
Abstract
Histologic chorioamnionitis is an inflammatory disorder of the placenta that commonly precedes preterm delivery. Preterm birth related to chorioamnionitis and fetal inflammation has been associated with a risk for serious inflammatory complications in infancy. In addition, preterm infants exposed to chorioamnionitis may be more susceptible to infection in the neonatal intensive care unit and possibly later in life. A significant body of work has established an association between chorioamnionitis and inflammatory processes. However, the potential consequences of this inflammation on postnatal immunity are less understood. In this review, we will discuss current knowledge regarding the effects of fetal exposure to inflammation on postnatal immune responses.
Similar content being viewed by others


Introduction
The diagnosis of suspected clinical chorioamnionitis is based on non-specific symptoms, such as maternal fever, leukocytosis, abdominal or uterine tenderness, or fetal tachycardia.1 However, the “gold standard” for confirmation of this diagnosis rests on placental evidence of acute histological chorioamnionitis (HCA), represented by the infiltration of inflammatory neutrophils in maternal or fetal placental tissues.2 A more updated but still controversial definition of chorioamnionitis, also referred to as intrauterine inflammation, infection, or both (“Triple I”), incorporates both clinical and histologic criteria.3 While clinical chorioamnionitis is commonly accompanied by HCA,4 the reverse situation may not be true. In fact, most cases of HCA occur without clinical symptoms in the mother or fetus and thus present “silently.”5,6 Despite the lack of clinical expression, however, asymptomatic placental inflammation is not innocuous even in the absence of infection.7 A diagnosis of HCA often precedes the delivery of extremely preterm infants5 and, like clinical chorioamnionitis, is associated with early-onset infection.8 Conversely, HCA was correlated with a decreased risk of late-onset neonatal infection with coagulase-negative staphylococci.9 HCA has also been closely linked to the pathogenesis of serious postnatal inflammatory disorders, including bronchopulmonary dysplasia, brain injury, retinopathy of prematurity, and necrotizing enterocolitis.10,11,12,13 Preterm infants born to mothers with clinically suspected chorioamnionitis are identified as being at higher risk for infection and are typically screened.14 In contrast, in the absence of maternal symptoms, the possibility that a preterm infant has been exposed to HCA and a consideration of its inherent inflammatory and infectious risks may not be addressed in a timely fashion or even at all. This is particularly true given that a diagnosis of HCA rests on microscopic examination of the delivered placenta, and resulting information may not be available for days to weeks after birth.
A variety of approaches to identify gestations affected by HCA have been studied. The expression patterns of biological markers in amniotic fluid and cord blood, such as interleukin-6 and C-reactive protein, have been assessed for their predictive value in HCA; however, sensitivity and specificity of these markers have not been consistent.15,16,17 Clinical prediction rules for HCA and funisitis have also been developed in order to identify newborns exposed to antenatal inflammation.18 The targeted clinical variables included the absence of pre-eclampsia, normal intrauterine growth, maternal or fetal evidence of clinical chorioamnionitis, prolonged premature rupture of membranes (PPROM), and vaginal delivery. Although these methods have shown clinical promise, to date none have been uniformly successful in identifying gestations with HCA.
The inflammatory complications associated with HCA have been well described.13,19,20,21,22,23,24 Less appreciated is that affected preterm infants also may be at risk for immune consequences in addition to or in combination with the adverse effects of HCA-mediated inflammation.25,26 Increasing evidence supports the concept that the ensuing neonatal immune dysfunction reflects the effects of inflammation on immune programming during critical developmental “windows.”26 The goals of the present review article are to summarize the following: (1) The effects of inflammation during pregnancy on the reconfiguration of neonatal inflammatory and immune responses; and (2) The implications of intrauterine inflammatory exposure for immunity in the neonatal period and beyond. Understanding how in utero inflammation programs the postnatal immune response may reveal novel approaches to reduce inflammatory injury and the risk for infection in preterm infants.
Effects of antenatal inflammation on neonatal immunity
Inflammation and immunity
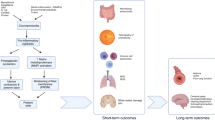
Inflammatory exposure during intrauterine life is a pathologic force that can drive alterations of postnatal innate and adaptive immunity (Fig. 1). A growing body of data implicates myriad environmental exposures during pregnancy, many associated with inflammation, on subsequent immunity (reviewed in ref. 25). Recent evidence of a stereotypic developmental pattern of converging immune responses in preterm and term infants in the first 3 months of life, but divergent responses in infants with inflammatory exposure, additionally credits the role of intrauterine exposure at critical developmental windows in shaping immunity.26

Potential effects of HCA on neonatal inflammatory and immune responses. Studies in humans and in animal models have linked HCA, a neutrophil (PMN)-driven placental disorder associated with increased Th17 responses, and exaggerated inflammatory responses of both innate and adaptive immune cells. Neutrophil (PMN) production and activation may be increased, along with the release of inflammatory cytokines and chemokines that promote PMN infiltration and injury to major organs. Experimental fetal inflammation can induce functional maturation and activation of monocytes (Mono) and macrophages (Macs) that can also heighten inflammatory responses. Fetal inflammation enhances the generation of inflammatory Th17 cells and IL-17+ Treg cells; while IL-17 is important to host protection, high levels can induce organ injury, particularly in the brain. Exaggerated inflammatory responses may lead to suppression of protective immune responses, which increase risk for infection. Neonatal infection in the context of HCA exposure has also been shown to increase risk for organ injury and has been linked to bronchopulmonary dysplasia
Studies in humans and in animal models have begun to define how inflammatory exposure can shape immune function in fetuses and in the newborn period; these are summarized in several recent reviews.27,28 Acute HCA associated with fetal inflammation is a risk factor for numerous adverse neonatal outcomes.13,19,20,21,22,23,24 Inflammatory injury related to HCA has been observed in both extremely preterm infants as well as in late preterm infants delivered after PPROM.29 However, HCA at the earliest gestations may more heavily influence neonatal immune responses, such as increased T helper type 17 (Th17) frequencies.30 This enhanced effect in very preterm newborns likely reflects the age-dependent “waves” of immune cell populations with inflammatory or regulatory function that are generated in the developing fetus.31
Immune priming and HCA
In utero “priming” or activation of the fetal immune system at critical developmental time points can lead to chronic inflammatory disorders as well as increased vulnerability to infection after birth.25 Maternal infections with chronic inflammation, such as human immunodeficiency virus (HIV) or malaria, during pregnancy were associated with fetal inflammation and alterations in infant B cell responses.32 Infants born to mothers with allergic disease had lower frequencies of T regulatory (Treg) cells, which in turn were impaired in their capacity to suppress effector T cells, particularly Th2 cells.33 This latter finding may be of particular relevance to exposed infants and future risk for asthma given its close association with Th2 polarization.34 Furthermore, even the low-grade systemic inflammation associated with maternal obesity was shown to induce placental and fetal inflammation.35
Emerging evidence also points to a critical role of activated fetal cells in driving intrauterine responses during chorioamnionitis. Gomez-Lopez et al. utilized DNA fingerprinting to show that predominance of inflammatory fetal neutrophils in the amniotic fluid of gestations with chorioamnionitis was highly associated with the delivery of extremely preterm neonates.36 Increased neonatal T cell activation has also been associated with preterm delivery.37 Frascoli et al. observed that activation of the fetal adaptive immune system suppressed maternal–fetal tolerance in the context of preterm labor.38 In that study, fetal blood showed early maturation of dendritic cells and enhanced maternal microchimerism in preterm relative to term gestations. In addition, preterm (but not term) fetal T cells were alloreactive to maternal antigens, and maternal antigen-specific stimulation induced the proliferation of fetal Th1-type cells. Furthermore, the cytokines (interferon-γ (IFNγ) or tumor necrosis factor-α) released by proliferating T cells directly increased myometrial contractility in an in vitro assay, suggesting a directive role of activated fetal T cells in preterm labor. Although naive T cells typically predominate in fetuses, high frequencies of memory (CD4+CD45+RO+RA−) T cells have been observed in association with preterm labor.37 This finding may be important given differing gene expression patterns and function in naive vs. memory T cells.39
The inflammatory processes induced by HCA also contribute to fetal immune activation. In a recent transcriptomic study, preterm infants exposed to HCA exhibited gene expression signatures indicative of immune priming.40 The most frequently upregulated genes in these neonates were associated with activation of innate and adaptive immune pathways. Notably, the microRNA, MiR-155, was shown to be a top upstream regulator. MiR-155 is a master modulator of inflammatory and immune responses, and its elevated expression in immune cells has been associated with chronic inflammatory states, including atopic dermatitis, multiple sclerosis, and rheumatoid arthritis (reviewed in ref. 41). Pertinently, iR-155, which is also expressed in activated CD4+ T cells, can promote pathogenic Th17-biased responses.42
Evidence of immune priming was also observed in a murine model of lipopolysaccharide (LPS)-induced antenatal inflammation followed by a postnatal “second hit” immune challenge.43 In pups exposed to antenatal inflammation, infection with Sendai virus (the murine counterpart to the respiratory syncytial virus (RSV) that causes bronchiolitis in human infants) triggered strong inflammatory responses not only in the lungs, the primary site of infection, but also in distal organs, such as the liver. This exaggerated correlation was not witnessed in infected control pups without antenatal LPS exposure. In addition, an inductive effect of maternal inflammation on lung CD4 T helper cell populations with a pro-inflammatory Th1 and Th17 phenotypes was most pronounced in exposed weanling pups relative to neonates. These findings suggested that the processes initiated in utero not only persisted but also were possibly amplified beyond the neonatal period. Interestingly, a similar enhancement of lung Th17 cells was observed following secondary RSV challenge in adult mice that had survived severe sepsis.44
Inflammatory innate immune responses in HCA
A number of studies have shown that experimentally induced antenatal inflammation leads to exaggerated inflammatory immune responses in exposed offspring. In ex vivo studies of preterm fetal sheep, experimental chorioamnionitis promoted the functional maturation of lung monocytes and hastened their capacity to produce inflammatory cytokines in response to stimulation.45 Preterm piglets born after several doses of intra-amniotic LPS had increased systemic and organ-specific (gut and lung) inflammatory responses at birth.46 In a murine model of antenatal inflammation, neonatal and weanling offspring of LPS-treated dams showed increased basal innate immune responses in the lungs and livers that were amplified following a “second hit” viral infection.43
The fetal inflammatory responses induced by HCA have also been shown to persist in newborn infants as systemic inflammation, much of it driven by neutrophils. As part of the ELGAN study, Chen et al. showed that the elevated levels of key neutrophil-associated inflammatory proteins (including myeloperoxidase, interleukin (IL)-1β, IL-8, intercellular adhesion molecule-3 and matrix metalloproteinase-9) in the cord blood of preterm infants born with funisitis (inflammation of cord blood vessels consistent with the fetal inflammatory response syndrome (FIRS)) remained high on postnatal day 7.24 Autopsies of human fetuses and newborn infants who died after severe chorioamnionitis also showed amplified neutrophil production (myelopoiesis) in hematopoietic organs.47,48 These observations are consistent with the excessive neutrophil responses associated with this perinatal condition36,49,50,51 as well as the neutrophil-driven inflammatory responses in neonatal lungs and other organs.52,53 Similar observations of neutrophil-driven inflammation have been observed in animal models. Antenatal inflammation was shown to promote neutrophil recruitment and the infiltration of organs, such as the lungs and brain.43,54,55 High expression levels of inflammatory cytokines, including IL-1β, IL-6, IL-8, and IL-17, in the blood, thymi, lungs, and/or intestinal tracts of fetal sheep, macaques, and piglets animals following experimental chorioamnionitis have been reported.46,56,57,58 In addition, altered DNA methylation profiles have been observed in placentas with HCA, reflecting activation of innate immunity and neutrophil increases.59
Antenatal fetal exposure has also been shown to induce inflammatory responses in the liver. In sheep studies of liver homeostasis and metabolism after LPS-induced chorioamnionitis, Vlassaks et al. found increased hepatic T lymphocytes and apoptotic hepatocytes in term newborns and increased liver triglycerides and cholesterol levels at 7 weeks of life, indicating long-lasting postnatal effects on lipid metabolism.60 Endotoxin-induced chorioamnionitis also caused hepatic damage associated with disturbed lipid and glucose metabolism, reduced antioxidant capacity, and elevated liver enzymes.61 The adverse hepatic effects of fetal inflammation may have specific relevance to neonatal immunity, given the increasingly appreciated role of the liver in directing immune function.62
Inflammatory adaptive immune responses after HCA
T helper cell subsets belong to the adaptive arm of the immune system and can promote or suppress inflammatory responses. In addition to the effects of fetal inflammation on innate immunity, recent studies have identified the robust involvement of pro-inflammatory T helper cell lymphocyte subsets, such as Th17 cells, in fetuses or preterm infants with antenatal inflammation. Th17 cells characteristically function to protect the host against extracellular pathogens.63,64,65 However, under certain inflammatory conditions, Th17 cells may become pathogenic and promote tissue injury.66 Th17 cells release the canonical cytokine, IL-17, which is also produced by other immune cells such as γδ T cells and pro-inflammatory Treg cells.67 IL-17 plays a critical role in processes involved in FIRS associated with HCA.58 The developing brain is particularly sensitive to inflammatory injury, and exposure to IL-17 at critical “windows” of immune development can induce microglial activation and white matter injury (reviewed in ref. 68). Furthermore, in addition to directly inducing tissue injury, Th17 cells can amplify inflammatory responses through cross-talk with neutrophils.69
While Th17 cells play an important biological role in normal pregnancy,70 increased frequencies of pathogenic Th17 cells have been observed in placentas of women with recurrent miscarriages71 and in gestations affected by chorioamnionitis.72 Higher circulating Th17 frequencies in mothers or in the cord blood of babies of preterm gestations with HCA have also been reported.30,73 The exact mechanism(s) that promote Th17 responses in the context of HCA remain enigmatic. However, the expression levels of several cytokines that are critical to the propagation of Th17 cells from naive CD4 cells, including IL-1β and IL-6,74 are also increased in the amniotic fluid in HCA.75,76 The finding that inflammatory neutrophils promote in vitro propagation of Th17 cells77 suggests their contribution to an intrauterine cytokine milieu that also modulates Th17 responses in HCA, as observed in the context of chronic inflammatory conditions, such as rheumatoid arthritis.78
In a recent human study, cord blood from preterm and term infants with HCA had increased frequencies of Th17 cells relative to unaffected controls.30 Th17 cells were highest in the cord blood of extremely preterm infants, who also exhibited increased T cells with an effector memory phenotype associated with Th17-type responses.79 In addition, the elevated circulating Th17 frequencies observed at birth in preterm neonates exposed to chorioamnionitis persisted in the first month of life.73 Increased Th17-type responses have been observed in the cord blood of human infants following both acute and chronic HCA72 and in animal models in the context of antenatal inflammation. Fetal macaques exposed to LPS-induced chorioamnionitis had increased splenic IL-17+ and IL-22+ Th17 cells,58 while weanling murine pups exposed to LPS exhibited increased lung Th17 responses.43
Treg cells constitute a T helper cell subset that typically functions to suppress activated cells and inflammatory responses, including those mediated by Th17 cells.80,81 Chorioamnionitis has been variably associated with decreased Treg cell frequencies or reduced Treg-suppressor function.82 Fetal rhesus monkeys and sheep exposed to experimental chorioamnionitis had an increased ratio of IL-17-producing cells to Treg cells in lymphoid organs.83 Exposure was also associated with decreased frequencies of circulating Treg cells in extremely preterm human neonates and in fetal macaques.30,58 However, the majority of Treg cells in these two studies also co-expressed the canonical Th17 transcription factor, RORγt, and/or IL-17, consistent with a pro-inflammatory rather than a regulatory phenotype.84,85 Pertinently, IL-17+ Treg cells can serve as a major source of IL-17 during inflammation.84
The enhanced Th17-type responses observed in conjunction with antenatal inflammation have been linked to inflammatory injury in the lungs or brain. Elevated frequencies of IL-17-producing cells in fetal rhesus monkeys with chorioamnionitis were associated with lung inflammation in neonates.83 When LPS-induced antenatal inflammation was combined with neonatal hypoxic–ischemic brain injury in a rat pup model, Th17-like lymphocytes migrated to the brain to direct neuroinflammatory responses.86 Th17 cells appear to be the major cell group mediating this inflammatory IL-17 effect; while γδ t cells also produce IL-17,87 experimental HCA did not measurably alter this lymphocyte population in exposed lambs.88
Other lymphocyte subsets may have the capacity to contribute to neonatal inflammatory responses in HCA that are not mediated by IL-17. A higher proportion of Th1 cells were determined in the umbilical cord blood of human neonates with clinical evidence of perinatal infection.89 A recently described subset of lymphocytes unique to cord blood produces IL-8/C-X-C chemokine motif ligand 8 and can activate neutrophils and γδ t cells,90 although whether and how HCA influences these lymphocytes is not clear.
Immune suppression and HCA
In contrast to the hyper-inflammatory responses associated with HCA exposure, protective immune responses may be suppressed. Immune-suppressive mechanisms in chorioamnionitis may be selectively quantitative. Human fetuses and neonates exposed to chorioamnionitis have been shown to exhibit both thymic involution and depletion of splenic T cells.91,92 Studies in fetal sheep affected by chorioamnionitis found reductions in CD8+ but not in CD4+ T cells in thymic cell populations.93
Intrauterine inflammatory exposure may also lead to qualitative alterations in neonatal innate or adaptive immune function. A relationship between an inflammatory antenatal environment and immune suppression is suggested by the enhanced HIV positivity observed in human infants born to HIV-affected mothers in the context of chorioamnionitis,94 possibly due to activated fetal lymphocytes.95 A recent study also showed suppressed transcriptional responses to Staphyloccoccus epidermidis in ex vivo monocytes from preterm human neonates with chorioamnionitis.96 Studies in animal models are supportive of this premise: Repetitive intrauterine LPS exposure in sheep induced “immune paralysis” of ex vivo fetal and neonatal monocytes following stimulation with LPS or other Toll-like receptor ligands.45,97 Similarly, chronic, but not acute, intra-amniotic infection with Ureaplasma parvum resulted in suppressed “second hit” LPS-induced cytokine responses in the fetal lung.98,99 This evidence further supports the idea that prenatal exposure to HCA-mediated inflammation, particularly if long-standing, can alter postnatal immune response patterns. Pertinently, septic human neonates have been observed to exhibit early hyper-inflammatory responses followed by suppressed immune responses,100 a pattern reminiscent of that observed in infants exposed to HCA. Similarly, Azizia et al. found a correlation between prematurity, neonatal sepsis, and reduced monocyte major histocompatibility complex class II expression associated with immune paralysis in HCA-exposed gestations, with an increased risk for sepsis and organ dysfunction.101
Potential mechanisms of immune suppression in HCA
The immune system in preterm infants is developmentally restricted in its capacity to protect the host against infection.102 The added burden of intrauterine inflammatory exposure during sensitive developmental “windows” to already impaired immune function also remain incompletely understood but may involve developmentally regulated epigenetic processes.103 In studies of short-term antenatal LPS exposure in preterm sheep, the role of timing rather than the specific inflammatory trigger was found to have a greater impact on abnormal neurological findings in the fetal brain.104 However, the mechanisms involved in the inflammation-induced immune suppression of infants exposed to HCA, like the immune dysfunction associated with neonatal sepsis,100 remain incompletely understood.105
A variety of quantitative and qualitative alterations of immune function that are biologically prevalent in preterm infants can contribute to processes that suppress immunity31,106 (Fig. 2). The characteristic limitations of neutrophil production and storage that are typical in preterm infants can lead to rapid depletion and severe neutropenia during periods of increased utilization, such as sepsis.107 In addition, neonatal neutrophils and monocytes exhibit intrinsic dysfunction, including hyporesponsiveness to stimulation and impaired antimicrobial capacity108,109 that may be additionally affected by inflammation-induced immune paralysis. HCA can induce excessive fetal neutrophil production (granulopoiesis), suggesting a fetal capacity to overcome or circumvent developmental restrictions under inflammatory conditions.110 However, the functionality of these newly minted neutrophils may also be impaired. Inflammation can lead to hypofunctional T cells through a process that downregulates the T cell receptor zeta-chain,111 although whether this functions as a suppressive mechanism in the context of HCA is unknown. Conversely, while neonatal immune cells are also at a developmental disadvantage in terms of generating protective cytokines, such as IFNγ, neonatal Th1 cell frequencies may be increased following HCA exposure.112

Potential mechanisms of suppressed protective immunity in neonates exposed to fetal inflammation associated with HCA. Experimental HCA has been associated with “immune paralysis” as suggested by decreased LPS responsiveness in fetal sheep monocytes. HCA has been variably associated with quantitative and qualitative defects in T cells. Conversely, increases in Th17 and inflammatory Treg cells promote IL-17 release. While IL-17 provides immune-protective function, it can also promote the generation of myeloid-derived suppressor cells (MDSCs), which adversely affect protective immunity. The increased expression of S100 proteins, particularly S100A8 and S100A9, may promote host protection; however, high levels can increase MDSC generation. Recent evidence also indicates an immunosuppressive role of CD71+ erythroid cells, which could potentially be increased with HCA
Cells with regulatory function may serve to further suppress immune function in newborns exposed to HCA (Fig. 2). Granulocytic myeloid-derived suppressor cells (Gr-MDSCs), an immature neutrophil subset with high frequencies in neonates, suppress T cell function.113,114 This action may occur through the reduction of L-arginine levels,115 which are biologically low in preterm infants.116 Pertinently, increased expression of arginase 1 and subsequent depletion of L-arginine were observed in exposed offspring in a rat model of LPS-induced chorioamnionitis.117 MDSCs are also important negative regulators of inflammatory responses.118,119 Elevated circulating frequencies of Gr-MDSCs have been reported in extremely preterm infants in association with clinical inflammation, though not specifically HCA.120 Importantly, these MDSCs persisted for several months beyond the immediate neonatal period, suggesting an immunosuppressive role in later infancy. Neonatal inflammatory neutrophils and monocytes also release the alarmins, S100A8 and S100A9, which may suppress hyper-inflammatory responses through the expansion of MDSCs.121,122 Pertinently, increased S100 protein expression levels in amniotic fluid have been observed in gestations with HCA.123
Lymphocytes with intrinsic suppressive function can also inhibit immune responses in preterm infants. Treg cells are critical to the suppression of T cell responses to self and maternal antigens that is necessary for maternal–fetal tolerance.124,125 Although Treg cells in preterm infants with HCA may exhibit a pro-inflammatory (Th17-like) phenotype,30 conversely their release of IL-17 could attract MDSCs to mediate immune suppression.126 Regulatory B cells, another type of immune cell, can modulate neonatal inflammatory responses127 and promote Th2 skewing in neonatal mice through suppressive actions on dendritic cells.128
Recent evidence also points to a role of a unique subset of CD71+ erythroid cells in modulating myeloid and T cell responses.129 Pertinently, these regulatory erythroid cells are found in high numbers in preterm but not in term neonates. CD71+ cells were shown to suppress protective immune responses to pertussis infection in neonatal mice, in part through actions mediated by arginase and the expression of programmed death ligand-1.130
Steroid-associated effects on immune responses in HCA
While current treatment guidelines for chorioamnionitis are institutionally varied, antenatal steroids (such as betamethasone) are commonly administered for preterm labor. A recent meta-analysis showed that steroid administration in the setting of HCA was associated with reduced mortality and incidence of respiratory distress, patent ductus arteriosus, intraventricular hemorrhage (IVH), and severe IVH; in the setting of clinical chorioamnionitis, steroid administration reduced severe IVH and periventricular leukomalacia.13 Although several studies suggest that antenatal steroids can dampen the inflammatory cascade, their effects on fetal inflammation are not well defined. Evidence of anti-inflammatory effects of steroid administration includes the inhibition of intrauterine transforming growth factor-β signaling associated with fetal lung inflammation and the partial prevention of the structural lung changes induced by LPS exposure.131,132
The antenatal timing of steroid administration may also influence inflammatory responses. Kuypers et al. showed that, while steroid administration prior to intrauterine LPS exposure reduced the adverse effects of inflammation on the brain in fetal sheep, conversely steroids aggravated inflammatory changes in the brain and thymus in the context of pre-existing inflammation.56,133 These observations suggest that, in the presence of chorioamnionitis, steroids could potentially amplify fetal injury in an organ-specific manner. In studies of fetal sheep exposed to intra-amniotic endotoxin and subsequently treated with steroids, inflammatory responses in ex vivo monocytes were initially suppressed but were followed by a later activation, possibly the result of steroid-induced functional maturation.134
Summary
HCA is a common disorder that is tightly linked to preterm delivery and dysregulated immune function. Inroads are being made toward better defining the immune effects of antenatal inflammatory exposure on the fetus and newborn, which includes a pattern of hyper-inflammation combined with immune suppression. However, much remains to be learned regarding the underlying mechanisms so that potential therapeutic targets can be identified.
Perinatal inflammation has clear implications for human health. Mounting evidence points to a negative impact of early inflammatory exposure of any origin on the developing immune program.135,136 Chorioamnionitis has been identified as a contributing factor in childhood asthma,137,138 possibly through a mechanism involving Th2 skewing.139 However, much remains to be learned in this regard. Numerous factors aside from microbial exposure have been shown to induce systemic maternal inflammation and/or chorioamnionitis, including nutritional and psychosocial factors (reviewed in ref. 135). Of great concern are the observations linking perinatal inflammation from various causes with immune dysfunction and abnormal stress responses, not only in the immediate postnatal period but also possibly throughout life or even into the next generation.140 Thus the importance of advancing knowledge of perinatal inflammation and its causes cannot be overstated.
References
Tita, A. T. & Andrews, W. W. Diagnosis and management of clinical chorioamnionitis. Clin. Perinatol. 37, 339–354 (2010).
Redline, R. W. Inflammatory response in acute chorioamnionitis. Semin. Fetal Neonatal Med. 17, 20–25 (2012).
Peng, C. C., Chang, J. H., Lin, H. Y., Cheng, P. J. & Su, B. H. Intrauterine inflammation, infection, or both (Triple I): a new concept for chorioamnionitis. Pediatr. Neonatol. 59, 231–237 (2018).
Suzuki, S. Association between clinical chorioamnionitis and histological funisitis at term. J. Neonatal Perinat. Med. 12, 37–40 (2019).
Lahra, M. M. & Jeffery, H. E. A fetal response to chorioamnionitis is associated with early survival after preterm birth. Am. J. Obstet. Gynecol. 190, 147–151 (2004).
Horvath, B., Lakatos, F., Toth, C., Bodecs, T. & Bodis, J. Silent chorioamnionitis and associated pregnancy outcomes: a review of clinical data gathered over a 16-year period. J. Perinat. Med. 42, 441–447 (2014).
Park, J. W., Park, K. H. & Jung, E. Y. Clinical significance of histologic chorioamnionitis with a negative amniotic fluid culture in patients with preterm labor and premature membrane rupture. PLoS ONE 12, e0173312 (2017).
Hillier, S. L. et al. A case-control study of chorioamnionic infection and histologic chorioamnionitis in prematurity. N. Engl. J. Med. 319, 972–978 (1988).
Strunk, T. et al. Histologic chorioamnionitis is associated with reduced risk of late-onset sepsis in preterm infants. Pediatrics 129, e134–e141 (2012).
Rocha, G. Chorioamnionitis and lung injury in preterm newborns. Crit. Care Res Pract. 2013, 890987 (2013).
Anblagan, D. et al. Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 6, 37932 (2016).
Villamor-Martinez, E. et al. Chorioamnionitis as a risk factor for retinopathy of prematurity: an updated systematic review and meta-analysis. PLoS ONE 13, e0205838 (2018).
Been, J. V., Lievense, S., Zimmermann, L. J., Kramer, B. W. & Wolfs, T. G. Chorioamnionitis as a risk factor for necrotizing enterocolitis: a systematic review and meta-analysis. J. Pediatr. 162, 236–242 (2013).
Sung, J. H., Choi, S. J., Oh, S. Y., Roh, C. R. & Kim, J. H. Revisiting the diagnostic criteria of clinical chorioamnionitis in preterm birth. BJOG 124, 775–783 (2017).
Tasci, Y. et al. The value of cord blood interleukin-6 levels for predicting chorioamnionitis, funisitis and neonatal infection in term premature rupture of membranes. Eur. J. Obstet. Gynecol. Reprod. Biol. 128, 34–39 (2006).
Yoon, B. H. et al. C-reactive protein in umbilical cord blood: a simple and widely available clinical method to assess the risk of amniotic fluid infection and funisitis. J. Matern. Fetal Neonatal Med. 14, 85–90 (2003).
Yoon, B. H. et al. The relationship among inflammatory lesions of the umbilical cord (funisitis), umbilical cord plasma interleukin 6 concentration, amniotic fluid infection, and neonatal sepsis. Am. J. Obstet. Gynecol. 183, 1124–1129 (2000).
Been, J. V. et al. A clinical prediction rule for histological chorioamnionitis in preterm newborns. PLoS ONE 7, e46217 (2012).
McElrath, T. F. et al. Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am. J. Epidemiol. 168, 980–989 (2008).
Gomez, R. et al. The fetal inflammatory response syndrome. Am. J. Obstet. Gynecol. 179, 194–202 (1998).
Dessardo, N. S. et al. Chorioamnionitis and chronic lung disease of prematurity: a path analysis of causality. Am. J. Perinatol. 29, 133–140 (2012).
Malaeb, S. & Dammann, O. Fetal inflammatory response and brain injury in the preterm newborn. J. Child Neurol. 24, 1119–1126 (2009).
Yoon, B. H. et al. Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am. J. Obstet. Gynecol. 177, 19–26 (1997).
Chen, M. L. et al. Placenta microbiology and histology and the risk for severe retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 52, 7052–7058 (2011).
Rychlik, K. A. & Sille, F. C. M. Environmental exposures during pregnancy: mechanistic effects on immunity. Birth Defects Res. 111, 178–196 (2019).
Olin, A. et al. Stereotypic immune system development in newborn children. Cell 174, 1277–1292 (2018).
Kallapur, S. G., Presicce, P., Rueda, C. M., Jobe, A. H. & Chougnet, C. A. Fetal immune response to chorioamnionitis. Semin. Reprod. Med. 32, 56–67 (2014).
Van Well, G. T. J., Daalderop, L. A., Wolfs, T. & Kramer, B. W. Human perinatal immunity in physiological conditions and during infection. Mol. Cell. Pediatr. 4, 4–0070 (2017).
Kim, S. A., Park, K. H. & Lee, S. M. Non-invasive prediction of histologic chorioamnionitis in women with preterm premature rupture of membranes. Yonsei Med. J. 57, 461–468 (2016).
Rito, D. C., Viehl, L. T., Buchanan, P. M., Haridas, S. & Koenig, J. M. Augmented Th17-type immune responses in preterm neonates exposed to histologic chorioamnionitis. Pediatr. Res. 81, 639–645 (2017).
Zhang, X., Zhivaki, D. & Lo-Man, R. Unique aspects of the perinatal immune system. Nat. Rev. Immunol. 17, 495–507 (2017).
Yeo, K. T. et al. HIV, cytomegalovirus, and malaria infections during pregnancy lead to inflammation and shifts in memory B cell subsets in Kenyan neonates. J. Immunol. 202, 1465–1478 (2019).
Meng, S. S. et al. Maternal allergic disease history affects childhood allergy development through impairment of neonatal regulatory T-cells. Respir. Res. 17, 114–0430 (2016).
Noutsios, G. T. & Floros, J. Childhood asthma: causes, risks, and protective factors; a role of innate immunity. Swiss Med. Wkly. 144, w14036 (2014).
Aye, I. L. et al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol. Reprod. 90, 129 (2014).
Gomez-Lopez, N. et al. Are amniotic fluid neutrophils in women with intraamniotic infection and/or inflammation of fetal or maternal origin? Am. J. Obstet. Gynecol. 217, 693 (2017).
Luciano, A. A., Yu, H., Jackson, L. W., Wolfe, L. A. & Bernstein, H. B. Preterm labor and chorioamnionitis are associated with neonatal T cell activation. PLoS ONE 6, e16698 (2011).
Frascoli, M. et al. Alloreactive fetal T cells promote uterine contractility in preterm labor via IFN-gamma and TNF-alpha. Sci. Transl. Med. 10, 10–438 (2018).
Booth, N. J. et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J. Immunol. 184, 4317–4326 (2010).
Weitkamp, J. H. et al. Histological chorioamnionitis shapes the neonatal transcriptomic immune response. Early Hum. Dev. 98, 1–6 (2016).
Mahesh, G. & Biswas, R. MicroRNA-155: a master regulator of inflammation. J. Interferon Cytokine Res. 39, 321–330 (2019).
Hu, R. et al. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J. Immunol. 190, 5972–5980 (2013).
Gleditsch, D. D. et al. Maternal inflammation modulates infant immune response patterns to viral lung challenge in a murine model. Pediatr. Res. 76, 33–40 (2014).
Mukherjee, S., Allen, R. M., Lukacs, N. W., Kunkel, S. L. & Carson, W. F. STAT3-mediated IL-17 production by postseptic T cells exacerbates viral immunopathology of the lung. Shock 38, 515–523 (2012).
Kramer, B. W. et al. Endotoxin-induced chorioamnionitis modulates innate immunity of monocytes in preterm sheep. Am. J. Respir. Crit. Care Med. 171, 73–77 (2005).
Nguyen, D. N. et al. Prenatal intra-amniotic endotoxin induces fetal gut and lung immune responses and postnatal systemic inflammation in preterm pigs. Am. J. Pathol. 188, 2629–2643 (2018).
Pfisterer, C., Faber, R. & Horn, L. C. Chorioamnionitis-induced changes of fetal extramedullar hematopoiesis in the second trimester of gestation. Is diagnosis from fetal autopsy possible? Virchows Arch. 446, 150–156 (2005).
Miranda, R. N. et al. Myelopoiesis in the liver of stillborns with evidence of intrauterine infection. Arch. Pathol. Lab. Med. 130, 1786–1791 (2006).
Sampson, J. E. et al. Fetal origin of amniotic fluid polymorphonuclear leukocytes. Am. J. Obstet. Gynecol. 176, 77–81 (1997).
Andrews, W. W. et al. The Alabama Preterm Birth study: polymorphonuclear and mononuclear cell placental infiltrations, other markers of inflammation, and outcomes in 23- to 32-week preterm newborn infants. Am. J. Obstet. Gynecol. 195, 803–808 (2006).
Howman, R. A. et al. Inflammatory and haematological markers in the maternal, umbilical cord and infant circulation in histological chorioamnionitis. PLoS ONE 7, e51836 (2012).
Cheah, F. C., Jobe, A. H., Moss, T. J., Newnham, J. P. & Kallapur, S. G. Oxidative stress in fetal lambs exposed to intra-amniotic endotoxin in a chorioamnionitis model. Pediatr. Res. 63, 274–279 (2008).
Hikino, S. et al. Tracheal aspirate gene expression in preterm newborns and development of bronchopulmonary dysplasia. Pediatr. Int. 54, 208–214 (2012).
Hudalla, H. et al. LPS-induced maternal inflammation promotes fetal leukocyte recruitment and prenatal organ infiltration in mice. Pediatr. Res. 84, 757–764 (2018).
Presicce, P. et al. IL-1 signaling mediates intrauterine inflammation and chorio-decidua neutrophil recruitment and activation. JCI Insight 3, 98306 (2018).
Kuypers, E. et al. Intraamniotic lipopolysaccharide exposure changes cell populations and structure of the ovine fetal thymus. Reprod. Sci. 20, 946–956 (2013).
Schmidt, A. F. et al. Intra-amniotic LPS modulates expression of antimicrobial peptides in the fetal sheep lung. Pediatr. Res. 76, 441–447 (2014).
Rueda, C. M. et al. Lipopolysaccharide-induced chorioamnionitis promotes IL-1-dependent inflammatory FOXP3+ CD4+ T cells in the fetal Rhesus macaque. J. Immunol. 196, 3706–3715 (2016).
Konwar, C. et al. DNA methylation profiling of acute chorioamnionitis-associated placentas and fetal membranes: insights into epigenetic variation in spontaneous preterm births. Epigenetics Chromatin 11, 63 (2018).
Vlassaks, E. et al. Antenatal exposure to chorioamnionitis affects lipid metabolism in 7-week-old sheep. J. Dev. Orig. Health Dis. 3, 103–110 (2012).
Bieghs, V. et al. Chorioamnionitis induced hepatic inflammation and disturbed lipid metabolism in fetal sheep. Pediatr. Res. 68, 466–472 (2010).
Kubes, P. & Jenne, C. Immune responses in the liver. Annu. Rev. Immunol. 36, 247–277 (2018).
Happel, K. I. et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 170, 4432–4436 (2003).
Huang, W., Na, L., Fidel, P. L. & Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 190, 624–631 (2004).
Ishigame, H. et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30, 108–119 (2009).
Lee, Y. et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999 (2012).
Jin, W. & Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2, e60 (2013).
Lawrence, S. M. & Wynn, J. L. Chorioamnionitis, IL-17A, and fetal origins of neurologic disease. Am. J. Reprod. Immunol. 79, e12803 (2018).
Pelletier, M. et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 115, 335–343 (2010).
Figueiredo, A. S. & Schumacher, A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology 148, 13–21 (2016).
Lee, S. K., Kim, J. Y., Lee, M., Gilman-Sachs, A. & Kwak-Kim, J. Th17 and regulatory T cells in women with recurrent pregnancy loss. Am. J. Reprod. Immunol. 67, 311–318 (2012).
Singh, A. M. et al. Fetal cord blood and tissue immune responses to chronic placental inflammation and chorioamnionitis. Allergy Asthma Clin. Immunol. 14, 66 (2018).
Jackson, C. M. et al. Pro-inflammatory immune responses in leukocytes of premature infants exposed to maternal chorioamnionitis or funisitis. Pediatr. Res. 81, 384–390 (2017).
Huang, G., Wang, Y. & Chi, H. Regulation of Th17 cell differentiation by innate immune signals. Cell Mol. Immunol. 9, 287–295 (2012).
Romero, R. et al. Interleukin 6 determination in the detection of microbial invasion of the amniotic cavity. Ciba Found. Symp. 167, 205–220 (1992). Discussion 220–223.
Baud, O. et al. Amniotic fluid concentrations of interleukin-1beta, interleukin-6 and TNF-alpha in chorioamnionitis before 32 weeks of gestation: histological associations and neonatal outcome. Br. J. Obstet. Gynaecol. 106, 72–77 (1999).
Lin, J. et al. Neonatal neutrophils stimulated by group B Streptococcus induce a proinflammatory T-helper cell bias. Pediatr. Res. 83, 739–746 (2018).
Wang, W. et al. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol. Int. 32, 887–893 (2012).
Liu, H. & Rohowsky-Kochan, C. Regulation of IL-17 in human CCR6+ effector memory T cells. J. Immunol. 180, 7948–7957 (2008).
Stewart, C. A. et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Invest. 123, 4859–4874 (2013).
Zhao, H., Liao, X. & Kang, Y. Tregs: where we are and what comes next? Front. Immunol. 8, 1578 (2017).
Rueda, C. M. et al. Effect of chorioamnionitis on regulatory T cells in moderate/late preterm neonates. Hum. Immunol. 76, 65–73 (2015).
Kallapur, S. G. et al. Intra-amniotic IL-1beta induces fetal inflammation in rhesus monkeys and alters the regulatory T cell/IL-17 balance. J. Immunol. 191, 1102–1109 (2013).
Pandiyan, P. & Zhu, J. Origin and functions of pro-inflammatory cytokine producing Foxp3+ regulatory T cells. Cytokine 76, 13–24 (2015).
Jung, M. K., Kwak, J. E. & Shin, E. C. IL-17A-producing Foxp3(+) regulatory T cells and human diseases. Immune Netw. 17, 276–286 (2017).
Yang, D. et al. Blocking lymphocyte trafficking with FTY720 prevents inflammation-sensitized hypoxic-ischemic brain injury in newborns. J. Neurosci. 34, 16467–16481 (2014).
Papotto, P. H., Ribot, J. C. & Silva-Santos, B. IL-17(+) gammadelta T cells as kick-starters of inflammation. Nat. Immunol. 18, 604–611 (2017).
Lee, A. J. et al. Fetal responses to lipopolysaccharide-induced chorioamnionitis alter immune and airway responses in 7-week-old sheep. Am. J. Obstet. Gynecol. 204, 364–24 (2011).
Matsuoka, T. et al. Increase of cord blood cytokine-producing T cells in intrauterine infection. Pediatr. Int. 43, 453–457 (2001).
Gibbons, D. et al. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat. Med. 20, 1206–1210 (2014).
Toti, P. et al. Acute thymic involution in fetuses and neonates with chorioamnionitis. Hum. Pathol. 31, 1121–1128 (2000).
Toti, P. et al. Spleen depletion in neonatal sepsis and chorioamnionitis. Am. J. Clin. Pathol. 122, 765–771 (2004).
Melville, J. M., Bischof, R. J., Meeusen, E. N., Westover, A. J. & Moss, T. J. Changes in fetal thymic immune cell populations in a sheep model of intrauterine inflammation. Reprod. Sci. 19, 740–747 (2012).
Lawn, S. D., Butera, S. T. & Folks, T. M. Contribution of immune activation to the pathogenesis and transmission of human immunodeficiency virus type 1 infection. Clin. Microbiol Rev. 14, 753–777 (2001).
Bernstein, H. B., Jackson, R. W., Anderson, J. & Kinter, A. L. The effect of elective cesarean delivery and intrapartum infection on fetal lymphocyte activation and susceptibility to HIV infection. Am. J. Obstet. Gynecol. 187, 1283–1289 (2002).
De Jong, E. et al. Exposure to chorioamnionitis alters the monocyte transcriptional response to the neonatal pathogen Staphylococcus epidermidis. Immunol. Cell Biol. 96, 792–804 (2018).
Kallapur, S. G. et al. Pulmonary and systemic endotoxin tolerance in preterm fetal sheep exposed to chorioamnionitis. J. Immunol. 179, 8491–8499 (2007).
Kallapur, S. G. et al. Chronic fetal exposure to Ureaplasma parvum suppresses innate immune responses in sheep. J. Immunol. 187, 2688–2695 (2011).
Snyder, C. C. et al. Modulation of lipopolysaccharide-induced chorioamnionitis by Ureaplasma parvum in sheep. Am. J. Obstet. Gynecol. 208, 399.e1–399.e8 (2013).
Hibbert, J. E., Currie, A. & Strunk, T. Sepsis-induced immunosuppression in neonates. Front. Pediatr. 6, 357 (2018).
Azizia, M., Lloyd, J., Allen, M., Klein, N. & Peebles, D. Immune status in very preterm neonates. Pediatrics 129, e967–e974 (2012).
Adkins, B., Leclerc, C. & Marshall-Clarke, S. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4, 553–564 (2004).
Adkins, B. Neonatal immunology: responses to pathogenic microorganisms and epigenetics reveal an “immunodiverse” developmental state. Immunol. Res. 57, 246–257 (2013).
Gussenhoven, R. et al. Chorioamnionitis, neuroinflammation, and injury: timing is key in the preterm ovine fetus. J. Neuroinflammation 15, 113–1149 (2018).
Gervassi, A. L. & Horton, H. Is infant immunity actively suppressed or immature? Virology (Auckl.) 2014, 1–9 (2014).
Koenig, J. M. & Yoder, M. C. Neonatal neutrophils: the good, the bad, and the ugly. Clin. Perinatol. 31, 39–51 (2004).
Melvan, J. N., Bagby, G. J., Welsh, D. A., Nelson, S. & Zhang, P. Neonatal sepsis and neutrophil insufficiencies. Int. Rev. Immunol. 29, 315–348 (2010).
Goenka, A. & Kollmann, T. R. Development of immunity in early life. J. Infect. 71(Suppl 1), S112–S120 (2015).
Kollmann, T. R. et al. Neonatal innate TLR-mediated responses are distinct from those of adults. J. Immunol. 183, 7150–7160 (2009).
Stallmach, T. & Karolyi, L. Augmentation of fetal granulopoiesis with chorioamnionitis during the second trimester of gestation. Hum. Pathol. 25, 244–247 (1994).
Baniyash, M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat. Rev. Immunol. 4, 675–687 (2004).
Kollmann, T. R., Kampmann, B., Mazmanian, S. K., Marchant, A. & Levy, O. Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity 46, 350–363 (2017).
Gervassi, A. et al. Myeloid derived suppressor cells are present at high frequency in neonates and suppress in vitro T cell responses. PLoS ONE 9, e107816 (2014).
Rieber, N. et al. Neutrophilic myeloid-derived suppressor cells in cord blood modulate innate and adaptive immune responses. Clin. Exp. Immunol. 174, 45–52 (2013).
Raber, P., Ochoa, A. C. & Rodriguez, P. C. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol. Invest. 41, 614–634 (2012).
Badurdeen, S., Mulongo, M. & Berkley, J. A. Arginine depletion increases susceptibility to serious infections in preterm newborns. Pediatr. Res. 77, 290–297 (2015).
Dedja, A. et al. Lipopolysaccharide-induced chorioamnionitis and postnatal lung injury: the beneficial effects of L-citrulline in newborn rats. Exp. Lung Res. 44, 226–240 (2018).
He, Y. M. et al. Transitory presence of myeloid-derived suppressor cells in neonates is critical for control of inflammation. Nat. Med. 24, 224–231 (2018).
Kostlin, N. et al. Granulocytic myeloid-derived suppressor cells from human cord blood modulate T-helper cell response towards an anti-inflammatory phenotype. Immunology 152, 89–101 (2017).
Schwarz, J. et al. Granulocytic myeloid-derived suppressor cells (GR-MDSC) accumulate in cord blood of preterm infants and remain elevated during the neonatal period. Clin. Exp. Immunol. 191, 328–337 (2018).
Ulas, T. et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat. Immunol. 18, 622–632 (2017).
Heinemann, A. S. et al. In neonates S100A8/S100A9 alarmins prevent the expansion of a specific inflammatory monocyte population promoting septic shock. FASEB J. 31, 1153–1164 (2017).
Buhimschi, C. S. et al. Proteomic biomarkers of intra-amniotic inflammation: relationship with funisitis and early-onset sepsis in the premature neonate. Pediatr. Res. 61, 318–324 (2007).
Mold, J. E. & McCune, J. M. Immunological tolerance during fetal development: from mouse to man. Adv. Immunol. 115, 73–111 (2012).
Mold, J. E. et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 322, 1562–1565 (2008).
Kong, X., Sun, R., Chen, Y., Wei, H. & Tian, Z. gammadeltaT cells drive myeloid-derived suppressor cell-mediated CD8+ T cell exhaustion in hepatitis B virus-induced immunotolerance. J. Immunol. 193, 1645–1653 (2014).
Esteve-Sole, A. et al. B regulatory cells: players in pregnancy and early life. Int. J. Mol. Sci. 19, E2099 (2018).
Sun, C. M., Deriaud, E., Leclerc, C. & Lo-Man, R. Upon TLR9 signaling, CD5+ B cells control the IL-12-dependent Th1-priming capacity of neonatal DCs. Immunity 22, 467–477 (2005).
Elahi, S. et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 504, 158–162 (2013).
Delyea, C. et al. CD71(+) erythroid suppressor cells promote fetomaternal tolerance through arginase-2 and PDL-1. J. Immunol. 200, 4044–4058 (2018).
Collins, J. J. et al. Antenatal glucocorticoids counteract LPS changes in TGF-beta pathway and caveolin-1 in ovine fetal lung. Am. J. Physiol. Lung Cell Mol. Physiol. 304, L438–L444 (2013).
Collins, J. J. et al. LPS-induced chorioamnionitis and antenatal corticosteroids modulate Shh signaling in the ovine fetal lung. Am. J. Physiol. Lung Cell Mol. Physiol. 303, L778–L787 (2012).
Kuypers, E. et al. Effects of intra-amniotic lipopolysaccharide and maternal betamethasone on brain inflammation in fetal sheep. PLoS ONE 8, e81644 (2013).
Kramer, B. W. et al. Antenatal betamethasone changes cord blood monocyte responses to endotoxin in preterm lambs. Pediatr. Res. 55, 764–768 (2004).
McDade, T. W. Early environments and the ecology of inflammation. Proc. Natl Acad. Sci. USA 109(Suppl 2), 17281–17288 (2012).
McDade, T. W., Hoke, M., Borja, J. B., Adair, L. S. & Kuzawa, C. Do environments in infancy moderate the association between stress and inflammation in adulthood? Initial evidence from a birth cohort in the Philippines. Brain Behav. Immun. 31, 23–30 (2013).
Getahun, D. et al. Effect of chorioamnionitis on early childhood asthma. Arch. Pediatr. Adolesc. Med. 164, 187–192 (2010).
Zhu, T., Zhang, L., Qu, Y. & Mu, D. Meta-analysis of antenatal infection and risk of asthma and eczema. Medicine (Baltimore) 95, e4671 (2016).
Belderbos, M., Levy, O. & Bont, L. Neonatal innate immunity in allergy development. Curr. Opin. Pediatr. 21, 762–769 (2009).
Kuzawa, C. W., Tallman, P. S., Adair, L. S., Lee, N. & McDade, T. W. Inflammatory profiles in the non-pregnant state predict offspring birth weight at Cebu: evidence for inter-generational effects of low grade inflammation. Ann. Hum. Biol. 39, 267–274 (2012).
Acknowledgements
This work was supported in part by funding from the Saint Louis University Department of Pediatrics, the Cardinal Glennon Foundation, the National Institutes of Health (AI094478, AI138096), and The Gerber Foundation (all to J.M.K.).
Author information
Authors and Affiliations
Contributions
Substantial contribution to conception and design and drafting the article or revising it critically for important intellectual content: both the authors. Final approval of the version to be published: J.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sabic, D., Koenig, J.M. A perfect storm: fetal inflammation and the developing immune system. Pediatr Res 87, 319–326 (2020). https://doi.org/10.1038/s41390-019-0582-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-019-0582-6
This article is cited by
-
Omics approaches: interactions at the maternal–fetal interface and origins of child health and disease
Pediatric Research (2023)
-
Safety of in utero exposure to maternal IBD pharmacotherapies
Nature Reviews Gastroenterology & Hepatology (2022)
-
A potent myeloid response is rapidly activated in the lungs of premature Rhesus macaques exposed to intra-uterine inflammation
Mucosal Immunology (2022)
-
Chorioamnionitis induces changes in ovine pulmonary endogenous epithelial stem/progenitor cells in utero
Pediatric Research (2021)
