
Abstract
Eph receptors, the largest subfamily of receptor tyrosine kinases, are linked with proliferative disease, such as cancer, as a result of their deregulated expression or mutation. Unlike other tyrosine kinases that have been clinically targeted, the development of therapeutics against Eph receptors remains at a relatively early stage. The major reason is the limited understanding on the Eph receptor regulatory mechanisms at a molecular level. The complexity in understanding Eph signalling in cells arises due to following reasons: (1) Eph receptors comprise 14 members, two of which are pseudokinases, EphA10 and EphB6, with relatively uncharacterised function; (2) activation of Eph receptors results in dimerisation, oligomerisation and formation of clustered signalling centres at the plasma membrane, which can comprise different combinations of Eph receptors, leading to diverse downstream signalling outputs; (3) the non-catalytic functions of Eph receptors have been overlooked. This review provides a structural perspective of the intricate molecular mechanisms that drive Eph receptor signalling, and investigates the contribution of intra- and inter-molecular interactions between Eph receptors intracellular domains and their major binding partners. We focus on the non-catalytic functions of Eph receptors with relevance to cancer, which are further substantiated by exploring the role of the two pseudokinase Eph receptors, EphA10 and EphB6. Throughout this review, we carefully analyse and reconcile the existing/conflicting data in the field, to allow researchers to further the current understanding of Eph receptor signalling.
Similar content being viewed by others

Eph receptors and ephrin ligands: an overview
Receptor tyrosine kinases (RTKs) are a major type of membrane receptors, which govern cell proliferation, differentiation and mobility [1]. Deregulation of RTK signalling pathways leads to many diseases, such as cancers and developmental disorders [2]. The erythropoietin-producing hepatoma (Eph) receptor subfamily is the largest amongst the RTKs with 14 members classified into type A and type B. Compared with other RTKs, Eph receptors share common functions in some disease states, as shown by their roles in cancer progression [3]. In addition, Eph receptors can govern tissue patterning and cell differentiation [4]. Their activation relies on the binding of their cognate membrane-tethered ligands, known as ephrins (Fig. 1). Human EphA receptors (EphA1–A8 and EphA10) preferentially bind to the ephrin A ligands, whereas human EphB receptors (EphB1–B4 and EphB6) bind to the ephrin B ligands. However, promiscuous binding of EphA receptors to ephrin-Bs, or EphB receptors to ephrin-As have been shown [5,6,7]. The specificity of the Eph receptors appears to be dictated by their ectodomains, as shown in a chimera experiment [8]. Thus, the promiscuous binding of type-A and type-B ephrin ligands can likely provide a broader range of signalling functions downstream of an ephrin-ligated Eph receptor.

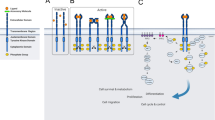
The architecture of Eph receptors and ephrins. Class A and B Eph receptors share a very similar architecture. The ligand-binding domains (LBD) of Eph receptors can bind to the receptor-binding domain (RBD) of the ephrin ligands. C-terminal to the LBD is the cysteine-rich domain (CRD) and two fibronectin III domains in tandem. Intracellularly, the Eph receptors consist of a juxtamembrane (JM) region, a kinase domain (KD), a SAM domain and a C-terminal PDZ domain-binding motif (PBM). Class A ephrins harbour a glycosylphosphatidylinositol (GPI) anchor tethering the RBD, whereas class B ephrins have a transmembrane domain and a PBM intracellularly
Both type A and type B ephrins have an extracellular receptor-binding domain (RBD) in their N-terminus. The primary difference between ephrin-As and ephrin-Bs is how their RBDs are tethered to the plasma membrane: ephrin-As are linked to the plasma membrane by a glycosylphosphatidylinositol (GPI) linker, whereas ephrin-Bs harbour a transmembrane domain and a C-terminal PDZ (an acronym from three proteins: PSD-95, Dlg1 and ZO-1) domain-binding motif (Fig. 1).
Due to their membrane-tethered nature, ephrin ligands account for the ability of the Eph receptors to initiate intracellular signalling events upon cell–cell contact. Although the signalling events mediated by Eph receptors are substantially dependent on cell types, common signalling pathways (e.g. Rho family GTPases-mediated cytoskeletal reorganisation) that principally govern developmental processes, including cell sorting, tissue patterning and cell migration, have been mapped [5].
Research has so far focused on studying how Eph receptors drive signalling via their catalytic activity. However, recent studies have highlighted that the Eph receptor family proteins also display non-catalytic functions. Two members of the family, EphA10 and EphB6, are classified as pseudokinases due to the absence of key amino acids known to catalyse phosphoryl transfer from ATP in conventional protein kinases (Fig. 2b) [9, 10]. The presence of these two catalytically-dead Eph receptors suggests a role for non-catalytic functions in regulating kinase-active Eph receptors. Emerging evidence suggests that EphB6 can be phosphorylated by other Eph receptors, such as EphB1 and EphB4, potentially leading to a reciprocal regulation of EphB6 through direct interaction with its kinase-active counterparts [11, 12]. Such a regulatory mechanism has been demonstrated for the receptor tyrosine pseudokinase ErbB3/HER3, where ErbB3 acts as an activator of EGFR upon heterodimerisation [13]. As per ErbB3, EphB6 and EphA10 have retained an intact ATP binding site [14]. The ability of EphB6 and EphA10 to bind nucleotide suggests that they may function as molecular switches, modulating the kinase activity of other kinase-active Eph receptors [15]. Importantly, recent studies have revealed that deregulated expression of EphA10 and EphB6 is associated with cancers, raising the prospect that, like other pseudokinases, these proteins may contribute to disease states [9, 16].

Sequence alignment of the juxtamembrane region and the kinase domain of Eph receptors. a Sequence alignment of the juxtamembrane (JM) region and the kinase domain (KD) of all Eph receptors. Grey shading indicates the conserved residues critical for kinase catalytic activity, and/or highlights residues that serve as docking sites for SH2 domain-containing proteins when phosphorylated. Bold highlights the catalytically critical residues that have diverged in EphB6 and EphA10. The predicted secondary structures are based on the EphB2 kinase domain crystal structure (PDB: 1JPA) and are annotated as follows: bars represent helix structures, arrows represent β-strands. b Summary of the key catalytic motifs necessary for kinase activity compared with the motifs found in EphA10 and EphB6
In this review, we carefully examine the current state of knowledge on the Eph receptor signalling by dissecting their structural features and molecular mechanisms of regulation. We focus on the importance of the non-catalytic functions of the kinase-active Eph receptors, and the roles of EphA10 and EphB6 pseudokinases in cancers.
Overall organisation and regulation of the Eph receptor intracellular domains
The Eph N-terminal ectodomain is connected by a single transmembrane α-helix, which is extended intracellularly to a juxtamembrane (JM) region that tethers a tyrosine kinase domain. The tyrosine kinase domain is connected by a linker to a sterile-alpha motif (SAM) domain and a PDZ domain-binding motif (Fig. 1) [1]. These additional protein–protein interaction domains imply that Eph receptors coordinate complex intracellular signalling pathways and we detail below the structural and functional characteristics of each of these intracellular domains.
The juxtamembrane region and the kinase domain
Structurally, a JM region is a 35–40 amino acid long peptide linker located N-terminal to the tyrosine kinase domain (Figs. 2 and 3). The role of the JM region is two-fold. One role is to regulate the intrinsic kinase activity of its adjacent kinase domain by locking the protein in an inactive conformation and therefore blocking the substrate and nucleotide access [1]. Mutations of the two conserved tyrosine residues in the JM region (termed JX1 and JX2) to phenylalanine has been shown to completely abolish the kinase activity of EphA4 [17], suggesting that phosphorylation of the JM region is required to unleash an active conformation. The second role is to provide binding sites to SH2 domain-containing proteins upon autophosphorylation, thereby stabilising the kinase domain in an active conformation and allowing the propagation of downstream signals.

Structural features of the Eph receptor kinase domain. a Key structural features of the Eph receptor kinase domain are highlighted in the EphA3 kinase domain crystal structure (PDB: 2QO2). Note that this kinase domain adopted an inactive conformation. b Superposition of the C-lobes of EphA3 (PDB: 2QO2, in cyan) and EphB2 (PDB: 1JPA, in brown) kinase domain structures shows a very similar alignment of the juxtamembrane regions. The distortion of the αC helix, coordinated by the unphosphorylated juxtamembrane region, leads to an inactive form of the kinase domain. c Superposition of the C-lobes from the inactive (PDB: 2QO2, in cyan) and the active (PDB: 2QO9, in pink) EphA3 kinase domain structures reveals the impact of unphosphorylated juxtamembrane region on the activation loop. The unphosphorylated juxtamembrane region (in cyan) of 2QO2 causes misalignment of the activation loop (in red), giving rise to an inactive conformation of the kinase domain. The phosphorylated juxtamembrane region of 2QO9 dislodges from the kinase domain and cannot be seen in the crystal structure. This results in a more ordered activation loop (in pink), which stabilises the kinase domain in its active conformation
The structure of EphB2 kinase domain (PDB: 1JPA) in the presence of the JM region, harbouring phenylalanine mutations in both JX1 and JX2 sites, clearly demonstrated the role that the JM region plays in stabilising an inactive conformation of the kinase domain (Fig. 3b). The JM region adopts a helix-turn-helix conformation and wraps around the αC helix, preventing its correct alignment to allow phosphotransfer activity [18]. The JX1 (Y→F) residue sits in close proximity to the αC helix in this structure, pointing toward the catalytic site, while the JX2 is located at the hinge of the helix-turn-helix and is solvent exposed (Fig. 3b), implying that it can serve as a binding site, once phosphorylated, for phosphotyrosine binding proteins. The structure of the EphA3 kinase domain connected to the JM region (PDB: 2QO2) also highlighted a similar positioning of the unphosphorylated JX1 (Fig. 3b) [19]. In addition to locking the αC helix conformation, the unphosphorylated JM region also plays a role in preventing the activation loop from fully adopting an active conformation (Fig. 3c) [19]. In agreement with this, Wiesner et al. used NMR spectroscopy to demonstrate that phosphorylation of JX1 and JX2 unleashed the JM region from the kinase domain, leading to an active form of the EphB2 kinase domain [17]. Similarly, in vitro kinase assays suggested that the JM region autophosphorylation is a sequential event, whereby autophosphorylation of JX2 preceded autophosphorylation of JX1 [20, 21]. The JM region therefore provides the first layer of the kinase activity regulation. Once phosphorylated and dislodged, Eph kinase activity relies on the phosphorylation of the conserved tyrosine residue in the activation loop (Fig. 2), a feature conserved in many RTKs.
The SAM domain linker and the SAM domain
C-terminal to the Eph receptor kinase domain is a protein–protein interaction domain called the SAM domain (Figs. 1c and 4). The modular SAM domain is highly conserved, comprising five helices that govern homo-/hetero-dimerisation or oligomerisation [22, 23] (Fig. 4b–d). The crystal structure of homo-dimeric EphA4 SAM (PDB: 1B0X) has identified the α1, α3 and the C-terminal segment of the α5 helices are the major structural elements engaged at the dimerisation interface (Fig. 4b). Mutagenesis studies revealed that substitution of the key residues at this interface, such as L940, M972 and M976 that are relatively conserved in other Eph receptors, disrupted SAM domain dimerisation (Fig. 4a) [22]. On the other hand, the crystal structure of EphB2 SAM domain (PDB: 1B4F) has identified a possible oligomerisation mechanism through two additional interaction interfaces (Fig. 4a, c). One oligomerisation interface, defined by the authors [23] as the “b-region interface”, is primarily composed of the loop connecting α3 and α4 helices, and the C-terminal segment of the α5 helix. The other oligomerisation interface, called the “arm-exchange interface”, consists of multiple conserved residues on the α1, α3 and the C-terminal segment of the α5 helices. Therefore, it appears that α1, α3 and the C-terminal α5 helices of the Eph receptor SAM domains are responsible for both homo-dimerisation and homo-oligomerisation.

Sequence alignment of the SAM domain linker and the SAM domain of Eph receptors. a The predicted secondary structures based on the EphB2 SAM domain crystal structure (PDB: 1B4F) are annotated: bars represent helix structures. The key amino acid residues for potential homo-dimerisation/-oligomerisation are highlighted in dark green. The key amino acid residues critical for interaction with downstream interactors are highlighted in yellow. Once phosphorylated, the conserved tyrosine residue highlighted in grey is a potential docking site of SH2 domain-containing proteins. The four C-terminal amino acids highlighted in green are predicted to be the PDZ-domain binding motif (PBM). Note that the protein sequence of EphB2 is based on the EphB2 isoform 2 from Uniprot, as it contains a conserved C-terminal PBM. b The key structural features mediating homo-dimerisation of the EphA4 SAM domain (PDB: 1B0X). c The key structural features facilitating homo-oligomerisation of the EphB2 SAM domain (PDB: 1B4F). The oligomeric EphB2 SAM domains harbour two interaction interfaces and the critical amino acid residues mediating oligomerisation are labelled. d Superposition of the Eph SAM domains of the heterodimeric structures (PDB: 5ZRX, 5ZRY and 5ZRZ, respectively) of the EphA2/SHIP2, EphA6/Odin and EphA5/SAMD5 SAM domains reveal that the key dimerising interface is mediated by the N-terminal residues of Eph receptor α5 helix. e The crystal structure of the EphA5 SAM domain (PDB: 5ZRZ, in purple) were colour coded to represent the interaction interfaces of homo- (the α1, α3 and the C-terminal segment of α5 helices, in pink) and hetero- (the N-terminal segment of the α5 helix, in green) dimerisation/oligomerisation of the Eph receptor SAM domains
In addition to acting as a direct protein–protein interaction domain, the Eph SAM domain can modulate the activity of its adjacent kinase domain and can facilitate recruitment of SH2 domain-containing proteins. Phosphorylation of the conserved tyrosine residue (Y928 of EphB1 and Y921 of EphA2) located in the α2 helix (Fig. 4a) of the SAM domain is able to recruit SH2 domain-containing proteins, such as the adapter proteins Grb7, Grb10 and low-molecular-weight protein tyrosine phosphatase [23,25,26,27]. Truncation of the SAM domain was shown to promote EphA2 and EphB2 homo-dimerisation and clustering at the plasma membrane, respectively [27,29,30], although opposing findings suggested that the presence of the SAM domain enhanced EphA3 dimerisation in cells [31]. Relevant to this, the truncation of the SAM domain induced autophosphorylation in the activation loop of EphA2 in cells [29, 32], whereas the truncation of the SAM domain in EphA3 exhibited decreased activation loop autophosphorylation [31], suggesting an intramolecular regulatory role of the Eph SAM domain. The increased receptor clustering and phosphorylation observed upon deletion of the SAM domain on EphA2 and EphB2 dimerisation [27,29,30] seems to be counter-intuitive as the SAM domain is a known dimerisation domain predicted to favour Eph receptor dimerisation/oligomerisation. One possibility to reconcile this discrepancy is that the SAM domain may impose steric hindrance on the kinase domain and the kinase domain could also be a major intracellular dimerisation determinant. The core kinase domain in other proteins has previously been demonstrated to act as a scaffold [15]. While we cannot rule out a role for the SAM domain in mediating dimerisation/oligomerisation, Wimmer-Kleikamp et al. clearly demonstrated that in addition to the ectodomains that contribute to Eph receptor clustering, the lateral homotypic recruitment of EphA3 is independent of its kinase activity [33], and the intracellular domains of the Eph receptors were also reported to contribute to the receptor hetero-clustering [34] Thus, the Eph tyrosine kinase domain may exhibit non-catalytic functions, including aiding the recruitment of other Eph receptors. Further investigation is required to consolidate this potential non-catalytic function of Eph receptors.
Attempts to determine the structure of the tandem EphA3 kinase and SAM domains by Davies et al. failed due to protein degradation and hence the structural understanding of how SAM domain modulates kinase activity remains unresolved [19]. Using bioinformatics, molecular dynamics simulation in conjunction with biochemical analyses, Kwon et al. recently demonstrated that the linker connecting the SAM domain and the kinase domain (termed the SAM domain linker) plays a major role in modulating the intrinsic tyrosine kinase activity (Figs. 3a and 4a). The interaction between the SAM domain linker and the kinase domain αF–αG loop, located away from the active site, was shown to impact on the autophosphorylation of the JM region. In addition, the SAM domain linker and the JM region have been demonstrated to collaboratively regulate the autophosphorylation of the conserved tyrosine residue in the activation loop (Y779 in EphA3) [20]. The JM region and the SAM domain linker therefore seem to be critical allosteric regulatory elements, with their spatial organisation dictating Eph receptor tyrosine kinase activation. In addition to the regulatory role of the SAM domain linker, it is very likely that the SAM domain itself directly impacts on the conformation of the kinase domain and hence regulates its kinase activity [28].
While the structural role of the SAM domain in fine tuning Eph receptor kinase activity is unclear and awaits the determination of the three-dimensional structure of a construct encompassing the JM, the kinase and the SAM domains, the interaction between the SAM domain of Eph receptors with SAM domains of other proteins has been clearly demonstrated. Disrupting such interaction abrogated the normal cell retraction response upon ephrin-A1 treatment [35]. The heterodimeric SAM domain crystal structures (PDB: 5ZRX, 5ZRY and 5ZRZ, respectively) have been solved for SHIP2, Odin and SAMD5 with EphA2, EphA6 and EphA5, respectively (Fig. 4d). This has provided fruitful structural insights, such as the identification of the key residues responsible for hetero-dimerisation, and a greater understanding of how downstream signalling effectors are recruited (Fig. 4d). Specifically, the residues of the Eph SAM domain responsible for downstream interactor hetero-dimerisation are predominantly concentrated on the N-terminus of the α5 helix, revealing a distinct interaction mechanism compared with the one driving Eph SAM homo-dimerisation/-oligomerisation (Fig. 4a, e).
Unique features of EphA10 and EphB6 pseudokinases
In contrast to the extensive studies of the kinase-active Eph receptors, very little is known about their pseudokinase counterparts, EphA10 and EphB6. To date, there are no three-dimensional protein structures solved for any domains of EphB6 and EphA10. Based on protein sequence alignments of kinase and pseudokinase domains (Fig. 2a), EphA10 and EphB6 have relatively high sequence conservation and the same domain organisation compared with other Eph receptors. Specifically, the closest homologue of EphA10 is EphA7, with a 53.80% identity in protein sequence, whereas EphB6 shares 49.17% identity with its closest homologue, EphB1 (Fig. 2). This close protein sequence similarity implies that EphA10 and EphB6 originated from gene duplication events [16]. Both EphA10 and EphB6 likely harbour non-catalytic regulatory functions (Fig. 2a), although the exact role they play in regulating Eph receptor signalling is unknown. Interestingly, the JM region of EphB6 retains the two conserved tyrosine residues (JX1 and JX2), whereas in EphA10 the corresponding residues are phenylalanine and cysteine, respectively (Fig. 2). This suggests a distinct regulation of the JM region for each of these two Eph receptor pseudokinases. Notably, half of the predicted activation loop of EphB6 is missing, as well as the conserved activation loop tyrosine residue (Fig. 2a), implying a distinct activation loop regulatory mechanism compared with other Eph receptors. By contrast, the activation loop of EphA10 has retained the conserved tyrosine residue, suggesting that EphA10 could adopt various conformational states dependent on its activation loop phosphorylation state. These unique features between EphB6 and EphA10 clearly underscore their mechanistic specificity in the Eph receptor signalling pathways. Future biochemical and structural studies of these two kinase-dead Eph receptors will shed light on the role of non-catalytic functions in Eph receptor signalling.
Eph receptor forward signalling
By the virtue that Eph receptors and ephrin ligands are tethered to the presenting cells, their predominant roles are in cell communication. Upon cell–cell contact, both Eph receptors and ephrin can initiate signal transduction in each of the ligand- and receptor-presenting cells. The signalling initiated by the ephrin-ligated Eph receptors is called “forward signalling”, whereas the signalling initiated by the Eph-bound ephrins is called “reverse signalling” (Fig. 5a). The Eph forward signalling has drawn greater attention as it is driven by the “canonical receptors” whose role is to transduce signals from ligand stimulation.

Dimerisation and oligomerisation of Eph receptors activate forward (and reverse) signaling. a Ephrin-induced dimerisation of Eph receptors triggers autophosphorylation of the Eph receptor tyrosine kinase domain. Autophosphory lation of the juxtamembrane region, the kinase domain and the SAM domain provides binding sites for recruitment of SH2 domain-containing proteins, which themselves may be substrates for phosphorylation by the Eph receptor tyrosine kinase domain. In cells expressing ephrins ligated to the Eph receptors, Src family kinases (SFKs)-mediated phosphorylation of the C-terminal tail of class B ephrins induces reverse signalling pathways. b Ephrin-induced dimerised Eph receptors can further oligomerise to form clustering signaling centres, from which more Eph receptors and downstream signalling proteins can be recruited, phosphorylated or activated. This clustered signalling centre amplifies forward signalling, and presumably also reverse signalling in the opposed cell. Both forward signalling and reverse signalling substantially rely on Src family kinases (SFKs), which further phosphorylate Eph receptor-interacting proteins or even Eph receptors themselves. The extensive number of phosphorylation sites from the clustered signalling centre facilitates recruitment of effectors for diverse signalling pathways, including the PI3K-Akt/PKB signalling axis
The role that Eph receptors play in regulating major pathways, such as via Rho/Rac GTPases, which control actin organisation, and Ras/MAPK, which controls proliferation, have been extensively reviewed elsewhere [36]. Thus, we will focus on the PI3K-Akt/PKB signalling axis downstream of Eph receptors as an example of Eph forward signalling. We will also illustrate how phosphorylation of the Eph receptors is able to relay the signals via the Src-homology 2 (SH2)-containing proteins, which are critical in both Eph forward and reverse signalling.
The Eph-mediated signalling pathways are largely receptor, cell type and context dependent, as suggested by a report describing that some Eph receptors, such as EphA2 [5], can promote both tumour progression and suppression. The tyrosine kinase domain of Eph receptors plays a central role in forward signalling, such that upon ligand stimulation, the ephrin-bound Eph receptors undergo dimerisation, which results in transphosphorylation and activation of the receptor kinase domains (Fig. 5a). The activated Eph kinase domain then phosphorylates downstream substrates, such as adaptor protein Nck1/2 [37]. The two most frequent autophosphorylation sites are located in the JM region, JX1 and JX2, as demonstrated by in vitro kinase assays and in cell proteomic mapping [38]. The phosphorylation of JX1 and JX2 is a sequential event, which further leads to the autophosphorylation of the activation loop, resulting in full kinase activity [20]. Once phosphorylated, these tyrosine residues also become potential docking sites for SH2 domain-containing signalling or adaptor proteins (Fig. 5a, b). The best characterised SH2 domain-containing proteins that interact with Eph receptors include the adaptor proteins Nck and CrkII [39] and Src family kinases (SFKs). For example, Src was demonstrated to bind to the phosphorylated tyrosine residue JX2 in the JM region of EphB2 [40]. SFKs recruited to the Eph receptors are thought to then phosphorylate downstream substrates, relaying signals from the Eph receptors. In these cases, once phosphorylated, Eph receptors execute their non-catalytic functions by acting as scaffold proteins.
The fact that Eph receptors can act as scaffold proteins has been well exemplified by the interaction with several guanine nucleotide exchange factors (GEFs). Sahin et al. demonstrated that, in fibroblasts, the activation of an upstream GEF of Rho small GTPases, ephexin1 requires EphA4 kinase activity, implying ephexin1 is an EphA4 substrate. Phosphorylated ephexin1 promoted RhoA activity, leading to the formation of stress fibres. However, the protein kinase Src rather than EphA4, appears to be the direct upstream kinase of ephexin1. This suggests that phosphorylated EphA4, presumably via its JM region, recruits Src kinase, which in turn phosphorylates the downstream substrates [41]. Similarly, other SH2 domain-containing proteins such as the GEFs Vav2 and Vav3 were shown to interact with the JM region phosphorylated tyrosine residues of EphA2, suggesting that an active kinase conformation is required to allow interactions with their binding partners. In addition, the interaction of SH2 domain-containing proteins with Eph receptors is not restricted to the JM region. In the same study, the interaction of the p85 subunit of phosphoinositide 3-kinase (PI3K) with EphA2 was disrupted, when tyrosine Y745 (the conserved tyrosine residue preceding the catalytic loop) of the kinase domain and Y930 of the SAM domain were mutated [42]. Together, these studies clearly highlight the scaffolding function of Eph receptors, a function dependent on their phosphorylation states.
PI3K has been known to interact with RTKs, such as platelet-derived growth factor receptor, via its SH2 domain(s) in the p85 subunit. Fang et al. demonstrated that the p85 subunit of PI3K interacts with EphA2 by immunoprecipitation [42]. Multiple studies have demonstrated that the Eph receptors modulate the PI3K-Akt/PKB signalling axis in cell migration. For example, Akt/PKB is phosphorylated and activated upon EphB2 binding to ephrin-B1 in HEK293 cells stably expressing the microtubule-associated protein tau. In addition, the sole overexpression of EphB2 without ephrin-B1 induction triggered PI3K-Akt/PKB signalling to a lesser extent [43].
On the other hand, conflicting evidence suggested that in non-small cell lung cancer (NSCLC) cell lines, ephrin-B1 activated EphB3 led to downregulated phosphorylation of the Ser/Thr residues on Akt/PKB that are required for activation, a mechanism mediated by phosphatase PP2A [44]. Similarly, dephosphorylation of the same Ser/Thr residues in Akt/PKB was observed downstream of EphA2 activation in prostate cancer cells [45]. In a glioblastoma (GBM) cell line, EphA2 can act both upstream (as a regulator) and downstream (as a substrate) of Akt/PKB in a ligand-dependent or -independent mode, respectively [46]. The crosstalk between Eph receptors and Akt/PKB was further demonstrated by Stallaert et al., who showed that activation of EphAs inhibited Akt/PKB activity, which hindered the endosomal trafficking of EGFR and reduced EGFR recycling to the plasma membrane. In line with this, EGF-induced cell migration was also suppressed upon EphA receptor activation [47]. These studies collectively suggest that distinct Eph receptors can elicit opposite signalling events under different cellular contexts.
Taken together, the Eph receptor-mediated forward signalling relies on autophosphorylation and activation of the tyrosine kinase domain, in turn creating binding sites for downstream adaptors or signalling proteins. Surprisingly, other than the Eph receptor itself, very little is known about the direct protein substrates of the Eph receptor tyrosine kinase domain. The main difficulty in identifying the Eph receptor kinase domain direct downstream substrates is due to the associated SH2 domain-containing SFKs, as both of them are tyrosine kinases with potential overlapping substrates. Nck1/2, an adaptor protein that is involved in cytoskeletal organisation, was recently identified to be a direct substrate of EphA4 in vitro and in cells [37]. The binding between the Nck SH3 domain and its interacting partners was abolished once the conserved tyrosine residue in the Nck SH3 domain is phosphorylated by EphA4. Nck was also reported to bind to the phosphorylated JM region of EphB1 via its SH2 domain [48, 49]. In addition, the adapter protein Caskin recruited by Nck was shown to be phosphorylated by EphB1 [49]. These results combined suggests that Eph receptor phosphorylation sites within the JM region, activation loop, the SAM domain can recruit adaptors or effectors, which then become potential substrates for Eph tyrosine kinase domain. Deciphering the Eph interactome will therefore be critical in order to fully appreciate the signalling network of Eph receptors.
Ephrin-independent Eph receptor activation
The Eph receptor-mediated signalling events are reliant on the phosphorylation status of the Eph receptors its intracellular domains. The activation mechanism of Eph receptors upon membrane-tethered ephrin ligation resembles those used by other RTKs: receptors undergo dimerisation upon binding to soluble ligands for activation. Normal Eph signalling initiated upon binding of ephrins to Eph receptors induces dimerisation, however, the ephrin-ligated Eph receptor can further oligomerise to form clusters, from which downstream signalling can be magnified (Fig. 5b) [50]. Interestingly, the cellular signalling events/phenotypic changes triggered by activation of dimeric or oligomeric Eph receptors appeared to be different [25]. On the other hand, aberrant expression of Eph receptors is commonly observed in cancers. This leads to the hypothesis that upregulated expression of Eph receptors can achieve Eph receptor-mediated signalling by dimerisation and higher order oligomerisation, independent of ephrin ligation. Moreover, the signalling pathway outputs resulting from ligand binding or from ephrin-independent Eph receptor dimerisation/oligomerisation can be distinct or even opposite as demonstrated in the case of EphA2—an apparent oncogene in the absence of ephrins, but a tumour suppressor when interacting with its ephrin ligands [45, 46, 51, 52].
Structurally, the ephrin induces an extensive dimerisation interface upon binding to the ectodomain of the Eph receptors via a highly conserved interface [53]. For example, a conserved polar bulky amino acid (Gln) at position 109 of ephrin-B2 forms a hydrogen bond to the conserved Thr38 in the ectodomain of EphB2, and very likely to those of other EphB receptors. The crystal structure of ephrin-B2 binding to the ligand-binding domain (LBD) of EphB2 revealed a tetrameric complex (Fig. 6a) [53]. However, as suggested by the crystal packing, the LBD of EphB2 seems to be able to tetramerise without the involvement of ephrins in the tetrameric complex structure (Fig. 6a) [53]. A similar Eph LBD–LBD interface is also seen in the crystal structures of heterotetrameric EphA2/ephrin-A5 and EphA4/ephrin-A5 [54,55,56,57]. In these EphA receptor structures, a second Eph–Eph interaction interface dictated by the cysteine-rich domain (CRD) was found, suggesting that in addition to the LBD, the CRD is another dimerising determinant allowing Eph receptors to assemble via Eph–Eph interaction (Fig. 6b) [54,55,56,57]. Both Eph LBD–LBD and CRD–CRD interfaces do not engage in ephrin binding (Fig. 6b, c). In the absence of ephrins, the EphA2 ectodomain crystal structure showed the presence of the same Eph CRD–CRD interface (Fig. 6c), suggesting a conserved mechanism of dimerisation driven by Eph receptor ectodomains. Mutations introduced to the Eph CRD–CRD interface amino acids (L223, L254 and V255) perturbed EphA2 dimerisation [58], which is in agreement with the argument that Eph receptors can dimerise/oligomerise in the absence of ephrin. Taken together, these data show that Eph receptors do not have to rely on ephrins to form dimers and potentially cluster to high-order oligomers if Eph receptor concentration in the plasma membrane is sufficient (Fig. 6d). Indeed, once the initial EphA3 and ephrin-A5 nucleating centre is formed, more EphA3 receptors can be laterally recruited without being directly associated with ephrin-A5 in cells [33]. Moreover, using fluorescence resonance energy transfer, EphA2 or EphA3 was shown to form dimers in the plasma membrane without binding to the ephrin ligands [29, 31]. These dimerised Eph receptors are constitutively active, as signified by autophosphorylation of the kinase domain activation loop [31], suggesting that when Eph receptors are highly expressed, they can exhibit basal activity at the plasma membrane even in the absence of ephrin stimulation.

Ephrin-independent Eph receptor dimerisation/oligomerisation. Dimerisation/oligomerisation of the Eph receptors can be ephrin independent, as Eph receptors harbour Eph–Eph interacting interfaces, as shown from both class A and class B Eph receptors. a The ephrin-B2 (shown in blue) ligated EphB2 LBD (shown in green) crystal structure has Eph–Eph crystal-packing interface indicated by black arrows (PDB: 1KGY). b The crystal structure comprising two heterotetramers of ephrin-A5 ligated the EphA2 ectodomains (LBD + CRD + N-terminal fibronectin domain III) reveals two Eph–Eph interacting interfaces (PDB: 3MX0). Highlighted in the purple circle is the Eph LBD–LBD interface (key residues including K116, T144 and P147, etc.), whereas highlighted in the green circle is the Eph CRD–CRD interface (key residues such as L223, L254 and V255, etc.). The G131 residue lies at the ephrin-induced Eph LBD–LBD interface, as circled in black. c In the absence of ephrin ligation, very similar Eph LBD–LBD and Eph CRD–CRD interfaces also exist, as shown in another EphA2 crystal structure (PDB: 3FL7). d The Eph receptor dimerisation/oligomerisation is hypothesized to be also regulated by the intracellular domains. The lateral extension of the Eph receptor to the clustered signalling centre can be potentially mediated by the non-catalytic functions of the kinase domain, and by the SAM domain
Although capable of functioning independent of ephrins, under physiological conditions, it is rare that Eph receptors exclude the engagement of ephrins. As illustrated in Fig. 6b, c, in the presence of ephrins, a simplified Eph receptor oligomer can be regarded as repeats of ephrin-dependent Eph receptor dimers (with an ephrin-induced Eph LBD–LBD interface, circled in black) and ephrin-independent Eph receptor dimers (with an Eph LBD–LBD interface, circled in purple, and an Eph CRD–CRD interface, circled in green). Interestingly, the major residues clustering on the Eph LBD–LBD interface (e.g. D104, K116, E117 and T144 of EphA2) and those clustering on the Eph CRD–CRD interface (e.g. L223, V255 of EphA2) are highly conserved across EphA and EphB receptors (Fig. 6b, c) [54,55,56,57], suggesting that both types of Eph receptors are able to undergo similar ephrin-independent dimerisation. This implies that dimerisation of the interclass Eph receptors can occur. In contrast, the EphA2 G131 residue located at the ephrin-induced Eph LBD–LBD interface is specifically conserved in EphA receptors, except for EphA4 (Fig. 6b). The G131Y EphA2 variant exhibited a reduced clustering propensity in a ephrin-dependent manner [57]. G131, however, appeared to be not essential for Eph receptor dimerisation/oligomerisation in the absence of ephrins [58]. Collectively, the ephrin-independent Eph receptor dimers/oligomers are predicted to be promiscuously composed of type A and B Eph receptors, whereas ephrin binding to Eph receptors is likely a mechanism to selectively produce homotypic dimers/oligomers of EphA, or EphB receptors.
Ephrin reverse signalling
The ligands of the Eph receptors, ephrins, are membrane tethered and are able to initiate downstream signalling in their expressing cells. To this end, they can also act as “receptors”. The reverse signalling mediated by ephrins is induced by their ligation to the Eph receptors and additional mechanisms [59]. Similar to Eph receptors, the intracellular tail of the B-type ephrins harbours conserved multiple tyrosine residues that can be phosphorylated. For example, ephrin-B1 can be phosphorylated following incubation with the ectodomain of EphB2 [59,60,61], which can be mediated by SFKs [62]. Similar to Eph receptors, tyrosine phosphorylation on the C-terminal tail of ephrin-Bs provides binding sites to SH2 domains of SFKs, thereby inducing subsequent signalling events [62]. Intriguingly, the cytosolic domain of ephrin-Bs was also reported to affect the Eph forward signalling events in trans in a phosphoproteomics study [63], but the molecular details remain unclear. In the same study, Jorgensen et al. demonstrated that co-incubation of EphB2- and ephrin-B1-expressing HEK293T cells led to asymmetric intracellular signalling with differential levels of substrate tyrosine phosphorylation [63]. This bias likely arises from the distinct intracellular architectures of Eph receptors and ephrins.
Fewer studies have examined ephrin-A mediated reverse signalling pathways compared with those driven by ephrin-Bs. Ephrin-As are tethered to cells solely via a GPI linker embedded in the plasma membrane. This precludes reverse signalling directly, although it was proposed that ephrin-As interact with other transmembrane proteins upon binding to Eph receptors [5].
The recombinant soluble ectodomain of ephrin is not sufficient to elicit the Eph forward signalling events, as it is required to be either membrane bound in the expressing cells or pre-clustered by antibodies [64]. A possible explanation is that ephrins exert both inhibitory and activating effects on Eph receptors. In trans, membrane-bound or pre-clustered ephrins can activate Eph receptors, but, when expressed in the same cell (in cis), ephrins bind and block Eph receptors from initiating forward signalling. Therefore, the soluble ectodomain of ephrins can potentially act in cis, failing to activate the Eph forward signalling [65]. Yin et al. showed that the cis-expression of ephrin-A2 inhibited the trans-binding of ephrin-A5 to the EphA4 receptor in HEK293 cells [66]. The competition of cis and trans binding of ephrin-As was also reflected in cis binding reducing EphA receptor phosphorylation levels, consistent with ephrin-As regulating Eph-mediated forward signalling. In agreement, multiple studies have independently confirmed the attenuated activation of trans-EphA receptor by interacting with ephrin-As in cis from both in vitro and in cells neuron growth cone models [67, 68].
To add to the complexity of this system, each EphA and EphB receptor has multiple ephrin-As and ephrin-Bs as ligands, respectively, resulting in additional cross-reactivity among EphA/ephrin-Bs and EphB/ephrin-As. Potentially, the overall expression pattern of ephrins and Eph receptors in one cell dictates which Eph receptors can be activated by an adjacent cell upon cell–cell contact. Nevertheless, such pleiotropy necessitates caution in interpreting studies, in which a single type of ephrin is used to activate Eph receptors, as several ephrins and Eph receptors usually coexist in each cell type.
The non-catalytic functions of alternative Eph receptor isoforms
While the tyrosine kinase activity of Eph receptors is central to many well-characterised signalling pathways, their kinase-independent functions have proved to be important in cancer development. Strikingly, almost all the Eph receptor genes yield at least one isoform that does not have kinase activity due to partial or complete truncation of the intracellular domains from alternative splicing. In addition to the roles in cancer, these Eph isoforms possess a wide array of functions in neuronal development [69, 70], cell reprogramming [71] and cell adhesion and repulsion [72]. Holmberg et al. discovered that expression of an EphA7 isoform that lacks the entire intracellular domain is able to shift cell repulsion to adhesion. The intracellular domain-truncated EphA7 isoform was able to block the full-length EphA7 autophosphorylation [72], which might be partially due to a dominant-negative effect, where co-clustering of the truncated form dilutes the cross-phosphorylation of Eph receptor intracellular domains [34]. The EphA7 isoform was later found to be able to inhibit the MAPK and SFK signalling pathways, likely through a mechanism in which EphA7 forms an inhibitory dimer with EphA2 that antagonises lymphoma development [73]. Similarly, EphA10 is a proposed oncogenic Eph receptot but the secreted EphA10 isoform, comprising only the LBD abd rge partial CBD, was shown to suppress breast tumour growth abdmetastasis in mice [74].
In addition to isoforms derived from alternative splicing, the proteolytic products of the full-length Eph receptors appear to serve additional functions. For example, the cleavage of EphA2 by membrane type-1 matrix metalloproteinase induced an ephrin-independent EphA2 activation. The resulting activation of small Rho GTPases led to enhanced cancer cell invasion in vivo [75, 76]. More detailed consequences of the proteolysis of the Eph receptors and ephrins have been reviewed elsewhere [77]. Collectively, the physiological and pathological roles of these truncated Eph isoforms and proteolytically cleaved forms provide strong evidence for the importance of the non-catalytic and ephrin-independent functions of Eph receptors.
Eph compositions in signalling clusters and their non-catalytic functions
One of the major difficulties of studying Eph signalling is to precisely define the genuine functions of each Eph receptor family member in cells. This is mainly due to the forward signalling output representing the integrated input of all the stimulated Eph receptors in a clustered signalling centre. While phosphatase activity driven by the protein tyrosine phosphatase receptor type O (Ptpro) and other protein tyrosine phosphatases can negate Eph receptor activation [78, 79], co-expressed ephrins may also exert potential cis-inhibitory effects on Eph receptors. Not surprisingly, one outstanding question is how the interaction among Eph receptors dictates the overall downstream signalling events. Another outstanding question is: to what extent can the Eph non-catalytic functions govern the signalling pathways in an Eph receptor clustering centre? As reviewed elsewhere, different pools of Eph receptors/ephrins in cells can confer opposing functions on a given Eph receptor [36]. The non-catalytic functions of the Eph receptors are likely to be responsible for this observation.
The preferential binding of Eph receptors to their corresponding type of ephrins provides a useful avenue to demonstrate that a cell expresses a mixture of Eph receptors, and is able to respond distinctly upon binding to type A or type B ephrins. Astin et al. observed that treatment with ephrin-A1 or ephrin-A5 rendered contact inhibition of locomotion (CIL) in the PC3 prostate cancer cell line [80]. On the contrary, ephrin-B2 promoted PC3 cells to migrate. The authors further demonstrated that PC3 cells exhibited homotypic CIL among themselves, but failed to undergo CIL, when contacting normal cells, such as fibroblasts and endothelial cells. By comparing the Eph/ephrin expression level vs. the corresponding phenotypic changes, they concluded that the differential expression of Eph receptors/ephrins in PC3 cells, fibroblasts and endothelial cells was responsible for the distinct CIL responses. The abundance of ephrins in the activating cells (fibroblasts and endothelial cells) dictate the composition of the Eph receptors in the signalling cluster of the receiving cells (PC3 cells), thereby evoking cell retraction or invasiveness. More importantly, in prostate cancer patient specimens, the surrounding stroma cells of the prostate cancer cells expressed relatively high levels of ephrin-B2, implicating Eph-ephrin signalling in the cancer microenvironment, potentially facilitating cancer metastasis [80].
An Eph receptor cluster does not have to be homotypic, adding to the complexity of the signalling platform. EphA receptors can intermingle with EphB receptors in a clustered signalling centre as exemplified by EphA3 and EphB2, regardless of the EphA3 kinase activity [34]. This heterotypic binding was mainly mediated by their ectodomains, although the intracellular domains also partially accounted for the interaction. Importantly, expression of wild-type EphA3 elevated the EphB2 kinase activity and collaboratively induced cell rounding. In addition, aberrant cell segregation due to the truncation of the intracellular domains of EphB2 was able to be restored by wild-type EphA3 expression. This observation has two implications: [1] EphA3 can compensate for EphB2 kinase activity and/or [2] EphA3 is able to reconstitute the potential EphB2 non-catalytic functions by recruiting EphB2 interacting partners. Surprisingly, a kinase-dead EphA3 mutant suppressed EphB2 kinase activity, reinforcing the idea that Eph receptors harbour non-catalytic regulatory functions. This is thought to work through a dominant-negative effect, where co-clustering of the truncated form effectively dilutes the cross-phosphorylation of intracellular domains. Thus, Eph receptor signalling clusters can contain a wide array of different Eph receptor species, with the combination of species dictating the forward signalling output following ephrin stimulation.
Accumulating evidence suggests that Eph receptors exhibit non-catalytic functions. The kinase-inactive EphA2 mutant can induce similar chemotaxis compared with wild-type EphA2, when overexpressed in HEK293 cells [46]. In another study using breast cancer cell lines, the kinase-dead EphA2 mutant exerted a dominant-negative effect that mitigated ephrin-A1 mediated EphA2 phosphorylation and contributed to tumour suppression [81]. EphA3 was reported to regulate the protease activity of the transmembrane A-Disintegrin-And-Metalloprotease 10 (ADAM10), independently of the EphA3 intrinsic tyrosine kinase activity [82]. The proximity of the inactive EphA3 kinase domain to the plasma membrane prevented ADAM10-mediated cleavage through steric hindrance, until binding of ephrin-A5 and ensuing receptor clustering and phosphorylation reorganised the EphA3 JM region and the kinase domain, and enabled access of ADAM10. This example illustrates one of the Eph receptor non-catalytic functions, in which EphA3 can block ADAM10 activity, and is reminiscent of the functions that pseudoenzymes perform as protein–protein interaction domains to regulate signalling [15]. More broadly, it is unlikely that all Eph receptors within a cluster solely rely on their kinase activity to transduce signals. Instead, some of the Eph receptors primarily perform their non-catalytic functions, exerting regulatory properties via scaffolding, recruiting or competing with other Eph receptors. The best evidence supporting this argument is the existence of the two kinase-deficient Eph receptor members, EphA10 and EphB6, and their emerging roles in cancers.
The roles of EphA10 and EphB6 in cancers
Endocrinologically, EphB6 expression is correlated with catecholamine biosynthesis and secretion [83,84,85]. Its role in cancer, however, has attracted more attention. For example, EphB6 was reported to modulate cell death by anoikis in cancer cells by interacting with another Eph receptor, EphA2, via their ectodomains [86]. Nonetheless, the molecular mechanisms underpinning EphB6 functions remain to be fully determined. Expression of EphB6 in MDA-MB-231 triple-negative breast cancer cells, which reportedly do not express the receptor endogenously, lowered cadherin 17 protein expression and altered MEK2 and β-catenin expression [87]. Using HEK293T cells, overexpression of EphB6 allowed Matsuoka et al. to identify SFKs as constitutive interactors of EphB6 [88]. In addition, upon binding to ephrin-B2, tyrosine phosphorylation of EphB6 was increased. Nonetheless, it is inconclusive whether the phosphorylation was catalysed by the associated SFKs, or it was carried out by other kinase-active Eph receptors, such as EphB4 and EphB1 (Fig. 7) [12, 13].

The potential roles of EphA10 and EphB6 pseudokinases. While very little is known about the EphA10 interactors, EphB1 and EphB4 were reported to phosphorylate EphB6. EphB6 phosphorylation may potentially perturb the binding of constitutive EphB6 interactors, such as Fyn kinase [88] and Cbl E3 ligase [12]. It is postulated that EphB6 can regulate the kinase activity of EphB1 and EphB4 via heterodimerisation, resulting in changes to downstream phospho-signalling events
Downregulation of EphB6 mRNA resulting from promoter hypermethylation has been found in NSCLC and breast cancer cells with invasive characteristics or tendency [89, 90]. The protein expression level of EphB6 was also decreased in multiple cancers, including breast cancer [89], NSCLC [90] and colorectal cancer [91]. Moreover, downregulation of EphB6 promoted cancer metastasis [92,93,94], whereas restoration of EphB6 expression suppressed metastasis [90, 94, 95]. Thus, EphB6 has been proposed as a tumour suppressor. Interestingly, while playing a role as a potential metastasis suppressor, EphB6 was recently reported to accelerate cell proliferation in triple-negative breast cancer (TNBC) cell lines [95]. These studies imply that at different stages of cancer progression, EphB6 can perform distinct functions, although other factors, such as cell types or receptor expression level, cannot be excluded.
Very little is known about EphA10, but upregulated expression of EphA10 is associated with multiple cancers [96,97,98]. EphA10 expression is rarely seen in normal tissues except for testes [99]. Owing to the prevalence of EphA10 expression in cancers, researchers have proposed to use EphA10 as a marker for identifying cancers.
Eph receptors: oncogenes or tumour suppressors
In 2010, the landmark review article by Elena Pasquale clearly described Eph receptors as (paradoxical) tumour suppressors or oncogenes, depending on their different expression pattern and the cellular contexts [3]. The versatile activation mechanisms displayed by the well-characterised Eph receptor, EphA2, is an excellent example that illustrates the biphasic role of Eph receptors in cancer [3]. The seemingly controversial cancer-agonising and -antagonising actions downstream of EphA2 receptor activation are due to different degrees of contribution from other Eph receptors and ephrin ligands, and the ensuing signalling events. In cancer biology, the tumour microenvironment (TME) has proved to be essential for the development of cancer cells. Studying the functions of Eph receptors using cancer cells in culture can be restricted by the lack of interaction with Eph receptor-expressing stromal and endothelial cells. Thus, below, we carefully examine the recent studies on understanding the Eph receptor functions in cancer in mouse models. In particular, we emphasise the Eph receptor non-catalytic functions in vivo. The sequences of the human and mouse Eph receptors share very high similarity, implying a resemblance of the functionalities, and making mouse models more likely to recapitulate the human Eph receptor functions. In spite of the complexity of Eph receptor regulation, certain Eph receptors have been categorised as either oncogenes or tumour suppressors based on their predominant function shown in the mouse model.
EphA7 was proposed to be a tumour suppressor in follicular lymphoma, in which downregulated expression due to hypermethylation of the gene promoter was found. The survival rate of the follicular lymphoma mouse model significantly decreased, when EphA7 expression was suppressed, equivalent to the effect exhibited by knocking down the tumour suppressor, p53. The EphA7 tumour-antagonising effect is independent of its intracellular domains, as restoration of the EphA7 ectodomains in the human lymphoma cells xenografted into mice was sufficient to exhibit a profound antiproliferative and apoptosis-inducing effects [73]. In another study, EphA7 was also shown to be a tumour suppressor in prostate cancer. Nonetheless, only the full-length EphA7 with intact kinase activity was able to contribute to tumour shrinkage in prostate tumour xenografted mice [100].
The kinase-dependent and kinase-independent functions of Eph receptors can sometimes determine whether an Eph receptor is a tumour suppressor or an oncogene. For example, the tumour growth was significantly suppressed in EphA3 knockout mice, where a primary human GBM cell line was injected. The orthotopic xenograft model further confirmed that the survival rate of mice transplanted with high EphA3-expressing patient sample was much lower than the low expression counterparts [101]. Interestingly, the oncogenic effect upon EphA3 expression appeared to be not kinase activity dependent, as the EphA3 from human GBM specimens and primary cell lines remained constitutively unphosphorylated. Similarly, the oncogenic properties of EphA3 were observed in a prostatic tumour xenograft mouse setting: expression of EphA3 in the stromal and vascular regions benefited tumour formation [102]. These effects could be countered by an activating EphA3-specific antibody, which reduced the tumour burden in of both GMB and prostatic cancer mouse models by increasing apoptosis [101, 102]. It therefore appears that EphA3 promotes oncogenesis in a kinase-independent manner in some contexts, and exerts a kinase-dependent tumour suppressive effect in others.
The epigenetic silencing of Eph receptors such as EphA5, EphA7, EphB6 and others that contribute to downregulated protein expression has been linked to cancer development [3, 73, 103,104,105], suggesting their potential roles as tumour suppressors. Interestingly, in different cancers, they can somehow function in an opposite manner. For example, overexpression of EphA5 has been found in lung cancers [106]. Neutralisation and degradation of EphA5 proteins by an EphA5-specific antibody sensitised the lung tumour bearing in vivo models to irradiation. Also, inhibition of EphA5 kinase activity was able to suppress the progression of hepatocellular carcinoma in mice [107]. Although more detailed studies are required, it appears that the oncogenic property of EphA5 is dependent on its catalytic activity.
Depletion of EphA2 was shown to exhibit an antiproliferative effect in NSCLC and TNBC in in vivo models [108, 109]. Consistently, in an oncogenic KRasK12D mutant-driven NSCLC mouse model, the tumour cells grown in the EphA2−/− mice underwent significant apoptosis. In addition, the administration of a relatively selective EphA2 kinase inhibitor was able to reduce the tumour size in the NSCLC xenografted mice, by inducing apoptosis, suggesting that EphA2 is oncogenic in NSCLC [110]. Interestingly, Yeddula et al. reported that targeting EphA2 in a tissue-specific manner can lead to an opposite outcome: knockout of EphA2 from the lung adenocarcinoma tissue driven by KRasK12D in vivo deteriorated cancer development. The increased tumour burden shown in lung tissue-specific EphA2-deficient mice suggested that EphA2 is a tumour suppressor. The EphA2 tumour suppressive effect was likely due to an inhibition of the MAPK pathway downstream of EphA2 activation by binding to its ephrin ligand, ephrinA1, as shown in analysis at the cellular level [111]. These results collectively suggested that whether EphA2, and other Eph receptors, are oncogenic or tumour suppressive in a cancer cell, is determined by its surrounding environment, namely, the tumour microenvironment.
Concluding remarks
The Eph receptors have been identified as critical players that control tissue development. Aberrant expression or mutation of Eph receptors is a hallmark of many diseases, including cancers. In contrast to other RTKs, the uniqueness of the Eph receptor-mediated signalling pathways includes: [1] ephrin ligand-dependent or -independent Eph receptor activation; [2] Eph receptor oligomerisation, a mechanism allowing the amplification of the downstream signal transduction; [3] the presence of the two kinase-dead members, EphA10 and EphB6, whose roles remain to be determined. To further advance the understanding of how Eph receptors function, in-depth investigation of their structural details, the spatiotemporal mechanism of oligomerisation in the plasma membrane and their intracellular signalling pathways is required.
Owing to the nature of membrane proteins, full-length three-dimensional structures of the Eph receptors will not likely be solved by the traditional techniques, such as NMR and X-ray crystallography. The emerging state-of-the-art cryo-electron microscopy may provide a feasible solution to examine how ephrin binding to the ectodomains of the Eph receptor can convey extracellular cues to intracellular effectors. Recent advances in microscopy techniques, such as total internal reflection fluorescence (TIRF), are likely to cast light on the dynamic oligomerisation process of the Eph receptors in the plasma membrane. Two recent studies used TIRF to study the kinetics of ephrin-induced Eph receptor oligomerisation in the plasma membrane [50, 112]. Ephrin-bound Eph receptor oligomerisation was thus described as a process of nucleation, polymerisation and condensation [50]. Furthermore, the amplitude of the Eph receptor oligomerisation correlated with their intracellular autophosphorylation [50], indicating the oligomerisation state is a key parameter in controlling Eph receptor forward signalling. Most unknowns about Eph receptor functions lie in their downstream signalling pathways. Specifically, how EphA10 and EphB6, the two Eph receptors without tyrosine kinase activity, are able to modulate the Eph receptor signalling is an intriguing question, and is illustrative of the importance of catalysis-independent functions more broadly within the Eph family. An enhanced molecular level understanding of the non-catalytic family members, EphA10 and EphB6, and how they act as molecular switches to regulate their kinase-active Eph receptor counterparts is essential to target the non-catalytic functions of Eph receptors therapeutically, including in cancers.
References
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
McDonell LM, Kernohan KD, Boycott KM, Sawyer SL. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum Mol Genet. 2015;24(R1):R60–6.
Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–80.
Wilkinson DG. Regulation of cell differentiation by Eph receptor and ephrin signaling. Cell Adhes Migr. 2014;8:339–48.
Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5:a009159–a.
Dai D, Huang Q, Nussinov R, Ma B. Promiscuous and specific recognition among ephrins and Eph receptors. Biochim Biophys Acta. 2014;1844:1729–40.
Noberini R, Rubio de la Torre E, Pasquale EB. Profiling Eph receptor expression in cells and tissues: a targeted mass spectrometry approach. Cell Adhes Migr. 2012;6:102–12.
Rohani N, Parmeggiani A, Winklbauer R, Fagotto F. Variable combinations of specific ephrin ligand/Eph receptor pairs control embryonic tissue separation. PLoS Biol. 2014;12:e1001955.
Jacobsen AV, Murphy JM. The secret life of kinases: insights into non-catalytic signalling functions from pseudokinases. Biochem Soc Trans. 2017;45:665–81.
Reiterer V, Eyers PA, Farhan H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol. 2014;24:489–505.
Truitt L, Freywald T, DeCoteau J, Sharfe N, Freywald A. The EphB6 receptor cooperates with c-Cbl to regulate the behavior of breast cancer cells. Cancer Res. 2010;70:1141–53.
Freywald A, Sharfe N, Roifman CM. The kinase-null EphB6 receptor undergoes transphosphorylation in a complex with EphB1. J Biol Chem. 2002;277:3823–8.
Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal. 2014;7:ra114.
Murphy JM, Zhang Q, Young SN, Reese ML, Bailey FP, Eyers PA, et al. A robust methodology to subclassify pseudokinases based on their nucleotide-binding properties. Biochem J. 2014;457:323–34.
Murphy JM, Mace PD, Eyers PA. Live and let die: insights into pseudoenzyme mechanisms from structure. Curr Opin Struct Biol. 2017;47:95–104.
Murphy JM, Lucet IS. A structural perspective of the pseudokinome: defining the targetable space. In: Ward RA, Goldberg FW, editors. Kinase drug discovery: modern approaches. Royal Society of Chemistry; 2018. pp. 359–80. www.rsc.org.
Wiesner S, Wybenga-Groot LE, Warner N, Lin H, Pawson T, Forman-Kay JD, et al. A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 2006;25:4686–96.
Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–57.
Davis TL, Walker JR, Loppnau P, Butler-Cole C, Allali-Hassani A, Dhe-Paganon S. Autoregulation by the juxtamembrane region of the human ephrin receptor tyrosine kinase A3 (EphA3). Structure. 2008;16:873–84.
Kwon A, John M, Ruan Z, Kannan N. Coupled regulation by the juxtamembrane and sterile alpha motif (SAM) linker is a hallmark of ephrin tyrosine kinase evolution. J Biol Chem. 2018;293:5102–16.
Singla N, Erdjument-Bromage H, Himanen JP, Muir TW, Nikolov DB. A semisynthetic Eph receptor tyrosine kinase provides insight into ligand-induced kinase activation. Chem Biol. 2011;18:361–71.
Stapleton D, Balan I, Pawson T, Sicheri F. The crystal structure of an Eph receptor SAM domain reveals a mechanism for modular dimerization. Nat Struct Biol. 1999;6:44–9.
Thanos CD, Goodwill KE, Bowie JU. Oligomeric structure of the human EphB2 receptor SAM domain. Science. 1999;283:833–6.
Stein E, Cerretti DP, Daniel TO. Ligand activation of ELK receptor tyrosine kinase promotes its association with Grb10 and Grb2 in vascular endothelial cells. J Biol Chem. 1996;271:23588–93.
Stein E, Lane AA, Cerretti DP, Schoecklmann HO, Schroff AD, Van Etten RL, et al. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–78.
Han DC, Shen TL, Miao H, Wang B, Guan JL. EphB1 associates with Grb7 and regulates cell migration. J Biol Chem. 2002;277:45655–61.
Borthakur S, Lee H, Kim S, Wang BC, Buck M. Binding and function of phosphotyrosines of the Ephrin A2 (EphA2) receptor using synthetic sterile alpha motif (SAM) domains. J Biol Chem. 2014;289:19694–703.
Shi X, Hapiak V, Zheng J, Muller-Greven J, Bowman D, Lingerak R, et al. A role of the SAM domain in EphA2 receptor activation. Sci Rep. 2017;7:45084.
Singh DR, Ahmed F, Paul MD, Gedam M, Pasquale EB, Hristova K. The SAM domain inhibits EphA2 interactions in the plasma membrane. Biochim Biophys Acta Mol Cell Res. 2017;1864:31–8.
Schaupp A, Sabet O, Dudanova I, Ponserre M, Bastiaens P, Klein R. The composition of EphB2 clusters determines the strength in the cellular repulsion response. J Cell Biol. 2014;204:409–22.
Singh DR, Cao Q, King C, Salotto M, Ahmed F, Zhou XY, et al. Unliganded EphA3 dimerization promoted by the SAM domain. Biochem J. 2015;471:101–9.
Shi X, Hapiak V, Zheng J, Muller-Greven J, Bowman D, Lingerak R, et al. A role of the SAM domain in EphA2 receptor activation. Sci Rep. 2017;7:p45084.
Wimmer-Kleikamp SH, Janes PW, Squire A, Bastiaens PIH, Lackmann M. Recruitment of Eph receptors into signaling clusters does not require ephrin contact. J Cell Biol. 2004;164:661–6.
Janes PW, Griesshaber B, Atapattu L, Nievergall E, Hii LL, Mensinga A, et al. Eph receptor function is modulated by heterooligomerization of A and B type Eph receptors. J Cell Biol. 2011;195:1033–45.
Wang Y, Shang Y, Li J, Chen W, Li G, Wan J, et al. Specific Eph receptor-cytoplasmic effector signaling mediated by SAM–SAM domain interactions. ELife. 2018;7:e35677.
Nievergall E, Lackmann M, Janes PW. Eph-dependent cell-cell adhesion and segregation in development and cancer. Cell Mol Life Sci. 2012;69:1813–42.
Dionne U, Chartier FJM, Lopez de Los Santos Y, Lavoie N, Bernard DN, Banerjee SL, et al. Direct phosphorylation of SRC homology 3 domains by tyrosine kinase receptors disassembles ligand-induced signaling networks. Mol Cell. 2018;70:995–1007 e11.
Kalo MS, Pasquale EB. Multiple in vivo tyrosine phosphorylation sites in EphB receptors. Biochemistry. 1999;38:14396–408.
Lawrenson ID, Wimmer-Kleikamp SH, Lock P, Schoenwaelder SM, Down M, Boyd AW. et al. Ephrin-A5 induces rounding, blebbing and de-adhesion of EphA3-expressing 293T and melanoma cells by CrkII and Rho-mediated signalling. J Cell Sci. 2002;115:1059–72.
Zisch AH, Kalo MS, Chong LD, Pasquale EB. Complex formation between EphB2 and Src requires phosphorylation of tyrosine 611 in the EphB2 juxtamembrane region. Oncogene. 1998;16:2657–70.
Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, et al. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204.
Fang WB, Brantley-Sieders DM, Hwang Y, Ham A-JL, Chen J. Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase. J Biol Chem. 2008;283:16017–26.
Jiang J, Wang ZH, Qu M, Gao D, Liu XP, Zhu LQ, et al. Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt-mediated inactivation of glycogen synthase kinase-3beta. Sci Rep. 2015;5:11765.
Li G, Ji X-D, Gao H, Zhao J-S, Xu J-F, Sun Z-J, et al. EphB3 suppresses non-small-cell lung cancer metastasis via a PP2A/RACK1/Akt signalling complex. Nat Commun. 2012;3:667.
Yang N-Y, Fernandez C, Richter M, Xiao Z, Valencia F, Tice DA, et al. Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell Signal. 2011;23:201–12.
Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20.
Stallaert W, Bruggemann Y, Sabet O, Baak L, Gattiglio M, Bastiaens PIH. Contact inhibitory Eph signaling suppresses EGF-promoted cell migration by decoupling EGFR activity from vesicular recycling. Sci Signal. 2018;11:eaat0114.
Stein E, Huynh-Do U, Lane AA, Cerretti DP, Daniel TO. Nck recruitment to Eph receptor, EphB1/ELK, couples ligand activation to c-Jun kinase. J Biol Chem. 1998;273:1303–8.
Pesti S, Balazs A, Udupa R, Szabo B, Fekete A, Bogel G, et al. Complex formation of EphB1/Nck/Caskin1 leads to tyrosine phosphorylation and structural changes of the Caskin1 SH3 domain. Cell Commun Signal. 2012;10:36.
Ojosnegros S, Cutrale F, Rodriguez D, Otterstrom JJ, Chiu CL, Hortiguela V, et al. Eph-ephrin signaling modulated by polymerization and condensation of receptors. Proc Natl Acad Sci USA. 2017;114:13188–93.
Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–6.
Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–9.
Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor-ephrin complex. Nature. 2001;414:933–8.
Seiradake E, Harlos K, Sutton G, Aricescu AR, Jones EY. An extracellular steric seeding mechanism for Eph-ephrin signaling platform assembly. Nat Struct Mol Biol. 2010;17:398–402.
Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, et al. Architecture of Eph receptor clusters. Proc Natl Acad Sci USA. 2010;107:10860–5.
Xu K, Tzvetkova-Robev D, Xu Y, Goldgur Y, Chan YP, Himanen JP, et al. Insights into Eph receptor tyrosine kinase activation from crystal structures of the EphA4 ectodomain and its complex with ephrin-A5. Proc Natl Acad Sci USA. 2013;110:14634–9.
Seiradake E, Schaupp A, del Toro Ruiz D, Kaufmann R, Mitakidis N, Harlos K, et al. Structurally encoded intraclass differences in EphA clusters drive distinct cell responses. Nat Struct Mol Biol. 2013;20:958–64.
Singh DR, Kanvinde P, King C, Pasquale EB, Hristova K. The EphA2 receptor is activated through induction of distinct, ligand-dependent oligomeric structures. Commun Biol. 2018;1:15.
Bruckner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275:1640–3.
Kalo MS, Yu HH, Pasquale EB. In vivo tyrosine phosphorylation sites of activated ephrin-B1 and ephB2 from neural tissue. J Biol Chem. 2001;276:38940–8.
Lim BK, Matsuda N, Poo MM. Ephrin-B reverse signaling promotes structural and functional synaptic maturation in vivo. Nat Neurosci. 2008;11:160–9.
Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, et al. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell. 2002;9:725–37.
Jorgensen C, Sherman A, Chen GI, Pasculescu A, Poliakov A, Hsiung M, et al. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009;326:1502–9.
Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, et al. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816–9.
Locard-Paulet M, Lim L, Veluscek G, McMahon K, Sinclair J, van Weverwijk A, et al. Phosphoproteomic analysis of interacting tumor and endothelial cells identifies regulatory mechanisms of transendothelial migration. Sci Signal. 2016;9:ra15.
Yin Y, Yamashita Y, Noda H, Okafuji T, Go MJ, Tanaka H. EphA receptor tyrosine kinases interact with co-expressed ephrin-A ligands in cis. Neurosci Res. 2004;48:285–96.
Carvalho RF, Beutler M, Marler KJ, Knoll B, Becker-Barroso E, Heintzmann R, et al. Silencing of EphA3 through a cis interaction with ephrinA5. Nat Neurosci. 2006;9:322–30.
Kao TJ, Kania A. Ephrin-mediated cis-attenuation of Eph receptor signaling is essential for spinal motor axon guidance. Neuron. 2011;71:76–91.
Kullander K, Mather NK, Diella F, Dottori M, Boyd AW, Klein R. Kinase-dependent and kinase-independent functions of EphA4 receptors in major axon tract formation in vivo. Neuron. 2001;29:73–84.
Zhao J, Boyd AW, Bartlett PF. The identification of a novel isoform of EphA4 and ITS expression in SOD1(G93A) mice. Neuroscience. 2017;347:11–21.
Lee J, Nakajima-Koyama M, Sone M, Koga M, Ebisuya M, Yamamoto T, et al. Secreted ephrin receptor A7 promotes somatic cell reprogramming by inducing ERK activity reduction. Stem Cell Rep. 2015;5:480–9.
Holmberg J, Clarke DL, Frisen J. Regulation of repulsion versus adhesion by different splice forms of an Eph receptor. Nature. 2000;408:203–6.
Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147:554–64.
Li Y, Jin L, Ye F, Ma Q, Yang Z, Liu D, et al. Isoform expression patterns of EPHA10 protein mediate breast cancer progression by regulating the E-Cadherin and beta-catenin complex. Oncotarget. 2017;8:30344–56.
Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiainen M, Gstaiger M, et al. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol. 2013;201:467–84.
Koshikawa N, Hoshino D, Taniguchi H, Minegishi T, Tomari T, Nam SO, et al. Proteolysis of EphA2 converts it from a tumor suppressor to an oncoprotein. Cancer Res. 2015;75:3327–39.
Atapattu L, Lackmann M, Janes PW. The role of proteases in regulating Eph/ephrin signaling. Cell Adhes Migr. 2014;8:294–307.
Shintani T, Ihara M, Sakuta H, Takahashi H, Watakabe I, Noda M. Eph receptors are negatively controlled by protein tyrosine phosphatase receptor type O. Nat Neurosci. 2006;9:761–9.
Wimmer-Kleikamp SH, Nievergall E, Gegenbauer K, Adikari S, Mansour M, Yeadon T, et al. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood. 2008;112:721–32.
Astin JW, Batson J, Kadir S, Charlet J, Persad RA, Gillatt D, et al. Competition amongst Eph receptors regulates contact inhibition of locomotion and invasiveness in prostate cancer cells. Nat Cell Biol. 2010;12:1194–204.
Fang WB, Brantley-Sieders DM, Parker MA, Reith AD, Chen J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859–68.
Janes PW, Wimmer-Kleikamp SH, Frangakis AS, Treble K, Griesshaber B, Sabet O, et al. Cytoplasmic relaxation of active Eph controls ephrin shedding by ADAM10. PLoS Biol. 2009;7:e1000215.
Luo H, Wu Z, Tremblay J, Thorin E, Peng J, Lavoie JL, et al. Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J Biol Chem. 2012;287:6819–29.
Wang Y, Shi W, Blanchette A, Peng J, Qi S, Luo H, et al. EPHB6 and testosterone in concert regulate epinephrine release by adrenal gland chromaffin cells. Sci Rep. 2018;8:842.
Shi W, Wang Y, Peng J, Qi S, Vitale N, Kaneda N, et al. EPHB6 controls catecholamine biosynthesis by up-regulating tyrosine hydroxylase transcription in adrenal gland chromaffin cells. J Biol Chem. 2019;294:6871–87.
Akada M, Harada K, Negishi M, Katoh H. EphB6 promotes anoikis by modulating EphA2 signaling. Cell Signal. 2014;26:2879–84.
Bhushan L, Tavitian N, Dey D, Tumur Z, Parsa C, Kandpal RP. Modulation of liver-intestine cadherin (Cadherin 17) expression, ERK phosphorylation and WNT signaling in EPHB6 receptor-expressing MDA-MB-231 cells. Cancer Genom Proteom. 2014;11:239–49.
Matsuoka H, Obama H, Kelly ML, Matsui T, Nakamoto M. Biphasic functions of the kinase-defective Ephb6 receptor in cell adhesion and migration. J Biol Chem. 2005;280:29355–63.
Fox BP, Kandpal RP. Transcriptional silencing of EphB6 receptor tyrosine kinase in invasive breast carcinoma cells and detection of methylated promoter by methylation specific PCR. Biochem Biophys Res Commun. 2006;340:268–76.
Yu J, Bulk E, Ji P, Hascher A, Tang M, Metzger R, et al. The EPHB6 receptor tyrosine kinase is a metastasis suppressor that is frequently silenced by promoter DNA hypermethylation in non-small cell lung cancer. Clin Cancer Res. 2010;16:2275–83.
Peng L, Tu P, Wang X, Shi S, Zhou X, Wang J. Loss of EphB6 protein expression in human colorectal cancer correlates with poor prognosis. J Mol Histol. 2014;45:555–63.
Muller-Tidow C, Diederichs S, Bulk E, Pohle T, Steffen B, Schwable J, et al. Identification of metastasis-associated receptor tyrosine kinases in non-small cell lung cancer. Cancer Res. 2005;65:1778–82.
Cho WCS, Bulk E, Yu J, Hascher A, Koschmieder S, Wiewrodt R, et al. Mutations of the EPHB6 receptor tyrosine kinase induce a pro-metastatic phenotype in non-small cell lung cancer. PLoS ONE. 2012;7:e44591.
Mateo-Lozano S, Bazzocco S, Rodrigues P, Mazzolini R, Andretta E, Dopeso H, et al. Loss of the EPH receptor B6 contributes to colorectal cancer metastasis. Sci Rep. 2017;7:43702.
Toosi BM, El Zawily A, Truitt L, Shannon M, Allonby O, Babu M, et al. EPHB6 augments both development and drug sensitivity of triple-negative breast cancer tumours. Oncogene. 2018;37:4073–93.
Nagano K, Kanasaki S-i, Yamashita T, Maeda Y, Inoue M, Higashisaka K, et al. Expression of Eph receptor A10 is correlated with lymph node metastasis and stage progression in breast cancer patients. Cancer Med. 2013;2:972–7.
Nagano K, Maeda Y, Kanasaki S, Watanabe T, Yamashita T, Inoue M, et al. Ephrin receptor A10 is a promising drug target potentially useful for breast cancers including triple negative breast cancers. J Control Release. 2014;189:72–9.
Nagano K, Yamashita T, Inoue M, Higashisaka K, Yoshioka Y, Abe Y, et al. Eph receptor A10 has a potential as a target for a prostate cancer therapy. Biochem Biophys Res Commun. 2014;450:545–9.
Aasheim HC, Patzke S, Hjorthaug HS, Finne EF. Characterization of a novel Eph receptor tyrosine kinase, EphA10, expressed in testis. Biochim Biophys Acta. 2005;1723:1–7.
Li S, Wu Z, Ma P, Xu Y, Chen Y, Wang H, et al. Ligand-dependent EphA7 signaling inhibits prostate tumor growth and progression. Cell Death Dis. 2017;8:e3122.
Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, et al. EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23:238–48.
Vail ME, Murone C, Tan A, Hii L, Abebe D, Janes PW, et al. Targeting EphA3 inhibits cancer growth by disrupting the tumor stromal microenvironment. Cancer Res. 2014;74:4470–81.
Fox BP, Kandpal RP. Transcriptional silencing of EphB6 receptor tyrosine kinase in invasive breast carcinoma cells and detection of methylated promoter by methylation specific PCR. Biochem Biophys Res Commun. 2006;340:268–76.
Fu DY, Wang ZM, Wang BL, Chen L, Yang WT, Shen ZZ, et al. Frequent epigenetic inactivation of the receptor tyrosine kinase EphA5 by promoter methylation in human breast cancer. Hum Pathol. 2010;41:48–58.
Li S, Zhu Y, Ma C, Qiu Z, Zhang X, Kang Z, et al. Downregulation of EphA5 by promoter methylation in human prostate cancer. BMC Cancer. 2015;15:18.
Staquicini FI, Qian MD, Salameh A, Dobroff AS, Edwards JK, Cimino DF, et al. Receptor tyrosine kinase EphA5 is a functional molecular target in human lung cancer. J Biol Chem. 2015;290:7345–59.
Wang X, Zhang M, Ping F, Liu H, Sun J, Wang Y, et al. Identification and therapeutic intervention of coactivated anaplastic lymphoma kinase, fibroblast growth factor receptor 2, and ephrin type-A receptor 5 kinases in hepatocellular carcinoma. Hepatology. 2019;69:573–86.
Song W, Ma Y, Wang J, Brantley-Sieders D, Chen J. JNK signaling mediates EPHA2-dependent tumor cell proliferation, motility, and cancer stem cell-like properties in non-small cell lung cancer. Cancer Res. 2014;74:2444–54.
Song W, Hwang Y, Youngblood VM, Cook RS, Balko JM, Chen J, et al. Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene. 2017;36:5620–30.
Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H, et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J Clin Investig. 2014;124:2037–49.
Yeddula N, Xia Y, Ke E, Beumer J, Verma IM. Screening for tumor suppressors: loss of ephrin receptor A2 cooperates with oncogenic KRas in promoting lung adenocarcinoma. Proc Natl Acad Sci USA. 2015;112:E6476–85.
Dong M, Spelke DP, Lee YK, Chung JK, Yu CH, Schaffer DV, et al. Spatiomechanical modulation of EphB4-Ephrin-B2 signaling in neural stem cell differentiation. Biophys J. 2018;115:865–73.
Acknowledgements
This work is supported by the Australian Cancer Research Foundation (to LYL, OP and IL), and the Cancer Council of Victoria (to PWJ). IL acknowledges support from the Walter and Eliza Hall Institute. LYL is supported by Melbourne Research Scholarship. JMM is grateful to the NHMRC for fellowship support (1105754). We acknowledge the NHMRC IRIISS and the Victorian State Government Operational Infrastructure Support Scheme.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, LY., Patel, O., Janes, P.W. et al. Eph receptor signalling: from catalytic to non-catalytic functions. Oncogene 38, 6567–6584 (2019). https://doi.org/10.1038/s41388-019-0931-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0931-2
This article is cited by
-
Co-clustering of EphB6 and ephrinB1 in trans restrains cancer cell invasion
Communications Biology (2024)
-
Eph receptors and ephrins in cancer progression
Nature Reviews Cancer (2024)
-
Targeting EphA2: a promising strategy to overcome chemoresistance and drug resistance in cancer
Journal of Molecular Medicine (2024)
-
EphrinA5 regulates cell motility by modulating Snhg15/DNA triplex-dependent targeting of DNMT1 to the Ncam1 promoter
Epigenetics & Chromatin (2023)
-
Recurring EPHB1 mutations in human cancers alter receptor signalling and compartmentalisation of colorectal cancer cells
Cell Communication and Signaling (2023)
