
Abstract
Acute graft-versus-host disease (aGVHD) is a common complication of allogeneic hematopoietic stem cell transplantation (alloHCT) and is a major cause of morbidity and mortality. Systemic steroid therapy is the first-line treatment for aGVHD, although about half of patients will become refractory to treatment. As the number of patients undergoing alloHCT increases, developing safe and effective treatments for aGVHD will become increasingly important, especially for those whose disease becomes refractory to systemic steroid therapy. This paper reviews current treatment options for patients with steroid-refractory aGVHD and discusses data from recently published clinical studies to outline emerging therapeutic strategies.
Similar content being viewed by others

Introduction
Acute graft-versus-host disease (aGVHD) is a common complication of allogeneic hematopoietic stem cell transplantation (alloHCT), occurring in ~30–50% of patients, with 14–36% developing severe aGVHD [1]. Onset of aGVHD classically occurs within 100 days of transplant; however, late-onset aGVHD may present after 100 days [2]. aGVHD is a major cause of morbidity, and opportunistic infections are prevalent [3, 4]. Mortality is also high, with only 25–30% of patients with grade III aGVHD and 1–2% of patients with grade IV aGVHD surviving long term (>2 years) [5].
Systemic steroid therapy is the standard first-line treatment for aGVHD [6,7,8]. However, in ~35–50% of patients, aGVHD becomes refractory to systemic steroid therapy [9, 10]. Mortality is high and quality of life is poor in these patients, and only one treatment is currently approved for steroid-refractory aGVHD (SR-aGVHD) [7, 11]. This review focuses on the unmet need and current and emerging therapies for patients with SR-aGVHD.
Overview of aGVHD
aGVHD occurs primarily in the skin, gastrointestinal tract, and liver and can occur in alloHCT recipients despite prophylaxis [3]. Patients usually present with a maculopapular rash, nausea, vomiting, profuse watery diarrhea and abdominal cramping, and hyperbilirubinemia with jaundice [2, 11]. The incidence and severity of aGVHD depend on a variety of risk factors, but it occurs more frequently with increased severity after alloHCT from HLA-nonidentical or unrelated donors than from HLA-matched sibling donors [8, 12]. Non-HLA risk factors associated with aGVHD include older patient and/or donor age, use of female donor for male recipient, use of peripheral blood as stem cell source, nature of GVHD prophylaxis, and recipient seropositivity for cytomegalovirus [11, 13].
Diagnosis and staging
aGVHD is diagnosed clinically after laboratory analysis, imaging, and/or endoscopic examination to exclude potential differential diagnoses. Biopsy may help confirm the diagnosis but lacks sensitivity and specificity. Following diagnosis, aGVHD severity is graded from mild (grade I) to very severe (grade IV) [5]. The modified Glucksberg staging system is considered the standard in the field [5]; however, clinical staging of aGVHD differs between hospitals, making comparisons between clinical studies difficult and possibly contributing to the reason that promising treatments often fail to show benefit in randomized, multicenter clinical trials [14]. However, differences in staging seen across studies occur because of the interpretation and application of the staging system, not the staging system itself. For example, clinicians may have alternative explanations for skin rash, diarrhea, or raised bilirubin level; subjective differences exist in area estimation of skin rash by the rule of nines; and various means are used to stage transaminitis for liver-only aGVHD in the absence of raised bilirubin level. In addition, biopsy of the affected organ may not be immediately feasible due to center and patient factors. Hence, even though guidelines for aGVHD staging based on symptom severity are well established, quantification of the severity of symptoms has not been standardized across centers.
To improve reproducibility, guidelines have been developed through international expert consensus to standardize clinical trial data collection of aGVHD symptoms for use in a large international GVHD research consortium (Mount Sinai Acute GVHD International Consortium [MAGIC]). These new guidelines provide accurate information on the absolute quantification of symptoms that should be included in clinical staging of aGVHD to facilitate future retrospective studies [14] and could be used as a tool to standardize data collection for research. Furthermore, the Transplant Complications Working Party of the European Society for Blood and Marrow Transplantation and the US National Institutes of Health developed an electronic tool, the eGVHD App, based on MAGIC criteria, to assist healthcare professionals in the assessment of GVHD in clinical practice [15]. Although validation of these guidelines is still needed, increased standardization of aGVHD symptom quantification may lead to enhanced clinical study reproducibility. In addition, consensus is growing for adoption of the day 28 response assessment as a short-term primary endpoint in aGVHD studies [16].
Pathophysiology
The pathophysiology of aGVHD is typically described in 3 phases (Fig. 1). During phase I, conditioning treatment damages patient tissues and causes release of inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and others that lead to activation of host antigen-presenting cells [13, 17]. During phase II, antigen-presenting cells activate mature donor cells through IL-12 and IL-23 to produce T helper cell type 1 (Th1) cytokines, such as IL-2, IL-6, and interferon γ [13, 17]. T cells migrate to target tissues and cause tissue destruction during phase III. Th1 cells then promote proliferation and differentiation of cytotoxic T lymphocytes and stimulate natural killer cells, which induce apoptosis [13, 17]. The process is also regulated by other parts of the immune system, such as regulatory T cells (Tregs) and invariant natural killer T cells [18,19,20].

During phase I, conditioning chemotherapy or radiation damages tissues and causes release of inflammatory cytokines; during phase II, donor T cells are activated; and in phase III, T cells migrate to target tissues and cause apoptosis. APC antigen-presenting cell, CTL cytotoxic T lymphocyte, IDO indoleamine 2,3-dioxygenase, IFN interferon, IL interleukin, LPS lipopolysaccharide, NK natural killer cell, NOD2 nucleotide-binding oligomerization domain–containing protein 2, TNF tumor necrosis factor, Treg regulatory T cell. From Harris et al. Br J Haematol. 2013;160:288–302. © 2012 Blackwell Publishing Ltd.
A deeper understanding of the pathophysiology of aGVHD has already resulted in the development of targeted therapies, and new findings may lead to the development of even more. For instance, some therapies target T-cell activation by using antibodies against the IL-2 receptor (IL-2R) [21], whereas others target the third phase of aGVHD, using antibodies against various markers on T lymphocytes [22]. A number of studies also demonstrated that gut microbiota dysbiosis is associated with SR-aGVHD [23,24,25,26,27]. These studies validated that gut microbiota may modulate systemic alloimmune responses and showed that after alloHCT, the abnormal gut microbiota damages gastrointestinal mucosa and ultimately collapses the diversity of intestinal microbiota. A promising new therapy, fecal microbiota transplant (FMT), may restore the microbiota system [23,24,25,26,27]. Another study found that continued tissue damage in patients with intestinal aGVHD led to increased enterocyte proliferation and significant telomere loss [28]. Telomere loss has been associated with replicative exhaustion and tissue failure and might contribute to treatment failure in intestinal aGVHD, providing a basis for stem cell–protective therapies. An additional study suggested that endothelial cell damage also may play a role in the pathogenesis of steroid resistance in aGVHD [29]. Pretransplant levels of angiopoietin-2, a mediator of endothelial vulnerability, correlated with the risk of developing SR-GVHD in patients with ongoing GVHD. The measurement of pretransplant markers of endothelial vulnerability could help determine the transplantation strategy in patients with high risk for developing GVHD.
Steroid-refractory aGVHD
The recommended first-line treatment for aGVHD is systemic steroid therapy [7, 30]; however, ~35–50% of patients become refractory to steroid therapy [10, 31]. SR-aGVHD can be defined as a clear progression after 3–5 days of treatment or no response after 5–7 days of treatment; [6, 7] however, the exact definition can vary by center. SR-aGVHD is associated with a high mortality risk [8, 10, 32, 33]. The weighted average 6-month survival estimate across 25 studies was 49%; [8] another study reported an estimated 2-year survival rate of 17% [10]. Patients with SR-aGVHD also had higher nonrelapse mortality at 18 months (63% vs 34%) and 2 years (65% vs 35%) than patients who responded to steroid treatment [10]. Likewise, infection-related mortality was high in patients with SR-aGVHD. In a retrospective study of 127 adults, the 1-year incidence of bacterial, viral, and fungal infections was 74%, 65%, and 14%, respectively. Infection-related mortality and overall survival (OS) at 4 years were 46% and 15%, respectively. Bacterial infection was the most common infection leading to death [32].
Treatments for SR-aGVHD
There is no accepted standard-of-care treatment for SR-aGVHD [11, 34]. This is due to most studies in SR-aGVHD being retrospective, single-arm, phase 2 studies [8, 11, 35], which cannot be easily compared with current patient populations due to significant changes in not only supportive care but also prophylaxis of GVHD. In addition, there are a lack of standardized endpoints, small numbers of enrolled patients, and decreased survival rates, making it difficult to compare data across studies [6, 8]. Consequently, treatment choices are based on physicians’ experiences, taking into consideration GVHD prophylaxis as well as the risks of potential toxicity and exacerbation of preexisting comorbidities [11, 34].
The joint working group established by the British Committee for Standards in Hematology and the British Society for Bone Marrow Transplantation outlined the following options for therapy in SR-aGVHD based on critical review of available literature: extracorporeal photopheresis (ECP), anti–TNF-α antibodies, mechanistic target of rapamycin kinase inhibitors, mycophenolate mofetil, methotrexate, or anti–IL-2R antibodies [6]. They recommended that patients who experience failure of one second-line therapy try another before moving on to third-line options.
The American Society for Blood and Marrow Transplantation (ASBMT) reviewed various therapies (e.g., methotrexate, mycophenolate mofetil, ECP, anti–IL-2R antibodies, alemtuzumab, antithymocyte globulin [ATG], etanercept, and infliximab) and also developed recommendations for treatment of SR-aGVHD [8]. The ASBMT concluded that the choice of a second-line therapy should be based on the effects of any previous treatment and consideration of potential toxicity with treatments. In addition, they supported participation in well-designed clinical studies. The following sections review current treatments for SR-aGVHD (Table 1).
ECP
ECP is widely used for treating SR-aGVHD and consists of exposing peripheral blood mononuclear cells to photoactivated 8-methoxypsoralen, followed by reinfusion of treated cells [36]. ECP is generally considered a safe and effective method for treating SR-GVHD and was associated with superior survival in patients with grade II SR-aGVHD (hazard ratio [HR], 4.6; P = 0.016) compared with anti-cytokine therapy; grade > II SR-aGVHD at onset of salvage therapy was associated with inferior survival (HR, 9.4; P < 0.001) [37].
A meta-analysis of prospective clinical trials evaluating ECP in patients with SR-aGVHD reported an overall response rate (ORR) and complete response (CR) rate of 71% each [38]. The pooled response rates for skin, liver, and gut SR-aGVHD were 86%, 60%, and 68%, respectively. However, evidence was insufficient to assess benefit due to limited enrollment (N = 121). The authors concluded that ECP could be an effective therapy for skin SR-aGVHD. Results were also promising for liver and gut SR-aGVHD, but large double-blind clinical trials are needed to verify these findings.
ATG
ATG is frequently used for SR-aGVHD, in part due to the Minnesota study [39] and a randomized study comparing prednisone with ATG + prednisone [40]. However, several studies have suggested that it may not be an effective option in SR-aGVHD. For instance, a study assessing long-term survival of patients with grade III/IV aGVHD treated after 2000 found that 41% of steroid-refractory patients (14/34) treated with ATG had a response at 4 weeks, but this decreased to 21% at 12 weeks; 4 patients (12%) were alive at the time of the analysis (range, 3.6–12.8 years posttransplant) [41]. Similar findings were reported in a 20-year (1992–2012) retrospective study [42]. An overall response was observed in 43% of patients; however, OS was poor (5.5 months), with high mortality rates due to infection. These studies suggest that although ATG leads to an initial response, especially in patients with skin-only GVHD, that does not translate to prolonged OS.
More recent studies have reported better outcomes with ATG, which may reflect improvements in contemporary prophylaxis and care regimens. In a prospective, randomized study evaluating ATG vs ABX-CBL, an antibody targeting CD147, the ORR with ATG was 57%, and survival at 18 months was 45% [43]. A phase 3, randomized trial comparing inolimomab (an anti–IL-2R antibody) with ATG found a 1-year survival rate of 40% in the ATG arm [35, 44]. In addition, a recent, small, retrospective study (N = 11) reported positive outcomes with low-dose ATG with gradual dose escalation based on response (response-guided ATG therapy). The overall improvement at day 28 was 55%, with rates of OS and transplant-related mortality at 1 year of 55% and 45%, respectively, suggesting that this approach may be necessary when treating patients with ATG [45]. The use of ATG for SR-aGVHD in patients who received ATG as part of their conditioning therapy should be studied to determine the effects of therapy.
Antibodies against IL-2R: daclizumab
Retrospective studies showed that daclizumab, a humanized monoclonal antibody against the α chain of IL-2R, had activity in SR-aGVHD (ORR, 51–67%), especially in patients with skin or mild gut aGVHD, but was associated with an increase in infectious complications and poor long-term outcomes [46, 47]. In a phase 2 study of daclizumab in SR-aGVHD (N = 62), 69% of patients had a CR on day 30, and 21% had a partial response (PR); the 4-year event-free survival was 55% [48]. Lower event-free survival was seen in patients with grade ≥III SR-aGVHD, in those with ≥2 involved organs at baseline, and those who were >18 years old [48]. Similar results were reported in other studies, including in pediatric patients; [46, 49, 50] however, one study reported poor efficacy (17%), possibly due to GVHD severity [51]. Despite encouraging responses, rates of infectious complications were high (up to 95% in one study), and long-term survival was poor [48,49,50,51,52].
Findings from a small, retrospective study suggest that daclizumab may lead to a better response if administered in combination with other agents and with aggressive infection prophylaxis [50]. In this study, all patients treated with daclizumab alone (n = 6) or in combination with ATG or infliximab (n = 6) achieved a CR and had a Kaplan–Meier probability of survival at 100 days of 100% [50]. Other studies also found the combination therapy to be effective (ORR, 47–86%) [53, 54], but survival was not always improved. No randomized studies with daclizumab in GVHD are currently ongoing.
Antibodies against IL-2R: basiliximab
Basiliximab, a chimeric monoclonal antibody against the α chain of IL-2R, was first reported to be effective and safe for the treatment of SR-aGVHD in a study of 17 patients with skin, liver, and/or intestinal SR-aGVHD [55]. Treatment with basiliximab led to an ORR of 71%, with 53% CR (median follow-up, 123 days). No bacterial or fungal infections were observed; 5 of 17 patients had a cytomegalovirus reactivation. A prospective phase 2 study (N = 23) also reported a high ORR (83%); however, the rate of CR was lower (18%) [56]. Infections occurred in 65% of patients in this study; 48% of patients were alive after a mean follow-up of 2 years. Several subsequent studies also showed high ORRs (82–92%), including in pediatric patients; however, ~50–70% of patients experienced GVHD recurrence, and rates of infectious complications were high [21, 57,58,59]. Kaplan–Meier-estimated probabilities were 48% for 3-year event-free survival and 20% for 5-year OS [21, 57].
Antibodies against IL-2R: inolimomab
Retrospective studies suggested that inolimomab, a monoclonal antibody that inhibits the α chain of IL-2R, might be an effective therapy for patients with SR-aGVHD, particularly those without gastrointestinal involvement [60, 61]. The ORR in these studies ranged from 58 to 63%, and OS was 30% at 1 year and 26% at 3 years. No drug-associated toxicity was reported, although 93% of patients had ≥1 infectious event in one study [61], and 14% of patients died due to infections in another [60]. Inolimomab was recently evaluated in a phase 3, randomized, open-label, multicenter trial that compared inolimomab (n = 49) with ATG (n = 51) in adult patients with SR-aGVHD [35]. The primary endpoint of therapy success, defined as OS at 1 year without changing baseline therapy, was not met and was reached by 14 patients treated with inolimomab (29%) and 11 patients (22%) treated with ATG (adjusted HR, 0.722; P = 0.188); 53% and 60% of patients died in the inolimomab and ATG groups, respectively. A long-term follow-up analysis of this study (median follow-up, 58.4 months) showed a clinical benefit associated with inolimomab compared with ATG (31% vs 20% survival; adjusted HR, 0.572 [95% CI, 0.346–0.947]; two-sided P = 0.030) [44]. The number of deaths related to infection was two times lower in patients treated with inolimomab than with ATG (12% vs 24%). Although findings are promising, further studies are needed to determine the benefits of inolimomab compared with other therapies.
Anti–TNF-α antibodies: infliximab
Infliximab has shown mixed results for the treatment of SR-aGVHD [62, 63]. In a retrospective study (N = 68; 51 patients [75%] with grade III/IV), 41 patients (60%) showed response to infliximab therapy at day 7, and 31 patients (46%) showed response at day 28 [62]. Twenty-four patients (35%) achieved the composite endpoint of 6 months’ freedom from treatment failure, and 34% of patients were alive at 24 months. However, infections occurred in 61 patients (90%) and infections led to death in 17 patients (33%). The main cause of treatment failure within 6 months was death (31 patients). In a small study, nine of ten patients responded to treatment; however, four patients died of sepsis [64]. A recent study showed that infliximab therapy in SR-aGVHD was associated with a modest, poorly sustained response along with an increased risk of severe infection [63]; other studies showed similar results [65].
Anti–TNF-α antibodies: etanercept
Early studies evaluating etanercept, a second anti–TNF-α antibody, in combination with other agents for the treatment of SR-aGVHD reported high response rates of 67 [66] and 81% [67]. In the first retrospective study to evaluate etanercept monotherapy in SR-aGVHD, etanercept led to a clinical response rate of 46% (6/13) in patients with SR-aGVHD, with the highest response rates seen in patients with gastrointestinal involvement (64%; 7/11). A survival benefit was also observed; 69% of patients were alive at a median follow-up of 10.6 months. Common infections were cytomegalovirus reactivation (48%), bacterial infections (14%), and fungal infections (19%) [68]. Subsequent studies found response rates to be similar (50–55%) [32, 69, 70], although a retrospective study with a median follow-up of 74 months reported a clinical response rate of 28% [33]. Despite inducing response, etanercept showed little to no improvement in OS in these studies (0–28%) [32, 33, 69, 70]. However, these were all small (<30 patients treated with etanercept in each), retrospective studies, which may have affected results.
Although the rate of infectious complications was high in these studies (up to 87%), etanercept is usually associated with lower rates of infection compared with infliximab [32, 68,69,70]. This may be due to the different mechanisms of action of the two agents, in particular the cytolytic activity observed with infliximab but not etanercept; however, no direct comparison between etanercept and infliximab has been performed [68].
Combination of anti–TNF-α and anti–IL-2R antibodies
Basiliximab in combination with etanercept was assessed in a prospective, multicenter clinical trial of 65 patients with severe (grades III–IV) SR-aGVHD [22]. The ORR at day 28 was 91%; 75% of patients had a CR. The 2-year OS rate was 55% [22], suggesting that combination therapy may further improve outcomes in patients with SR-aGVHD. However, this clinical benefit may be limited to basiliximab plus etanercept, given that a retrospective study of combination therapy with inolimomab plus etanercept only showed an ORR of 48% at day 28 and a 2-year OS of 10% [71]. Similarly, a study of basiliximab plus infliximab led to lower response rates (ORR, 76%; CR, 43%) and worse survival (24% at 1 year) than basiliximab alone [65].
Third-line options
Treatments with fewer data available are considered to be third-line treatment options and include alemtuzumab (anti–CD52 receptor antibody), pentostatin, mesenchymal stem cells (MSC), and methotrexate [6]. However, studies have suggested that MSC and methotrexate are promising therapies. Two recent studies of MSC reported positive outcomes [72, 73]. One retrospective study of 46 patients with grade III/IV SR-aGVHD reported a 50% response rate (3 patients had a CR, 14 had a PR, and 6 had a transient PR), and the estimated probability of survival at 2 years was 17% [73]. The other, a prospective study of 69 patients (51 children, 18 adults), reported an ORR of 83% at day 28 (22 patients had a CR, 35 patients had a PR), and the 6-month OS probability was 71% [72]. However, the use of MSC may be difficult in some countries because of regulatory issues.
Similarly, in a retrospective study of low-dose methotrexate for SR-aGVHD, 7 of 12 patients (58%) responded (5 had CR) [74]. Eight patients had grade III/IV SR-aGVHD, with five patients responding to low-dose methotrexate. Seven patients died of disease progression (nonresponders, four patients; responders, three patients). In a pooled data analysis, the observed overall response was 70% (79/113 patients), with a CR in 59% of patients and a PR in 11% [75]. Pooled data suggested the potential use of methotrexate for SR-aGVHD; however, randomized controlled studies are needed. In both studies, methotrexate toxicity was low [74, 75].
The use of other therapies has been reported; however, evidence is insufficient to support recommending their use in the management of SR-aGVHD [6]. These agents include rituximab (anti-CD20), visilizumab (anti-CD3), ABX-CBL (anti-CD147), thalidomide (immunomodulatory drug), azathioprine (purine analog), intra-arterial methylprednisolone, and Tregs.
Novel therapies evaluated in recent clinical studies
Ruxolitinib
Ruxolitinib is a Janus kinase (JAK) 1/JAK2 inhibitor that became the first JAK inhibitor approved for the treatment of myelofibrosis [76]. JAKs are intracellular signaling molecules and are important effectors of all three phases of the pathogenesis of aGVHD [77,78,79,80]. Ruxolitinib’s inhibition of JAK1/JAK2 influences a wide range of immune system components, both adaptive (dendritic cells and CD4+ T cells) and innate (natural killer cells and neutrophils) [81]. In preclinical studies, ruxolitinib reduced the severity of GVHD and prolonged survival in animal models of GVHD while preserving the graft-versus-leukemic effect through inhibition of the production of proinflammatory cytokines, suppression of T-cell expansion, and promotion of Treg proliferation (Table 2) [77, 79].
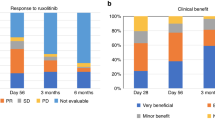
In retrospective clinical studies, ruxolitinib resulted in fair to high response rates, prolonged survival in patients with SR-aGVHD, and demonstrated a favorable safety profile in these patients [78, 80, 82, 83]. A retrospective survey evaluated outcomes of 95 patients with SR-GVHD (54 with aGVHD, 41 with chronic GVHD) who received ruxolitinib as second-line therapy [80]. The ORR in the SR-aGVHD group was 82% (CR, 46%). The estimated 6-month survival and relapse rate were 79% and 7%, respectively [80]. Long-term follow-up at a median of 19 months showed that 41% of patients had an ongoing response and were free of immunosuppression, with a 1-year OS rate of 62% [83].
A retrospective study of 13 pediatric patients who received ruxolitinib as salvage therapy for SR-aGVHD evaluated response rates after 4 weeks of therapy [82]. Of 11 evaluable patients, one achieved a CR, four had PR, and two had no response; treatment failed in four patients [82]. Seven patients were alive at long-term follow-up at a median of 401 days [82].
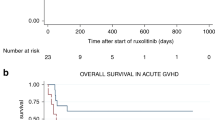
Three ongoing studies are evaluating ruxolitinib in SR-aGVHD [78]. The first is the Ruxolitinib in Patients With Refractory GVHD After Allogeneic Stem Cell Transplantation 1 (REACH1; NCT02953678) study, which is an open-label, single-cohort, multicenter, phase 2 study to assess the combination of ruxolitinib with steroids for the treatment of SR-aGVHD (grades II–IV); the primary endpoint is ORR at day 28 [78, 84]. A total of 71 patients were enrolled (median age, 58 years); 68% had grade III/IV GVHD at baseline. The study met its primary endpoint, with an ORR of 55% at day 28 and a best overall response at any time of 73% (CR, 56%). Median duration of response with ≥6 months’ follow-up was 345 days in both day 28 responders and patients who had a best overall response at any time during treatment. In addition, most patients achieved a sustained reduction in steroid dose. The most common hematologic treatment-emergent adverse events (AEs) were anemia (65%), thrombocytopenia (62%), and neutropenia (48%). Infections included cytomegalovirus (13%), sepsis (13%), and bacteremia (10%). Fatal treatment-related AEs were sepsis and pulmonary hemorrhage (1 patient each) and were attributed to both ruxolitinib and steroid treatment. On the basis of this study, ruxolitinib recently became the first US Food and Drug Administration-approved treatment for SR-aGVHD in adult and pediatric patients ≥12 years old [76].
Ruxolitinib is also being assessed in the REACH2 study (NCT02913261), an open-label, multicenter, phase 3 crossover study comparing ruxolitinib with best available treatment (BAT) for SR-aGVHD; the study met its primary endpoint of ORR at day 28 [78, 85]. The third study, Ruxolitinib in GVHD (RIG; NCT02396628), is an open-label, multicenter, prospective, randomized, phase 2 study comparing the efficacy of ruxolitinib plus BAT vs BAT in SR-aGVHD [86].
Fecal microbiota transplant
FMT is a therapy that reestablishes the microbiota system through infusing a fecal suspension from a healthy donor into a patient’s gastrointestinal tract [87, 88]; three case reports of its use in patients with SR-aGVHD have been published (Table 2) [23,24,25, 27]. A pilot study of four patients (three with gastrointestinal SR-aGVHD; one with steroid-dependent gastrointestinal aGVHD) evaluated the safety and efficacy of FMT [24]. All four patients responded to treatment (three had a CR; one had a PR). All AEs were mild and transient. The authors noted that peripheral effector Treg counts were increased during the response to FMT. In addition, temporal dynamics of microbiota appeared to be associated with the health of patients’ gastrointestinal tracts. The authors presented another case report of a patient with gastrointestinal SR-aGVHD that also demonstrated restoration of bacterial diversity with no safety concerns following administration of FMT capsules [23]; other case reports showed similar results [25, 27]. A pilot study conducted in eight patients with gastrointestinal SR-aGVHD who were treated with FMT (NCT03148743) [26] showed that all patients’ symptoms improved and bacterial diversity was restored. No severe AEs were reported; four patients died, but the deaths were unrelated to FMT. Progression-free survival improved in the patients who received FMT when the authors retrospectively compared findings with those of patients with gastrointestinal SR-aGVHD who did not receive FMT (P = 0.003); however, no difference in OS was observed. Six studies are ongoing to evaluate the efficacy and safety of FMT in patients with gastrointestinal SR-aGVHD (NCT03819803, NCT03549676, NCT03492502, NCT03812705, NCT03359980, and NCT03214289).
α1-Antitrypsin
α1-Antitrypsin (AAT), a circulating protease inhibitor produced by the liver, is implicated in several aspects of immune regulation: It inactivates serine proteases from neutrophils and macrophages, induces IL-10, and suppresses plasma proinflammatory cytokines (Table 2) [89]. In murine models, AAT reduced the severity of aGVHD, reducing inflammatory cytokines and increasing the ratio of Tregs to effector T cells—this led to a phase 2 clinical trial in patients with SR-aGVHD (NCT01700036). Forty patients received 60 mg/kg AAT intravenously every 4 days for up to 4 weeks. At day 28, ORR and CR rates were 65% and 35%, respectively [89]. There is an ongoing early-access clinical study (NCT03172455), and AAT is also being investigated as prophylaxis for SR-aGVHD (NCT03459040).
Anti-CD3/CD7 immunotoxin
An immunotoxin combination, consisting of a mixture of anti-CD3 and anti-CD7 antibodies separately conjugated to recombinant ricin A (CD3/CD7-IT) induces in vivo depletion of T cells and natural killer cells and suppresses T-cell receptor activation (Table 2) [90]. In a phase 1/2 trial of CD3/CD7-IT in SR-aGVHD, 20 patients were given 4-h intravenous infusions of 4 mg/m2 CD3/CD7-IT; 4 infusions were given, administered at 48-h intervals. At day 28, ORR and CR rates were 60% and 50%, respectively [90]. CD3/CD7-IT was recently given fast-track designation for the treatment of SR-aGVHD by the US Food and Drug Administration, and a phase 3 trial is ongoing (NCT04128319).
Vedolizumab
Vedolizumab is a monoclonal antibody that recognizes the integrin α4β7 present on circulating lymphocytes and inhibits their relocation to the gastrointestinal tract [91, 92]. Recent case series have reported mixed results for vedolizumab in gastrointestinal SR-aGVHD (Table 2) [91,92,93,94].
Fløisand et al. described a case series of six patients with SR-aGVHD with single-site-involvement, in which all patients responded to second-line treatment with vedolizumab; four patients were alive at a median follow-up of 10 months [92]. However, another case series reported only two PRs from five patients with multiple-site-involvement treated with vedolizumab in the third or fourth line [93].
Another retrospective case series also described a high mortality rate following vedolizumab therapy for gastrointestinal SR-aGVHD: Of nine patients with multiple-site- involvement, six died before the 4-week follow-up, and only one patient survived past 2 months. However, patients surviving past the 4-week follow-up did experience some clinical response [91].
The discrepancies in outcomes with vedolizumab treatment for SR-aGVHD may be related to its mechanism of action: Preventing the migration of lymphocytes to the gastrointestinal tract may be effective only at an early stage and not when GVHD is established. A retrospective case series of 29 patients reported significantly better outcomes in patients treated with vedolizumab in the second vs third or higher line (ORR, 13/13 [100%] vs 10/16 [63%]; P = 0.012 and CR, 7/13 [54%] vs 1/16 [6%]; P = 0.005) [94]. Nevertheless, a recent dose-finding, prospective, phase 2 study of vedolizumab in the second line (NCT02993783) was terminated early for lack of efficacy [95]. The ORR at day 28 was 50% in patients treated at 300 mg (n = 8) and 22% in patients treated at 600 mg (n = 9); 12% and 0% of patients, respectively, achieved CR. Consequently, vedolizumab development has been abandoned in the setting of SR-aGVHD. Of note, given vedolizumab’s mechanism of action, development now focuses on GVHD prophylaxis (NCT03657160).
Other therapies for treating SR-aGVHD
Other promising therapies have also been evaluated in the SR-aGVHD setting but have not shown positive results. Brentuximab vedotin, a CD30-directed antibody drug conjugate, showed promising activity in a phase 1 study in patients with SR-aGVHD [96]. The ORR at day 28 was 38% (CR, 15%); seven additional patients achieved CR by day 56. The OS rate was 41% (95% CI, 25–57%) at 6 months and 38% (95% CI, 22–54%) at 12 months. However, all clinical studies of brentuximab vedotin for the treatment of SR-aGVHD have been terminated or withdrawn. It is currently being evaluated in the prophylaxis of GVHD (NCT01700751).
Begelomab, an antibody targeting CD26 on CD4+ T lymphocytes, was in phase 2/3 development for SR-aGVHD. In one study of 28 patients, 75% of patients achieved a response at day 28 compared with 41% of matched controls (P = 0.004) [97]. The OS at 1 year was 50% vs 31% in the control group (P = 0.06). Although these findings led to further assessment in a phase 3, randomized study (NCT02411084), the study was terminated due to a low accrual rate.
Conclusions
aGVHD is a major cause of morbidity and mortality in patients undergoing alloHCT. As the number of patients undergoing alloHCT increases, it will become imperative to determine safe and effective treatment options for these patients, especially those who become refractory to systemic steroid therapy. Understanding of the pathophysiology of the disease will continue to be important in determining new targets. As new therapies are developed, demonstrating their efficacy and safety—including long-term effects, such as the incidence of relapse, chronic GVHD, and infections—in large, randomized clinical studies will be imperative. Several emerging therapies are currently being evaluated in phase 3 randomized studies, and first results are eagerly awaited.
References
Al-Kadhimi Z, Gul Z, Chen W, Smith D, Abidi M, Deol A, et al. High incidence of severe acute graft-versus-host disease with tacrolimus and mycophenolate mofetil in a large cohort of related and unrelated allogeneic transplantation patients. Biol Blood Marrow Transpl. 2014;20:979–85.
Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transpl. 2005;11:945–56.
Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–61.
Holtan SG, Pasquini M, Weisdorf DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood. 2014;124:363–73.
Cahn JY, Klein JP, Lee SJ, Milpied N, Blaise D, Antin JH, et al. Prospective evaluation of 2 acute graft-versus-host (GVHD) grading systems: a joint Société Française de Greffe de Moëlle et Thérapie Cellulaire (SFGM-TC), Dana Farber Cancer Institute (DFCI), and International Bone Marrow Transplant Registry (IBMTR) prospective study. Blood. 2005;106:1495–1500.
Dignan FL, Clark A, Amrolia P, Cornish J, Jackson G, Mahendra P, et al. Diagnosis and management of acute graft-versus-host disease. Br J Haematol. 2012;158:30–45.
Ruutu T, Gratwohl A, de Witte T, Afanasyev B, Apperley J, Bacigalupo A, et al. Prophylaxis and treatment of GVHD: EBMT-ELN working group recommendations for a standardized practice. Bone Marrow Transpl. 2014;49:168–73.
Martin PJ, Rizzo JD, Wingard JR, Ballen K, Curtin PT, Cutler C, et al. First- and second-line systemic treatment of acute graft-versus-host disease: recommendations of the American Society of Blood and Marrow Transplantation. Biol Blood Marrow Transpl. 2012;18:1150–63.
Axt L, Naumann A, Toennies J, Haen SP, Vogel W, Schneidawind D, et al. Retrospective single center analysis of outcome, risk factors and therapy in steroid refractory graft-versus-host disease after allogeneic hematopoietic cell transplantation. Bone Marrow Transpl. 2019;54:1805–14.
Westin JR, Saliba RM, De Lima M, Alousi A, Hosing C, Qazilbash MH, et al. Steroid-refractory acute GVHD: predictors and outcomes. Adv Hematol. 2011;2011:601953.
Zeiser R, Blazar BR. Acute graft-versus-host disease—biologic process, prevention, and therapy. N Engl J Med. 2017;377:2167–79.
Han LJ, Wang Y, Fan ZP, Huang F, Zhou J, Fu YW, et al. Haploidentical transplantation compared with matched sibling and unrelated donor transplantation for adults with standard-risk acute lymphoblastic leukaemia in first complete remission. Br J Haematol. 2017;179:120–30.
Harris AC, Ferrara JL, Levine JE. Advances in predicting acute GVHD. Br J Haematol. 2013;160:288–302.
Harris AC, Young R, Devine S, Hogan WJ, Ayuk F, Bunworasate U, et al. International, multicenter standardization of acute graft-versus-host disease clinical data collection: a report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transpl. 2016;22:4–10.
Schoemans HM, Goris K, Van Durm R, Fieuws S, De Geest S, Pavletic SZ, et al. The eGVHD App has the potential to improve the accuracy of graft-versus-host disease assessment: a multicenter randomized controlled trial. Haematologica. 2018;103:1698–707.
Inamoto Y, Martin PJ, Storer BE, Mielcarek M, Storb RF, Carpenter PA. Response endpoints and failure-free survival after initial treatment for acute graft-versus-host disease. Haematologica. 2014;99:385–91.
He FC, Holtan SG. Biomarkers in graft-versus-host disease: from prediction and diagnosis to insights into complex graft/host interactions. Curr Hematol Malig Rep. 2018;13:44–52.
Leclerc M, Naserian S, Pilon C, Thiolat A, Martin GH, Pouchy C, et al. Control of GVHD by regulatory T cells depends on TNF produced by T cells and TNFR2 expressed by regulatory T cells. Blood. 2016;128:1651–9.
Mavers M, Maas-Bauer K, Negrin RS. Invariant natural killer T cells as suppressors of graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Front Immunol. 2017;8:900.
Malard F, Labopin M, Chevallier P, Guillaume T, Duquesne A, Rialland F, et al. Larger number of invariant natural killer T cells in PBSC allografts correlates with improved GVHD-free and progression-free survival. Blood. 2016;127:1828–35.
Wang JZ, Liu KY, Xu LP, Liu DH, Han W, Chen H, et al. Basiliximab for the treatment of steroid-refractory acute graft-versus-host disease after unmanipulated HLA-mismatched/haploidentical hematopoietic stem cell transplantation. Transpl Proc. 2011;43:1928–33.
Tan Y, Xiao H, Wu D, Luo Y, Lan J, Liu Q, et al. Combining therapeutic antibodies using basiliximab and etanercept for severe steroid-refractory acute graft-versus-host disease: a multi-center prospective study. Oncoimmunology. 2017;6:e1277307.
Kaito S, Toya T, Yoshifuji K, Kurosawa S, Inamoto K, Takeshita K, et al. Fecal microbiota transplantation with frozen capsules for a patient with refractory acute gut graft-versus-host disease. Blood Adv. 2018;2:3097–101.
Kakihana K, Fujioka Y, Suda W, Najima Y, Kuwata G, Sasajima S, et al. Fecal microbiota transplantation for patients with steroid-resistant acute graft-versus-host disease of the gut. Blood. 2016;128:2083–8.
Kakihana K. Fecal microbiota transplantation for acute graft-versus-host disease of the gut. Rinsho ketsueki (Jpn J Clin Hematol). 2017;58:499–505.
Qi X, Li X, Zhao Y, Wu X, Chen F, Ma X, et al. Treating steroid refractory intestinal acute graft-vs.-host disease with fecal microbiota transplantation: a pilot study. Front Immunol. 2018;9:2195.
Spindelboeck W, Schulz E, Uhl B, Kashofer K, Aigelsreiter A, Zinke-Cerwenka W, et al. Repeated fecal microbiota transplantations attenuate diarrhea and lead to sustained changes in the fecal microbiota in acute, refractory gastrointestinal graft-versus-host-disease. Haematologica. 2017;102:e210–e213.
Hummel S, Ventura Ferreira MS, Heudobler D, Huber E, Fahrenkamp D, Gremse F, et al. Telomere shortening in enterocytes of patients with uncontrolled acute intestinal graft-versus-host disease. Blood. 2015;126:2518–21.
Dietrich S, Falk CS, Benner A, Karamustafa S, Hahn E, Andrulis M, et al. Endothelial vulnerability and endothelial damage are associated with risk of graft-versus-host disease and response to steroid treatment. Biol Blood Marrow Transpl. 2013;19:22–27.
Mielcarek M, Furlong T, Storer BE, Green ML, McDonald GB, Carpenter PA, et al. Effectiveness and safety of lower dose prednisone for initial treatment of acute graft-versus-host disease: a randomized controlled trial. Haematologica. 2015;100:842–8.
MacMillan ML, Robin M, Harris AC, DeFor TE, Martin PJ, Alousi A, et al. A refined risk score for acute graft-versus-host disease that predicts response to initial therapy, survival, and transplant-related mortality. Biol Blood Marrow Transpl. 2015;21:761–7.
Garcia-Cadenas I, Rivera I, Martino R, Esquirol A, Barba P, Novelli S, et al. Patterns of infection and infection-related mortality in patients with steroid-refractory acute graft versus host disease. Bone Marrow Transpl. 2017;52:107–13.
Xhaard A, Rocha V, Bueno B, de Latour RP, Lenglet J, Petropoulou A, et al. Steroid-refractory acute GVHD: lack of long-term improved survival using new generation anticytokine treatment. Biol Blood Marrow Transpl. 2012;18:406–13.
Martin PJ, Inamoto Y, Flowers ME, Carpenter PA. Secondary treatment of acute graft-versus-host disease: a critical review. Biol Blood Marrow Transpl. 2012;18:982–8.
Socie G, Vigouroux S, Yakoub-Agha I, Bay JO, Furst S, Bilger K, et al. A phase 3 randomized trial comparing inolimomab vs usual care in steroid-resistant acute GVHD. Blood. 2017;129:643–9.
de Waure C, Capri S, Veneziano MA, Specchia ML, Cadeddu C, Di Nardo F, et al. Extracorporeal photopheresis for second-line treatment of chronic graft-versus-host diseases: results from a health technology assessment in Italy. Value Health. 2015;18:457–66.
Jagasia M, Greinix H, Robin M, Das-Gupta E, Jacobs R, Savani BN, et al. Extracorporeal photopheresis versus anticytokine therapy as a second-line treatment for steroid-refractory acute GVHD: a multicenter comparative analysis. Biol Blood Marrow Transpl. 2013;19:1129–33.
Zhang H, Chen R, Cheng J, Jin N, Chen B. Systematic review and meta-analysis of prospective studies for ECP treatment in patients with steroid-refractory acute GVHD. Patient Prefer Adherence. 2015;9:105–11.
Dugan MJ, DeFor TE, Steinbuch M, Filipovich AH, Weisdorf DJ. ATG plus corticosteroid therapy for acute graft-versus-host disease: predictors of response and survival. Ann Hematol. 1997;75:41–46.
Cragg L, Blazar BR, Defor T, Kolatker N, Miller W, Kersey J, et al. A randomized trial comparing prednisone with antithymocyte globulin/prednisone as an initial systemic therapy for moderately severe acute graft-versus-host disease. Biol Blood Marrow Transpl. 2000;6:441–7.
Jamani K, Russell JA, Daly A, Stewart D, Savoie L, Duggan P, et al. Prognosis of grade 3-4 acute GVHD continues to be dismal. Bone Marrow Transpl. 2013;48:1359–61.
Ozen M, Bozdag SC, Cakmak G, Topcuoglu P, Eroglu AH, Gunduz M, et al. Antilymphocyte/thymocyte globulin for the treatment of steroid-refractory acute graft-versus-host disease: 20-year experience at a single center. Int J Hematol Oncol. 2015;25:236–44.
Macmillan ML, Couriel D, Weisdorf DJ, Schwab G, Havrilla N, Fleming TR, et al. A phase 2/3 multicenter randomized clinical trial of ABX-CBL versus ATG as secondary therapy for steroid-resistant acute graft-versus-host disease. Blood. 2007;109:2657–62.
Socie G, Milpied N, Yakoub-Agha I, Bay JO, Furst S, Bilger K, et al. Long-term follow-up of a phase 3 clinical trial of inolimomab for the treatment of primary steroid refractory aGVHD. Blood Adv. 2019;3:184–6.
Nishimoto M, Nakamae H, Koh H, Nakamae M, Hirose A, Hayashi Y, et al. Response-guided therapy for steroid-refractory acute GVHD starting with very-low-dose antithymocyte globulin. Exp Hematol. 2015;43:177–9.
Przepiorka D, Kernan NA, Ippoliti C, Papadopoulos EB, Giralt S, Khouri I, et al. Daclizumab, a humanized anti-interleukin-2 receptor alpha chain antibody, for treatment of acute graft-versus-host disease. Blood. 2000;95:83–89.
Willenbacher W, Basara N, Blau IW, Fauser AA, Kiehl MG. Treatment of steroid refractory acute and chronic graft-versus-host disease with daclizumab. Br J Haematol. 2001;112:820–3.
Bordigoni P, Dimicoli S, Clement L, Baumann C, Salmon A, Witz F, et al. Daclizumab, an efficient treatment for steroid-refractory acute graft-versus-host disease. Br J Haematol. 2006;135:382–5.
Miano M, Cuzzubbo D, Terranova P, Giardino S, Lanino E, Morreale G, et al. Daclizumab as useful treatment in refractory acute GVHD: a paediatric experience. Bone Marrow Transpl. 2009;43:423–7.
Srinivasan R, Chakrabarti S, Walsh T, Igarashi T, Takahashi Y, Kleiner D, et al. Improved survival in steroid-refractory acute graft versus host disease after non-myeloablative allogeneic transplantation using a daclizumab-based strategy with comprehensive infection prophylaxis. Br J Haematol. 2004;124:777–86.
Hui CH, Sia H, Mangos H, Horvath N, Lee H, Lewis I, et al. Daclizumab has poor efficacy in steroid-refractory severe acute graft-versus-host disease: a single centre experience with 12 allograft patients. Bone Marrow Transpl. 2008;41:409–10.
Perales MA, Ishill N, Lomazow WA, Weinstock DM, Papadopoulos EB, Dastigir H, et al. Long-term follow-up of patients treated with daclizumab for steroid-refractory acute graft-vs-host disease. Bone Marrow Transpl. 2007;40:481–6.
Rager A, Frey N, Goldstein SC, Reshef R, Hexner EO, Loren A, et al. Inflammatory cytokine inhibition with combination daclizumab and infliximab for steroid-refractory acute GVHD. Bone Marrow Transpl. 2011;46:430–5.
Rao K, Rao A, Karlsson H, Jagani M, Veys P, Amrolia PJ. Improved survival and preserved antiviral responses after combination therapy with daclizumab and infliximab in steroid-refractory graft-versus-host disease. J Pediatr Hematol Oncol. 2009;31:456–61.
Massenkeil G, Rackwitz S, Genvresse I, Rosen O, Dörken B, Arnold R. Basiliximab is well tolerated and effective in the treatment of steroid-refractory acute graft-versus-host disease after allogeneic stem cell transplantation. Bone Marrow Transpl. 2002;30:899–903.
Schmidt-Hieber M, Fietz T, Knauf W, Uharek L, Hopfenmuller W, Thiel E, et al. Efficacy of the interleukin-2 receptor antagonist basiliximab in steroid-refractory acute graft-versus-host disease. Br J Haematol. 2005;130:568–74.
Funke VA, de Medeiros CR, Setubal DC, Ruiz J, Bitencourt MA, Bonfim CM, et al. Therapy for severe refractory acute graft-versus-host disease with basiliximab, a selective interleukin-2 receptor antagonist. Bone Marrow Transpl. 2006;37:961–5.
Tang FF, Cheng YF, Xu LP, Zhang XH, Yan CH, Han W, et al. Basiliximab as treatment for steroid-refractory acute graft-versus-host disease in pediatric patients after haploidentical hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2020;26:351–7.
Chakupurakal G, Garcia-Marquez MA, Shimabukuro-Vornhagen A, Theurich S, Holtick U, Hallek M, et al. Immunological effects in patients with steroid-refractory graft-versus-host disease following treatment with basiliximab, a CD25 monoclonal antibody. Eur J Haematol. 2016;97:121–7.
Bay JO, Dhedin N, Goerner M, Vannier JP, Marie-Cardine A, Stamatoullas A, et al. Inolimomab in steroid-refractory acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation: retrospective analysis and comparison with other interleukin-2 receptor antibodies. Transplantation. 2005;80:782–8.
Piñana JL, Valcarcel D, Martino R, Moreno ME, Sureda A, Briones J, et al. Encouraging results with inolimomab (anti-IL-2 receptor) as treatment for refractory acute graft-versus-host disease. Biol Blood Marrow Transpl. 2006;12:1135–41.
Nygaard M, Andersen NS, Moser CE, Olesen G, Schjodt IM, Heilmann C, et al. Evaluation of infliximab as second-line treatment of acute graft versus host disease -validating response on day 7 and 28 as predictors of survival. Bone Marrow Transpl. 2018;53:844–51.
Yalniz FF, Hefazi M, McCullough K, Litzow MR, Hogan WJ, Wolf R, et al. Safety and efficacy of infliximab therapy in the setting of steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transpl. 2017;23:1478–84.
Nogueira MC, Azevedo AM, Pereira SC, Ferreira JL, Lerner D, Lobo AM, et al. Anti-tumor necrosis factor-a for the treatment of steroid-refractory acute graft-versus-host disease. Braz J Med Biol Res. 2007;40:1623–9.
Nadeau M, Perreault S, Seropian S, Foss F, Isufi I, Cooper DL. The use of basiliximab-infliximab combination for the treatment of severe gastrointestinal acute GvHD. Bone Marrow Transpl. 2016;51:273–6.
Wolff D, Roessler V, Steiner B, Wilhelm S, Weirich V, Brenmoehl J, et al. Treatment of steroid-resistant acute graft-versus-host disease with daclizumab and etanercept. Bone Marrow Transpl. 2005;35:1003–10.
Kennedy GA, Butler J, Western R, Morton J, Durrant S, Hill GR. Combination antithymocyte globulin and soluble TNFalpha inhibitor (etanercept) +/− mycophenolate mofetil for treatment of steroid refractory acute graft-versus-host disease. Bone Marrow Transpl. 2006;37:1143–7.
Busca A, Locatelli F, Marmont F, Ceretto C, Falda M. Recombinant human soluble tumor necrosis factor receptor fusion protein as treatment for steroid refractory graft-versus-host disease following allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2007;82:45–52.
Park JH, Lee HJ, Kim SR, Song GW, Lee SK, Park SY, et al. Etanercept for steroid-refractory acute graft versus host disease following allogeneic hematopoietic stem cell transplantation. Korean J Intern Med. 2014;29:630–6.
De Jong CN, Saes L, Klerk CPW, Van der Klift M, Cornelissen JJ, Broers AEC. Etanercept for steroid-refractory acute graft-versus-host disease: a single center experience. PLoS One. 2017;12:e0187184.
van Groningen LF, Liefferink AM, de Haan AF, Schaap NP, Donnelly JP, Blijlevens NM, et al. Combination therapy with inolimomab and etanercept for severe steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transpl. 2016;22:179–82.
Bader P, Kuci Z, Bakhtiar S, Basu O, Bug G, Dennis M, et al. Effective treatment of steroid and therapy-refractory acute graft-versus-host disease with a novel mesenchymal stromal cell product (MSC-FFM). Bone Marrow Transpl. 2018;53:852–62.
Dotoli GM, De Santis GC, Orellana MD, de Lima Prata K, Caruso SR, Fernandes TR, et al. Mesenchymal stromal cell infusion to treat steroid-refractory acute GvHD III/IV after hematopoietic stem cell transplantation. Bone Marrow Transpl. 2017;52:859–62.
de Lavallade H, Mohty M, Faucher C, Furst S, El-Cheikh J, Blaise D. Low-dose methotrexate as salvage therapy for refractory graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation. Haematologica. 2006;91:1438–40.
Nassar A, Elgohary G, Elhassan T, Nurgat Z, Mohamed SY, Aljurf M. Methotrexate for the treatment of graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. J Transpl. 2014;2014:980301.
Jakafi [package insert]. Wilmington, DE: Incyte Corporation; 2019.
Schroeder MA, Choi J, Staser K, DiPersio JF. The role of Janus kinase signaling in graft-versus-host disease and graft versus leukemia. Biol Blood Marrow Transpl. 2018;24:1125–34.
Jagasia M, Zeiser R, Arbushites M, Delaite P, Gadbaw B, Bubnoff NV. Ruxolitinib for the treatment of patients with steroid-refractory GVHD: an introduction to the REACH trials. Immunotherapy. 2018;10:391–402.
Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123:3832–42.
Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder SK, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia. 2015;29:2062–8.
Elli EM, Baratè C, Mendicino F, Palandri F, Palumbo GA. Mechanisms underlying the anti-inflammatory and immunosuppressive activity of ruxolitinib. Front Oncol. 2019;9:1186.
Khandelwal P, Teusink-Cross A, Davies SM, Nelson AS, Dandoy CE, El-Bietar J, et al. Ruxolitinib as salvage therapy in steroid-refractory acute graft-versus-host disease in pediatric hematopoietic stem cell transplant patients. Biol Blood Marrow Transpl. 2017;23:1122–7.
Zeiser R, Burchert A, Lengerke C, Verbeek M, Maas-Bauer K, Metzelder S, et al. Long-term follow-up of patients with corticosteroid-refractory graft-versus-host disease treated with ruxolitinib. Blood. 2016;128:4561 [abstract 722].
Jagasia M, Perales MA, Schroeder MA, Ali H, Shah NN, Chen YB, et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): a multicenter, open-label, phase 2 trial. Blood. 2020 [Epub ahead of print].
Incyte announces that the REACH2 pivotal trial of ruxolitinib (Jakafi®) meets primary endpoint in patients with steroid-refractory acute graft-versus-host disease. News release. Incyte; October 16, 2019. https://investor.incyte.com/news-releases/news-release-details/incyte-announces-reach2-pivotal-trial-ruxolitinib-jakafir-meets. Accessed Jan 2020.
von Bubnoff N, Ihorst G, Grishina O, Rothling N, Bertz H, Duyster J, et al. Ruxolitinib in GvHD (RIG) study: a multicenter, randomized phase 2 trial to determine the response rate of Ruxolitinib and best available treatment (BAT) versus BAT in steroid-refractory acute graft-versus-host disease (aGvHD) (NCT02396628). BMC Cancer. 2018;18:1132.
Shouval R, Geva M, Nagler A, Youngster I. Fecal microbiota transplantation for treatment of acute graft-versus-host disease. Clin Hematol Int. 2019;1:28–35.
Malard F, Gasc C, Plantamura E, Dore J. High gastrointestinal microbial diversity and clinical outcome in graft-versus-host disease patients. Bone Marrow Transpl. 2018;53:1493–7.
Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, Pawarode A, et al. α1-Antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood. 2018;131:1372–9.
Groth C, van Groningen LFJ, Matos TR, Bremmers ME, FWMB Preijers, Dolstra H, et al. Phase I/II trial of a combination of anti-CD3/CD7 immunotoxins for steroid-refractory acute graft-versus-host disease. Biol Blood Marrow Transpl. 2019;25:712–9.
Coltoff A, Lancman G, Kim S, Steinberg A. Vedolizumab for treatment of steroid-refractory lower gastrointestinal acute graft-versus-host disease. Bone Marrow Transpl. 2018;53:900–4.
Fløisand Y, Lundin KEA, Lazarevic V, Kristiansen JD, Osnes LTN, Tjønnfjord GE, et al. Targeting integrin α4β7 in steroid-refractory intestinal graft-versus-host disease. Biol Blood Marrow Transpl. 2017;23:172–5.
Bukauskas A, Griskevicius L, Peceliunas V. Lessons learned from early experiences with vedolizumab for steroid-refractory acute graft- versus-host disease with gastrointestinal involvement. Biol Blood Marrow Transpl. 2017;23:1597.
Danylesko I, Bukauskas A, Paulson M, Peceliunas V, Gedde-Dahl DYT, Shimoni A, et al. Anti-α4β7 integrin monoclonal antibody (vedolizumab) for the treatment of steroid-resistant severe intestinal acute graft-versus-host disease. Bone Marrow Transpl. 2019;54:987–93.
ClinicalTrials.gov. A dose-finding study of vedolizumab for treatment of steroid-refractory acute intestinal graft-versus-host disease (GvHD) in participants who have undergone allogeneic hematopoietic stem cell transplantation (allo-HSCT). https://clinicaltrials.gov/ct2/show/NCT02993783. Accessed Jan 2020.
Chen YB, Perales MA, Li S, Kempner M, Reynolds C, Brown J, et al. Phase 1 multicenter trial of brentuximab vedotin for steroid-refractory acute graft-versus-host disease. Blood. 2017;129:3256–61.
Bacigalupo A, Deeg HJ, Caballero D, Gualandi F, Raiola AM, Varaldo R, et al. Treatment of patients with steroid refractory acute graft vs host disease (SR-GvHD: a matched paired analysis of anti-CD26 (begelomab) compared to other treatment. Blood. 2016;128:671.
Hart JW, Shiue LH, Shpall EJ, Alousi AM. Extracorporeal photopheresis in the treatment of graft-versus-host disease: evidence and opinion. Ther Adv Hematol. 2013;4:320–34.
Mohty M. Mechanisms of action of antithymocyte globulin: T-cell depletion and beyond. Leukemia. 2007;21:1387–94.
Najar M, Raicevic G, Fayyad-Kazan H, Bron D, Toungouz M, Lagneaux L. Mesenchymal stromal cells and immunomodulation: a gathering of regulatory immune cells. Cytotherapy. 2016;18:160–71.
Acknowledgements
Medical writing assistance was provided Karen Chinchilla, PhD (ArticulateScience LLC, Hamilton, NJ).
Funding
Editorial support was funded by Novartis; authors retained full control of the content of this review, uninfluenced by Novartis.
Author information
Authors and Affiliations
Contributions
FM, X-JH, and JPYS wrote this review and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Malard, F., Huang, XJ. & Sim, J.P.Y. Treatment and unmet needs in steroid-refractory acute graft-versus-host disease. Leukemia 34, 1229–1240 (2020). https://doi.org/10.1038/s41375-020-0804-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-020-0804-2
This article is cited by
-
Expansion of human bone marrow-derived mesenchymal stromal cells with enhanced immunomodulatory properties
Stem Cell Research & Therapy (2023)
-
Acute graft-versus-host disease
Nature Reviews Disease Primers (2023)
-
Acute Graft-versus-Host Disease: An Update on New Treatment Options
Drugs (2023)
-
Glucocorticoid and glycolysis inhibitors cooperatively abrogate acute graft-versus-host disease
Science China Life Sciences (2023)
-
Ruxolitinib Treatment of Steroid-Refractory Graft-versus-Host Disease in Children: A Case Series and Review of the Literature
Pediatric Drugs (2023)
