
Abstract
Chronic stress may precipitate psychiatric disorders including anxiety. We reported that Repeated Social Defeat (RSD) in mice increased accumulation of inflammatory monocytes within the brain vasculature, which corresponded with increased interleukin (IL)-1 Receptor 1-mediated activation of endothelia, and augmented anxiety-like behavior. One unknown, however, is the role of immune-activated endothelia in regulating the physiological and behavioral responses to social stress. Thus, we sought to determine the RNA profile of activated endothelia and delineate the pathways by which these endothelia communicate within the brain to influence key responses to social stress. First, endothelial-specific RiboTag mice were exposed to RSD and brain endothelial mRNA profiles from the whole brain and prefrontal cortex were determined using RNAseq. RSD increased expression of cell adhesion molecules (Icam1), inflammatory genes (Lrg1, Lcn2, Ackr1, Il1r1), and cyclooxygenase-2 (Ptgs2/COX-2). In studies with IL-1R1KO mice, there was clear dependence on IL-1R1 on endothelia-associated transcripts including Lrg1, Icam1, Lcn2. Moreover, prostaglandin (PG)E2 was increased in the brain after RSD and Ptgs2 was localized to endothelia, especially within the hypothalamus. Next, a selective COX-2 inhibitor, Celecoxib (CCB), was used with social stress. RSD increased PGE2 in the brain and this was abrogated by CCB. Moreover, CCB reduced RSD-induced Hypothalamic–Pituitary–Adrenal (HPA) axis activation with attenuation of hypothalamic paraventricular neuron activation, hypothalamic Crh expression, and corticosterone in circulation. Production, release, and accumulation of inflammatory monocytes after RSD was COX-2 independent. Nonetheless, CCB blocked anxiety-like behavior in response to RSD. Collectively, social stress stimulated specific endothelia RNA profiles associated with increased cell adhesion, IL-1 and prostaglandin signaling, HPA axis activation, and anxiety.
Similar content being viewed by others


Introduction
Stress-related psychiatric disorders are the most common mental health conditions, with up to 33% of the population experiencing anxiety over the course of their lifetimes [1]. In humans, chronic psychosocial stressors, including low socioeconomic status, caregiver stress, and low perceived social status, increase the risk of developing anxiety and the frequency of anxious symptoms [2,3,4,5]. Moreover, inflammation after stress is associated with these behavioral deficits [6]. Prolonged stress exposure promotes inflammation evident in increased circulating inflammatory cytokines (interleukin(IL)-6, IL-1β), pro-inflammatory transcriptome of blood monocytes, glucocorticoid resistance, and hypothalamic–pituitary–adrenal (HPA) axis dysfunction [7,8,9,10,11]. These behavioral and immunological alterations with chronic stress are recapitulated in mice exposed to Repeated Social Defeat (RSD). RSD causes activation of neurons, endothelia, microglia; increases production of pro-inflammatory and glucocorticoid-insensitive monocytes; increases HPA axis activation and accumulation of monocytes in the brain; and induces anxiety-like behaviors [12,13,14,15].
We previously reported region-specific activation of microglia and neurons after RSD in the prefrontal cortex, hippocampus, hypothalamus, amygdala, lateral septum, and bed nucleus of the stria terminalis [15]. These are regions associated with fear, threat appraisal, and behavior changes [16, 17]. Additionally, RSD induced a reactive brain endothelium with increased expression of cell adhesion molecules (ICAM-1, VCAM-1), selectins (E-selectin, P-selectin), and IL-1 Receptor (IL-1R1) [17, 18]. The reactive endothelium responded to inflammatory signals (i.e., IL-1β) from peripheral monocytes through IL-1R1 and propagated these signals to cells of the CNS [19, 20]. The interaction between inflammatory monocytes (IL-1β) and brain endothelia (IL-1R1) was critical for development of anxiety after RSD [17, 19]. Nonetheless, the pathways induced in immune-activated brain endothelia that regulate the physiological and behavioral responses to social stress are unclear.
One potential mechanism is that monocytic IL-1β induces cyclooxygenase-2 (COX-2) in brain endothelia. COX-2 synthesizes prostaglandin (PG)H2 from membrane arachidonic acid that is converted to other prostaglandins (PGD2, PGE2, PGF2), prostacyclin (PGI2), or thromboxane A2 [21]. Previous reports indicate COX-2 was induced in brain endothelia following peripheral challenge with LPS or IL-1β and the resulting prostaglandins mediated sickness responses and HPA axis activation [22,23,24,25]. Stressors including crowding, isolation, chronic unpredictable stress, and social defeat, increased COX-2 and prostaglandins across several brain regions (prefrontal cortex, hippocampus, hypothalamus, subcortical regions) [26,27,28,29]. COX-2 inhibition during restraint and chronic unpredictable stress also prevented anxiety- and depressive-like behaviors [27, 30]. Thus, we sought to determine the RNA profile of activated endothelia and delineate the pathways by which these endothelia communicate within the brain to influence physiological and behavioral responses to social stress.
Materials and methods
Mice
C57BL/6 and CD-1 mice were obtained from Charles River Laboratories. Male C57BL/6 mice were housed in cohorts of three, while CD-1 aggressors were individually housed. Transgenic lines (Tie2-Rpl22-HA, Cdh5-Rpl22-HA, Il1r1loxP/loxP, and Il1r1r/r) were bred in-house. All procedures were in accordance with NIH Guidelines and were approved by OSU-IACUC.
Repeated social defeat
Mice were subjected to RSD as described in Supplementary Materials [31, 32]. CD-1 aggressor mice were placed into the home cages of experimental mice for 2 h (16:00–18:00) per night for six consecutive nights. During each cycle, submissive behaviors (upright posture, fleeing, crouching) were observed to ensure defeat.
RiboTag HA-immunoprecipitation
Immunoprecipitation of polysome-bound mRNA from brain endothelia was performed in Tie2-Rpl22-HA and Cdh5-Rpl22-HA mice as described [33, 34] and outlined in Supplementary Materials. Tissue was homogenized and HA-ribosomes were immunoprecipitated using Protein A/G magnetic beads with anti-HA. RNA was eluted with high salt buffer and isolated using the RNeasy Micro-Kit.
RNA sequencing
Quality control was performed using PicoAssay and samples with RIN > 8.0 were processed for sequencing. cDNA libraries were generated with the Ovation SoLo RNA-Seq System and sequenced at 40 M single-end bp100 reads. Data was aligned to the mouse mm9 reference genome and differential expression was analyzed using DESeq2 [35,36,37]. Pathway analyses were performed using Ingenuity Pathway Analysis, Cytoscape EnrichmentMap, and PANTHER Overrepresentation analysis.
Gene expression analysis
Total RNA was extracted with Tri-Reagent and reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit. Quantitative real-time (q)-PCR was performed using the TaqMan Gene Expression Assay.
RNAscope
RNAscope labeling of Ptgs2 and Cdh5 was performed using the RNAscope Multiplex Fluorescent Reagent Kit v2 (ACDBio). Brains were fixed with 4% formaldehyde, saturated in sucrose, and sectioned (10 μm). Sections were heated, post-fixed, dehydrated, and subjected to antigen retrieval. Probes (Ptgs2-C1 and Cdh5-C2) were hybridized. Probe amplification was performed according to manufacturer’s instructions. Probe signals were developed using Opal dyes (Ptgs2-Opal 690; Cdh5-Opal 520) and counterstained with DAPI. Images were collected using a Leica SP8 confocal microscope at 40× magnification and were quantified using ImageJ.
Celecoxib treatment
The dosage of Celecoxib (CCB) was selected based on previous reports [38,39,40]. Mice received 10 mg/kg CCB or vehicle i.p. 30 min before each cycle of RSD to ensure CCB concentration peaked during RSD [41].
PGE2 ELISA
Mice were euthanized with CO2 asphyxiation immediately after the final cycle of RSD. PGE2 concentration was determined in clarified brain lysates by ELISA (Cayman). ELISA values were normalized to total protein concentration measured via Bradford assay.
Immunohistochemistry for ΔFosB
Mice were transcardially perfused with PBS and 4% formaldehyde. Brains were post-fixed, saturated in sucrose, sectioned (30 μm), and labeled for ΔFosB. Slides were imaged at 20× magnification and the number of ΔFosB+ cells was determined using ImageJ.
Corticosterone ELISA
Mice were euthanized with CO2 asphyxiation immediately after the final cycle of RSD. Plasma corticosterone was assessed using the corticosterone ELISA kit (Enzo) according to manufacturer’s instructions.
Flow cytometry
Labeling of cell surface antigens in the bone marrow, blood, and brain was performed as described [42]. In brief, Fc receptors were blocked, cells were washed, and cells incubated with select antibodies. Antigen expression was determined using a FACSCalibur cytometer with Cytek nine-color upgrade. Data were analyzed using FlowJo software and positive labeling for each antibody was determined based on isotype-labeled controls.
Pain sensitivity
Tactile mechanical sensitivity was determined using von Frey filaments 14 h after RSD. Mice were placed on a mesh platform in a compartment (8 × 12 × 5.5 cm) and habituated. Filaments were applied to the mid-plantar area of the hind paw and measurements were determined by the up-down method [43]. Withdrawal threshold was determined by averaging the results from the left and right hind paws. Logarithmic transformation of the 50% withdrawal threshold was performed before statistical analysis [44].
Anxiety-like behavior
Anxiety-like behavior was determined using the open field test as described [17, 45] 14 h after the 6th cycle of RSD. In brief, mice were placed in the corner of the test apparatus (40 × 40 × 25 cm Plexiglas box) and activity was recorded for 5 min. Behavior was recorded and analyzed using Fusion software (Omnitech).
Social avoidance
Social avoidance was performed as described 14 h after the 6th cycle of RSD [20, 46]. First, an empty 18 × 8 × 8 cm metal wire enclosure was placed on one side of the open field arena and experimental mice were permitted to explore the enclosure (2.5 min). Then, an unfamiliar CD-1 mouse was placed inside the wire enclosure. Experimental mice were placed into the arena and activity was recorded (2.5 min). The duration of social investigation of the caged mouse was determined using Fusion.
Statistical analysis
GraphPad Prism software was used for analysis of variance (ANOVA) of histological and behavioral data. One-way or two-way ANOVA were used to determine main effects and interactions between factors. Tukey’s HSD was used for post-hoc analysis when main effects and/or interactions were determined.
Results
RSD induced a unique RNA profile in brain endothelia consistent with increased cell adhesion, leukocyte migration, and prostaglandin synthesis
IL-1R1 signaling in endothelia (from monocytes) is critical for prolonged anxiety after RSD [17,18,19]. This study investigated if immune-activated endothelia regulate physiological and behavioral responses to stress. Here, endothelial-specific RiboTag mice were generated by crossing Rpl22-StoploxP/loxP-Hemagglutinin (HA) mice with endothelial-specific Tie2-Cre mice (Fig. 1A). HA expression was confirmed in CD31+ endothelia of Cre+ mice (Fig. 1B). Next, Tie2-Rpl22-HA mice were exposed to RSD and endothelial-specific mRNA from the brain was sequenced (Fig. 1C). RSD caused differential expression of 355 genes (DEGs; 176 decreased & 179 increased) in brain endothelia (Fig. 1D, padj < 0.05). Notably, RSD increased Icam1 (intercellular adhesion molecule 1), Lrg1 (leucine rich alpha-2-glycoprotein 1), Lcn2 (lipocalin 2), Ackr1 (atypical chemokine receptor 1), and Ptgs2 (prostaglandin-endoperoxide synthase 2) in endothelia (Fig. 1E–I, p < 0.02, for each).

A Diagram shows generation of endothelial RiboTag (Tie2-Rpl22-HA) mice. B Representative images of HA (green) and CD31 (red) labeling in the hippocampus of Cre- and Cre+ RiboTag mice. C Experimental design with male Cre+ Tie2-Rpl22-HA mice subjected to Repeated Social Defeat (RSD) or left undisturbed as controls (CON). 14 h following the final cycle of social defeat, the brain was collected, polysome-bound endothelial mRNA was immunoprecipitated, and RNA was sequenced (n = 3; litters = 3). D Volcano plot of differentially expressed genes in brain endothelia between CON and RSD. The gray triangles represent genes that were significantly different between CON and RSD. The blue triangles represent genes that were significantly decreased with |log2FoldChange| >1. The red triangles represent genes that were significantly increased with |log2FoldChange| > 1. From the same RNAseq data set, normalized endothelial mRNA counts of E Icam1, F Lrg1, G Lcn2, H Ackr1, I and Ptgs2 were assessed. Values represent the mean ± SEM, and individual data points are provided. Values with (*) are different (p < 0.05) from controls. J Ingenuity Pathway Analysis of Canonical Pathways inhibited or activated by RSD in endothelia (Z-score). K Ingenuity Pathway Analysis of Upstream Regulators inhibited or activated (Z-score) in endothelia by RSD. L Gene Ontology Biological Processes determined by Cytoscape EnrichmentMap (CEM) that were increased in brain endothelia after RSD. Circle size represents the number of genes within the category and the color of the circle represents the relative strength of connections. (M) Heat map of differentially expressed genes organized by Gene Ontology Biological Processes determined by PANTHER Overrepresentation Analysis. Individuals within stress groups are represented in the heatmap.
Ingenuity Pathway Analysis was used to assess Canonical Pathways and Upstream Regulators in endothelia after RSD. Canonical Pathways decreased by RSD included Integrin-Linked Kinase, Platelet-Derived Growth Factor, and Integrin signaling. Pathways increased by RSD included Endothelin-1, Oxidative Phosphorylation, and Chemokine Signaling (Fig. 1J; p < 0.05, for each). For predicted upstream regulators, α-catenin was decreased by RSD and myriad inflammatory regulators (IKBKB, IKBKG, CCR2, CHUK) and Insulin-Like Growth Factor 1 Receptor were increased (Fig. 1K; p < 0.05, for each). Next, DEGs increased by stress were input into Cytoscape EnrichmentMap to visualize Gene Ontology (GO) Biological Processes. Pathways associated with adhesion, vascular remodeling, and lipid biosynthetic processes (Fig. 1L) were increased in endothelia following RSD. DEGs were also used in PANTHER overrepresentation analysis and highly enriched GO Biological Processes were represented in a heat map (Fig. 1M). RSD influenced biological processes in endothelia associated with regulation of cell migration (Ptgs2, Lcn2), cellular respiration (Atp5k, Cycs), cell–cell adhesion (Cd200, Nrcam), leukocyte migration (Icam1, Cxcl12), and insulin stimulus (Gpld1, Igf2) (p < 0.05, for each). Overall, activation of brain endothelia after RSD was associated with increased leukocyte adhesion and migration, cellular respiration, and prostaglandin synthesis.
The Prefrontal Cortex (PFC) is a key region for stress-induced endothelial IL-1R1 expression and recruitment of peripheral monocytes [17]. In parallel experiments, endothelial-specific (inducible) RiboTag mice were generated by crossing Rpl22-StoploxP/loxP-HA mice and Cdh5-CreERT2 mice to prevent nonspecific HA expression during development [47,48,49] (Fig. S1A). HA expression was confirmed in CD31+ endothelia of Cre+ mice after tamoxifen injections (Fig. S1B). Next, Cdh5-Rpl22-HA mice were exposed to RSD and endothelial-specific mRNA from the PFC was sequenced (Fig. S1C). In the PFC, there was differential expression of 86 genes with RSD (9 increased and 77 decreased, Fig. S1D). Ackr1, Lrg1, Lcn2, and Il1r1 (Interleukin-1 Receptor Type 1) were increased by RSD (Fig. S1E–H, padj < 0.05, for each). Mfsd2a and Cldn5 (Claudin-5) were decreased by RSD (Fig. S1I, J, padj < 0.05, for each). PANTHER Overrepresentation Analysis showed that the DEGs after RSD were associated with Response to Cytokines, Cell Migration, and Regulation of Endothelial Proliferation pathways (Fig. S1K, p < 0.05). Thus, RSD elicited an RNA profile in PFC-endothelia consistent with increased cytokine signaling, cell migration, and IL-1R1 signaling.
Immune-induced endothelial activation after RSD was dependent on IL-1R1
Endothelial-selective profiling after RSD showed a unique profile associated with immune activation, cell adhesion, and cell migration. This profile was regulated by activators of the transcription factor NF-κB (Figs. 1 and S1), which is key target of IL-1R1 signaling [50]. Thus, the influence of IL-1R1 signaling on endothelial genes induced by RSD in the brain and PFC was investigated. Here, Il1r1WT and Il1r1KO mice (Fig. 2A) were exposed to RSD (Fig. 2B) and the PFC, HPC, and HYP were collected. Expression of several endothelial genes associated with stress in RiboTag mice (Cldn5, Lrg1, Icam1, Lcn2, Ptgs2) were determined in Il1r1WT and Il1r1KO mice. Cldn5 was not influenced by RSD or IL-1R1 in the PFC (Fig. 2C), but was increased by RSD in the HPC (Fig. 2D; F(1,16) = 5.687, p < 0.03). Cldn5 was increased by RSD in the HYP and this was IL-1R1-dependent (interaction, F(1,18) = 7.005, p < 0.02). Lrg1 expression was increased by RSD in the PFC, HPC, and HYP (Fig. 2F–H; F(1,17) = 21.0, p < 0.001, for each) and was IL-1R1-dependent only in the HPC (interaction, F(1,16) = 9.0, p < 0.01). Icam1 and Lcn2 in the PFC, HPC, and HYP were increased by RSD (Fig. 2I–N; F(1,18) = 12.7, p < 0.003, for each) and were IL-1R1-dependent (interaction, F(1,18) = 4.2, p < 0.06, for each). Finally, Ptgs2 was increased by RSD in the PFC and HYP (Fig. 2O–Q, F(1,11) = 6.987, p < 0.03). This was IL-1R1-dependent only in the HYP (interaction, F(1,13) = 47.93, p < 0.001). Overall, most genes induced by RSD were region- and IL-1R1-dependent.

A Diagram of “wild-type IL-1R1” (IL-1R1loxP/loxP) and IL-1R1KO (IL-1R1r/r) mice. Phenotypic WT controls have loxP sites inserted into the Il1r1 gene but express Il1r1 in all cells. IL-1R1KO mice have a disruption sequence in exon 9 of the Il1r1 gene leading to functional knockout of Il1r1 in all cells. B IL-1R1WT and IL-1R1KO mice were subjected to Repeated Social Defeat (RSD) or left undisturbed as controls (CON) (litters = 2 per genotype). 14 h following the final cycle of social defeat, the brain was microdissected and RNA was collected from the prefrontal cortex (PFC), hippocampus (HPC), and hypothalamus (HYP). RNA levels of C–E Cldn5, F–H Lrg1, I–K Icam1, L–N Lcn2, O–Q and Ptgs2 were determined from the prefrontal cortex, hippocampus, and hypothalamus (n = 3). Data are represented as fold change from the WT-CON group. Bars represent the mean ± SEM, and individual data points are provided. Letters (a,b,c) represent post-hoc analysis of multiple comparisons (Tukey’s HSD) and denote statistically different groups (p < 0.05). Italicized letters (b) denote groups that tended to be different from WT-CON (p < 0.10).
PGE2 increased in the brain after RSD and Ptgs2 was increased in the PFC and PVN
Endothelial-selective profiling after RSD indicated that prostaglandin synthesis was increased (Fig. 1). PGE2 is a key prostaglandin generated by cyclooxygenase-2 (COX-2 or Ptgs2) and is associated with fever, edema, leukocyte recruitment, inflammation, and pain [25, 51,52,53]. Here, C57BL/6 mice were subjected to RSD and PGE2 concentration was determined in whole brain homogenates immediately after 1, 3, and 6 cycles of RSD. PGE2 was increased in the brain after 6 cycles of RSD (Fig. 3B; F(3,17) = 7.703, p < 0.002).

A Experimental design with male C57BL/6 mice subjected to Repeated Social Defeat (RSD) or left undisturbed as controls (CON). Immediately following the final cycle of social defeat, brain samples were collected for prostaglandin (PGE2) and RNA analyses. B PGE2 concentration (μg/mg total protein) in whole brain homogenates collected after RSD. Graph contains pooled data from three separate cohorts (1, 3, 6 cycles; n = 3, 3, 5 respectively). C Representative images of RNAscope labeling of Cdh5 RNA (red) and Ptgs2 RNA (green) in the prefrontal cortex (40×). Arrows and inset show Cd5h+/Ptgs2+ (yellow) cells in the PFC. D Percent area of Ptgs2 RNA labeling in the PFC (n = 4). E Representative images of RNAscope labeling of Cdh5 RNA (red) and Ptgs2 RNA (green) and F percent area of Ptgs2 RNA labeling in the DG (n = 4). G Representative images of RNAscope labeling of Cdh5 RNA (red) and Ptgs2 RNA (green) in the PVN. Arrows and inset show Cdh5+/Ptgs2+ (yellow) cells in the PVN. H Percent area of Ptgs2 RNA labeling in the PVN (n = 4). Bars represent the mean ± SEM, and individual data points are provided. Values with (*) are different (p < 0.05) from CON.
Next, RNAscope was used to assess region-specific expression of Ptgs2. C57BL/6 mice were subjected to RSD and Ptgs2 and Cdh5 (endothelial marker [48, 49]) was determined in three stress-responsive brain regions (PFC, HPC, and HYP). In controls, Ptgs2 was expressed in Cdh5− cells of the PFC and dentate gyrus (DG). Ptgs2 was increased by RSD in the PFC (Fig. 3C, D; p < 0.02). Increased PFC Ptgs2 labeling was evident in both and Cdh5- and Cdh5+ cells (inset). In the DG, Ptgs2 was highly expressed (Cdh5-, non-endothelia) at baseline and was not influenced by RSD (Fig. 3E, F). In the Paraventricular Nucleus (PVN) of the HYP, baseline expression of Ptgs2 was minimal. RSD increased Ptgs2 expression in the PVN only in Cdh5+ endothelia (Fig. 3G, H, p < 0.05). Overall, stress increased prostaglandin production and enhanced endothelial expression of Ptgs2 in the PFC and HYP.
COX-2 inhibition with Celecoxib attenuated RSD-induced PGE2 signaling and HPA axis activation
Ptgs2 encodes COX-2, the enzyme that generates PGE2 in the brain. Thus, we investigated if inhibition of COX-2 with Celecoxib (CCB) blocked the physiological and behavioral responses to social stress. C57BL/6 mice were injected (i.p.) with CCB (10 mg/kg) 30 min prior to each cycle of RSD (Fig. 4A). RSD increased PGE2 concentration in the brain immediately after RSD (Fig. 4B; F(1,35) = 14.2, p < 0.001) that tended to be CCB-dependent (interaction, F(1,35) = 3.3, p = 0.08). Post-hoc analysis confirmed that PGE2 was highest in the RSD-Veh group (p < 0.002) compared to all groups (p < 0.02). Thus, brain PGE2 induction after RSD was attenuated by CCB.

A Experimental design with male C57BL/6 mice receiving injections of vehicle (0.25% carboxymethylcellulose) or 10 mg/kg celecoxib (CCB) 30 min prior to each cycle of social defeat. In the first experiment, the brain was collected for PGE2 and RNA analyses and plasma was collected for corticosterone immediately after the final cycle of RSD. B PGE2 concentration (μg/mg of total protein) in whole brain homogenates normalized to Con-Veh group collected immediately after RSD (n = 9). In a parallel study, the brain was microdissected and RNA was collected from the hypothalamus (HYP). RNA levels of C Ptgs2 and D Crh were determined in the HYP (n = 6). E Concentration of corticosterone was determined in the plasma after RSD (n = 6). F Representative images of ΔFosB labeling (20×) in the prefrontal cortex (PFC) and G number of positive ΔFosB cells in the PFC (n = 4). H Representative images of ΔFosB labeling and I number of positive ΔFosB cells in the DG (n = 4). J Representative images of ΔFosB labeling and K number of positive ΔFosB cells in the PVN (n = 3). Bars represent the mean ± SEM, and individual data points are provided. Letters (a,b) represent post-hoc analysis of multiple comparisons (Tukey’s HSD) and denote statistically different groups (p < 0.05).
Ptgs2 was increased in PVN endothelia after stress (Fig. 3G). The PVN is critical in HPA axis activation by releasing corticotrophin releasing hormone (CRH) during stress. Ptgs2 and Crh levels were determined in the HYP. RSD increased Ptgs2 in the HYP (F(1,20) = 31.3, p < 0.001) which was unaffected by COX-2 inhibition (Fig. 4C). RSD also increased Crh in the HYP (F(1,20) = 8.171, p < 0.01) which was COX-2-dependent (Fig. 4D; interaction, F(1,20) = 10.07, p < 0.005). HPA axis activation results in release of glucocorticoids into circulation. Thus, corticosterone was determined in plasma after RSD. Plasma corticosterone was increased by RSD (F(1,19) = 8.4, p < 0.01) and tended to be COX-2-dependent (interaction, F(1,19) = 3.1, p = 0.1). Post-hoc analysis confirmed that RSD-Veh mice had the highest amount of plasma corticosterone compared to all other groups (Fig. 4E; p < 0.05).
Next, neuronal activation in stress-responsive brain regions was assessed. RSD increased the number of ΔFosB+ cells in the PFC (Fig. 4G, H; F(1,17) = 13.8, p < 0.003) and DG (Fig. 4I, J; F(1,16) = 17.2, p < 0.002) independent of COX-2. ΔFosB+ cells in the PVN were also increased by RSD and this induction was COX-2-dependent (Fig. 4K, L; interaction, F(1,15) = 9.9, p < 0.008). Overall, inhibition of COX-2 with CCB prevented stress-induced activation of PVN neurons which was associated with reduced Crh and plasma corticosterone.
COX-2 inhibition with Celecoxib attenuated RSD-induced anxiety
RSD increases microglial activation and recruitment of monocytes to the brain. This stress-dependent enhancement of neuroinflammation influences behavioral, pain, and cognitive processes [15, 17, 20, 46, 54]. In general, social avoidance after RSD was monocyte/IL-1β-independent [55], pain was microglia-dependent [54], and anxiety was monocyte/IL-1β/eIL-1R1-dependent [17, 19]. Thus, these interactions were investigated with COX-2 inhibition by CCB. Mice were injected with CCB prior to each cycle of RSD (Fig. 5A) and monocyte production, release, and accumulation were determined. RSD increased production of myeloid cells in the bone marrow including neutrophils (F(1,19) = 32.5, p < 0.001) and monocytes (F(1,19) = 16.3, p < 0.001, Fig. 5B, C). In parallel, RSD reduced B-cells and red blood cells (RBC) (Fig. 5B). All these bone marrow changes with RSD were COX-2-independent. RSD also increased inflammatory (Ly6Chi) monocytes in circulation (F(1,15) = 16.9, p < 0.002) and brain (F(1,14) = 11.84, p < 0.004). Monocyte accumulation in the blood and brain after RSD were both COX-2-independent. Overall, monocyte production, release, and brain accumulation after RSD did not require COX-2.

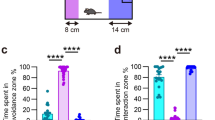
A Experimental design with male C57BL/6 mice receiving injections of vehicle (0.25% carboxymethylcellulose) or 10 mg/kg celecoxib (CCB) 30 min prior to each cycle of social defeat. In the first experiment, percentages of Ly6Chi monocytes were determined in the bone marrow, blood and brain (n = 6). B Percentage of B-cells (Ter119−/CD11b−/B220+), neutrophils (Ter119−/CD11b+/Ly6G+), and red blood cells (Ter119+) in the bone marrow. C Percentage of LyC6hi monocytes (Ter119-/CD11b+/Ly6Chi) in the bone marrow. D Percentage of CD11b+/Ly6Chi monocytes in the blood (n = 4) E Representative bivariate dot plots and gating strategy for inflammatory monocytes in the brain. F Percentage of inflammatory monocytes (CD11b+/CD45hi/Ly6Chi) in the brain (of total CD11b+ cells). Bars represent the mean ± SEM, and individual data points are provided. G In a separate study, mechanical allodynia was assessed (n = 6). H Body weights of mice assessed in Von Frey. I Normalized Von Frey withdrawal threshold after 0, 1, 6 cycles of RSD (logarithmic transformation). J In a separate study, social avoidance was assessed in a two-trial social interaction paradigm. Gray areas indicate interaction and corner zones. The duration within the interaction zone during K empty trial and L during social trial. Parallel to these experiments, anxiety-like behavior in the open field was assessed (n = 25). M Representative heat maps of duration in zones during open field test. N Distance traveled in the open field. O Total duration in center zone of open field. Bars represent the mean ± SEM. Letters (a,b,c) represent post-hoc analysis of multiple comparisons (Tukey’s HSD) and denote statistically different groups (p < 0.05). Italicized letters (b) denote groups that tended to be different from CON-Veh (p = 0.10).
RSD increased mechanical allodynia that was dependent on inflammation [54]. Because PGE2 is a mediator of pain [52], we assessed if COX-2 inhibition influenced pain sensitivity after RSD (Fig. 5G). Baseline (0 cycle) withdrawal threshold was determined. Next, C57BL/6 mice were injected with CCB prior to each cycle of RSD and mechanical allodynia was determined 14 h after the 1st and 6th cycle. There were no differences in body weight between groups (Fig. 5H). There was an exposure-dependent increase in pain sensitivity after RSD (F(6,40) = 4.046, p < 0.003) that was independent of COX-2 (Fig. 5I).
Next, C57BL/6 mice were injected with CCB prior to each cycle of RSD and social avoidance and anxiety-like behavior were determined 14 h later (Fig. 5A). Social avoidance was determined using a two-trial paradigm (Fig. 5J). During the empty trial, all mice explored the empty cage and spent most time within the interaction zone (Fig. 5K). During the social trial with the CD-1 mouse, however, time spent in the interaction zone was reduced by RSD (F(1,107) = 15.8, p < 0.001, Fig. 5L), independent of COX-2 inhibition. In the open field, representative heat maps of duration are provided (Fig. 5M). There were no differences in total locomotion across groups (Fig. 5N). Time spent in the center of the open field tended to be decreased by RSD (F(1,85) = 2.971, p = 0.09) and was prevented by COX-2 inhibition (Fig. 5O; interaction, F(1,85) = 5.2, p < 0.03). Post-hoc analysis confirmed that the RSD-Veh group had the least amount of center time compared to all other groups (p < 0.05). Overall, of the monocyte, pain, and social behavior related-parameters examined here, only anxiety in the open field required the COX-2/PGE2 pathway.
Discussion
Repeated Social Defeat (RSD) increases the accumulation of inflammatory monocytes within the brain vasculature, which corresponds with increased IL-1R1-mediated activation of endothelia, neuronal activation, and anxiety-like behavior [17, 19, 20]. Here, we focused on understanding immune-activated brain endothelia in regulating key responses to RSD (Fig. S2). RNA profiling of endothelia after RSD revealed an active role of endothelia in leukocyte adhesion (Lrg1, Lcn2), leukocyte recruitment (Icam1), IL-1R1 signaling (Il1r1) and prostaglandin synthesis (Ptgs2). These data are consistent with previous reports using global brain assessments [17, 18, 56]. Pathway analysis revealed these gene changes were mediated by activators of NF-κB, a key transcription factor associated with IL-1R1-activation. Furthermore, endothelial-associated transcripts induced by stress including Icam1, Lrg1, Lcn2, and Ptgs2 were IL-1R1-dependent. Notably, a previous study with social defeat showed reduced blood-brain barrier (BBB) integrity with decreased expression of the tight junction protein Claudin-5 (Cldn5). Decreased BBB integrity in the Nucleus Accumbens was associated with increased IL-6 and social avoidance [57]. Here, Cldn5 expression was decreased in the PFC after RSD (endothelial RNA-sequencing). The results from qPCR studies of PFC, HYP, and HPC were less consistent, but these evaluations were not cell-specific. Based on the presence of inflammatory monocytes, the endothelial profile, and previous evidence for decreased integrity, RSD may elicit localized BBB permeability. For example, Lcn2 is associated with glial activation, neuronal death, and BBB leakage after LPS challenge and stroke induction [58,59,60,61] and was increased in endothelia after RSD. One limitation was that only male mice were used. Nonetheless, RSD in female mice increased monocyte trafficking, IL-1β production in the brain, and anxiety [42]. Therefore, we predict that RSD in female elicits similar IL-1R1-mediated changes in brain endothelia. In fact, a recent study using female mice showed that social defeat increased expression of inflammatory mediators (Il18, Ccl19, Ccr5, Hsp90b1) in brain endothelia [62]. Overall, there was a unique RNA profile in brain endothelia that developed after RSD consistent with increased cell adhesion, leukocyte migration, IL-1R1 signaling, and reduced BBB integrity.
Another relevant finding was that Ptgs2/COX-2 was induced in brain endothelia by RSD. Previous studies reported increased COX-2 expression in homogenates of the PFC and HPC with social isolation and chronic unpredictable stress [27, 28]. We extend these findings by showing vascular (Cdh5+) and non-vascular (Cdh5−, likely neuronal) induction of COX-2 after RSD. COX-2 induction was region-dependent and increased in the PFC (endothelia and neuronal) and PVN (endothelia only), but not the DG. Notably, neurons in the cortex and HPC express COX-2 constitutively with expression levels coupled to excitatory neuronal activity [63, 64]. Thus, increased neuronal activity in the PFC after RSD likely promoted COX-2 expression in these neurons [15, 17, 46]. Here, COX-2 induction in the HYP was dependent on IL-1R1. This is consistent with a previous report that (i.v.) IL-1β injection caused robust cerebrovascular expression of COX-2 [23]. Collectively, IL-1R1 promoted vascular COX-2 expression after RSD in the HYP and PFC while non-vascular COX-2 expression in the PFC is likely mediated by neuronal activity.
One novel finding was COX-2 inhibition attenuated stress-induced activation of the HPA axis. CCB prevented hypothalamic expression of Crh and increased corticosterone in circulation after RSD. These data are consistent with studies showing vascular COX-2 in the PVN mediates HPA axis activation through PGE2 signaling [65,66,67,68]. Neuronal activation (ΔFosB) after stress was prevented in the PVN by COX-2 inhibition. Moreover, COX-2 expression was localized to endothelia in the PVN after stress. This stress-associated neuronal activation in the PVN also indicates that COX-2 activation of the HPA axis is important to the response to social stressors.
CCB did not prevent production, release, trafficking, and accumulation of inflammatory (Ly6Chi) monocytes in the brain after stress. We previously reported that monocyte recruitment to the brain with RSD depended on microglia and IL-1R1 signaling [17, 19, 20]. Thus, the current findings indicate that COX-2 activity was downstream of monocyte-to-endothelial signaling via IL-1R1. Likewise, we previously reported that corticosterone influenced monocyte release from the bone marrow into circulation and subsequent recruitment to the brain [14]. Both pharmacologic inhibition of corticosterone synthesis and adrenalectomy before stress prevented monocyte release into circulation [14]. Here, COX-2 inhibition also reduced corticosterone levels in plasma after RSD, but monocyte accumulation in the brain was still evident. Explanations for these differences are related to level of inhibition and timing. In previous experiments, our interventions ablated corticosterone throughout RSD, while COX-2 inhibition with CCB attenuated corticosterone at one specific time point (immediately after the last cycle). The presence of monocytes was time-dependent and requires at least 3 repeated cycles of RSD [69]. In this study, PGE2 levels in the brain were increased after 6 cycles of RSD, but not 1 cycle. Collectively, inflammatory monocytes reinforced HPA activation and corticosterone release via a COX-2-dependent pathway.
Another key finding was RSD-induced anxiety-like behavior in the open field was prevented by CCB. These results indicate that prostaglandins were critical in promoting RSD-induced anxiety. These data are consistent with studies showing COX-2 inhibition modulated calcium currents in the amygdala and prevented anxiety-like behavior after restraint stress and chronic corticosterone administration [30, 70]. Furthermore, blocking PGE2 signaling with an EP1 receptor knockout prevented anxiety-like behavior after social defeat [71]. Moreover, recent clinical studies indicate that Celecoxib was an effective adjunct therapy for anxiety disorders in both men and women [72, 73]. Our data connect these findings to show that social stress-induced PGE2 synthesis via endothelial COX-2 modulated neuronal activity and HPA activation and was essential for anxiety, providing support for the effectiveness of CCB in humans with anxiety. These data are also consistent with previous data that anxiety-like behavior after stress was blocked by endothelial IL-1R1 knockdown [19], microglial inhibition/elimination, and in mice with caspase-1 deficient monocytes [17]. These interventions affected monocyte recruitment to the brain or their ability to communicate with endothelia through IL-1β/IL-1R1. The extension here is that CCB blocked COX-2 activity, presumably in brain endothelia, preventing endothelial interpretation of IL-1R1 activation and release of prostaglandins. Thus, anxiety was the only behavior tested here that was COX-2-dependent. For example, mechanical allodynia was increased by RSD, but was COX-2-independent. While COX-2 inhibitors are common pain relievers [74], the effects of CCB on RSD-induced anxiety were unrelated to pain. Next, CCB did not influence neuronal activation in the PFC and HPC after RSD, suggesting that threat appraisal and fear after social defeat were independent of COX-2. As such, RSD-induced social avoidance of an unfamiliar CD-1 was independent of CCB. This finding is in line with previous reports that social avoidance developed after 1 cycle of RSD, was independent of monocyte accumulation and communication with endothelia, and was COX-2-independent [17, 55, 71]. In conclusion, brain endothelia and COX-2 regulate neuroinflammation, neuroendocrine activity, and anxiety-like responses to RSD.
References
Bandelow B, Michaelis S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin Neurosci. 2015;17:327–35.
Dura JR, Stukenberg KW, Kiecolt-Glaser JK. Anxiety and depressive disorders in adult children caring for demented parents. Psychol Aging. 1991;6:467–73.
Bjorkenstam E, Burstrom B, Vinnerljung B, Kosidou K. Childhood adversity and psychiatric disorder in young adulthood: an analysis of 107,704 Swedes. J Psychiatr Res. 2016;77:67–75. https://doi.org/10.1016/j.jpsychires.2016.02.018.
Scott KM, Al-Hamzawi AO, Andrade LH, Borges G, Caldas-de-Almeida JM, Fiestas F, et al. Associations between subjective social status and DSM-IV mental disorders: results from the World Mental Health surveys. JAMA Psychiatry. 2014;71:1400–8. https://doi.org/10.1001/jamapsychiatry.2014.1337.
Cole SW. Social regulation of human gene expression. Curr Dir Psychol Sci. 2009;18:132–7. https://doi.org/10.1111/j.1467-8721.2009.01623.x.
Salim S, Chugh G, Asghar M. Inflammation in anxiety. Adv protein Chem Struct Biol. 2012;88:1–25. https://doi.org/10.1016/B978-0-12-398314-5.00001-5.
Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, et al. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA. 2013;110:16574–9. https://doi.org/10.1073/pnas.1310655110.
Brydon L, Edwards S, Jia H, Mohamed-Ali V, Zachary I, Martin JF, et al. Psychological stress activates interleukin-1beta gene expression in human mononuclear cells. Brain, Behav, Immun. 2005;19:540–6. https://doi.org/10.1016/j.bbi.2004.12.003.
Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. 2002;21:531–41. https://doi.org/10.1037//0278-6133.21.6.531.
Gouin JP, Glaser R, Malarkey WB, Beversdorf D, Kiecolt-Glaser J. Chronic stress, daily stressors, and circulating inflammatory markers. Health Psychol. 2012;31:264–8. https://doi.org/10.1037/a0025536.
Thoma MV, Gianferante D, Hanlin L, Fiksdal A, Chen X, Rohleder N. Stronger hypothalamus-pituitary-adrenal axis habituation predicts lesser sensitization of inflammatory response to repeated acute stress exposures in healthy young adults. Brain, Behav, Immun. 2017;61:228–35. https://doi.org/10.1016/j.bbi.2016.11.030.
Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP. Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology. 2012;37:1491–505. https://doi.org/10.1016/j.psyneuen.2012.02.003.
Avitsur R, Stark JL, Sheridan JF. Social stress induces glucocorticoid resistance in subordinate animals. Hormones Behav. 2001;39:247–57. https://doi.org/10.1006/hbeh.2001.1653.
Niraula A, Wang Y, Godbout JP, Sheridan JF. Corticosterone production during repeated social defeat causes monocyte mobilization from the bone marrow, glucocorticoid resistance, and neurovascular adhesion molecule expression. J Neurosci. 2018;38:2328–40. https://doi.org/10.1523/JNEUROSCI.2568-17.2018.
Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–88. https://doi.org/10.1523/JNEUROSCI.0450-11.2011.
Robinson OJ, Pike AC, Cornwell B, Grillon C. The translational neural circuitry of anxiety. J Neurol Neurosurg Psychiatry. 2019;90:1353–60. https://doi.org/10.1136/jnnp-2019-321400.
McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. 2018;23:1421–31. https://doi.org/10.1038/mp.2017.64.
Sawicki CM, McKim DB, Wohleb ES, Jarrett BL, Reader BF, Norden DM, et al. Social defeat promotes a reactive endothelium in a brain region-dependent manner with increased expression of key adhesion molecules, selectins and chemokines associated with the recruitment of myeloid cells to the brain. Neuroscience. 2015;302:151–64. https://doi.org/10.1016/j.neuroscience.2014.10.004.
Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci. 2014;34:2583–91. https://doi.org/10.1523/JNEUROSCI.3723-13.2014.
DiSabato DJ, Nemeth DP, Liu X, Witcher KG, O’Neil SM, Oliver B, et al. Interleukin-1 receptor on hippocampal neurons drives social withdrawal and cognitive deficits after chronic social stress. Mol Psychiatry. 2020. https://doi.org/10.1038/s41380-020-0788-3.
Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–73.
Liu X, Yamashita T, Chen Q, Belevych N, McKim DB, Tarr AJ, et al. Interleukin 1 type 1 receptor restore: a genetic mouse model for studying interleukin 1 receptor-mediated effects in specific cell types. J Neurosci. 2015;35:2860–70. https://doi.org/10.1523/JNEUROSCI.3199-14.2015.
Matsuwaki T, Eskilsson A, Kugelberg U, Jonsson JI, Blomqvist A. Interleukin-1beta induced activation of the hypothalamus-pituitary-adrenal axis is dependent on interleukin-1 receptors on non-hematopoietic cells. Brain, Behav, Immun. 2014;40:166–73. https://doi.org/10.1016/j.bbi.2014.03.015.
Murray CL, Skelly DT, Cunningham C. Exacerbation of CNS inflammation and neurodegeneration by systemic LPS treatment is independent of circulating IL-1beta and IL-6. J Neuroinflammation. 2011;8:50 https://doi.org/10.1186/1742-2094-8-50.
Ivanov AI, Romanovsky AA. Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci. 2004;9:1977–93. https://doi.org/10.2741/1383.
Gadek-Michalska A, Bugajski AJ, Bugajski J. Prostaglandins and interleukin-1beta in the hypothalamic-pituitary-adrenal response to systemic phenylephrine under basal and stress conditions. J Physiol Pharm. 2008;59:563–75.
Song Q, Fan C, Wang P, Li Y, Yang M, Yu SY. Hippocampal CA1 betaCaMKII mediates neuroinflammatory responses via COX-2/PGE2 signaling pathways in depression. J Neuroinflammation. 2018;15:338 https://doi.org/10.1186/s12974-018-1377-0.
Zlatkovic J, Todorovic N, Boskovic M, Pajovic SB, Demajo M, Filipovic D. Different susceptibility of prefrontal cortex and hippocampus to oxidative stress following chronic social isolation stress. Mol Cell Biochem. 2014;393:43–57. https://doi.org/10.1007/s11010-014-2045-z.
Nie X, Kitaoka S, Shinohara M, Kakizuka A, Narumiya S, Furuyashiki T. Roles of Toll-like receptor 2/4, monoacylglycerol lipase, and cyclooxygenase in social defeat stress-induced prostaglandin E2 synthesis in the brain and their behavioral relevance. Sci Rep. 2019;9:17548 https://doi.org/10.1038/s41598-019-54082-5.
Morgan A, Gaulden A, Altemus M, Williford K, Centanni S, Winder D, et al. Cyclooxygenase-2 inhibition prevents stress induced amygdala activation and anxiety-like behavior. Brain, Behav, Immun. 2020;89:513–7. https://doi.org/10.1016/j.bbi.2020.07.046.
Avitsur R, Stark JL, Dhabhar FS, Sheridan JF. Social stress alters splenocyte phenotype and function. J Neuroimmunol. 2002;132:66–71.
Avitsur R, Stark JL, Dhabhar FS, Kramer KA, Sheridan JF. Social experience alters the response to social stress in mice. Brain Behav Immun. 2003;17:426–37. https://doi.org/10.1016/s0889-1591(03)00034-5.
Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA. 2009;106:13939–44. https://doi.org/10.1073/pnas.0907143106.
Rakers C, Schleif M, Blank N, Matuskova H, Ulas T, Handler K, et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia. 2019;67:619–33. https://doi.org/10.1002/glia.23544.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. https://doi.org/10.1093/bioinformatics/bts635.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550 https://doi.org/10.1186/s13059-014-0550-8.
Weber MD, McKim DB, Niraula A, Witcher KG, Yin W, Sobol CG, et al. The influence of microglial elimination and repopulation on stress sensitization induced by repeated social defeat. Biol Psychiatry. 2018. https://doi.org/10.1016/j.biopsych.2018.10.009.
Guo JY, Li CY, Ruan YP, Sun M, Qi XL, Zhao BS, et al. Chronic treatment with celecoxib reverses chronic unpredictable stress-induced depressive-like behavior via reducing cyclooxygenase-2 expression in rat brain. Eur J Pharmacol. 2009;612:54–60. https://doi.org/10.1016/j.ejphar.2009.03.076.
Maciel IS, Silva RB, Morrone FB, Calixto JB, Campos MM. Synergistic effects of celecoxib and bupropion in a model of chronic inflammation-related depression in mice. PloS One. 2013;8:e77227. https://doi.org/10.1371/journal.pone.0077227.
EM EL, Zeid MS, Kawy HS, Hendawy N, Baher W. Celecoxib attenuates depressive-like behavior associated with immunological liver injury in C57BL/6 mice through TNF-alpha and NF-kappab dependent mechanisms. Life Sci. 2016;163:23–37. https://doi.org/10.1016/j.lfs.2016.08.027.
Paulson SK, Zhang JY, Breau AP, Hribar JD, Liu NW, Jessen SM, et al. Pharmacokinetics, tissue distribution, metabolism, and excretion of celecoxib in rats. Drug Metab Dispos. 2000;28:514–21.
Yin W, Gallagher NR, Sawicki CM, McKim DB, Godbout JP, Sheridan JF. Repeated social defeat in female mice induces anxiety-like behavior associated with enhanced myelopoiesis and increased monocyte accumulation in the brain. Brain, Behav, Immun. 2019;78:131–42. https://doi.org/10.1016/j.bbi.2019.01.015.
Laumet G, Edralin JD, Dantzer R, Heijnen CJ, Kavelaars A. CD3(+) T cells are critical for the resolution of comorbid inflammatory pain and depression-like behavior. Neurobiol Pain. 2020;7:100043 https://doi.org/10.1016/j.ynpai.2020.100043.
Mills C, Leblond D, Joshi S, Zhu C, Hsieh G, Jacobson P, et al. Estimating efficacy and drug ED50’s using von Frey thresholds: impact of weber’s law and log transformation. J Pain. 2012;13:519–23. https://doi.org/10.1016/j.jpain.2012.02.009.
Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain, Behav, Immun. 2007;21:458–66. https://doi.org/10.1016/j.bbi.2006.11.001.
McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP. Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J Neurosci. 2016;36:2590–604. https://doi.org/10.1523/JNEUROSCI.2394-15.2016.
Tang Y, Harrington A, Yang X, Friesel RE, Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48:563–7. https://doi.org/10.1002/dvg.20654.
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–47. https://doi.org/10.1523/JNEUROSCI.1860-14.2014.
Dejana E, Bazzoni G, Lampugnani MG. Vascular endothelial (VE)-cadherin: only an intercellular glue. Exp Cell Res. 1999;252:13–9. https://doi.org/10.1006/excr.1999.4601.
Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019. https://doi.org/10.1016/j.immuni.2018.12.012.
Umamaheswaran S, Dasari SK, Yang P, Lutgendorf SK, Sood AK. Stress, inflammation, and eicosanoids: an emerging perspective. Cancer Metastasis Rev. 2018;37:203–11. https://doi.org/10.1007/s10555-018-9741-1.
Hori T, Oka T, Hosoi M, Aou S. Pain modulatory actions of cytokines and prostaglandin E2 in the brain. Ann NY Acad Sci. 1998;840:269–81. https://doi.org/10.1111/j.1749-6632.1998.tb09567.x.
Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin E receptors. Biochim Biophys Acta. 2015;1851:414–21. https://doi.org/10.1016/j.bbalip.2014.07.008.
Sawicki CM, Kim JK, Weber MD, Faw TD, McKim DB, Madalena KM, et al. Microglia promote increased pain behavior through enhanced inflammation in the spinal cord during repeated social defeat stress. J Neurosci. 2019;39:1139–49. https://doi.org/10.1523/JNEUROSCI.2785-18.2018.
Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2014;75:970–81. https://doi.org/10.1016/j.biopsych.2013.11.029.
Stankiewicz AM, Goscik J, Majewska A, Swiergiel AH, Juszczak GR. The effect of acute and chronic social stress on the hippocampal transcriptome in mice. PloS One. 2015;10:e0142195 https://doi.org/10.1371/journal.pone.0142195.
Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20:1752–60. https://doi.org/10.1038/s41593-017-0010-3.
Jin M, Jang E, Suk K. Lipocalin-2 acts as a neuroinflammatogen in lipopolysaccharide-injected mice. Exp Neurobiol. 2014;23:155–62. https://doi.org/10.5607/en.2014.23.2.155.
Jin M, Kim JH, Jang E, Lee YM, Soo Han H, Woo DK, et al. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2014;34:1306–14. https://doi.org/10.1038/jcbfm.2014.83.
Shin HJ, Jeong EA, Lee JY, An HS, Jang HM, Ahn YJ, et al. Lipocalin-2 deficiency reduces oxidative stress and neuroinflammation and results in attenuation of kainic acid-induced hippocampal cell death. Antioxidants. 2021;10. https://doi.org/10.3390/antiox10010100.
Wang G, Weng YC, Chiang IC, Huang YT, Liao YC, Chen YC, et al. Neutralization of lipocalin-2 diminishes stroke-reperfusion injury. Int J Mol Sci. 2020;21. https://doi.org/10.3390/ijms21176253.
Dion-Albert L, Cadoret A, Doney E, Kaufmann FN, Dudek KA, Daigle B, et al. Vascular and blood-brain barrier-related changes underlie stress responses and resilience in female mice and depression in human tissue. Nat Commun. 2022;13:164 https://doi.org/10.1038/s41467-021-27604-x.
Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–86. https://doi.org/10.1016/0896-6273(93)90192-t.
Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–70.
Watanobe H, Takebe K. Effects of intravenous administration of interleukin-1-beta on the release of prostaglandin E2, corticotropin-releasing factor, and arginine vasopressin in several hypothalamic areas of freely moving rats: estimation by push-pull perfusion. Neuroendocrinology. 1994;60:8–15. https://doi.org/10.1159/000126714.
Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. https://doi.org/10.1152/physrev.1999.79.1.1.
Furuyashiki T, Narumiya S. Roles of prostaglandin E receptors in stress responses. Curr Opin Pharm. 2009;9:31–8. https://doi.org/10.1016/j.coph.2008.12.010.
Lacroix S, Rivest S. Effect of acute systemic inflammatory response and cytokines on the transcription of the genes encoding cyclooxygenase enzymes (COX-1 and COX-2) in the rat brain. J Neurochem. 1998;70:452–66. https://doi.org/10.1046/j.1471-4159.1998.70020452.x.
Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–33. https://doi.org/10.1523/JNEUROSCI.1671-13.2013.
Morgan A, Kondev V, Bedse G, Baldi R, Marcus D, Patel S. Cyclooxygenase-2 inhibition reduces anxiety-like behavior and normalizes enhanced amygdala glutamatergic transmission following chronic oral corticosterone treatment. Neurobiol Stress. 2019;11:100190 https://doi.org/10.1016/j.ynstr.2019.100190.
Tanaka K, Furuyashiki T, Kitaoka S, Senzai Y, Imoto Y, Segi-Nishida E, et al. Prostaglandin E2-mediated attenuation of mesocortical dopaminergic pathway is critical for susceptibility to repeated social defeat stress in mice. J Neurosci. 2012;32:4319–29. https://doi.org/10.1523/JNEUROSCI.5952-11.2012.
Elnazer HY, Sampson AP, Baldwin DS. Effects of celecoxib augmentation of antidepressant or anxiolytic treatment on affective symptoms and inflammatory markers in patients with anxiety disorders: exploratory study. Int Clin Psychopharmacol. 2021;36:126–32. https://doi.org/10.1097/YIC.0000000000000356.
Akhondzadeh S, Jafari S, Raisi F, Nasehi AA, Ghoreishi A, Salehi B, et al. Clinical trial of adjunctive celecoxib treatment in patients with major depression: a double blind and placebo controlled trial. Depression Anxiety. 2009;26:607–11. https://doi.org/10.1002/da.20589.
Dionne RA, Khan AA, Gordon SM. Analgesia and COX-2 inhibition. Clin Exp Rheumatol. 2001;19(6 Suppl 25):S63–S70.
Acknowledgements
We would like to thank Dr. Ning Quan (Florida Atlantic University) for providing access to his IL-1R1r/r mouse line which was a valuable tool in these studies. Illustrations included in this publication were created with BioRender.com.
Funding
This research was supported by NIMH R01-MH-119670 and NIMH R56-MH-116670 (to JPG and JFS). WY was supported by National Institute of Neurological Disorders and Stroke Training Grant T32-NS-105864 (to JPG) and F30-MH-125524 (to WY). The authors declare no competing financial interests.
Author information
Authors and Affiliations
Contributions
WY, JFS, and JPG contributed to study conception and design. WY, SPS, RGB, EJG, and NRG contributed to data collection. WY, SPS, and EJG contributed to analysis and interpretation of results. WY, NRG, JFS, and JPG contributed to manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yin, W., Swanson, S.P., Biltz, R.G. et al. Unique brain endothelial profiles activated by social stress promote cell adhesion, prostaglandin E2 signaling, hypothalamic–pituitary–adrenal axis modulation, and anxiety. Neuropsychopharmacol. 47, 2271–2282 (2022). https://doi.org/10.1038/s41386-022-01434-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01434-x
This article is cited by
-
Enhanced fear memory after social defeat in mice is dependent on interleukin-1 receptor signaling in glutamatergic neurons
Molecular Psychiatry (2024)
-
Exosomal PGE2 from M2 macrophages inhibits neutrophil recruitment and NET formation through lipid mediator class switching in sepsis
Journal of Biomedical Science (2023)
