
Abstract
Sensing invasive cytosolic DNA is an integral component of innate immunity. cGAS was identified in 2013 as the major cytosolic DNA sensor that binds dsDNA to catalyze the synthesis of a special asymmetric cyclic-dinucleotide, 2′3′-cGAMP, as the secondary messenger to bind and activate STING for subsequent production of type I interferons and other immune-modulatory genes. Hyperactivation of cGAS signaling contributes to autoimmune diseases but serves as an adjuvant for anticancer immune therapy. On the other hand, inactivation of cGAS signaling causes deficiency to sense and clear the viral and bacterial infection and creates a tumor-prone immune microenvironment to facilitate tumor evasion of immune surveillance. Thus, cGAS activation is tightly controlled. In this review, we summarize up-to-date multilayers of regulatory mechanisms governing cGAS activation, including cGAS pre- and post-translational regulations, cGAS-binding proteins, and additional cGAS regulators such as ions and small molecules. We will also reveal the pathophysiological function of cGAS and its product cGAMP in human diseases. We hope to provide an up-to-date review for recent research advances of cGAS biology and cGAS-targeted therapies for human diseases.
Similar content being viewed by others

Introduction
Germline-encoded pattern-recognition receptors (PRRs) are key players for human innate immunity. Depending on the source of products, PRRs are divided into two groups, including pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).1,2,3 Pathogen-derived nucleic acids including DNA and RNA are detected by PRRs that subsequently trigger downstream innate-immune responses.2,3,4,5,6 Over the past two decades, a variety of PRRs have been identified, including Toll-like receptors (TLRs), C-type lectin-like receptors (CLRs), retinoic acid-inducible gene I–like receptors (RLRs), NOD-like receptors (NLRs), and the absent in melanoma 2 (AIM2)-like receptors (ALRs).7,8
TLRs and CLRs are membrane-associated receptors, while RLRs, NLRs, and ALRs are cytosolic nucleotide sensors. Cytosolic DNA derived from either pathogens (non-self-DNA, including viral and bacterial DNA) or host genome (self-DNA, including damaged mitochondrial DNA (mtDNA), leaked/damaged nuclear DNA from chromosome instability (CIN), cytosolic DNA in micronuclei and from cell debris), are powerful activators for the innate-immune system. There are four major ALRs all belonging to the PYHIM family members identified in human, including AIM2,9,10,11 interferon-inducible protein 16 (IFI16),12 interferon-inducible protein X (IFIX),13 and myeloid nuclear differentiation antigen (MNDA).14 In addition, other candidates have also been proposed to sense cytosolic DNA, including DNA-dependent activator of IRFs (DAI),15 LRR binding FLII interacting protein 1 (LRRFIP1),16 RNA polymerase III,17,18 Ku heterodimers (Ku70 and Ku80),19,20 DExD/H box helicases (DDX41),21 meiotic recombinations 11 homolog A (MRE11),22 and others. Notably, distinct from other DNA sensors that stimulate interferon production, AIM2 activation in macrophages triggers the formation of a multiprotein complex named inflammasome, leading to activation of the protease procaspase 1 that cleaves pro-IL-1β and pro-IL-18 in triggering proptosis,23 a process antagonized by p202.24 These PRRs are cell-type or DNA-sequence specific,25,26 thus excluding their function as a universal cytosolic DNA sensor. In 2013, the cyclic GMP–AMP (cGAMP) synthase (cGAS) was identified as one of the most important cytosolic DNA sensors,27 given that cGAS recognizes and responds to cytosolic DNA in a DNA-sequence-independent but DNA length-dependent manner in various cell types. Since its discovery, it quickly draws extensive attention from researchers. Within only 8 years, our understanding of cGAS structure, regulation, and function in human diseases has been significantly advanced due to contributions from many intriguing studies. These include but not limited to the identification of cGAS as an essential cytosolic DNA sensor for DNA viruses, RNA viruses, damaged mitochondrial and genomic DNA, illustrations of cGAS activation mechanisms by both structural and biochemical analyses, regulatory mechanisms controlling cGAS activation by cGAS modifications, binding partners and ions, pathophysiological roles of cGAS in biological processes and human diseases, nuclear cGAS function in regulating DNA damage repair and tethering with chromatin, as well as regulations and function of the cGAS enzymatic product cGAMP (Fig. 1).

Due to a large amount of work on this topic in the past 8 years, we cannot include all major findings in this time table and we sincerely apologize for colleagues whose important work are not mentioned in this figure due to space constraints
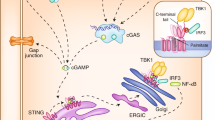
cGAS (also known as C6orf150, or male abnormal 21 domain containing 1 (MAB21D1)) is located on chromosome 6q13 and encodes a protein with 522 amino acids in human. Mechanistically, cGAS recognizes cytosolic dsDNA in a DNA length-dependent but DNA-sequence-independent manner. Interestingly, oxidized self-DNA (8-OHG), although resistant to TREX-1 degradation, could still be recognized by cGAS to promote cGAS activation and innate-immune recognition.28 Short dsDNA (<20 bp) binds but fails to activate cGAS due to its inability to induce cGAS dimerization.29,30 Longer dsDNA (>20 bp) activates cGAS through promoting cGAS dimerization by forming a 2:2 DNA/cGAS complex,31 allowing for rearrangement of the cGAS catalytic pocket for subsequent binding of cGAS substrates adenosine triphosphate (ATP) and guanosine triphosphate (GTP)32 to induce the synthesis of 2′3′-cGAMP.33,34 cGAMP then binds STING (also known as TMEM173, MPYS, ERIS, and MITA) on ER membrane35,36,37 to further recruit TBK1 that facilitates IRF3 phosphorylation and subsequent interferon β (IFNβ) production to trigger inflammation, adaptive immunity38,39,40,41 and expression of other co-regulated genes (Fig. 2).42 STING as an endoplasmic reticulum (ER) localized protein is composed of four N-terminal transmembrane helices and one globular C-terminal cytosolic domain (CTD).38 STING activation is regulated by a number of candidate DNA sensors, including cGAS, IFI16, DDX41, MRE11, and Lsm14A. STING recruits TBK1 to phosphorylate STING on Ser366 residue that further recruits IRF3, where TBK1 phosphorylates IRF3 for its nuclear translocation, dimerization, and activation, which is necessary for IFNβ transcription.34

DNA is a pathogen-associated molecular pattern when delivered to the host cytoplasm by viral or microbial infection, and a danger-associated molecular pattern when leaked into the cytoplasm from damaged mitochondria or nucleus. cGAS is the cytosolic DNA sensor that recognizes and binds cytosolic DNA in a DNA-sequence-independent manner that subsequently triggers cGAS dimerization and production of a special dinucleotide messenger, 2′3′-cGAMP from ATP and GTP. 2′3′-cGAMP binds STING localized on ER, through trafficking to Golgi to recruit and activate IKK and TBK1. TBK1 phosphorylates STING, which in turn recruits IRF3 for phosphorylation by TBK1. Phosphorylated IRF3 dimerizes and enters the nucleus, where it cooperates with NF-κB signaling to turn on transcription of type I IFNs and other immunomodulatory genes
In addition, cGAS dimmers form higher orders of oligomers31 and undergo liquid phase separation43 that significantly enrich local concentrations of cGAS/DNA to further boost cGAS activation. A recent study reveals that cGAS phase transition precludes TREX-1 from degrading dsDNA, thus sustaining DNA-induced cGAS activation.44 Interestingly, although ssDNA and dsRNA can bind cGAS, they fail to rearrange the cGAS catalytic pocket for cGAS activation. The cGAS protein is composed of an unstructured and not well-conserved N-terminus (amino acid residues 1–160) and a highly conserved C terminus (161–522).45 The cGAS N-terminal fragment is highly disordered with a number of K/R residues that have been predicted to bind DNA, albeit the detailed mechanism(s) remains to be elucidated.46 This unstructured N-terminus may also play a role for cGAS plasma membrane attachment to restrain cGAS activation.47 The cGAS C-terminal domain contains two strongly conserved motifs, including a nucleotidyltransferase (NTase) core domain (160–330) and a Mab21 domain with zinc-ribbon insertion (213–513).31,48 The NTase domain is indispensable for cGAS enzyme activity.27 The conserved ZnF motif is vital for DNA binding, enzymatic activity, and downstream innate-immune signaling activation. Notably, the cGAS C-terminal domain contains a strong DNA-binding site A, a weaker DNA-binding site B,31,48 and an additional DNA-binding site C49 that facilitates cGAS activation and phase transition.
cGAS shares significant sequence similarity to the RNA sensor oligoadenylate synthase 1 (OAS1), indicating that cGAS and OAS1 may be derived from an evolutionarily related family of enzymes involved in host immune response.50,51 Interestingly, analyses of cGAS homologs in different species suggest that cGAS protein sequence is ancestral, and its cytosolic DNA sensor function might be conserved during evolution.52 Given to the unique role of cGAS in sensing cytosolic DNA regardless of its origin (including both exogenous and endogenous DNA) and sequence, cGAS exerts remarkably diverse regulatory functions in a variety of cellular progresses including DNA damage response, tissue fibrosis, senescence, inflammation, cell death, autophagy, and tumorigenesis.53,54,55,56,57,58,59,60,61,62,63,64 Hyperactivation of cGAS/STING signaling plays an indispensable role in the development of autoimmune diseases such as Aicardi–Goutieres Syndrome (AGS) and systemic lupus erythematosus (SLE),65,66,67 while on the other hand helps to establish an immune-friendly microenvironment by promoting T-cell infiltrations into tumors.68 In this scenario, a tightly controlled and balanced cGAS activation is necessary to maintain proper cell physiology and function. In this review, we summarize up-to-date knowledge for cGAS regulatory mechanisms, including cGAS expression regulations at DNA, RNA, and protein levels, cGAS activation controls by protein post-translational modification and binding proteins, as well as ions and small molecules. We will also review cGAS function in human diseases and current knowledge and trials in targeting cGAS, or its enzymatic product, 2′3′-cGAMP for disease treatment.
Regulatory mechanisms for cGAS activity control
cGAS gene alternations are rarely observed in human diseases
Although cGAS plays critical pathophysiological roles in autoimmune diseases, aging, and cancer, the cGAS gene has not been reported to be amplified/mutated/deleted in these human diseases. Querying TCGA datasets led us to find that the cGAS gene is mutated (13 in 20 cases) in SCLC (small cell lung cancer) patients and deleted (23 in 237 cases) in metastatic breast cancer patients (the MBC project). Thus, it is plausible that cGAS gene alternations may contribute to cancer phenotypes; however, more in-depth investigations are warranted to examine this concept. These observations suggest that alternations in cGAS gene may not be a major route through which cGAS activity is deregulated in human diseases.
Transcriptional and post-transcriptional regulations of cGAS expression
The region (−414 to +76) next to the transcription start site (TSS) in the cGAS gene was found to be critical as a promoter for maintaining cGAS transcription.69 Mutating Sp1 and CREB-binding motifs in this region led to reduced cGAS transcription,69 suggesting that both transcription factors govern cGAS transcription. In addition, an epigenetic cofactor NCOA3 was observed to maintain basal cGAS expression,70 while the identity of responsible transcription factors remains to be determined. In microglia, HDAC3, as a member of histone deacetylases, was found to be crucial for cGAS transcription by deacetylating p65 to enhance p65 association with cGAS promoter to transcriptionally potentiate cGAS expression.71
MicroRNAs (miRNAs) downregulate their target gene expression at post-transcriptional levels through binding the 3′ untranslated regions (3′UTRs) of the target messenger RNA (mRNA).70 It has been documented that a number of miRNAs are involved in DNA-sensing-related immune defense72,73 and are pivotal for presenting antigens and secreting immuno-cytokines. Hypoxia-responsive miRNAs including miR-93 and miR-25 were reported to remarkably downregulate cGAS expression in the immunosuppressive tumor microenvironment (TME) through targeting NCOA3 for suppression.70 Thus elevated expression of miR-93/25 observed in breast cancer facilitates tumor evasion from immune surveillance and destruction partially through downregulating cGAS expression. In addition to miRNAs, an HSV-1 tegument protein UL41 was also observed to utilize its endoribonuclease activity to degrade cGAS mRNA, through which UL41 negatively regulates DNA sensing to facilitate HSV-1 infection by escaping from immune surveillance.74
Post-translational regulations of cGAS activation
In addition to genetic and transcriptional controls, cGAS activity is also modulated at post-translational levels. Prior work identified multiple regulatory mechanisms governing cGAS activation, including regulations by post-translational modifications and binding proteins. In this section, we summarize major findings in these aspects. Notably, given that both human and mouse cGAS molecules have been used in these studies, and <60% cGAS protein sequence is shared between these two species, to clarify the exact amino acid(s) being modified, in this section we will label human and mouse cGAS proteins as hcGAS and mcGAS, respectively.
Regulation of cGAS activation by cGAS post-translational modifications
Control of cGAS activation by ubiquitin
As a reversible post-translational modification, protein ubiquitination or deubiquitination plays an indispensable and evolutionarily conserved function in both eukaryotes and prokaryotes75,76 in regulating a diversity of cellular processes, including degradation of unwanted proteins, cell cycle progression, DNA damage response, vesicle transport, endocytosis, signal transduction, and others.77,78 Protein ubiquitination is largely carried out by a cascade of enzymatic reactions governed by E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases. Among all these three categories of enzymes, it is the E3 ligase that determines ubiquitin substrate specificity. There are ~600 E3 ubiquitin ligases encoded in human genome that fall into three families: RING (really interesting new gene), HECT (homologous to E6AP carboxyl terminus), and RBR (RING-between-RING).78 Poly-ubiquitin chains, established by E3 ligase(s), can be removed by deubiquitinases (DUBs). E3 ligases and DUBs are diverse in structure and function with a myriad of distinct mechanistic features. Compared with many E3 ligases, there are only ~100 DUBs, suggesting that DUBs are less selective towards substrates. DUBs are currently classified into six families, including ubiquitin C-terminal hydrolases (UCHs), ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), Josephins, JAB1/MPN/Mov34 metalloenzymes (JAMMs), and motif interacting with Ub-containing novel DUB family (MINDYs).79 Although these DUBs are structurally unrelated, they all interact with a common hydrophobic patch on ubiquitin. Emerging evidence reveals roles of dysregulated E3 and DUB signaling in contributing to human diseases including cancer,80 immune diseases,81,82 brain diseases,83 and others. Agents modulating DUB activities84 or impeding E3-ligase interactions with substrates85 have been developed and tested in clinics, and PROTAC (proteolysis-targeting chimera)86 has emerged as a novel and powerful tool in this regard as a promising treatment direction utilizing E3-ligase-mediated protein-degradation processes. It is not surprising that cGAS is also under regulation by ubiquitin modifications.
The presence of seven lysine residues in each ubiquitin molecule assigns the possibility for the formation of diverse poly-ubiquitin chain linkages with distinct topologies and physiological functions.87 cGAS undergoes ubiquitin-mediated modifications including monoubiquitination and polyubiquitination that differentially modulate cGAS activation and function (Fig. 3).

cGAS is dynamically regulated by various post-translational modifications in responding to DNA insults, such as monoubiquitination, polyubiquitination, SUMOylation, glutamylation, phosphorylation, acetylation, and deamidation in cells. This figure illustrates up-to-date reported post-translational modifications occurring on cGAS proteins, including the modified residues and modifying enzymes. Please note that all human cGAS residues are labeled in pink while all mouse cGAS residues are labeled in orange with “m” inserted in front of the residue
Monoubiquitination of cGAS promotes cGAS activation
Two E3 ubiquitin ligases including Tripartite-motif containing (TRIM) E3 ligases TRIM56 and TRIM41 (RINCK) have been found to be responsible for cGAS monoubiquitination. TRIM56 was identified as a cGAS-binding protein in a proteomics study and was found to promote mcGAS monoubiquitination on Lys335,88 which led to enhanced cGAS dimerization, DNA binding, and cGAMP production. TIRM41 was also identified as a cGAS interactor by proteomics and shown to be able to promote cGAS monoubiquitination in biochemical assays.89 Although the modifying lysine residue(s) by TRIM41 remains unknown, this cGAS monoubiquitination is indispensable for cGAS activation upon DNA challenge to exert its full activities.89 The exact molecular details for how monoubiquitination of cGAS facilitates cGAS dimerization and DNA binding remain unclear. Although the mono-ubiquitin moiety does not directly bind DNA,90 given that the Lys335 residue is located to the close proximity of DNA-binding region in mcGAS (PDB 4LEZ), it is plausible that this monoubiquitination modification may help stabilize the ternary complex formed by cGAS dimers with two DNA molecules.
cGAS polyubiquitination in controlling cGAS activation and function
In addition to monoubiquitination, cGAS is also modified by polyubiquitination. The RING finger (RNF) containing E3 ubiquitin ligase RNF185 located on ER induces an accumulation of K27-linked poly-ubiquitin chains on Lys173/Lys384 residues in mcGAS, through an interaction mediated by the RING domain of RNF185 and the C-terminal domain of cGAS. K27-linked polyubiquitination of cGAS enhanced cGAS enzymatic activity both in vitro and in cells for cGAMP synthesis.91 Given K27-linked polyubiquitination of STING recruits TBK1 binding92 and K27-linked polyubiquitination of NEMO promotes Rhbdd3 binding,93 we speculate if cGAS recognizes K27-linked poly-ubiquitin chains so that K27-linked polyubiquitination of cGAS promotes cGAS dimerization or oligomerization. This requires further experimental evidence to support or reject the hypothesis. Interestingly, HSV-1 infection promotes the co-localization of RNF185 and cGAS in cells,91 suggesting that cells may utilize K27-linked ubiquitination to promote cGAS activation responding to DNA viral infection.
TRIM14 is a member of the tripartite-motif (TRIM) E3 ubiquitin ligase family with identified roles in facilitating sensing RNA viruses as a mitochondria adaptor.94 Although as a noncanonical TRIM lacking a RING domain, TRIM14 was found to be able to stabilize cGAS proteins by recruiting USP14 to cleave K48-linked poly-ubiquitin chains at the hcGAS-Lys414 residue, given that K48-linked polyubiquitination of hcGAS-Lys414 primes cGAS for p62 binding that targets cGAS for lysosomal degradation.95 In another study, TRIM14 was reported to bind cGAS and TBK1, and bridge TBK1 binding to STAT3 to promote STAT3 phosphorylation in the synthesis of ISGs (interferon-stimulated genes) that facilitate detection and clearance of Mycobacterium tuberculosis infection.96 Interestingly, TRIM14 itself is a transcriptional target for interferon signaling,97 which suggests that there might be a positive regulatory loop for augmenting innate immunity. Notably, the identity of the E3 ligase(s) that targets cGAS protein for ubiquitination and degradation remains elusive to date.
cGAS deubiquitination in controlling cGAS activation and function
Among all DUBs, 79 of them exert enzymatic activity. Members of the ubiquitin-specific peptide (USP) family of DUBs have emerged as vital molecules in regulating antiviral immunity, either as direct regulation factors of viral replication or as regulators of host nucleic acid-sensing pathways.98 It was reported that upon DNA virus infection, USP14 was recruited by TRIM14 to remove K48-linked ubiquitin chains at the Lys414 site in hcGAS, leading to cGAS stabilization to promote antiviral innate immunity.95 Similarly, USP27X99 and USP29100 have also been reported to stabilize cGAS proteins by cleaving K48-linked poly-ubiquitin chains from cGAS, and both of them serve as positive regulators in activating innate immunity to fight against DNA viral infection.
Regulation of cGAS activation by SUMOylation
Similar to ubiquitination, protein SUMOylation can rapidly regulate protein fate and function. SUMO (small ubiquitin-like modifier) is predominantly found in the nucleus with important roles in regulating various pathophysiological processes,101 including DNA damage response,102 cancer, Huntington’s disease, Alzheimer’s disease, Parkinson’s disease103 and innate immunity.104 Previously, it was reported that the E3 ubiquitin ligase TRIM38 negatively regulates TLR-mediated immune signaling by ubiquitinating TRIF and promoting TAB2/3 degradation.105 On the other hand, TRIM38 was reported to positively regulate RLR-induced innate immunity by SUMOylating MDA5106 and RIG-I107 for their stabilization through antagonizing ubiquitination-mediated protein degradation. Recently, TRIM38 was also shown to be able to modulate cytosolic DNA sensing and cGAS activation. Instead of regulating cGAS ubiquitination, at the resting state or early infection stages, TRIM38 maintains SUMOylation at Lys217 and Lys464 residues in mcGAS (corresponding to Lys231 and Lys479 in hcGAS), which prevents K48-linked cGAS polyubiquitination that directs cGAS for protein destruction.108 Thus, through stabilizing cGAS proteins, TRIM38 ensures cGAS availability in responding to DNA viral infection. Together, these observations suggest that TRIM38 may exert function in suppressing immune response as an E3 ubiquitin ligase, while potentiate DNA or RNA virus-induced innate immunity as a SUMOylase. At later stages of viral infection, the deSUMOylating enzyme Sentrin/SUMO-specific protease (SENP) 2 removes SUMO modifications from cGAS, subsequently promoting K48-linked ubiquitination at mcGAS-Lys464 residue (corresponding to Lys479 in hcGAS) that primes cGAS for degradation by the ubiquitin–proteasome pathway to attenuate the antiviral response.108 On the other hand, SUMOylation of mcGAS at Lys335/Lys372/Lys382 residues by unknown enzymes suppresses DNA binding, cGAS oligomerization, and cGAS nucleotidyltransferase activity, while SENP7 alleviates SUMO-mediated suppression of cytosolic DNA sensing by removing cGAS SUMOylation and potentiates cGAS activity.109 Notably, increased expression of either TRIM38108 or SENP7109 has been observed in patients with SLE (systemic lupus erythematosus), further revealing the pathophysiological significance of cGAS deSUMOylation in activating cGAS governed cytosolic DNA-sensing signaling. Taken together, cGAS SUMOylation can either potentiate cGAS activation through stabilizing cGAS proteins by antagonizing cGAS ubiquitination-mediated degradation or suppress cGAS activation by impeding cGAS dimer formation or DNA binding, depending on the modified lysine residues.
Regulation of cGAS activation by glutamylation
Protein glutamylation is a type of ATP-dependent post-translational modifications (PTM) vital for regulating bacterial and viral infection that involves the conjugation of glutamate side chains to the γ-carboxyl groups of glutamic acid residues in target proteins.110 Glutamylation is usually catalyzed by glutamylases and removed by carboxypeptidases.111 Recently, cGAS was reported to undergo glutamylation modifications. Specifically, TTLL4 (tubulin tyrosine ligase-like 4) and TTLL6 catalyzed mono-glutamylation and poly-glutamylation of mcGAS at Glu302 and Glu272 residues, respectively. TTLL4-mediated mono-glutamylation of cGAS inhibits cGAS enzymatic activity and TTLL6-governed cGAS poly-glutamylation attenuates DNA binding, both leading to reduced synthase activity of cGAS for cGAMP production.112 On the other hand, carboxypeptidases CCP5 and CCP6 removed mono- and poly-glutamylation modifications on mcGAS-Glu302 and Glu272, respectively, leading to cGAS activation.112 These findings provide additional insights into the fine-tune mechanisms for cGAS activity regulations that have been evolved during evolution to ensure cells adapt and respond to external invasive cues in an acute and regulatable manner.
Regulation of cGAS activation by phosphorylation
Protein phosphorylation is one of the most extensively studied protein modifications in dynamically regulating protein functions in a plethora of biological processes, including cell cycle regulation, apoptosis, DNA damage response, tumorigenesis, and immunity.113,114 Small-molecule kinase inhibitors (SMKIs) have been widely developed and tested as promising targeted therapeutics.115 The first post-translational modification reported on cGAS was AKT-mediated mcGAS phosphorylation at Ser291 (corresponding to Ser305 in hcGAS).116 AKT-mediated cGAS phosphorylation occurs in the carboxyl-terminal enzymatic domain of cGAS and results in suppression of cGAS enzymatic activity in cGAMP synthesis to alleviate immune response upon viral infection.116 Given that hyperactivation of the PI3K/AKT signaling has been widely observed in human cancers117 and evading immune surveillance and destruction is a hallmark of cancer,118 it is plausible that tumors hijack AKT/cGAS signaling to inactivate cGAS in creating an immune environment favored by tumors. In addition, recently the CDK1-cyclin B kinase complex was reported to phosphorylate hcGAS at Ser305 residue (Ser291 in mcGAS) as well, which inhibits its ability to synthesize cGAMP in mitotic cells.119 This process can be antagonized by the protein phosphatase 1 (PP1) through dephosphorylating cGAS upon mitotic exit to enable its DNA-sensing ability.119 Considering that Akt activity is also controlled in a cell cycle-dependent manner with peak activity at S/G2,120 it is plausible that Akt and CDK1 govern cGAS phosphorylation in S and M phases, respectively, to ensure cGAS activation is properly controlled during critical cell cycle phases with DNA replication (S) and DNA separation (M) accompanied by increased chances of cGAS exposure to genomic DNA. Recent studies reported that protein phosphatase 6 (PP6) dephosphorylates mcGAS at Ser420 (Ser435 in hcGAS) to restrain its substrate-binding ability and suppress cGAMP synthesis.121 Moreover, Tyr215 in hcGAS has been reported to be phosphorylated by BLK (B lymphocyte kinase) that retains cGAS in the cytoplasm,64 where cGAS is primed for cytosolic DNA sensing. Considering that deregulation of BLK,122 PP1, and PP6123 has been reported in various cancers and other human diseases, if and how these enzymes contribute to pathological processes through regulating cGAS modifications remains an interesting question.
Regulation of cGAS activation by acetylation
Protein acetylation occurs on lysine residues and is a result of a balanced action by acetyl-transferases and deacetylases.124 Acetylation events at the N-terminus of histones have been well-documented as key events for epigenetic regulation of gene transcription. hcGAS was found to be acetylated at Lys384/Lys394/Lys414 residues at resting states to restrain cGAS activation. Upon DNA challenge, cGAS was deacetylated on these sites allowing for cGAS activation.125 Interestingly, aspirin could directly acetylate cGAS to inhibit cGAS activation, which reveals a promise in applying aspirin in treating autoimmune diseases such as Aicardi–Goutieres syndrome (AGS).125 Recently, the lysine acetyl-transferase KAT5 was reported to acetylate Lys47/Lys56/Lys62/Lys83 residues located in the N-terminal unstructured region in hcGAS, and these acetylation events led to increased cGAS binding with DNA to promote cGAS activation in response to DNA challenge.126 Therefore, depending on the acetylation sites, acetylation of cGAS could either positively or negatively regulate cGAS activation and it is possible that acetylation occurring at different stages of infection may have a distinct function in cGAS activity control.
Notably, a recent proteomics effort in examining cGAS post-translational modifications upon HSV-1 infection from both human primary fibroblasts and HEK293T cells revealed new PTMs occurring on cGAS, including phosphorylation at Ser37, Ser116, Ser201, Ser221, Ser263, and acetylation at Lys198, Lys285, Lys355, and Lys414 in hcGAS.127 Further functional validation suggests that acetylation at Lys414 suppresses, while acetylation at Lys198 promotes hcGAS activation. Interestingly, hcGAS-Lys198 acetylation was found to be decreased by quantitative proteomics upon infection by either HSV-1 or HCMV (human cytomegalovirus), suggesting that these DNA viruses might hijack this acetylation regulation to targetedly inactivate cGAS to evade innate-immune surveillance.127 The detailed mechanism(s) mediating acetylation-dependent cGAS activity control on these sites remain unclear.
Taken together, a variety of post-translational modifications have been identified in regulating cGAS activity and function, including phosphorylation, acetylation, ubiquitination, and SUMOylation. Some modifications such as hcGAS acetylation at Lys384/Lys394/Lys414 occur at the resting state as a mechanism to ensure cGAS remains at a low activity until encountering DNA challenges. Some other modifications are triggered upon DNA challenge as means to acutely activate cGAS to ensure timely DNA sensing such as monoubiquitination of mcGAS-K335 or K27-linked polyubiquitination of mcGAS-Lys173/Lys384 that facilitates DNA binding and cGAS dimer formation. AKT and CDK1-mediated cGAS phosphorylation and inactivation of cGAS may serve as a mechanism to ensure cGAS is not aberrantly activated in cell cycle phases with exposures to naked DNA, including S/G2 and M phases. Thus, different modifications orchestra to fine-tune cGAS activation under distinct conditions and a tight control of cGAS activity through these modifications are crucial to maintain a proper innate-immune response. It is not surprising to find these modifications cross-talk with each other; however, given most of the studies focus on only one type of modifications, how tempo- and spatially these cGAS modifications occur and are regulated remain to be determined.
Regulation of cGAS activation by cGAS-binding proteins
In addition to post-translational modifications directly occurring on cGAS proteins, various cGAS-binding partners, including both host proteins and viral/bacterial proteins, have been identified to modulate cGAS activation in order to regulate the innate-immune response. Generally, binding of host proteins to cGAS can either enhance cGAS activation to facilitate detection and clearance of cytosolic DNA derived from viral/bacterial infection (foreign-DNA) and damaged genome or mitochondrial DNA (self-DNA), or restrain cGAS activity at resting state to avoid unnecessary cGAS activation; while viral/bacterial proteins usually bind and inactivate cGAS to escape from innate-immune surveillance. The regulatory function of host proteins in facilitating cGAS activation can be due to their ability to bind cGAS to enhance DNA binding or bind DNA to bridge DNA for cGAS binding (Table 1).
Host proteins binding to cGAS in promoting cGAS activation
For example, the host protein G3BP1 (GTPase-activating protein SH3 domain-binding protein 1) binds cGAS to potentiate cGAS binding with DNA, formation of cGAS oligomers, and activation of cGAS.128,129 Moreover, PCBP1 (Poly(rC)-binding protein 1) as a member of the heterogeneous nuclear ribonucleoprotein family, was reported to directly interact with cGAS in a viral infection-dependent manner to enhance cGAS binding to viral DNA, thus elevating cGAS activity.130 PQBP1 (polyglutamine binding protein 1) directly bridges reversely transcribed HIV-1 DNA and cGAS to trigger cGAS activation upon HIV infection in DCs (dendritic cells).131 ZCCHC3 directly interacts with dsDNA, leading to increased binding of dsDNA to cGAS and subsequent cGAS activation following viral infection.132 In addition, TRIM21 promotes proteasomal destruction of antibody-opsonized virions, leading to exposure of viral genome for cGAS detection and activation, thus indirectly facilitating cGAS activation.133
Host proteins binding to DNA in suppressing cGAS activation
The autophagy protein Beclin-1 directly interacted with cGAS and inhibited the synthesis of cGAMP by negatively regulating cGAS activity, thus increasing the autophagy-mediated degradation of cytosolic bacterial DNA to mediate innate antimicrobial immune response.62 Oligoadenylate synthetase family protein OASL directly bound to cGAS independent of dsDNA to non-competitively suppress cGAMP synthesis and subsequent type I IFN production during infection by DNA viruses such as vaccinia, herpes simplex, and adenovirus.134 Recently, it was reported that TRIM14 directly bound cGAS and acted as a scaffold protein between TBK1 and STAT3 to facilitate STAT3 phosphorylation in order to turn off STAT3 signaling and ISG expression during infection by M. tuberculosis.96 SAMHD1 has been observed as a restriction factor for HIV-1 infection by suppressing cGAS activity to limit innate and adaptive immune responses.135 Mechanistically, SAMHD1 facilitates clearance of naked DNA at stalled replication forks.136 In SAMHD1-deficient cells, cytosolic DNA accumulates to trigger the production of interferons, and this depends on MRE11 and RECQ1.136 Considering that SAMHD1 is frequently mutated in AGS and some cancer, it is plausible that inhibition of MRE11 and/or RECQ1 would attenuate cGAS activation through reducing the production of DNA fragments.
Sensing self-DNA should be tightly inhibited to avoid hyperactivation of innate-immune signaling leading to autoimmune diseases. Although a large portion of cGAS is present in the nucleus, its activation is suppressed by at least two mechanisms contributed by cGAS binding proteins. The first mechanism for inhibiting nuclear cGAS activation is achieved by binding cGAS to the chromatin-binding protein BAF (barrier-to-autointegration factor 1). As a natural opponent of cGAS activation, BAF binding to cGAS competes with DNA binding, thus restraining cGAS in an inactive state.137 The second mechanism is achieved by tight tethering of cGAS to chromatin,138 which is mediated by interactions between cGAS Arg anchors and H2A/H2B acidic patch residues, through which nucleosome binding to cGAS restrains cGAS binding to DNA for cGAS activation.139,140,141,142,143,144 In addition to cGAS binding proteins in restraining cGAS activation in the nucleus, cGAS phosphorylation by AKT and CDK1 also play a role in suppressing cGAS activation during cell cycle progression as discussed in the last section.
Viral/bacterial proteins binding to cGAS in modulating cGAS activation
Given to the essential function of cGAS in sensing infection by DNA viruses, RNA viruses (such as HIV-1, dengue, Zika, CHIKV, and others, detailed below), and bacteria, these human pathogens evolve mechanisms to disable cGAS cytosolic DNA sensor function. One of such mechanisms includes utilizing viral/bacterial proteins to directly bind cGAS to suppress cGAS activation. For example, the cytoplasmic isoforms of LANA (latency-associated nuclear antigen) from Kaposi sarcoma herpesvirus (KSHV) were identified as a cytosolic cGAS binding partner, and LANA binding to cGAS inhibited cGAS activation and subsequent phosphorylation of TBK1 and IRF3 that induces KSHV reactivation from latency.145 However, how LANA binding restrains cGAS activation remains to be determined. In addition, ORF52 is a gamma-herpesvirus tegument protein and was found to interact with both cGAS and DNA to suppress the antiviral immune response.146 Although ORF52 binding to cGAS does not affect cGAS binding to DNA, ORF52 binding to the cGAS/DNA complex impeded cGAS activation, presumably through blocking proper cGAS protein conformational changes necessary for its enzymatic activation. In addition, three human cytomegalovirus (HCMV) tegument proteins were found to bind cGAS and modulate cGAS activation. Specifically, UL31 was reported to bind cGAS to dissociate DNA binding, therefore leading to reduced cGAS activation for innate-immune evasion.147 Similarly, UL42 bound to both cGAS and STING, and UL42 binding to cGAS suppressed DNA binding, cGAS oligomerization, and cGAS enzymatic activity.148 UL83 only interacted with cGAS but not STING, and binding of UL83 to cGAS impeded cGAS activation upon HCMV infection, while how UL83 inhibits cGAS function remains unclear.149 Moreover, three HSV-1 (herpes simplex virus 1) tegument proteins were also observed to bind cGAS in suppressing cGAS activation. UL37 is an HSV-1 tegument protein and a deaminase bound to cGAS to deamidate hcGAS on Asn210, Asn389, Gln451, and Gln454 residues, leading to a suppression of intrinsic cGAS enzymatic activity without affecting cGAS binding to DNA, cGAS dimerization or substrate nucleotides.150 Another HSV-1 protein UL41 utilized its endoribonuclease activity to degrade cGAS mRNA, through downregulating cGAS protein expression to suppress innate-immune sensing.74 VP22 was also found to bind and suppress cGAS activation upon HSV-1 infection, while the detailed molecular mechanism for this suppression remains to be determined.151
F17, a protein encoded by poxviruses, exerted a crucial role in evading host antiviral responses by promoting cGAS degradation through elevating mTORC2 activity by sequestering Raptor and/or Rictor from mTORC1 during infection.152 The mammalian nuclear protein NONO directly bound HIV capsid in the nucleus to facilitate cGAS association with HIV DNA, thus playing an indispensable role in promoting cGAS-mediated innate-immune activation upon HIV infection.153 In addition, the DS2B protease cofactor from an RNA DENV virus (dengue virus) targeted cGAS for lysosomal-dependent degradation to evade cGAS detection of damaged/leaked mitochondrial DNA upon DENV infection.154 Similarly, the CHIKV (Chikungunya virus) capsid protein suppressed transcription of IFN-β during CHIKV infection through inducing autophagy-dependent cGAS degradation.155 Furthermore, the NSP1 protein from the RNA virus Zika stabilized caspase 1 to enhance cGAS cleavage and inactivation, leading to evasion of innate-immune sensing.156 Together, these viral proteins through binding cGAS to guide cGAS for degradation (through proteasome, lysosome, and autophagy) or cleavage as an approach to facilitate evasion of host detection. If any bacterial protein plays a similar function remains unknown.
In addition to viral proteins, recently our group identified a bacterial protein streptavidin in directly binding and activating cGAS through promoting cGAS association with DNA and subsequent cGAS liquid phase transition and enzymatic activation.157 Given that streptavidin is currently widely used as a tool in biotechnology to monitor biotin-tagged molecules, and a drug delivery vehicle with immune adjuvant activity in clinic, our studies horn an alarm to use streptavidin in these settings with caution due to its ability in facilitating activation of innate immunity.
Regulation of cGAS activation by cGAS proteolytic cleavage
Proteolytic cleavages by protein proteases play key roles in regulating protein structure and function, especially targeted and limited proteolysis158 including activation of growth factors such as TGFβ by proteases159. Caspase 1 cleaves Asp140 and Asp157 residues,160 while caspase 3 cleaves the Asp319 residue161 in hcGAS to restrain cGAS activation upon DNA or RNA viral infection. Caspase 4, 5, and 11 activated during noncanonical inflammasome formation were also shown to be able to cleave cGAS in the cGAS N-terminal R/K rich region to facilitate viral infection by inactivating cGAS sensing.160 In addition, caspase 3/7 or caspase 9 has also been shown to cleave cGAS at unknown residues to suppress mtDNA-induced cGAS activation and IFN production.162 Interestingly, the Zika virus NSP1 protein stabilized caspase 1 to enhance cGAS cleavage and inactivation, leading to evasion of innate-immune sensing,156 highlighting the pathophysiological significance of cGAS proteolytic cleavage in facilitating viral infection. In addition to viral infection, if cGAS cleavage by caspases occurs in physiological conditions or developmental stages, and if cGAS can be cleaved by non-caspase proteases remain interesting questions to answer.
Regulation of cGAS activation by ions
Interestingly, it has been observed that the cGAS enzyme activity was different between physiological buffers and low-salt buffers, and this difference was caused by ions present in buffers.43 Specifically, similar to many other enzymes, the inclusion of Zn2+, Mn2+, or Co2+ ions in physiological buffers facilitated cGAS activation.43 The reason for Zn2+ in promoting cGAS activation is largely due to the fact that Zn2+ induces cGAS phase transition in the presence of DNA.43 In addition, Mn2+ could also augment cGAS enzymatic activity by enhancing its sensitivity to low doses of dsDNA usually nonstimulatory,163 or directly activate cGAS independent of dsDNA to synthesize cGAMP,164 presumably through activating monomeric cGAS without dsDNA.165 Notably, there is new evidence demonstrating that K+ efflux might suppress cGAS activation in decreasing the type I interferon responses,166 while the underlying mechanistic insights warrant further investigations. Interestingly, some copper (Cu2+) complexes used in anticancer therapy inhibit topoisomerase 1 and 2, leading to DNA breaks and DNA fragments to activate cGAS signaling.167 Notably, transporters for these ions have been observed dysregulated in cancer,168 and it is plausible altered ion transport in cancer may facilitate cancer evasion of immune surveillance by modulating cGAS signaling. Although there is no direct evidence to support this speculation, it definitely warrants further investigations.
Regulation of cGAS activity by small molecules
Given the essential function of cGAS in cytosolic DNA sensing and various pathological conditions including both autoimmune diseases and cancer, research efforts have also been devoted to developing small molecules that modulate cGAS activity. RU.521 has been developed as a cGAS inhibitor by competitive binding to cGAS catalytic pocket with cGAS substrates ATP/GTP.169 Through screening a compound library using a luciferase-based platform, G140/G150 was found as potent cGAS inhibitors.170 In another fluorescence polarization assay utilizing recombinant active human cGAS proteins in vitro, PF-06928215 was characterized to be able to inhibit cGAS activity.171 Interestingly, a subsequent virtual compound screen based on a structure derived from PF-06928215 binding to cGAS led to the identification of additional compounds S2 and S3 as potent cGAS inhibitors.172 A virtual drug screen combined with further medicinal chemistry studies based on a solved structure of mouse cGAS led to the development of Cu-32 and Cu-76 as cGAS inhibitors by binding to cGAS DNA-binding pocket to prevent cGAS dimerization and subsequence activation.173 Through establishing compounds that stereo-chemically distinct from traditional compounds, SI-56 was found to be able to inhibit cGAS-induced IFN production in cells.174 In addition to these newly identified compounds, a handful of previously commercially available compounds have also been found to suppress cGAS activity. This includes hydroxychloroquine (HQC),175 X6 (used to treat malaria),176 and suramin177 that block cGAS binding to DNA, as well as aspirin125 that inhibits cGAS activity through directly acetylating cGAS proteins. Notably, to date, there is no cGAS activator that has been successfully developed.
Regulation of cGAS activation by lipids
The N-terminal unstructured region in cGAS has been shown to be necessary for DNA binding. A recent study uncovered a novel role of this N-terminus in directly binding phosphoinositide that positions cGAS at the plasma membrane.47 This ensures the resting state of cGAS is suppressed and separated from detecting cytosolic DNA, serving as an additional layer of regulatory mechanism.
Regulation of the cGAS enzymatic product 2′3′-cGAMP
2′3′-cGAMP, a special cyclic-dinucleotide (CDN) was characterized as the major cGAS enzymatic product.27 Cyclic dinucleotides are conserved secondary messengers in both prokaryotes and eukaryotes. In the former, CDNs regulate various cellular processes while in latter CDNs largely activate innate-immune response.178 Cyclic di-GMP and cyclic di-AMP are second messengers in gram-negative and gram-positive bacteria, respectively. Asymmetric cyclic AMP-GMP was identified in Vibrio cholerae as a virulence signaling molecule, synthesized by DncV, a dinucleotide cyclase with sequence similarity to eukaryotic OSA1 (oligoadenylate synthetase).179 The cGAS enzymatic product 2′3′-cGAMP was synthesized from GTP and ATP, with AMP-2′-GTP180 or 5′-pppGpG169 as intermediates. Although these intermediate products have been observed, how long they can stay in cells and if any of these intermediates exerts physiological function remains unknown.
As a potent STING agonist, 2′3′-cGAMP displays an exciting function in triggering innate immunity to facilitate anticancer treatment through promoting tumor-infiltrating T cells. Administration of 2′3′-cGAMP directly into mice showed a synergy with anti-immune-checkpoint blockade (anti-PD-L1) in suppressing melanoma growth.68 Although asymmetric, 2′3′-cGAMP was efficiently transferred inside or outside of cells, which was largely mediated by various characterized cGAMP importer SLC19A1181,182 or transporter LRRC8A:C/E heteromeric channels.183,184 Increased expression of SLC19A1 was observed in SLE (systemic lupus erythematosus) patients185 that might promote innate immunity activation. 2′3′-cGAMP was found to be steadily released from cells186 but degraded by extracellular ENPP1 (ecto-nucleotide pyrophosphatase/phosphodiesterase).187 Similar to 2′3′-cGAMP administration, inhibiting ENPP1 that leads to 2′3′-cGAMP accumulation synergized with IR (ionizing radiation) to impede tumor growth.186 Interestingly, 2′3′-cGAMP was reported to exert a bystander activity that cGAMP producing cells can inter-cellularly transfer cGAMP into bystander cells that rapidly amplify antiviral immunity signals188 to enhance antiviral immunity.183 2′3′-cGAMP could also be transferred from epithelial cells into co-cultured macrophages to transactivate STING signaling in a connexin-dependent manner.189 On the other hand, brain metastatic cancer cells could utilize the carcinoma-astrocyte gap junctions to transfer 2′3′-cGAMP into astrocytes, where 2′3′-cGAMP promotes inflammatory cytokine secretion as paracrine signaling to fuel tumor growth.190 Thus, there are both problems and benefits for 2′3′-cGAMP in regulating innate immunity and anticancer immunity. Although ENPP1 was identified as the major enzyme degrading extracellular 2′3′-cGAMP, the identities of the host-specific intracellular 2′3′-cGAMP degrading enzyme(s) remain unknown. Nonetheless, recently viral and metazoan poxins (poxvirus immune nucleases) were reported to cleave intracellular 2′3′-cGAMP (into linear Gp[2’-5’]Ap[3’]) to restrain STING activation as an approach to evade innate-immune detection.191
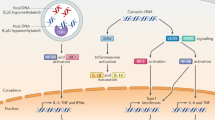
Roles of cGAS in human autoimmune and inflammation diseases
cGAS/STING innate-immune signaling plays a crucial role in many inflammation-related diseases, such as cardiovascular disease, neurodegenerative disease, inflammatory bowel disease, diabetes, fibrosis, lupus, arthritis, and psoriasis.192 (Table 2) For example, in Alzheimer’s disease (AD)-associated cardiac dysfunction, mutations of AD genes PS1 (presenilin 1), and PS2 suppressed mitophagy and contractile function through downregulating the cGAS/STING signaling.193
In ALS (amyotrophic lateral sclerosis), cytoplasmic accumulation of the nuclear DNA/RNA binding protein TDP-43 was reported to trigger mtDNA release through mPTP (mitochondrial permeability transition pore) that subsequently activates cGAS/STING signaling to induce neuroinflammation through activating NF-κB and promoting type I interferon (IFN) production.194 Strikingly, in mouse models, cGAS deletion greatly rescued autoimmune-disease phenotypes caused by Trex-1 loss such as AGS65 and SLE.66 These observations strongly support a critical role of cGAS signaling in contributing to autoimmune diseases and advocate for applying cGAS inhibitors in alleviating these disease symptoms in clinic. Hyperactivation of cGAS/STING signaling followed by an overproduction of harmful pro-inflammatory cytokines contributed to the pathogenesis of not only autoimmune diseases59,66,195 but also acute pancreatitis196 and insulin resistance.197 In addition, emerging evidence suggests that cGAS also promoted TH2 allergic inflammation likely via regulating airway epithelial GM-CSF production and might play an important role in immune responses of asthma pathophysiology.198 In mouse models, deletion of cGAS in airway epithelial cells reduced allergic airway inflammation.198 In another study, activation of cGAS signaling by mtDNA was observed to prohibit the YAP-mediated endothelial cell proliferation program to promote inflammatory injury.199
In addition, several studies have revealed that cGAS activation by self-DNA derived from chromatin instability (CIN), damaged mitochondria, micronuclei, or cell debris was connected to more familial and complex diseases, including myocardial infarction (MI) and age-related macular degeneration (AMD).57 MI has a high fatality rate in human, concomitant with increased inflammation and immune responses. This disease is caused by massively increased type I IFN production from heart macrophages caused by hyperactivation of cGAS–STING signaling, leading to massive death of cardiomyocytes. The importance of cGAS activation in this disease is also supported by the observation that knockout of cGAS, IRF3, and IFNAR1 in MI-related mouse models remarkably increased early survival.57,200 In mice receiving TAC (transverse aortic constriction) to induce heart failure or sham operation, downregulation of cGAS was found to be able to improve early survival, maintain left ventricular contractile function and attenuate cardiac hypertrophy or apoptosis induced by TAC, presumably through reducing inflammatory cytokine production.201 In addition, the ischemic myocardial injury was reported to activate cGAS signaling by inducing the release of nucleic acids that subsequently trigger inflammatory programs to promote macrophage transformation and regulate post-injury cardiac repair.202 In addition, cGAS signaling may also be indispensable for radiation-induced cardiovascular diseases given that radiation-induced DNA damage activates cGAS signaling and induces inflammation.203 Due to the fact that the heart is an organ relying on ATP consumption with abundant mitochondria, mitochondria dysfunction (leading to leakage of mtDNA and increased mtROS) triggers cGAS activation that contributes to cardiac inflammation.204 Thus, suppression of cGAS/STING signaling might serve as a therapeutic direction for treating these inflammation-related heart diseases through reducing inflammation signals.
Roles of cGAS in cancer
The cGAS signaling also plays important roles in regulating tumor immunity and tumor oncogenicity through modulating both tumor immune microenvironment and intrinsic tumorigenesis programs (such as cell senescence and DNA damage response). First, cGAS signaling is indispensable for the establishment of an anti-tumor immune environment given that type I IFNs stimulated by cGAS signaling bridge innate and adaptive immunity. Specifically, the tumorigenesis process is usually accompanied by DNA damage and leakage of damaged genomic or mitochondrial DNA in cancer cells or uptake of damaged cancer cells by dendritic cells (DCs).205 These increased levels of cytosolic DNA activate cGAS signaling to promote IFN production in cancer cells to prime tumor-specific T-cell infiltrations, and in DCs to lead to DC maturation.206 Subsequently, mature dendritic cells present tumor-associated antigens to activate CD8 + T cells in order to eradicate cancer cells through the immune system. In lung adenocarcinoma patients, decreased cGAS activity has been observed associated with poor patient survival by bioinformatics analyses;207 while in a recent study overexpression of cGAS has been observed to promote lung tumor growth through inhibiting homologous recombination.64 Consistent with a tumor-suppressive role of cGAS, expression of two hypoxia-responsive microRNAs miR-25 and miR-93 that suppressed cGAS activity through downregulating cGAS expression has been observed significantly increased in cohorts of glioma, colorectal carcinoma, and breast carcinoma patients accompanied by poor prognosis.70 Consistent with this notion, cGAMP administration synergized with anti-PD-L1 immune checkpoint blockade to suppress melanoma growth in a mouse model, through boosting innate immunity.68 Blocking extracellular cGAMP degradation by ENPP1 inhibitors also synergized with radiation to induce anti-tumor immunity.186 Recently, in tumors with dMMR (DNA mismatch repair deficiency) with increased neoantigen loads, cGAS signaling in sensing cytosolic DNA was shown to be indispensable for an effective response to anti-PD-1 therapy.203 Mechanistically, loss of the MutLα subunit MLH1 observed in half of dMMR cancer triggered the accumulation of cytosolic DNA due to loss of MLH1-dependent control of Exo1 (exonuclease 1) in DNA repair, leading to increased DNA excision by Exo1.204 In addition to modulating tumor immune environment, cGAS activation has been also observed to modulate intrinsic cellular programs. For example, cGAS was observed to be indispensable to antagonize cellular senescence, and cGAS loss facilitated the primary cell immortalization process.55
On the other hand, cGAMP transfer seems to promote brain metastasis. Specifically, the protein connexin 43 and proto-cadherin 7 induced cGAMP transfer from cancer cells migrated to and colonized in the brain to astrocytes via gap intersections. This promoted NF-κB signaling activation and production of inflammatory cytokines in astrocytes, which could function in a paracrine manner to fuel cancer cell growth to increase brain metastasis.208 In another study, chromosomal instability (CIN) that is commonly observed in cancer was observed to promote tumor metastasis to distal organs through generating cytosolic DNA, activating cGAS signaling, and subsequent noncanonical NF-κB activation.209 Notably, the extracellular cGAMP hydrolysis enzyme ENPP1 was found to be able to promote tumor metastasis by degrading cGAMP into adenosine to facilitate tumor immune evasion, and ENPP1 inhibition increased responses to immune checkpoint blockade.210 Thus, it seems cGAMP may exert distinct function in cancer and bystander cells in a cellular context-dependent manner. If and how cGAS signaling in cancer and host cells communicate to modulate both intrinsic cellular programs and tumor microenvironment to modulate cancer growth and metastasis warrant further in-depth investigations.
Concluding remarks and future perspectives
As a key component of PRRs (PAMP and DAMP) related host defense system through specifically sensing cytosolic DNA, since its discovery in 2013, cGAS has drawn extensive attention from the research community, not only due to its canonical roles in governing innate immunity211 and novel function in directly regulating cellular processes such as DNA damage64 but also its potential as a drug target in treating both autoimmune diseases and cancer. On one hand, cGAS signaling is indispensable to sense invasive cytosolic DNA to clear viral/bacterial infection. Cancer also hijacks a variety of mechanisms to inactivate cGAS signaling to evade immune surveillance. As a result, activating cGAS signaling, or administration of cGAMP benefits cancer treatment.68 On the other hand, hyperactivation of cGAS signaling leads to various human autoimmune diseases such as SLE and AGS, which can be treated by cGAS inhibition.65,66 Thus, both cGAS agonists and antagonists might be therapeutic directions for treating cancer and autoimmune diseases, respectively.
Analyses of the evolution route of cGAS suggest that DNA sensing and cGAMP synthesis are not only restricted in vertebrates. Recent studies identified cGAMP-synthesizing enzymes in non-vertebrate species such as bacteria212 and sea anemone species Nematostella vectensi.213 Bacterial cGAS-like enzymes could synthesize a variety of cyclic-dinucleotides that play important roles in anti-phage defense.214 cGAMP production was also observed in bacteria.179 In Gram-negative bacteria Virbrio cholerae 3’3’-cGAMP synthesized by DncV activated a phospholipase CapV (cGAMP-activated phospholipase in Vibrio) to cause degradation of the bacterial inner membrane.215 Thus, it seems cGAS/cGAMP/STING signaling is an evolutionarily conserved defense system with an ancient evolutionary root. Among vertebrates, the essential function of cGAS in sensing cytosolic DNA to produce type I IFN was more studied in birds and mammals,216 which might be due to the presence of common pathogens in these species that shapes up similar selection pressure for cGAS evolution.217 It remains unclear but interesting to explore if there are other cGAS-like enzymes in vertebrates beyond mammals and birds that play essential roles in sensing cytosolic DNA, and what is the function of ancestral cGAS homologs.
Given that both overphysiologically reduced and increased cGAS activation may cause human diseases, cGAS activation is tightly controlled spatially and temporally at multiple levels. This includes acute responses by post-translational modifications of cGAS and cGAS binding proteins and relatively long-term responses by cGAS transcriptional regulations. As a rapidly developing research field, there are still many interesting questions that remain to be answered. For example, if there are cross-talks among different cGAS PTMs, if certain PTMs could be recognized by cGAS binding partners and if cytosolic and nuclear cGAS are differentially modified and recognized by different binding partners. These multiple cGAS regulatory PTMs also raise a crucial question that how these cGAS PTMs acutely and accurately respond to infection in a coordinated manner at both spatial and temporal aspects. Furthermore, the tight control of cGAS activation is also achieved by restraining cGAS in certain cellular compartments (e.g., restraining cGAS on the plasma membrane through binding PI(4,5)P2218 or in the nucleus by association with nucleosomes/BAF), in a cell cycle-dependent manner by AKT and CDK1, and fine tuning cGAS activation by ions. Considering that dysregulation in suppressing cGAS sensing of nuclear DNA leads to Aicardi–Goutières syndrome (AGS)219 and Aujeszky’s disease,220 it is plausible that genetic alternations on cGAS or cGAS regulatory proteins contribute to pathological conditions. These complex and integrated signaling pathway analyses rely on more systematic approaches to tackle in the future.
Small molecules to inhibit cGAS activation have been successfully developed in treating autoimmune diseases, however, side-effects in suppressing the immune microenvironment that favors tumorigenesis would need to be taken into consideration. On the other hand, there is no cGAS activators have been identified that might be due to the vital role of DNA in cGAS activation. Nevertheless, administration of cGAMP, the major cGAS enzymatic product has shown effects in restoring immune-friendly microenvironment in facilitating anti-immune checkpoint blockage therapies68 and increasing concentrations of extracellular cGAMP levels through inhibiting cGAMP degrading enzyme ENPP1 has been reported to suppress tumor metastasis.210 Due to the cost for cGAMP synthesis, as well as the concern that cGAMP is actively degraded by extracellular protease ENPP1,187 small-molecule cGAS activators, together with ENPP1 inhibitors, would be a more cost-effective and robust approach as an adjuvant to convert “immune-cold” tumors into “immune-hot” tumors by inducing effective CD8 + T-cell infiltrates.
In addition to canonical cGAS function in sensing cytosolic DNA to initiate innate immunity, cGAS has also been observed to exert physiological function in regulating intrinsic cellular processes such as triggering senescence,55,56 modulating DNA damage responses through binding DNA damage repair factors57,64,221 and suppressing genomic instability through decelerating replication forks.222 In the past 8 years since the discovery of cGAS as a cytosolic DNA sensor, many researchers have dedicated efforts in elucidating cGAS regulations and functions through genetic, biochemical, and biophysical aspects that laid a solid foundation for future studies (Fig. 1). To this end, identification and characterization of the additional pathophysiological functions of cGAS would further advance our understanding of cGAS biology, and provide insights into new treatment modalities targeting cGAS signaling for human diseases.
References
Hu, M. M. & Shu, H. B. Cytoplasmic mechanisms of recognition and defense of microbial nucleic acids. Annu. Rev. Cell Dev. Biol. 34, 357–379 (2018).
Akira, S., Uematsu, S. & Takeuchi, O. Pathogen recognition and innate immunity. Cell 124, 783–801 (2006).
Atianand, M. K. & Fitzgerald, K. A. Molecular basis of DNA recognition in the immune system. J. Immunol. 190, 1911–1918 (2013).
Holm, C. K., Paludan, S. R. & Fitzgerald, K. A. DNA recognition in immunity and disease. Curr. Opin. Immunol. 25, 13–18 (2013).
Kagan, J. C. Signaling organelles of the innate immune system. Cell 151, 1168–1178 (2012).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Man, S. M. & Kanneganti, T. D. Regulation of inflammasome activation. Immunol. Rev. 265, 6–21 (2015).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Fernandes-Alnemri, T. et al. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
Ding, Y. et al. Antitumor activity of IFIX, a novel interferon-inducible HIN-200 gene, in breast cancer. Oncogene 23, 4556–4566 (2004).
Briggs, J. A., Burrus, G. R., Stickney, B. D. & Briggs, R. C. Cloning and expression of the human myeloid cell nuclear differentiation antigen: regulation by interferon alpha. J. Cell Biochem. 49, 82–92 (1992).
Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Yang, P. et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat. Immunol. 11, 487–494 (2010).
Chiu, Y. H., Macmillan, J. B. & Chen, Z. J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591 (2009).
Ablasser, A. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072 (2009).
Zhang, X. et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545 (2011).
Sui, H. et al. STING is an essential mediator of the Ku70-mediated production of IFN-lambda1 in response to exogenous DNA. Sci. Signal. 10, eaah5054 (2017).
Zhang, Z. et al. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965 (2011).
Kondo, T. et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl Acad. Sci. USA 110, 2969–2974 (2013).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Roberts, T. L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Rathinam, V. A. & Fitzgerald, K. A. Innate immune sensing of DNA viruses. Virology 411, 153–162 (2011).
Keating, S. E., Baran, M., Bowie, A. G. & Cytosolic, D. N. A. sensors regulating type I interferon induction. Trends Immunol. 32, 574–581 (2011).
Sun, L. et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013).
Andreeva, L. et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 (2017).
O’Neill, L. A. Immunology. Sensing the dark side of DNA. Science 339, 763–764 (2013).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031 (2013).
Zhang, X. et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014).
Gao, P. et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Xiao, T. S. & Fitzgerald, K. A. The cGAS-STING pathway for DNA sensing. Mol. Cell 51, 135–139 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Diner, E. J. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361 (2013).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Jin, L. et al. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol. 28, 5014–5026 (2008).
Zhong, B. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008).
Sun, W. et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl Acad. Sci. USA 106, 8653–8658 (2009).
Tanaka, Y. & Chen, Z. J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 5, ra20 (2012).
Du, M. & Chen, Z. J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018).
Zhou, W., Mohr, L., Maciejowski, J. & Kranzusch, P. J. cGAS phase separation inhibits TREX1-mediated DNA degradation and enhances cytosolic DNA sensing. Mol. Cell 81, 739–755 e737 (2021).
Kuchta, K. et al. Comprehensive classification of nucleotidyltransferase fold proteins: identification of novel families and their representatives in human. Nucleic Acids Res. 37, 7701–7714 (2009).
Kranzusch, P. J. & Vance, R. E. cGAS dimerization entangles DNA recognition. Immunity 39, 992–994 (2013).
Barnett, K. C. et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell 176, 1432–1446 e1411 (2019).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
Xie, W. et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl Acad. Sci. USA 116, 11946–11955 (2019).
Hartmann, R. et al. Crystal structure of the 2’-specific and double-stranded RNA-activated interferon-induced antiviral protein 2’-5’-oligoadenylate synthetase. Mol. Cell 12, 1173–1185 (2003).
Donovan, J., Dufner, M. & Korennykh, A. Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. Proc. Natl Acad. Sci. USA 110, 1652–1657 (2013).
Margolis, S. R., Wilson, S. C. & Vance, R. E. Evolutionary origins of cGAS-STING signaling. Trends Immunol. 38, 733–743 (2017).
Mackenzie, K. J. et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 (2017).
Schuliga, M. et al. Self DNA perpetuates IPF lung fibroblast senescence in a cGAS-dependent manner. Clin. Sci. 134, 889–905 (2020).
Yang, H. et al. cGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA 114, E4612–E4620 (2017).
Gluck, S. et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 (2017).
Li, T. & Chen, Z. J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018).
Wu, Y., Wei, Q. & Yu, J. The cGAS/STING pathway: a sensor of senescence-associated DNA damage and trigger of inflammation in early age-related macular degeneration. Clin. Interv. Aging 14, 1277–1283 (2019).
Wang, Y. et al. Cyclic GMP-AMP synthase is required for cell proliferation and inflammatory responses in rheumatoid arthritis synoviocytes. Mediators Inflamm. 2015, 192329 (2015).
Zierhut, C. et al. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178, 302–315 e323 (2019).
Ablasser, A. & Chen, Z. J. cGAS in action: expanding roles in immunity and inflammation. Science 363, eaat8657 (2019).
Liang, Q. et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 15, 228–238 (2014).
Liang, Q. et al. Autophagy side of MB21D1/cGAS DNA sensor. Autophagy 10, 1146–1147 (2014).
Liu, H. et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 (2018).
Gray, E. E., Treuting, P. M., Woodward, J. J. & Stetson, D. B. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J. Immunol. 195, 1939–1943 (2015).
Gao, D. et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl Acad. Sci. USA 112, E5699–E5705 (2015).
Xiao, N. et al. cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J. Autoimmun. 100, 84–94 (2019).
Wang, H. et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl Acad. Sci. USA 114, 1637–1642 (2017).
Chen, H. Y. et al. Transcriptional regulation of human cyclic GMP-AMP synthase gene. Cell. Signal. 62, 109355 (2019).
Wu, M. Z. et al. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat. Cell Biol. 19, 1286–1296 (2017).
Liao, Y. et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 10, 9644–9662 (2020).
Aguilar, C., Mano, M. & Eulalio, A. MicroRNAs at the host-bacteria interface: host defense or bacterial offense. Trends Microbiol. 27, 206–218 (2019).
Foster, P. S. et al. The emerging role of microRNAs in regulating immune and inflammatory responses in the lung. Immunological Rev. 253, 198–215 (2013).
Su, C. & Zheng, C. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J. Viol. 91, e02414-16 (2017).
Hu, H. & Sun, S. C. Ubiquitin signaling in immune responses. Cell Res. 26, 457–483 (2016).
Cai, J., Culley, M. K., Zhao, Y. & Zhao, J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell 9, 754–769 (2018).
Mevissen, T. E. T. & Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86, 159–192 (2017).
Zheng, N. & Shabek, N. Ubiquitin ligases: structure, function, and regulation. Annu Rev. Biochem 86, 129–157 (2017).
Komander, D., Clague, M. J. & Urbe, S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (2009).
Deng, L. et al. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target Ther. 5, 11 (2020).
Sun, S. C. Deubiquitylation and regulation of the immune response. Nat. Rev. Immunol. 8, 501–511 (2008).
Malynn, B. A. & Ma, A. Ubiquitin makes its mark on immune regulation. Immunity 33, 843–852 (2010).
Lim, K. H., Joo, J. Y. & Baek, K. H. The potential roles of deubiquitinating enzymes in brain diseases. Ageing Res. Rev. 61, 101088 (2020).
Harrigan, J. A., Jacq, X., Martin, N. M. & Jackson, S. P. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78 (2018).
Wu, L. et al. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol. 19, 1515–1524 (2012).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Yau, R. & Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 (2016).
Seo, G. J. et al. TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun. 9, 613 (2018).
Liu, Z. S. et al. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 8, 35 (2018).
Liu, P. et al. K63-linked polyubiquitin chains bind to DNA to facilitate DNA damage repair. Sci. Signal. 11, eaar8133 (2018).
Wang, Q. et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 13, e1006264 (2017).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Liu, J. et al. Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat. Immunol. 15, 612–622 (2014).
Zhou, Z. et al. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl Acad. Sci. USA 111, E245–E254 (2014).
Chen, M. et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. cell 64, 105–119 (2016).
Hoffpauir, C. T. et al. TRIM14 is a key regulator of the type I IFN response during Mycobacterium tuberculosis infection. J. Immunol. 205, 153–167 (2020).
Cui, J. et al. TRIM14 expression is regulated by IRF-1 and IRF-2. FEBS Open Bio 9, 1413–1420 (2019).
Isaacson, M. K. & Ploegh, H. L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5, 559–570 (2009).
Guo, Y. et al. Cutting edge: USP27X deubiquitinates and stabilizes the DNA sensor cGAS to regulate cytosolic DNA-mediated signaling. J. Immunol. 203, 2049–2054 (2019).
Zhang, Q. et al. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 30, 914–927 (2020).
Eifler, K. & Vertegaal, A. C. O. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem. Sci. 40, 779–793 (2015).
Su, S., Zhang, Y. & Liu, P. Roles of ubiquitination and SUMOylation in DNA damage response. Curr. Issues Mol. Biol. 35, 59–84 (2020).
Schwan, H. P. Early history of bioelectromagnetics. Bioelectromagnetics 13, 453–467 (1992).
Hannoun, Z., Maarifi, G. & Chelbi-Alix, M. K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 29, 3–16 (2016).
Hu, M. M. et al. TRIM38 negatively regulates TLR3/4-mediated innate immune and inflammatory responses by two sequential and distinct mechanisms. J. Immunol. 195, 4415–4425 (2015).
Fu, J. et al. MDA5 is SUMOylated by PIAS2beta in the upregulation of type I interferon signaling. Mol. Immunol. 48, 415–422 (2011).
Mi, Z., Fu, J., Xiong, Y. & Tang, H. SUMOylation of RIG-I positively regulates the type I interferon signaling. Protein Cell 1, 275–283 (2010).
Hu, M. M. et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569 (2016).
Cui, Y. et al. SENP7 potentiates cGAS activation by relieving SUMO-mediated inhibition of cytosolic DNA sensing. PLoS Pathog. 13, e1006156 (2017).
Zhou, Y., He, C., Wang, L. & Ge, B. Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 47, 1414–1426 (2017).
Janke, C. et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 308, 1758–1762 (2005).
Xia, P. et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016).
Esser, D. et al. Protein phosphorylation and its role in archaeal signal transduction. FEMS Microbiol. Rev. 40, 625–647 (2016).
Day, E. K., Sosale, N. G. & Lazzara, M. J. Cell signaling regulation by protein phosphorylation: a multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 40, 185–192 (2016).
Wu, P., Nielsen, T. E. & Clausen, M. H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today 21, 5–10 (2016).
Seo, G. J. et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 13, 440–449 (2015).
Yang, J. et al. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol. Cancer 18, 26 (2019).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Zhong, L. et al. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 6, 26 (2020).
Liu, P. et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545 (2014).
Li, M. & Shu, H. B. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell 11, 584–599 (2020).
Zhang, H. et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat. Genet. 44, 861–871 (2012).
Ruvolo, P. P. Role of protein phosphatases in the cancer microenvironment. Biochim Biophys. Acta Mol. Cell Res. 1866, 144–152 (2019).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. cell Biol. 20, 156–174 (2019).
Dai, J. et al. Acetylation blocks cGAS activity and inhibits Self-DNA-induced autoimmunity. Cell 176, 1447–1460 e1414 (2019).
Song, Z. M. et al. KAT5 acetylates cGAS to promote innate immune response to DNA virus. Proc. Natl Acad. Sci. USA 117, 21568–21575 (2020).
Song, B. et al. The DNA sensor cGAS is decorated by acetylation and phosphorylation modifications in the context of immune signaling. Mol. Cell Proteom. 19, 1193–1208 (2020).
Liu, Z. S. et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 20, 18–28 (2019).
Hu, S. et al. PKR-dependent cytosolic cGAS foci are necessary for intracellular DNA sensing. Sci. Signal. 12, eaav7934 (2019).
Liao, C. Y., Lei, C. Q. & Shu, H. B. PCBP1 modulates the innate immune response by facilitating the binding of cGAS to DNA. Cell. Mol. Immunol. (2020).
Yoh, S. M. et al. PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell 161, 1293–1305 (2015).
Lian, H. et al. ZCCHC3 is a co-sensor of cGAS for dsDNA recognition in innate immune response. Nat. Commun. 9, 3349 (2018).
Watkinson, R. E. et al. TRIM21 promotes cGAS and RIG-I sensing of viral genomes during infection by antibody-opsonized virus. PLoS Pathog. 11, e1005253 (2015).
Ghosh, A. et al. Oligoadenylate-synthetase-family protein OASL inhibits activity of the DNA sensor cGAS during DNA virus infection to limit interferon production. Immunity 50, 51–63 e55 (2019).
Maelfait, J. et al. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep. 16, 1492–1501 (2016).
Coquel, F. et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018).
Guey, B. et al. BAF restricts cGAS on nuclear DNA to prevent innate immune activation. Science 369, 823–828 (2020).
Volkman, H. E., Cambier, S., Gray, E. E. & Stetson, D. B. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife 8, e47491 (2019).
Cao, D. et al. Structural basis for nucleosome-mediated inhibition of cGAS activity. Cell Res. 30, 1088–1097 (2020).
Boyer, J. A. et al. Structural basis of nucleosome-dependent cGAS inhibition. Science 370, 450–454 (2020).
Kujirai, T. et al. Structural basis for the inhibition of cGAS by nucleosomes. Science 370, 455–458 (2020).
Pathare, G. R. et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature 587, 668–672 (2020).
Zhao, B. et al. The molecular basis of tight nuclear tethering and inactivation of cGAS. Nature 587, 673–677 (2020).
Michalski, S. et al. Structural basis for sequestration and autoinhibition of cGAS by chromatin. Nature 587, 678–682 (2020).
Zhang, G. et al. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl Acad. Sci. USA 113, E1034–E1043 (2016).
Li, W. et al. Kaposi’s sarcoma-associated herpesvirus inhibitor of cGAS (KicGAS), encoded by ORF52, is an abundant tegument protein and is required for production of infectious progeny viruses. J. Virol. 90, 5329–5342 (2016).
Huang, Z. F. et al. Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 24, 69–80 e64 (2018).
Fu, Y. Z. et al. Human cytomegalovirus protein UL42 antagonizes cGAS/MITA-mediated innate antiviral response. PLoS Pathog. 15, e1007691 (2019).
Biolatti, M. et al. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 92, e01774-17 (2018).
Zhang, J. et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24, 234–248 e235 (2018).
Huang, J. et al. Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J. Virol. 92, e00841-18 (2018).
Meade, N. et al. Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2 regulatory circuit. Cell 174, 1143–1157 e1117 (2018).
Lahaye, X. et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell 175, 488–501 e422 (2018).
Aguirre, S. et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2, 17037 (2017).
Webb, L. G. et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 16, e1008999 (2020).
Zheng, Y. et al. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 37, e99347 (2018).
Zhang, Y. et al. Streptavidin promotes DNA binding and activation of cGAS to enhance innate immunity. iScience 23, 101463 (2020).
Klein, T. et al. Proteolytic cleavage-mechanisms, function, and “Omic” approaches for a near-ubiquitous posttranslational modification. Chem. Rev. 118, 1137–1168 (2018).
Yu, Q. & Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 14, 163–176 (2000).
Wang, Y. et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017).
Ning, X. et al. Apoptotic caspases suppress type i interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31 e17 (2019).
White, M. J. et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014).
Wang, C. et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 48, 675–687 e677 (2018).
Zhao, Z. et al. Mn(2+) directly activates cGAS and structural analysis suggests Mn(2+) induces a noncanonical catalytic synthesis of 2'3’-cGAMP. Cell Rep. 32, 108053 (2020).
Hooy, R. M. et al. Allosteric coupling between Mn2+ and dsDNA controls the catalytic efficiency and fidelity of cGAS. Nucleic Acids Res. 48, 4435–4447 (2020).
Banerjee, I. et al. Gasdermin D restrains type i interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 49, 413–426 e415 (2018).
Molinaro, C., Martoriati, A., Pelinski, L. & Cailliau, K. Copper complexes as anticancer agents targeting topoisomerases I and II. Cancers 12, 2863 (2020).
Pan, Z. et al. Zinc transporters and dysregulated channels in cancers. Front Biosci. 22, 623–643 (2017).
Vincent, J. et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 8, 750 (2017).
Lama, L. et al. Development of human cGAS-specific small-molecule inhibitors for repression of dsDNA-triggered interferon expression. Nat. Commun. 10, 2261 (2019).
Hall, J. et al. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS ONE 12, e0184843 (2017).
Zhao, W. et al. In silico screening-based discovery of novel inhibitors of human cyclic GMP-AMP synthase: a cross-validation study of molecular docking and experimental testing. J. Chem. Inf. Model 60, 3265–3276 (2020).
Padilla-Salinas, R. et al. Discovery of small-molecule cyclic GMP-AMP synthase inhibitors. J. Org. Chem. 85, 1579–1600 (2020).
Huffman, B. J. et al. Electronic complementarity permits hindered butenolide heterodimerization and discovery of novel cGAS/STING pathway antagonists. Nat. Chem. 12, 310–317 (2020).
An, J. et al. Cutting edge: antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol. 194, 4089–4093 (2015).
An, J. et al. Inhibition of cyclic GMP-AMP synthase using a novel antimalarial drug derivative in Trex1-deficient mice. Arthritis Rheumatol. 70, 1807–1819 (2018).
Wang, M. et al. Suramin potently inhibits cGAMP synthase, cGAS, in THP1 cells to modulate IFN-beta levels. Future Med. Chem. 10, 1301–1317 (2018).
Danilchanka, O. & Mekalanos, J. J. Cyclic dinucleotides and the innate immune response. Cell 154, 962–970 (2013).
Davies, B. W., Bogard, R. W., Young, T. S. & Mekalanos, J. J. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370 (2012).
Hall, J. et al. The catalytic mechanism of cyclic GMP-AMP synthase (cGAS) and implications for innate immunity and inhibition. Protein Sci. 26, 2367–2380 (2017).
Ritchie, C. et al. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 75, 372–381 e375 (2019).
Luteijn, R. D. et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438 (2019).
Zhou, C. et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral. Immunity Immunity 52, 767–781 e766 (2020).
Lahey, L. J. et al. LRRC8A:C/E heteromeric channels are ubiquitous transporters of cGAMP. Mol. Cell 80(578-591), e575 (2020).
Yu, J. et al. Expression of the cGAMP transporter SLC19A1 is altered in systemic lupus erythematosus [abstract]. Arthritis Rheumatol. 72 (Suppl 10), (2020).
Jacqueline, A. et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Cancer 1, 184–196 (2020).
Li, L. et al. Hydrolysis of 2'3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol. 10, 1043–1048 (2014).
Ablasser, A. et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534 (2013).
Pepin, G. et al. Connexin-dependent transfer of cGAMP to phagocytes modulates antiviral responses. mBio. 11, e03187-19 (2020).
Chen, Q. et al. Corrigendum: carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 544, 124 (2017).
Eaglesham, J. B., Pan, Y., Kupper, T. S. & Kranzusch, P. J. Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 566, 259–263 (2019).
Chen, Q., Sun, L. & Chen, Z. J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149 (2016).
Wang, S. et al. ALDH2 contributes to melatonin-induced protection against APP/PS1 mutation-prompted cardiac anomalies through cGAS-STING-TBK1-mediated regulation of mitophagy. Signal Transduct. Target. Ther. 5, 119 (2020).
Yu, C. H. et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell 183, 636–649 (2020).
An, J. et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. 69, 800–807 (2017).
Zhao, Q. et al. STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology 154, 1822–1835 e1822 (2018).
Bai, J. et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl Acad. Sci. USA 114, 12196–12201 (2017).
Han, Y. et al. Airway epithelial cGAS is critical for induction of experimental allergic airway inflammation. J. Immunol. 204, 1437–1447 (2020).
Huang, L. S. et al. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52, 475–486 e475 (2020).
Kerur, N. et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat. Med. 24, 50–61 (2018).
Hu, D. et al. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 318, H1525–H1537 (2020).
Cao, D. J. et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137, 2613–2634 (2018).
Philipp, J. et al. Radiation response of human cardiac endothelial cells reveals a central role of the cGAS-STING pathway in the development of inflammation. Proteomes 8, 30 (2020).
Nakayama, H., Otsu, K. & Mitochondrial, D. N. A. as an inflammatory mediator in cardiovascular diseases. Biochem J. 475, 839–852 (2018).
Khoo, L. T. & Chen, L. Y. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 19, e46935 (2018).
Fuertes, M. B. et al. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 34, 67–73 (2013).
Ma, F. et al. LncRNA NEAT1 interacted with DNMT1 to regulate malignant phenotype of cancer cell and cytotoxic T cell infiltration via epigenetic inhibition of p53, cGAS, and STING in lung cancer. Front. Genet. 11, 250 (2020).
Chen, Q. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Li, J. et al. Metastasis and immune evasion from extracellular cGAMP hydrolysis. Cancer Discov. CD-20-0387 (2020).
Li, X. D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Whiteley, A. T. et al. Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199 (2019).
Kranzusch, P. J. et al. Ancient origin of cGAS-STING reveals mechanism of universal 2’,3’ cGAMP signaling. Mol. Cell 59, 891–903 (2015).
Cohen, D. et al. Cyclic GMP-AMP signalling protects bacteria against viral infection. Nature 574, 691–695 (2019).
Severin, G. B. et al. Direct activation of a phospholipase by cyclic GMP-AMP in El Tor Vibrio cholerae. Proc. Natl Acad. Sci. USA 115, E6048–E6055 (2018).
Vitak, N. et al. Induction of interferon and cell death in response to cytosolic DNA in chicken macrophages. Dev. Comp. Immunol. 59, 145–152 (2016).
Hancks, D. C. et al. Overlapping patterns of rapid evolution in the nucleic acid sensors cGAS and OAS1 suggest a common mechanism of pathogen antagonism and escape. PLoS Genet. 11, e1005203 (2015).
Langereis, M. A. et al. Knockout of cGAS and STING rescues virus infection of plasmid DNA-transfected cells. J. Virol. 89, 11169–11173 (2015).
Pokatayev, V. et al. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med. 213, 329–336 (2016).
Wang, J. et al. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 16, e1008429 (2020).
Jiang, H. et al. Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 38, e102718 (2019).
Chen, H. et al. cGAS suppresses genomic instability as a decelerator of replication forks. Sci. Adv. 6, eabb8941 (2020).
Visan, I. Mn potentiates cGAS. Nat. Immunol. 19, 511 (2018).
Su, J. et al. HIV-2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS-STING-mediated NF-kappaB signalling. Nat. Microbiol. 4, 2552–2564 (2019).
Bai, J. & Liu, F. The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes 68, 1099–1108 (2019).
Gratia, M. et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 216, 1199–1213 (2019).
Keskitalo, S. et al. Novel TMEM173 mutation and the role of disease modifying alleles. Front. Immunol. 10, 2770 (2019).
Coll-Bonfill, N., Cancado de Faria, R., Bhoopatiraju, S. & Gonzalo, S. Calcitriol prevents RAD51 loss and cGAS-STING-IFN response triggered by progerin. Proteomics 20, e1800406 (2019).
Sharma, M. et al. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl Acad. Sci. USA 117, 15989–15999 (2020).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Weindel, C. G. et al. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. eLife 9, e51071 (2020).
Acknowledgements
We sincerely apologize to all colleagues whose important work could not be cited in this review owing to space limitations, especially many prominent and pioneer work in the cGAS/cGAMP/STING field. This work was supported by an NIH grant (R01CA244825, P.L.), the Breast Cancer Alliance Young Investigator Grant (P.L.), the Gabrielle’s Angel Foundation Medical Research Award (P.L.), and the UNC University Cancer Research Fund (P.L.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, L., Liu, P. Cytosolic DNA sensing by cGAS: regulation, function, and human diseases. Sig Transduct Target Ther 6, 170 (2021). https://doi.org/10.1038/s41392-021-00554-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00554-y
This article is cited by
-
Reversible conjugation of a CBASS nucleotide cyclase regulates bacterial immune response to phage infection
Nature Microbiology (2024)
-
Downstream STING pathways IRF3 and NF-κB differentially regulate CCL22 in response to cytosolic dsDNA
Cancer Gene Therapy (2024)
-
Phage defence system CBASS is regulated by a prokaryotic E2 enzyme that imitates the ubiquitin pathway
Nature Microbiology (2024)
-
Replicative senescence and high glucose induce the accrual of self-derived cytosolic nucleic acids in human endothelial cells
Cell Death Discovery (2024)
-
Mitochondria, Autophagy and Inflammation: Interconnected in Aging
Cell Biochemistry and Biophysics (2024)
