al. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol. Res. 5, 9–16 (2017).
Weinberg, S. E. et al. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499 (2019).
Kunisada, Y. et al. Attenuation of CD4(+)CD25(+) regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine 25, 154–164 (2017).
OuYang, L. Y. et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 13, 47 (2015).
Wei, J., Zhang, M. & Zhou, J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. 228, 695–701 (2015).
Lin, X. et al. Oxidative stress in malignant melanoma enhances tumor necrosis factor-α secretion of tumor-associated macrophages that promote cancer cell invasion. Antioxid. Redox Signal. 19, 1337–1355 (2013).
Ma, X. et al. CD36-mediated ferroptosis dampens intratumoral CD8 T cell effector function and impairs their antitumor ability. Cell Metab. 33, 1001–1012.e1005 (2021).
Xu, S. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8 T cells in tumors. Immunity 54, 1561–1577.e1567 (2021).
Poznanski, S. et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 33, 1205–1220.e1205 (2021).
Herber, D. et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886 (2010).
Rothe, T. et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J. Clin. Invest. 125, 1944–1954 (2015).
Ramakrishnan, R. et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunother. 192, 2920–2931 (2014).
Seifert, L. et al. Author Correction: The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 591, E28 (2021).
Ando, Y. et al. Necroptosis in pancreatic cancer promotes cancer cell migration and invasion by release of CXCL5. PLoS ONE 15, e0228015 (2020).
Jayakumar, A. & Bothwell, A. RIPK3-induced inflammation by I-MDSCs promotes intestinal tumors. Cancer Res. 79, 1587–1599 (2019).
Wang, W. et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 38, 585–590 (2020).
Hou, J. et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 10, 493 (2019).
Jiao, D. et al. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 28, 868–870 (2018).
Strilic, B. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218 (2016).
Grivennikov, S., Greten, F. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Liu, Z. et al. A class of viral inducer of degradation of the necroptosis adaptor RIPK3 regulates virus-induced inflammation. Immunity 54, 247–258.e247 (2021).
Newton, K., Dixit, V. & Kayagaki, N. Dying cells fan the flames of inflammation. Science 374, 1076–1080 (2021).
Conos, S. et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl Acad. Sci. USA 114, E961–E969 (2017).
Gutierrez, K. et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1β independently of gasdermin-D. J. Immunother. 198, 2156–2164 (2017).
Yang, Y. et al. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol. Sin. 34, 625–635 (2013).
Sotelo, J., Briceño, E. & López-González, M. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343 (2006).
Rojas-Puentes, L. et al. Phase II randomized, double-blind, placebo-controlled study of whole-brain irradiation with concomitant chloroquine for brain metastases. Radiat. Oncol. 8, 209 (2013).
Wolpin, B. et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 19, 637–638 (2014).
Zheng, Z. et al. Metformin activates AMPK/SIRT1/NF-κB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle 19, 1089–1104 (2020).
Wang, L. et al. Metformin induces human esophageal carcinoma cell pyroptosis by targeting the miR-497/PELP1 axis. Cancer Lett. 450, 22–31 (2019).
Dixon, S. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523 (2014).
Sun, J. et al. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 11, 87 (2017).
Mandal, P. et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253 (2010).
Harris, I. et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015).
Woo, J. et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 162, 441–451 (2015).
Ma, S., Henson, E., Chen, Y. & Gibson, S. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 7, e2307 (2016).
Chen, C. et al. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am. J. Transl. Res. 9, 5507–5517 (2017).
Shahsavari, Z., Karami-Tehrani, F., Salami, S. & Ghasemzadeh, M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: necroptosis as a desperate programmed suicide pathway. Tumor Biol. 37, 4479–4491 (2016).
McAfee, Q. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl Acad. Sci. USA 109, 8253–8258 (2012).
Rebecca, V. et al. A unified approach to targeting the lysosome’s degradative and growth signaling roles. Cancer Discov. 7, 1266–1283 (2017).
Rebecca, V. et al. PPT1 promotes tumor growth and is the molecular target of chloroquine derivatives in cancer. Cancer Discov. 9, 220–229 (2019).
Sanders, S. et al. Curation of the mammalian palmitoylome indicates a pivotal role for palmitoylation in diseases and disorders of the nervous system and cancers. PLoS Comput. Biol. 11, e1004405 (2015).
Egan, D. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59, 285–297 (2015).
Martin, K. et al. A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient. Stress. iScience 8, 74–84 (2018).
Pasquier, B. SAR405, a PIK3C3/Vps34 inhibitor that prevents autophagy and synergizes with MTOR inhibition in tumor cells. Autophagy 11, 725–726 (2015).
Pasquier, B. et al. Discovery of (2S)-8-[(3R)-3-methylmorpholin-4-yl]-1-(3-methyl-2-oxobutyl)-2-(trifluoromethyl)-3,4-dihydro-2H-pyrimido[1,2-a]pyrimidin-6-one: a novel potent and selective inhibitor of Vps34 for the treatment of solid tumors. J. Med. Chem. 58, 376–400 (2015).
Dyczynski, M. et al. Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 435, 32–43 (2018).
Fu, Y. et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 15, 295–311 (2019).
Chu, J. et al. ATG4B inhibitor FMK-9a induces autophagy independent on its enzyme inhibition. Arch. Biochem. Biophys. 644, 29–36 (2018).
Akin, D. et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 10, 2021–2035 (2014).
Chung, S. et al. ADIPOQ/adiponectin induces cytotoxic autophagy in breast cancer cells through STK11/LKB1-mediated activation of the AMPK-ULK1 axis. Autophagy 13, 1386–1403 (2017).
Zhang, P. et al. w09, a novel autophagy enhancer, induces autophagy-dependent cell apoptosis via activation of the EGFR-mediated RAS-RAF1-MAP2K-MAPK1/3 pathway. Autophagy 13, 1093–1112 (2017).
Qiao, L. et al. NETA induces pyroptosis of epithelial ovarian cancer cells through the GSDMD/caspase-4 pathway. FASEB J. 33, 12760–12767 (2019).
Hu, L. et al. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 11, 281 (2020).
Teng, J. et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers 12, 193 (2020).
Zhao, P. et al. Programming cell pyroptosis with biomimetic nanoparticles for solid tumor immunotherapy. Biomaterials 254, 120142 (2020).
Xie, Y. et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology 153, 1429–1443.e1425 (2017).
Zhang, M. et al. The roles of ROS and caspases in TRAIL-induced apoptosis and necroptosis in human pancreatic cancer cells. PLoS ONE 10, e0127386 (2015).
Geserick, P. et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limiting RIP1 kinase recruitment. J. Cell Biol. 187, 1037–1054 (2009).
Sonkusre, P. & Cameotra, S. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J. Nanobiotechnol. 15, 43 (2017).
Katifelis, H. et al. Ag/Au bimetallic nanoparticles trigger different cell death pathways and affect damage associated molecular pattern release in human cell lines. Cancers 14, 1546 (2022).
Hsieh, C. H. et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics 11, 7072–7091 (2021).
Anderson, C. & Reynolds, C. Synergistic cytotoxicity of buthionine sulfoximine (BSO) and intensive melphalan (L-PAM) for neuroblastoma cell lines established at relapse after myeloablative therapy. Bone Marrow Transplant. 30, 135–140 (2002).
Ongaro, A. et al. Enhancement of melphalan activity by buthionine sulfoximine and electroporation in melanoma cells. Anti-Cancer Drug. 26, 284–292 (2015).
Cramer, S. et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 23, 120–127 (2017).
Cao, K. et al. Glutathione-bioimprinted nanoparticles targeting of N6-methyladenosine FTO demethylase as a strategy against leukemic stem cells. Small 18, e2106558 (2022).
Hassannia, B. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 128, 3341–3355 (2018).
Socinski, M. et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 20, 1239–1251 (2019).
O’Donnell, J., Teng, M. & Smyth, M. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167 (2019).
Bear, A., Vonderheide, R. & O’Hara, M. Challenges and opportunities for pancreatic cancer immunotherapy. Cancer Cell 38, 788–802 (2020).
Starobinets, H. et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Invest. 126, 4417–4429 (2016).
Levy, J., Towers, C. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Noman, M. et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 6, eaax7881 (2020).
Yu, W. et al. Autophagy inhibitor enhance ZnPc/BSA nanoparticle induced photodynamic therapy by suppressing PD-L1 expression in osteosarcoma immunotherapy. Biomaterials 192, 128–139 (2019).
Shukla, S. et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 173, 624–633.e628 (2018).
Jiang, Q. et al. Platelet membrane-camouflaged magnetic nanoparticles for ferroptosis-enhanced cancer immunotherapy. Small 16, e2001704 (2020).
Guo, P. et al. In situintravesical immunostimulatory gel for triple therapy of bladder cancer. ACS Appl. Mater. Interfaces 12, 54367–54377 (2020).
Chen, J. & Galluzzi, L. Fighting resilient cancers with iron. Trends Cell Biol. 28, 77–78 (2018).
Roh, J., Kim, E., Jang, H. & Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262 (2017).
Kim, E. et al. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 432, 180–190 (2018).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Lu, C. et al. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol. Immunol. 122, 200–206 (2020).
Zhang, L. et al. Engineering multienzyme-mimicking covalent organic frameworks as pyroptosis inducers for boosting antitumor immunity. Adv. Mater. 34, e2108174 (2022).
Davila, M. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra225 (2014).
Vredevoogd, D. et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 180, 404–405 (2020).
Michie, J. et al. The immuno-modulatory effects of inhibitor of apoptosis protein antagonists in cancer immunotherapy. Cells 9, 207 (2020).
Smith, H. et al. RIPK1-mediated immunogenic cell death promotes anti-tumour immunity against soft-tissue sarcoma. EMBO Mol. Med. 12, e10979 (2020).
Beug, S. et al. Publisher Correction: Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nat. Commun. 9, 16231 (2018).
Dufva, O. et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 135, 597–609 (2020).
Van Hoecke, L. et al. Recombinant viruses delivering the necroptosis mediator MLKL induce a potent antitumor immunity in mice. Oncoimmunology 9, 1802968 (2020).
Petanidis, S. et al. Inhibition of kras-derived exosomes downregulates immunosuppressive BACH2/GATA-3 expression via RIP-3 dependent necroptosis and miR-146/miR-210 modulation. Biomed. Pharmacother. 122, 109461 (2020).
Galon, J. & Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218 (2019).
Pitroda, S., Chmura, S. & Weichselbaum, R. Integration of radiotherapy and immunotherapy for treatment of oligometastases. Lancet Oncol. 20, e434–e442 (2019).
Esteva, F., Hubbard-Lucey, V., Tang, J. & Pusztai, L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 20, e175–e186 (2019).
Matsumoto, H. et al. Pembrolizumab monotherapy versus pembrolizumab plus chemotherapy in patients with non-small-cell lung cancer: a multicenter retrospective trial. Thorac. Cancer 13, 228–235 (2021).
Inoue, H. & Tani, K. J. C. D. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 21, 39–49 (2014).
Aoto, K. et al. Immunogenic tumor cell death induced by chemotherapy in patients with breast cancer and esophageal squamous cell carcinoma. Oncol. Rep. 39, 151–159 (2018).
Zhang, J. et al. Immunostimulatory properties of chemotherapy in breast cancer: from immunogenic modulation mechanisms to clinical practice. Front. Immunol. 12, 819405 (2021).
Gao, Y. et al. Methotrexate-loaded tumour-cell-derived microvesicles can relieve biliary obstruction in patients with extrahepatic cholangiocarcinoma. Nat. Biomed. Eng. 4, 743–753 (2020).
Fan, J. X. et al. Epigenetics-based tumor cells pyroptosis for enhancing the immunological effect of chemotherapeutic nanocarriers. Nano Lett. 19, 8049–8058 (2019).
Shalapour, S. et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98 (2015).
Philippou, Y. et al. Harnessing the potential of multimodal radiotherapy in prostate cancer. Nat. Rev. Urol. 17, 321–338 (2020).
McLaughlin, M. et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217 (2020).
Liu, Y. et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis. 8, e2579 (2017).
Mohseni, G. et al. The function of cGAS-STING pathway in treatment of pancreatic cancer. Front. Immunol. 12, 781032 (2021).
Obeid, M. et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 14, 1848–1850 (2007).
Ko, A. et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 21, 92–99 (2014).
Chen, W. et al. Stereotactic body radiation combined with oncolytic vaccinia virus induces potent anti-tumor effect by triggering tumor cell necroptosis and DAMPs. Cancer Lett. (2021).
Ma, X. et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest. 124, 1406–1417 (2014).
Qin, X. et al. The Bcr-Abl inhibitor GNF-7 inhibits necroptosis and ameliorates acute kidney injury by targeting RIPK1 and RIPK3 kinases. Biochem. Pharmacol. 177, 113947 (2020).
Jiang, H. et al. Activating immune recognition in pancreatic ductal adenocarcinoma via autophagy inhibition, MEK blockade, and CD40 agonism. Gastroenterology 162, 590–603.e514 (2022).
Li, H. et al. The beneficial role of sunitinib in tumor immune surveillance by regulating tumor PD-L1. Adv. Sci. 8, 2001596 (2021).
Zhang, N. et al. SA-49, a novel aloperine derivative, induces MITF-dependent lysosomal degradation of PD-L1. EBioMedicine 40, 151–162 (2019).
Maher, C. M. et al. Small-molecule Sigma1 modulator induces autophagic degradation of PD-L1. Mol. Cancer Res. 16, 243–255 (2018).
Tu, X. et al. PD-L1 (B7-H1) competes with the RNA exosome to regulate the DNA damage response and can be targeted to sensitize to radiation or chemotherapy. Mol. Cell 74, 1215–1226.e1214 (2019).
Kheirandish, M. et al. Anti-cancer effects of metformin: recent evidences for its role in prevention and treatment of Cancer. Curr. Drug Metab. 19, 793–797 (2018).
Yue, E. et al. Anthocyanin is involved in the activation of pyroptosis in oral squamous cell carcinoma. Phytomedicine 56, 286–294 (2019).
Zhou, L. et al. Anti-tumor properties of anthocyanins from Lonicera caerulea ‘Beilei’ fruit on human hepatocellular carcinoma: In vitro and in vivo study. Biomed. Pharmacother. 104, 520–529 (2018).
Zhang, C. et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 24, 312–325 (2019).
Zhu, L. & Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell. Mol. Biol. Lett. 24, 40 (2019).
Erkes, D. et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov. 10, 254–269 (2020).
Fu, Z. et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 13, 580 (2013).
Wang, H. et al. Reprogramming tumor immune microenvironment (TIME) and metabolism via biomimetic targeting codelivery of shikonin/JQ1. Nano Lett. 19, 2935–2944 (2019).
Shen, L. et al. Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat. Rec. 304, 313–322 (2021).
Yin, H. et al. Resibufogenin suppresses growth and metastasis through inducing caspase-1-dependent pyroptosis via ROS-mediated NF-κB suppression in non-small cell lung cancer. Anat. Rec. 304, 302–312 (2021).
Gao, Y. et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J. Hepatol. 76, 148–159 (2021).
von Arx, C. et al. Effect of octreotide long-acting release on tregs and MDSC cells in neuroendocrine tumour patients: a pivotal prospective study. Cancers 12, 2422 (2020).
Pisanti, S. et al. Novel prospects of statins as therapeutic agents in cancer. Pharmacol. Res. 88, 84–98 (2014).
Qian, P. et al. Artesunate enhances γδ T-cell-mediated antitumor activity through augmenting γδ T-cell function and reversing immune escape of HepG2 cells. Immunopharmacol. Immunotoxicol. 40, 107–116 (2018).
Morikawa, N. et al. LY341495, an mGluR2/3 antagonist, regulates the immunosuppressive function of myeloid-derived suppressor cells and inhibits melanoma tumor growth. Biol. Pharm. Bull. 41, 1866–1869 (2018).
Oliver Metzig, M. et al. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-α-dependent necroptosis driven by RIP1 kinase and NF-κB. Oncogene 35, 3399–3409 (2016).
Wang, Y. et al. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 495, 1418–1425 (2018).
Zhao, D. et al. A rapid albumin-binding 5-fluorouracil prodrug with a prolonged circulation time and enhanced antitumor activity. Biomater. Sci. 5, 502–510 (2017).
Wang, S. et al. Iron and magnetic: new research direction of the ferroptosis-based cancer therapy. Am. J. Cancer Res. 8, 1933–1946 (2018).
Zhou, B. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 28, 1171–1185 (2018).
Mora, J. et al. Strategies to interfere with tumor metabolism through the interplay of innate and adaptive immunity. Cells 8, 445 (2019).
Yu, P. et al. Eukaryotic elongation factor-2 kinase regulates the cross-talk between autophagy and pyroptosis in doxorubicin-treated human melanoma cells in vitro. Acta Pharmacol. Sin. 40, 1237–1244 (2019).
Nam, J. et al. Chemo-photothermal therapy combination elicits anti-tumor immunity against advanced metastatic cancer. Nat. Commun. 9, 1074 (2018).
Guo, J. et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res. Treat. 50, 445–460 (2018).
Ursic, K. et al. Comparable effectiveness and immunomodulatory actions of oxaliplatin and cisplatin in electrochemotherapy of murine melanoma. Bioelectrochemistry 119, 161–171 (2018).
Author information
Authors and Affiliations
Contributions
Y.Y. conceived the manuscript. W.T. wrote the initial draft of the manuscript. X.Y. drew figures. Y.Z. and X.Q participated in the revision of the manuscript. All authors have read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
We had obtained ethical approval from the ethics committee.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, W., Wang, X., Zhou, Y. et al. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Sig Transduct Target Ther 7, 196 (2022). https://doi.org/10.1038/s41392-022-01046-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-022-01046-3
This article is cited by
-
Cilostazol protects against degenerative cervical myelopathy injury and cell pyroptosis via TXNIP-NLRP3 pathway
Cell Division (2024)
-
The application of nanoparticles-based ferroptosis, pyroptosis and autophagy in cancer immunotherapy
Journal of Nanobiotechnology (2024)
-
Inhibition of ferroptosis reverses heart failure with preserved ejection fraction in mice
Journal of Translational Medicine (2024)
-
Phase separation-mediated biomolecular condensates and their relationship to tumor
Cell Communication and Signaling (2024)
-
Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy
Journal of Hematology & Oncology (2024)
