
Abstract
In hypoxia and hyperglycemia, SET7/9 plays an important role in controlling HIF-1α methylation and regulating the transcription of HIF-1α target genes, which are responsible for angiogenesis and wound healing. Here, we report the Ir(III) complex Set7_1a bearing acetonitrile (ACN) ligands as a SET7/9 methyltransferase inhibitor and HIF-1α stabilizer. Interestingly, Set7_1a could engage SET7/9 and strongly inhibit SET7/9 activity, especially after preincubation with homocysteine (Hcy), which is elevated in diabetes. We hypothesize that Set7_1a exchanges ACN subunits for Hcy to disrupt the interaction between SET7/9 and SAM/SAH, which are structurally related to Hcy. Inhibition of SET7/9 methyltransferase activity by Set7_1a led to reduced HIF-1α methylation at the lysine 32 residue, causing increased HIF-1α level and recruitment of HIF-1α target genes that promote angiogenesis, such as VEGF, GLUT1, and EPO, in hypoxia and hyperglycemia. Significantly, Set7_1a improved wound healing in a type 2 diabetic mouse model by activating HIF-1α signaling and downstream proangiogenic factors. To our knowledge, this is the first Hcy-targeting iridium compound shown to be a SET7/9 antagonist that can accelerate diabetic wound healing. More importantly, this study opens a therapeutic avenue for the treatment of diabetic wounds by the inhibition of SET7/9 lysine methyltransferase activity.
Similar content being viewed by others


Introduction
Patients with diabetes have high amputation rates due to chronic and nonhealing wounds and the lack of available medical tools1. Becaplermin, which is recombinant platelet-derived growth factor-BB (rhPDGF-BB), is the first FDA-approved product for wound healing. However, the use of becaplermin has been linked with an enhanced risk of cancer mortality2. Bioengineered skin-care products, such as Apligraf and Dermagraft, have been approved by the FDA for diabetic foot ulcer (DFU) treatment3. However, compared with the standard of care in terms of complete wound closure, these products show limited superiority3. Thus, effectively treating diabetic wounds is still a major challenge in wound care4.
Hyperglycemia and hypoxia play key roles in the pathophysiological outcomes of diabetes due to the dysfunctional response of tissues to insufficient oxygen concentrations5. The transcription factor hypoxia-inducible factor-1 alpha (HIF-1α) is considered a key mediator of wound healing. HIF-1α stimulates angiogenesis by binding to the hypoxia response element (HRE) of target genes, including stromal cell-derived factor 1 (SDF-1), vascular endothelial growth factor (VEGF), and angiopoietin 2. This molecule also transcriptionally upregulates the expression of other key players in the wound healing process6. In external wounds, HIF-1α can also mediate the inflammatory response through its regulation of the glycolytic metabolic switch. The enhancement of angiogenesis by increased HIF-1α is critical for wound healing, as it contributes to the delivery of both oxygen and nutrients to the wound site. However, hyperglycemia suppresses HIF-1α levels, and only very low levels of HIF-1α protein are detected in diabetic wounds7. Hence, hyperglycemia-dependent destabilization and suppression of HIF-1α is a central pathogenic mechanism in diabetic wounds8. This finding suggests that stabilization of HIF-1α in hyperglycemia could be a potential strategy for improving defective wound healing in diabetic mellitus.
In normoxia, the hydroxylation of HIF-1α by prolyl hydroxylase domain proteins (PHDs) in the cytoplasm promotes HIF-1α degradation via the Cullin 2 (CUL2)-E3 ubiquitin ligase complex9. PHD inhibitors, including FG-2216, BAY-853934, FG-4592, ethyl 3,4-dihydroxybenzoate (EDHB), and GSK1278863, have hence been studied as stabilizers of HIF-1α10. Although PHD inhibitors have entered clinical trials for angiogenesis-related diseases, they are limited by low target selectivity and negative side effects11. For instance, FG-2216 was linked to liver abnormalities in a phase 2 clinical trial, and one patient developed fatal hepatic necrosis12. Alternatively, blocking the downstream interaction of HIF-1α and Von Hippel-Lindau tumor suppressor (VHL) protein, which is a negative regulator of HIF-1α, is another potential strategy to inhibit HIF-1α degradation. For example, VH298, the most effective in vitro VHL-HIF-1α protein-protein interaction (PPI) inhibitor described to date, has been reported to selectively stabilize hydroxylated HIF-α and promote enthesis healing and wound healing in vivo13,14,15. In addition to VH298, our group recently reported a small molecule VHL-HIF-1α PPI inhibitor as a promising agent for wound healing in vivo16. Although several small-molecule HIF-1α stabilizers for treating wound healing have been reported in the literatures, none have yet entered clinical trials for treating diabetic wounds. Notably, hyperglycemia was able to decrease HIF-1α stabilization even in the presence of the PHD inhibitor EDHB in both hypoxia and normoxia, suggesting that mechanisms other than proline hydroxylation may be involved in the regulation of HIF-1α protein turnover in diabetes17,18.
Histone methyltransferase (HMT) SET domain containing lysine methyltransferase 7 (SET7/9), also known as KMT7, SET7, SETD7, or SET9, was one of the first protein lysine methyltransferases to be discovered. In diabetes, enhanced activity of SET7/9 in peripheral blood mononuclear cells has been detected and was correlated with epigenetic modifications. Dysfunction of SET7/9 has therefore been implicated in defective wound healing, ischemia, chronic anemia, and cardiovascular disease. SET7/9 contributes to hyperglycemia-induced inflammation in cell culture and animal models19. Macrophages of diabetic mice displayed increased expression of inflammatory genes as well as enhanced SET7/9 recruitment20,21. Additionally, SET7/9 mediates glucose stimulation of vascular endothelial cells via both histone and nonhistone mechanisms22. More importantly, studies have shown that sustained SET7/9 vascular gene expression was enhanced in diabetes as a response to hyperglycemia in vascular endothelial cells23,24.
The histone methyltransferase SET7/9-dependent methylation and histone demethylase LSD1-dependent demethylation cycle regulates HIF-1α stability in both VHL-independent and proline hydroxylation manners, indicating that SET7/9 plays an important function in controlling the level of HIF-1α and regulating HIF-α transcriptional activity25,26. The methylation of HIF-1α by SET7/9 leads to protein degradation of HIF-1α, thus diminishing the transcription of HIF-1α target genes, including those responsible for angiogenesis and wound healing12. Therefore, the stabilization of HIF-1α by inhibiting SET7/9 function and antagonizing HIF-1α methylation is a possible strategy to enhance diabetic wound healing. To date, several attempts have been made to identify SET7/9 inhibitors, including S-adenosyl-L-methionine (SAM, also known as AdoMet) competitive inhibitors (e.g., AAM-1, DAAM-3, sinefungin)27,28, histone lysine competitive inhibitors (e.g., (R)-PFI-2)29, and other inhibitors (e.g., GSK343, GSK926, SETin-1, DC-S238, DC-S239)30,31,32. However, many of the reported SET7/9 inhibitors are multitarget inhibitors with low potencies31.
Metal-based compounds with high structural variety and easily adjustable electronic and steric characteristics possess several promising advantages as protein or enzyme inhibitors in comparison to organic compounds33,34,35,36,37,38,39. Metal complexes can participate in ligand exchange reactions with biomolecules to exert their biological effects40,41. Herein, we report the application of the reported Ir(III) complex Set7_1a as a potent inhibitor of SET7/9. Set7_1a induces the accumulation of HIF-1α, especially after preincubation with homocysteine (Hcy), an elevated factor in diabetes, to upregulate the expression of HIF-1α target proteins in cellulo and in vivo. Mechanistic studies suggested that Set7_1a bearing acetonitrile (ACN) ligands could undergo ligand exchange reactions with Hcy to disrupt the interaction between SET7/9 and SAM, which are structurally related to Hcy. This study not only provides critical mechanistic insight into the potential protective effect of Set7_1a against HIF-1α degradation by inhibiting SET7/9 function and antagonizing HIF-1α methylation in hypoxia and hyperglycemia but also validates the feasibility of targeting SET7/9 methyltransferase activity for treating diabetic wounds. We also anticipate that Hcy-targeting Set7_1a could be employed as a new scaffold for the future development of more selective and potent antagonists of SET7/9 as clinical therapeutics for diabetic wound healing.
Materials and methods
Materials, antibodies, and chemicals
SET7/9 polyclonal rabbit antibody (NBP2-99744), HIF-1α antibody (NB100-479) and VEGF antibody (NB100-664) were purchased from Novus Biologicals (Littleton, CO, USA). VHL antibody (GTX101087) was purchased from GeneTex (Alton Pkwy Irvine, CA, USA). PHD2 antibody (A300-322A) was purchased from Bethyl Laboratories, Inc. (Montgomery, Texas, USA). A VEGF ELISA kit (EHC108) was purchased from NeoBioscience (Shenzhen, Guangdong, China). The sequences of RNA interference oligonucleotides are as follows42:
SET7/9 siRNA: sense, 5’- GGGCACCUGGACGAUGACGGA-3’, antisense, 5’- UCCGUCAUCGUCCAGGUGCCC-3’.
Ctr siRNA: sense, 5’- UUCUCCGAACGUGUCACGU-3’, antisense, 5’- ACGUGACACGUUCGGAGAA-3’.
Fluorescence polarization
The fluorescence polarization assay was performed based on the instructions of the SET7/9 SAM-ScreenerTM Assay Kit. Briefly, 10 μL of SET7/9 (human recombinant) assay enzyme was incubated with 5 μL of positive control or negative control or complexes for 15 minutes at room temperature in a 384-well solid plate (low volume; black; 400093; Cayman Chemicals, Ann Arbor, MI, USA). Then, 5 μL of the reconstituted SAM-binding site probe was added to each well for 30 minutes at room temperature. The signals were recorded using a SpectraMax M5 microplate reader (Ex = 575 nm, Em = 620 nm). A two-sided t test was used to calculate P values.
Coimmunoprecipitation
Hyperglycemia-induced HUVECs (1 × 106 cells/well) were treated with the most promising complex, Set7_1a, or DMSO for 24 h under hypoxia. One hundred micrograms of the collected cell lysate was incubated with 10 μg of SET7/9 antibody in 500 μL of cell lysis buffer at 4 °C for 12 h. The proteins were immunoprecipitated using magnetic beads. The levels of the proteins coprecipitated by magnetic beads were analyzed with anti-SET7/9, anti-methyl lysine, and anti-HIF-1α antibodies and then visualized using ECL Western Blotting Detection Reagent (GE Healthcare, Madison, WI, USA).
In vitro scratch assay
Briefly, 5 × 105 hyperglycemia-induced HUVECs were seeded in 6-well plates under hypoxic conditions. Then, scratches were generated using a 200 μL plastic pipette tip when cells were grown to a confluent monolayer, and the cells were then washed with a medium containing 0.5% FBS three times. After treatment with compounds in hypoxia, images of the wounded monolayer of HUVECs were taken at 0, 12, 24, and 36 h using a bright-field inverted microscope (Nikon, Japan).
Animal experiments
Male C57BL/6 J mice were housed in the animal facility of the University of Macau and maintained at 23 ± 1 °C under a 12 h light/12 h dark cycle with 50% humidity and free access to water and food. Six- to eight-week-old mice were randomly divided into two groups. One group of mice (DM) was fed a high-fat diet (HFD, 60% calories from fat, Trophic Animal Feed High-Tech Co., Nantong, Jiangsu, China) for 8 weeks and then intraperitoneally injected with streptozotocin (STZ, 40 mg/kg body weight, 0.1 M citrate buffer, pH 4.5, Sigma-Aldrich, St. Louis, MO, USA) daily for 7 days. The other group of mice (NC) was fed a regular chow diet (Guangdong Medical Lab Animal Center, Guangzhou, Guangdong, China) for 8 weeks and then intraperitoneally injected with the same volume of citrate buffer. Three days after the last STZ injection, the mice in the DM group were fasted for 6 h, and then, the blood glucose levels were measured by a One-Touch Ultra glucometer (Lifescan, Milpitas, CA, USA). The mice with blood glucose between 15.0 and 28 mmol/L, accompanied by manifestations of polydipsia, polyuria and polyphagia, were considered to be mimic type 2 diabetic mice and used in the following experiments. The NC and DM mice were randomly allocated into two groups. The vehicle and Set7_1a groups were intraperitoneally injected with vehicle (PEG 400: distilled water = 6:4, v/v) and Set7_1a (50 mg/kg in PEG400 solution), respectively, every other day for 8 days.
ICP-MS study
Fifty microliters of the skin or plasma samples harvested after 8 days from the control and treatment groups were completely digested in 5 mL of a 68% HNO3:H2O2 (v/v = 4:1) solution. The iridium content in skin samples was determined by ICP-MS with an iridium standard solution (Sigma-Aldrich, St Louis, MO, USA) and calculated as pg[iridium]/μg[protein], and data from plasma samples were calculated as pg[iridium]/μL[plasma].
Assessment of skin perfusion
Regional skin perfusion at the wound area of mice was measured using a laser Doppler imager (PeriCam PSI System, Perimed, Sweden).
Statistical analysis
The number of samples or animals is specified in the figure for each experiment. One-way analysis of variance (ANOVA) was performed to assess the significant differences between groups unless otherwise noted. Significant differences were claimed when P < 0.05.
Detailed information about other experiments is included in the Supporting Information file.
Results
Screening of small molecules as SET7/9 inhibitors
As a protein lysine methyltransferase (PLMT/PKMT), SET7/9 catalyzes the methylation of the epsilon amino group in lysine residues using SAM as a cofactor, while SAM is converted into the Hcy derivative S-adenosyl-L-homocysteine (SAH). In this study, a fluorescence polarization (FP) assay was performed to investigate the binding of the transition metal complexes to SET7/9 in the presence of a small molecule fluorescent probe that selectively binds to the SAM-binding pocket in SET7/9 (Fig. 1a). An in-house library of cyclometalated Ir(III)/Rh(III) metal complexes (Set7_1–Set7_18) with varying ligands was initially utilized to explore their ability to inhibit SET7/9 (Fig. 1b). Among the 18 potential metal complexes, the Ir(III) complexes Set7_1 and Set7_2 showed significant displacement of the SAM binding site probe (Fig. 1c), with slightly lower potency than the positive control, sinefungin (5’-deoxy-5’-(1,4-diamino-4-carboxybutyl)adenosine) (Supplementary Fig. 1), a previously reported SET7/9 inhibitor28. The complexes Set7_1 and Set7_2 contain different C^N ligands, but both possess two ACN ligands, suggesting that the ACN group might play a role in inhibiting the interaction of SAM-SET7/9.

a Schematic diagram of the FP assay to monitor the binding ability of the complexes to SET7/9. b Chemical structures of the in-house Ir(III) and Rh(III) complexes. Complexes 1–2 are OTf– salts, and complexes 3–18 are PF6– salts. c Displacement of a SAM binding site probe from SET7/9 by complexes 1–18 and sinefungin (Sine) as determined by an FP assay. d Chemical structures of the focused library of metal complexes Set7_1a–Set7_1f (racemates) and the ACN and piq ligands. e Engagement of the SAM binding site in SET7/9 by complexes Set7_1a–Set7_1f and isolated ligands as determined by an FP assay. f Dose-dependent effect of the complex Set7_1a in the FP assay. IC50 = 6.31 μM. *P < 0.05, **P < 0.01 vs. the control (Ctr) group.
Inspired by the promising results from the complexes Set7_1 and Set7_2, an ACN-containing focused library of 5 cyclometalated Ir(III) complexes (Set7_1a–Set7_1e) (Fig. 1d) was designed and synthesized. The Ir(III) complex Set7_1a, bearing two 1-phenylisoquinoline (piq) C^N ligands, showed the highest displacement of the SAM binding site probe from SET7/9 (Fig. 1e). This result suggested that the piq ligand is superior to other C^N ligands, such as the 2-(p-tolyl)pyridine (tpy) (Set7_1b), 2-phenylquinoline (pq) (Set7_1c), 2-(2,4-difluorophenyl)pyridine (dFppy) (Set7_1d) and 2-methyl-6-phenylpyridine (mppy) (Set7_1e) ligands (Fig. 1e). Significantly, the Ir(III) complex Set7_1a also showed higher potency than the Rh(III) congener Set7_1 f, indicating that the metal ion is crucial for biological activity. Finally, the isolated N^N and C^N ligands of complex Set7_1a, piq and ACN, showed negligible engagement of the SAM binding site in SET7/9 in the FP assay. This finding indicates that the Ir(III) center plays a key role in organizing the ligands into a biologically active arrangement. To further investigate the potency of Set7_1a in modulating the SAM-SET7/9 interaction, we performed dose-response assays of Set7_1a (Fig. 1f and Supplementary Fig. 2). The results demonstrated that the complex Set7_1a exhibited dose-dependent displacement of the SAM binding site probe from SET7/9 with an IC50 value of approximately 6.31 μM, which was comparable to sinefungin (6.17 μM).
Set7_1a disrupts the SAM-SET7/9 interaction by engaging SET7/9
Target engagement, which describes the capability of a drug to bind to the target protein in vivo, is an important measure of drug efficiency. The cellular thermal shift assay (CETSA) evaluates the thermal stabilization of proteins upon compound binding, which could indicate the target engagement of the candidate compounds in cellulo43. In this study, the SET7/9 engagement of the most potent inhibitors was evaluated using the CETSA. After incubation in high glucose medium under hypoxia, human umbilical vein endothelial cell (HUVEC) lysates were collected and treated with Set7_1a (30 μM) for 30 min. Aliquots were heated to set temperatures, centrifuged to isolate the soluble protein fraction from the cell debris, and then analyzed by immunoblotting (IB) with an anti-SET7/9 antibody. A positive shift of approximately 5 °C in the melting curve of SET7/9, but not β-actin, was observed, indicating stabilization of SET7/9 by the complex Set7_1a in cell lysates (Figs. 2a, b). A fluorescence-based thermal shift (FTS) assay was also performed with recombinant SET7/9 in the presence of the complex Set7_1a to confirm whether the inhibitors directly bound to the isolated protein. In the presence of 10 μM Set7_1a, the Tm of SET7/9 was increased by approximately 5.7 °C (Fig. 2c), which further suggests that Set7_1a directly engages and stabilizes SET7/9. The interaction between Set7_1a and SET7/9 was characterized by isothermal titration calorimetry (ITC), which demonstrated a Kd value of 1.06 ± 0.53 μM between Set7_1a and recombinant SET7/9 protein with a 1:1 stoichiometry (N = 0.85), and the binding between Set7_1a and recombinant SET7/9 was driven by both enthalpic and entropic factors (ΔH = −7.19 kcal/mol, −TΔS = −0.964 kcal/mol, ΔG = −8.15 kcal/mol) (Fig. 2d and Supplementary Fig. 3). Moreover, biolayer interferometry (BLI) revealed Kd values of 1.40 ± 0.17 μM (steady-state fit) and 1.54 ± 0.03 μM (kinetic fit) (Fig. 2e).

a, b Western blot analysis to evaluate the effect of the Set7_1a complex (10 μM) on the stabilization of SET7/9 in cellulo. c FTS analysis to evaluate the stability of recombinant His-tagged SET7/9 protein in the presence or absence of Set7_1a (10 μM). d ITC titration of recombinant SET7/9 protein (500 μM) into Set7_1a (50 μM). e BLI sensorgrams of the interaction between Set7_1a and recombinant His-tagged SET7/9 protein. f Covalent binding of Hcy to Set7_1a was confirmed by mass spectrometry. The main adduct formed at 721.2821 (MS positive, apo-Set7_1a + Hcy). Apo = Apomictic. g The effect of Set7_1a with or without Hcy on the SAM-SET7/9 interaction in vitro was estimated using a fluorescence polarization assay. h The relative levels of SET7/9 in hyperglycemia-induced HUVECs after Ctr and SET7/9 siRNA treatment. i Effect of the Set7_1a complex on HIF-1α and SET7/9 levels in hyperglycemia-induced HUVECs with or without SET7/9 knockdown. *P < 0.05, **P < 0.01 vs. the Ctr group.
To further uncover the binding mechanism, we examined the UV/Vis absorption of the complex Set7_1a in DMSO and ACN solution. The results suggested that Set7_1a could exchange its ACN ligands for DMSO from the solvent (Supplementary Fig. 4a). Similar solvent exchange reactions have been reported for other complexes, such as complex Set7_1a40, NAMI-A, and KP101944. We also detected the stability of Set7_1a in DMSO solution by UV/Vis absorption spectroscopy (Supplementary Fig. 4b). The lack of significant shifts in the absorbance spectrum suggested that Set7_1a was stable in DMSO solution at 298 K for at least 48 h. Moreover, the absorbance of Set7_1a was not significantly changed after coincubation with SAM or SAH, suggesting that Set7_1a does not interact directly with SAM or SAH to inhibit SET7/9 activity (Supplementary Fig. 4c). Moreover, in the presence of 20 common amino acids, the complex Set7_1a displayed a selective response toward cysteine (Cys) with diminished luminescence at Imax = 625 nm (Supplementary Fig. 5a). However, a previous study showed that Cys blood levels in diabetes are reduced due to elevated glucagon concentrations, which leads to increased uptake of glutamine and glycine by the liver45,46. Additionally, SET7/9 methyltransferase does not have the Cys-rich regions in the postSET domain. These observations suggest that Set7_1a does not directly bind to Cys or Cys residues to regulate SET7/9 methyltransferase activity. Thus, we also evaluated the interaction of Set7_1a with Hcy, which is a homolog of Cys (Supplementary Fig. 5b). Hcy levels are increased in patients with diabetes, especially in those with type 2 diabetes, but also in prediabetic individuals with insulin resistance47. Interestingly, the complex Set7_1a emitted a comparable diminished luminescence response in the presence of Hcy. To validate the possible action mechanism of Set7_1a, we performed HRMS analysis on a mixture of the complex Set7_1a and Hcy after 30 min of incubation at room temperature (Fig. 2f). A fragment ion was detected at m/z = 600.98 (apo-Set7_1a), which corresponded to the loss of two can moieties from Set7_1a. Moreover, a new peak with m/z = 736.11 appeared, which matched the theoretical molecular weight of the apo-Set7_1a–Hcy complex. The ligand exchange with amino acids has also been previously observed with other metal complexes in the literature40,48.
To gauge the specificity of Set7_1a, we also measured its potency against PR-SET7 (also known as SET8, SETD8, KMT5A), which is structurally similar to SET7/949. The results showed that Set7_1a exhibited no significant effect on PR-SET7 thermal stability in the FTS assay (Supplementary Fig. 6a). Moreover, since Set7_1a interacted with Cys amino acids (Fig. 2f and Supplementary Fig. 5a), we also tested its interaction with two conserved Cys-rich proteins, Kelch-like ECH-associated protein 1 (Keap1) and 8-oxoguanine DNA glycosylase 1 (OGG1), which contain 27 and 8 Cys residues50,51, respectively. The results showed that Set7_1a did not significantly stabilize Keap1 (Supplementary Fig. 6b) or OGG1 (Supplementary Fig. 6c) in the FTS assay. Taken together, these data indicate that Set7_1a potently and selectively engages SET7/9.
We next evaluated the ability of Hcy to enhance the ability of Set7_1a to disrupt the interaction between the SAM probe and SET7/9 using the in vitro FP assay. As shown in Fig. 2g, preincubation of Set7_1a with Hcy further disrupted the interaction between the SAM probe and SET7/9 compared to Set7_1a alone. These results indicated that the covalent complex between Set7_1a and Hcy is even more effective at engaging the SAM binding pocket of SET7/9 compared to Set7_1a.
As the activity of SET7/9 reduces the stability of HIF-1α, we wanted to explore whether Set7_1a could stabilize HIF-1α in cellulo and investigate whether this stabilization was dependent on SET7/9. SET7/9 knockdown cells were prepared by treating hyperglycemia-induced HUVECs with SET7/9 siRNA. As depicted in Fig. 2h, treatment of the control siRNA cells with Set7_1a or sinefungin resulted in significant HIF-1α accumulation. However, the HIF-1α levels were relatively unaffected by Set7_1a or sinefungin in the SET7/9 knockdown cells (Fig. 2i). This finding suggests that the accumulation of HIF-1α induced by Set7_1a requires the presence of SET7/9 protein in hyperglycemia-induced HUVECs.
We also evaluated the selectivity of Set7_1a by evaluating its effect on other negative regulators of HIF-1α. The results showed that Set7_1a does not interfere with proteasome activity, oxygen consumption, PHD2, or VHL levels (Supplementary Fig. 7), indicating that SET7/9 is the main target of Set7_1a to activate the HIF signaling axis, which suggests that Set7_1a may function as a selective HIF-1α stabilizer.
Set7_1a inhibits SET7/9-mediated HIF-1α lysine methylation at the lysine-32 residue
A coimmunoprecipitation (co-IP) assay was performed to examine whether Set7_1a disrupted the interaction between HIF-1α and SET7/9 in cellulo. First, we demonstrated that hyperglycemia decreased the levels of HIF-1α under both hypoxic and normoxic conditions in HUVECs without affecting the level of SET7/9 (Fig. 3a). High glucose is known to promote SET7/9 nuclear localization in human endothelial cells but does not alter SET7/9 expression52. In the co-IP results, Set7_1a did not decrease the amount of HIF-1α coprecipitating with SET7/9 in hyperglycemia-induced HUVECs in hypoxia, indicating that it did not affect the interaction of SET7/9 and HIF-1α in the treated cells (Fig. 3b). Interestingly, we found that Set7_1a increased HIF-1α levels in a dose-dependent manner (Fig. 2i). Furthermore, cotreatment with Hcy, as described previously53, further increased HIF-1α levels in hyperglycemia-induced HUVECs compared to Set7_1a alone, demonstrating that Hcy could enhance the potency of Set7_1a to induce the stabilization of HIF-1α but not affecting the level of SET7/9 (Fig. 3c and Supplementary Fig. 8).

a Western blot analysis to evaluate the effect of glucose levels on the stabilization of HIF-1α in HUVECs under normoxic and hypoxic conditions. b The effect of Set7_1a on the interaction of SET7/9 and HIF-1α in cellulo using the co-IP assay. c Western blot analysis to evaluate the effect of the Set7_1a complex (10 μM) with or without Hcy on HIF-1α and SET7/9 levels in the hyperglycemia-induced HUVECs under hypoxia. Data are expressed as the mean ± SD (n = 3 independent experiments), *P < 0.05, **P < 0.01 vs. the Ctr group; ##P < 0.01, NSP > 0.05, the Set7_1a vs. Hcy + Set7_1a group. d Effect of Set7_1a on the HIF-1α methylation level in the hyperglycemia-induced HUVECs. e Effect of Set7_1a on the methylation of HIF-1α peptide mediated by Set7/9. Dot blot analysis of the synthesized HIF-1α peptide (WT) methylated by Set7/9 in vitro was performed with an anti-methyl lysine antibody. The HIF-1α peptide with lysine to arginine substitution (K32R) was used as a negative control for antibody detection. Protein domains of HIF-1α: bHLH, basic helix-loop-helix; PAS, Per-ARNT-Sim; N-TAD, NH2-terminal transactivation domain; ODDD, oxygen-dependent degradation domain; NLS, nuclear localization signal; C-TAD, COOH-terminal TAD. *P < 0.05, **P < 0.01 vs. the Ctr group.
HIF-1α possesses a consensus sequence that is recognized and methylated by SET7/9 near the lysine-32 site, leading to the ubiquitin-mediated degradation of HIF-1α54. To further study the mechanism by which Set7_1a prevents HIF-1α degradation, we performed immunoprecipitation with an anti-methyl lysine antibody in hyperglycemia-induced HUVECs treated with Set7_1a for 12 h under hypoxia. Afterward, IB analysis was performed with an anti-HIF-1α antibody. The results showed that HIF-1α lysine methylation was significantly decreased in a dose-dependent manner (Fig. 3d). To confirm whether Set7_1a could inhibit HIF-1α methylation specifically at the lysine-32 residue, we also conducted an in vitro methylation experiment using purified recombinant SET7/9 enzymes and synthesized HIF-1α wild-type (Supplementary Fig. 9) or K32R (Supplementary Fig. 10) peptides as substrates. Set7_1a significantly inhibited the lysine methylation level of HIF-1α wild-type (WT) peptides as shown by IB with an anti-methyl lysine antibody, while it had no effect on HIF-1α K32R peptides (Fig. 3e). These results suggested that Set7_1a could significantly inhibit SET7/9-mediated HIF-1α lysine methylation at the lysine-32 residue. This observation supports the hypothesis that Set7_1a (or apo-Set7_1a–Hcy) disrupts the interaction between SET7/9 and SAM/SAH rather than disrupting the interaction between SET7/9 and HIF-1α.
Set7_1a upregulates HIF-1α target protein expression and promotes angiogenesis in cellulo and in vivo
HIF-1α translocates into the nucleus, thus binding to HRE to promote a hypoxic response. In this study, the ability of Set7_1a to enhance the transcription of HIF-1α genes in cellulo was evaluated using a dual luciferase reporter assay. Set7_1a increased HRE-driven luciferase intensity (after normalization to Renilla luciferase activity as an internal control) in a dose-dependent manner (Fig. 4a). This result suggests that by inhibiting the methylation activity of SET7/9 in cellulo, HIF-1α is allowed to accumulate and translocate into the nucleus to activate gene expression.

a Effect of the Set7_1a complex on HRE luciferase activity in hyperglycemia-induced HUVECs under hypoxic conditions. b Effect of Set7_1a on the levels of VEGF protein in the hyperglycemia-induced HUVECs under hypoxic conditions. c The relative VEGF levels in the hyperglycemia-induced HUVECs under hypoxic conditions. VEGF concentration was determined by ELISAs. d Effect of the Set7_1a complex on the levels of EPO and GLUT1 proteins in the hyperglycemia-induced HUVECs under hypoxic conditions. e The effect of the Set7_1a complex on HUVEC migration was assessed using an in vitro wound-healing assay. Ninety percent confluent monolayers of HUVECs were scratch wounded and then incubated with Set7_1a (5 μM) for 24 h. f Microangiography of zebrafish embryo blood vessels. VRI was introduced to inhibit the angiogenesis of the zebrafish embryos. g Schematic model of the effect of Set7_1a on gene transcription. *P < 0.05, **P < 0.01 vs. the Ctr group.
HIF-1α activity drives the expression of several target genes involved in angiogenesis, including VEGF, EPO, and GLUT116,55. Therefore, the effect of Set7_1a on VEGF secretion from hyperglycemia-induced HUVECs under hypoxia was also investigated. Set7_1a treatment for 12 h in high-glucose medium under hypoxia increased VEGF secretion from HUVECs in a dose-dependent manner, as determined by Western blotting (Fig. 4b), which was also verified by ELISAs (Fig. 4c). Under similar conditions, the expression levels of GLUT1 and EPO were also increased by Set7_1a (Fig. 4d).
HIF-1α plays a key role in enhancing the hypoxic induction of epithelial cell migration during wound closure56. To evaluate the effect of Set7_1a on wound closure, we performed the scratch assay, a widely used experiment to assess wound healing in cellulo. HUVECs were grown to confluency as monolayers on plastic and scratched with a 200 μL pipette tip. Set7_1a (5 μM) treatment promoted the wound repair of HUVECs (Fig. 4e), as indicated by a significant decrease in the wound area versus the control. Additionally, although Set7_1a exhibited moderate cytotoxicity toward HUVECs, with an IC50 value of 29.5 μM under hypoxic conditions after 48 h (Supplementary Fig. 11), the concentrations of Set7_1a required to induce beneficial biological effects were much lower than its IC50 value. Moreover, the complex Set7_1a-stimulated migration of HUVECs was significantly enhanced in the presence of Hcy (100 μM) after 24 h (Supplementary Fig. 12). This result demonstrates that Hcy is a potential enhancer in Set7_1a-mediated functions in living HUVECs and supports the hypothesis that Set7_1a can exert enhanced beneficial biological effects at lower concentrations than its in vitro IC50 value.
HIF-1α expression is upregulated in hypoxia and is often associated with the induction of angiogenesis57. Given the promising ability of Set7_1a to promote HIF-1α target genes, we hypothesized that Set7_1a could potentially promote angiogenesis in vivo. Zebrafish is a commonly used angiogenesis model because it is fast-growing and optically transparent. In this study, zebrafish embryos at 20 hours post-fertilization (hpf) were pretreated with a known angiogenesis inhibitor, VEGFR tyrosine kinase inhibitor II (VRI), for 4 h and then incubated with Set7_1a or sinefungin for an additional 24 h. As expected, in the absence of Set7_1a, VRI strongly suppressed angiogenesis, with almost no blood vessel formation observed in the zebrafish trunk (Fig. 4f). Excitingly, Set7_1a (6.25 to 25 μM) could attenuate VRI-induced inhibition of angiogenesis in vivo in a dose-dependent manner, as revealed through an increase in blood vessel formation. Moreover, 6.25 μM of Set7_1a was more effective at promoting angiogenesis than 20 μM sinefungin. This result indicates that Set7_1a has the potential to be developed as a therapeutic agent to promote angiogenesis or wound healing. We hypothesize that the enhancement of angiogenesis or wound healing can be attributed, at least in part, to the ability of Set7_1a to inhibit the catalytic activity of SET7/9 and enhance HIF-1α stability, thus leading to an accumulation of HIF-1α-driven proangiogenic products (Fig. 4g).
Set7_1a accelerates wound healing in mimic type 2 diabetic mice
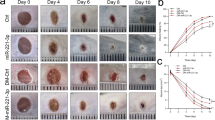
Finally, the role of Set7_1a in wound healing was evaluated in normal and type 2 mimic diabetic mice. The mice were fed a high-fat diet for 8 weeks, followed by low-dose STZ (40 mg/kg) injection daily for 1 week (Fig. 5a). Three days after the last STZ injection, the mice with 15 to 28 mmol/L fasting blood glucose levels were deemed mimic type 2 diabetic mice (DM) and recruited for wound healing experiments (Supplementary Fig. 13a). Vehicle (PEG 400:distilled water = 6:4, v/v) and Set7_1a (50 mg/kg in PEG 400 solution) were then intraperitoneally (i.p.) injected into the vehicle and Set7_1a groups, respectively, every other day for 8 days. Inductively-coupled plasma mass spectrometry (ICP-MS) experiments demonstrated the presence of iridium in skin and plasma specimens of treated mice, confirming the delivery of Set7_1a (Supplementary Fig. 14). Set7_1a treatment did not induce obvious adverse reactions or body weight changes over 8 days in either normal control (NC) or DM mice (Supplementary Fig. 13b). For analysis of wound healing, two full-thickness skin lesions were excised in the interscapular area of each animal, the wounds were monitored every other day, and wound closure rates were calculated for each group. Encouragingly, treatment with Set7_1a dramatically accelerated wound healing in both the NC and DM mice (Fig. 5b). In the NC groups, the Set7_1a-treated mice showed 36%, 56% and 74% closure at 2, 4 and 6 days postinjury and almost completed wound closure by Day 8 postinjury, whereas the vehicle-treated mice exhibited 23%, 42%, 59% and 78% closure after 2, 4, 6 and 8 days postinjury, respectively (Fig. 5b). In the DM groups, the wound closure rates in the Set7_1a-treated mice were 26%, 50%, 70% and 85% at 2, 4, 6 and 8 days postinjury, respectively, and the wound closure rates in the vehicle control mice were nearly 19%, 31%, 44% and 53% at 2, 4, 6 and 8 days postinjury, respectively (Fig. 5b). Taken together, these results indicated that Set7_1a accelerates wound healing in both normal and mimic type 2 diabetic mice.

a Timeline for the mouse animal study. b Representative images of wound (up) and wound closure rates (down). c H&E staining and Masson’s trichrome staining of dorsal skin sections (left) and skin thickness (right). Scale bar = 200 μm. d Representative images for each group (left) and baseline perfusion on the back skin of mice (right). e DAPI and CD31 staining in the wound bed. Scale bar = 50 μm. f IB (left) and quantitative (right) analyses of HIF-1α target genes in wound tissue. g The mRNA levels of proangiogenic genes involved in wound healing. Data are expressed as the mean ± SD (n = 5–8 mice). #P < 0.05, ##P < 0.01 DM vs. NC, *P < 0.05, **P < 0.01 the complex Set7_1a vs. vehicle.
H&E and Masson’s trichrome staining were further employed to assess the epithelial thickness of regenerated skin. The results showed that the dorsal skin thickness was greater in the Set7_1a-treated mice than in the vehicle control mice in both the NC and DM groups (Fig. 5c). Masson’s trichrome staining further showed increased collagen deposition in the Set7_1a complex-treated mice compared to the vehicle controls (Fig. 5c). Tissue angiogenesis plays a key role in wound healing. The skin perfusion pressure experiments revealed that Set7_1a enhanced the skin blood flow rate in both the NC and DM mice (Fig. 5d). Immunostaining for CD31, a specific marker of vascular vessels, further indicated that Set7_1a could significantly increase microvessel density in wounding areas in mice from both the NC and DM groups (Fig. 5e). The expression levels of HIF-1α, VEGF, EPO, and GLUT1 were significantly increased in the wound tissue from the Set7_1a-treated DM mice compared with those from the vehicle control DM mice (Fig. 5f). Moreover, HIF-1α products that are required for wound healing cell motility (HSP-90), angiogenesis (VEGF-R), and recruitment of CAG (Tie-2, SCF, and SDF-1) were also increased in the wound tissue from the Set7_1a treated DM mice (Fig. 5g and Supplementary Table 1). Taken together, Set7_1a accelerates wound healing in mimic type 2 diabetic mice by activating HIF-1α signaling and downstream proangiogenic factors.
Discussion
Patients with diabetes commonly have impaired wound healing that does not progress through the healing process in a timely manner, creating major challenges to healthcare systems worldwide58. Recent research has shown that dysfunctional cellular reactions to hypoxic conditions drive impaired wound healing in diabetes. HIF-1α is an important regulator of tissue repair; however, hyperglycemia destabilizes the HIF-1α protein in hypoxia59. Biopsies from individuals with DFU exhibit decreased HIF-1α levels compared to those from individuals with chronic venous ulcers60. As a protein lysine methyltransferase, SET7/9 plays an important role in controlling the HIF-1α level. HIF-1α methylation by SET7/9 leads to protein degradation of HIF-1α, thus diminishing the transcription of HIF-1α target genes, including those responsible for angiogenesis and wound healing. Recent studies showed that sustained vascular gene expression of methyltransferase SET7/9 was enhanced in diabetes as a response to elevated glucose in vascular endothelial cells24. Interestingly, enhanced levels of SET7/9 recruitment were uncovered in macrophages from diabetic mice, and SET7/9-induced epigenetic transitions have been linked to vascular dysfunction in type 2 diabetic patients61. In this context, HIF-1α stabilization by inhibiting SET7/9 function and antagonizing HIF-1α methylation thus becomes a possible strategy to promote angiogenesis to enhance wound healing in diabetes.
Here, we discovered that the Ir(III) complex Set7_1a bearing ACN ligands is a potent inhibitor of SET7/9 activity. This complex strongly inhibits SET7/9 activity, especially after preincubation with Hcy, an elevated factor in diabetes. Cell-based and cell-free assays suggest that Set7_1a (or apo-Set7_1a–Hcy) engages SET7/9 to disrupt its interaction with SAM. Moreover, the Set7_1a complex effectively induced HIF-1α accumulation and upregulated HIF-1α target protein expression involved in angiogenesis. Importantly, Set7_1a was able to improve wound healing in diabetic mice by activating HIF-1α signaling and downstream proangiogenic factors (Fig. 6). This study demonstrates a Hcy-targeting iridium compound as a SET7/9 inhibitor for accelerating diabetic wound healing and, more importantly, opens a novel avenue for the treatment of diabetic wounds by the inhibition of SET7/9 lysine methyltransferase activity.

This work reports the discovery of a homocysteine-targeting complex Set7_1a as a SET7/9 methyltransferase inhibitor that regulates the methylation-dependent degradation of HIF-1α, an important target protein for improving diabetic wound healing. Set7_1a could improve wound healing in mimic type 2 diabetic mice by activating HIF-1α signaling, opening an avenue for treating diabetic wounds via SET7/9 inhibition using homocysteine-targeting compounds.
Elevated levels of Hcy, a nonessential sulfur-containing amino acid, have been reported as a risk factor in diabetes62,63. Thus, an effective treatment for lowering elevated Hcy in chronic wound patients may promote the return of normal healing processes. Interestingly, our study demonstrates a treatment strategy using the selective SET7/9 inhibitor Set7_1a, which could interact with Hcy via ligand exchange, enhance HIF-1α stabilization of HUVECs in the presence of Hcy, and promote the expression of angiogenic proteins, such as VEGF, GLUT1 and EPO, in hypoxia and hyperglycemia. Hence, this complex may have the potential to correct elevated Hcy levels via covalent binding in a patient with impaired wound healing.
HIF-1α target genes (e.g., VEGF, EPO, and GLUT1) mediated by Set7_1a are factors that are responsible for angiogenesis and diabetic wound healing. For example, VEGF, as an endothelial cell mitogen and inducer of vascular permeability, has been identified as a crucial factor for the altered production of new blood vessels, which is one of the compromised phases in diabetic wound healing64,65. As a hematopoietic factor regulating the proliferation and differentiation of erythroid precursor cells, EPO was successfully used to improve diabetes-impaired wound healing by stimulating macrophage function and activity, which is responsible for the production of cytokines and growth factors that are essential for the healing cascade66. Apart from VEGF and EPO, GLUT1, as a mediator of a limiting step in glucose metabolism, enhances the wound repair process by regulating glycolysis and ROS production67,68. Moreover, other pathways independent of Set7_1a-mediated activation of these target genes, such as the Wnt/β-catenin, Notch and JAK/STAT signaling pathways, may be involved in promoting the wound healing process.
Notably, our previously published VHL-HIF-1α interaction blocker also showed HIF-1α stabilization and wound healing effects similar to the SET7/9 inhibitor Set7_1a reported in this study. However, Set7_1a could promote HIF-1α stability by disrupting the interaction between SET7/9 and SAM/SAH and blocking HIF-1α lysine methylation rather than disrupting the interaction between SET7/9 and HIF-1α, which is different from the mechanism of the VHL-HIF-1α interaction blocker. These results indicate that the combination of complexes Set7_1a and 1a may be a more potent strategy to induce the stabilization of HIF-1α by simultaneously blocking methylation-dependent degradation (HIF-1α lysine methylation) and hydroxylation-dependent degradation (VHL-HIF-1α interaction), which could open a new research direction for treating diabetic wound healing in the future.
References
Sharma, R., Sharma, S. K., Mudgal, S. K., Jelly, P. & Thakur, K. Efficacy of hyperbaric oxygen therapy for diabetic foot ulcer, a systematic review and meta-analysis of controlled clinical trials. Sci. Rep. 11, 2189 (2021).
Tang, X. et al. Wound healing driver gene and therapeutic development: political and scientific hurdles. Adv. Wound Care (N. Rochelle) 10, 415–435 (2021).
Matoori, S., Veves, A. & Mooney, D. J. Advanced bandages for diabetic wound healing. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.abe4839 (2021).
Jeffcoate, W. J., Vileikyte, L., Boyko, E. J., Armstrong, D. G. & Boulton, A. J. Current challenges and opportunities in the prevention and management of diabetic foot ulcers. Diabetes Care 41, 645–652 (2018).
Dascalu, A. M. et al. In Type 2 Diabetes - From Pathophysiology to Cyber Systems, (eds Stoian, A. P.) Ch. 10 (IntechOpen, United Kingdom, 2021).
Paskal, W. et al. Single dose of N-Acetylcysteine in local anesthesia increases expression of HIF1α, MAPK1, TGFβ1 and growth factors in rat wound healing. Int. J. Mol. Sci. 22, 8659 (2021).
Gabryelska, A., Karuga, F. F., Szmyd, B. & Bialasiewicz, P. HIF-1alpha as a mediator of insulin resistance, T2DM, and its complications: potential links with obstructive sleep apnea. Front. Physiol. 11, 1035 (2020).
Gao, Y. et al. LRG1 promotes keratinocyte migration and wound repair through regulation of HIF-1α stability. J. Investig. Dermatol. 140, 455–464 (2020).
Zurlo, G., Guo, J., Takada, M., Wei, W. & Zhang, Q. New insights into protein hydroxylation and its important role in human diseases. Biochim. Biophys. Acta Rev. Cancer 1866, 208–220 (2016).
Provenzano, R. et al. Oral hypoxia–inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clin. J. Am. Soc. Nephrol. 11, 982–991 (2016).
Schodel, J. & Ratcliffe, P. J. Mechanisms of hypoxia signalling: new implications for nephrology. Nat. Rev. Nephrol. 15, 641–659 (2019).
Haase, V. H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. 21, S110–S124 (2017).
Frost, J. et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun. 7, 13312 (2016).
Qiu, S. et al. Von Hippel-Lindau (VHL) protein antagonist, VH298, promotes functional activities of tendon-derived stem cells and accelerates healing of entheses in rats by inhibiting ubiquitination of hydroxy-HIF-1α. Biochem. Biophys. Res. Commun. 505, 1063–1069 (2018).
Qiu, S. et al. Von Hippel-Lindau (VHL) protein antagonist VH298 improves wound healing in streptozotocin-induced hyperglycaemic rats by activating hypoxia-inducible factor- (HIF-) 1 signalling. J. Diabetes Res. 2019, 1897174 (2019).
Li, G. et al. A small molecule HIF-1α stabilizer that accelerates diabetic wound healing. Nat. Commun. 12, 3363 (2021).
Catrina, S.-B., Okamoto, K., Pereira, T., Brismar, K. & Poellinger, L. Hyperglycemia regulates hypoxia-inducible factor-1α protein stability and function. Diabetes 53, 3226–3232 (2004).
Johar, D. et al. Protein networks linking Warburg and reverse Warburg effects to cancer cell metabolism. Biofactors https://doi.org/10.1002/biof.1768 (2021).
Chokpaisarn, J., Urao, N., Voravuthikunchai, S. P. & Koh, T. J. Quercus infectoria inhibits Set7/NF-κB inflammatory pathway in macrophages exposed to a diabetic environment. Cytokine 94, 29–36 (2017).
Sharma, N., Sankrityayan, H., Kale, A. & Gaikwad, A. B. Role of SET7/9 in the progression of ischemic renal injury in diabetic and non-diabetic rats. Biochem. Biophys. Res. Commun. 528, 14–20 (2020).
Natarajan, R. Epigenetic mechanisms in diabetic vascular complications and metabolic memory: the 2020 Edwin Bierman award lecture. Diabetes 70, 328–337 (2021).
Karimnia, N. et al. Current perspectives in Set7 mediated stem cell differentiation. Noncoding RNA 2, 14 (2016).
Khullar, M., Cheema, B. S. & Raut, S. K. Emerging evidence of epigenetic modifications in vascular complication of diabetes. Front. Endocrinol. 8, 237 (2017).
Lopez-Diez, R. et al. Diabetes and cardiovascular complications: the epidemics continue. Curr. Cardiol. Rep. 23, 1–11 (2021).
Luo, W. & Wang, Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-α protein stability and activity. Cell. Mol. Life Sci. 75, 1043–1056 (2018).
Koyasu, S., Kobayashi, M., Goto, Y., Hiraoka, M. & Harada, H. Regulatory mechanisms of hypoxia‐inducible factor 1 activity: two decades of knowledge. Cancer Sci. 109, 560–571 (2018).
Niwa, H. et al. Structures of histone methyltransferase SET7/9 in complexes with adenosylmethionine derivatives. Acta Crystallogr. D: Struct. Biol. 69, 595–602 (2013).
Sasaki, K. et al. Inhibition of set domain–containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J. Am. Soc. Nephrol. 27, 203–215 (2016).
Barsyte-Lovejoy, D. et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl Acad. Sci. USA. 111, 12853–12858 (2014).
Verma, S. K. et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096 (2012).
Meng, F. et al. Discovery and optimization of novel, selective histone methyltransferase SET7 inhibitors by pharmacophore-and docking-based virtual screening. J. Med. Chem. 58, 8166–8181 (2015).
Kashyap, S., Sandler, J., Peters, U., Martinez, E. J. & Kapoor, T. M. Using ‘biased-privileged’scaffolds to identify lysine methyltransferase inhibitors. Bioorg. Med. Chem. 22, 2253–2260 (2014).
Li, G. et al. A bioactive ligand-conjugated iridium (III) metal-based complex as a Keap1–Nrf2 protein-protein interaction inhibitor against acetaminophen-induced acute liver injury. Redox Biol. 48, 102129 (2021).
Chen, J. C. et al. Investigation of inducing apoptosis in human lung cancer A549 cells and related mechanism of a ruthenium(II) polypyridyl complex. Inorg. Chem. Commun. 69, 35–39 (2016).
Ma, D.-L., Wu, C., Li, G., Yung, T.-L. & Leung, C.-H. Transition metal complexes as imaging or therapeutic agents for neurodegenerative diseases. J. Mater. Chem. B 8, 4715–4725 (2020).
Li, G., Wu, C., Ma, D.-L. & Leung, C.-H. Drug screening strategies using metal-based luminescent probes. Trends Analyt. Chem. https://doi.org/10.1016/j.trac.2021.116270 (2021).
Suh, J.-M., Kim, G., Kang, J. & Lim, M. H. Strategies employing transition metal complexes to modulate amyloid-β aggregation. Inorg. Chem. 58, 8–17 (2018).
Liu, W. et al. Orientation-inspired perspective on molecular inhibitor of tau aggregation by curcumin conjugated with ruthenium (II) complex scaffold. J. Phys. Chem. B 124, 2343–2353 (2020).
Ott, I. Metal N-heterocyclic carbene complexes in medicinal chemistry. Adv. Inorg. Chem. 75, 121–148 (2020).
Zhong, H.-J. et al. An iridium (III)-based irreversible protein–protein interaction inhibitor of BRD4 as a potent anticancer agent. Chem. Sci. 6, 5400–5408 (2015).
Sullivan, M. P. et al. Probing the Paradigm of Promiscuity for N‐Heterocyclic Carbene Complexes and their Protein Adduct Formation. Angew. Chem. Int. Ed. 60, 19928–19932 (2021).
Liu, X. et al. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc. Natl Acad. Sci. USA. 108, 1925–1930 (2011).
Li, G. et al. Aurone derivatives as Vps34 inhibitors that modulate autophagy. Acta Pharm. Sin. B. 9, 537–544 (2019).
Wall, B. J. et al. Importance of hydrogen bonding: structure–activity relationships of ruthenium (III) complexes with pyridine-based ligands for alzheimer’s disease therapy. J. Med. Chem. 64, 10124–10138 (2021).
Jain, S. K. et al. Vitamin D and L-cysteine levels correlate positively with GSH and negatively with insulin resistance levels in the blood of type 2 diabetic patients. Eur. J. Clin. Nutr. 68, 1148–1153 (2014).
Kanikarla-Marie, P., Micinski, D. & Jain, S. K. Hyperglycemia (high-glucose) decreases L-cysteine and glutathione levels in cultured monocytes and blood of Zucker diabetic rats. Mol. Cell. Biochem. 459, 151–156 (2019).
Lei, X. W. et al. Association between homocysteine level and the risk of diabetic retinopathy: a systematic review and meta-analysis. Diabetol. Metab. Syndr. 10, 1–8 (2018).
Chao, D. B. & Zhang, Y. X. Aggregation enhanced luminescent detection of homocysteine in water with terpyridine-based Cu2+ complexes. Sens. Actuators B Chem. 245, 146–155 (2017).
Qian, C. & Zhou, M. M. SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell. Mol. Life Sci. 63, 2755–2763 (2006).
Saito, R. et al. Characterizations of three major cysteine sensors of keap1 in stress response. Mol. Cell. Biol. 36, 271–284 (2016).
Wang, K., Maayah, M., Sweasy, J. B. & Alnajjar, K. S. The role of cysteines in the structure and function of OGG1. J. Biol. Chem. 296, 100093 (2021).
Okabe, J. et al. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ. Res. 110, 1067–1076 (2012).
Wu, X. et al. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 20, 46–59 (2019).
Kim, Y. et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat. Commun. 7, 10347 (2016).
Ma, Z. et al. Targeting hypoxia-inducible factor-1, for cancer treatment: Recent advances in developing small-molecule inhibitors from natural compounds. Semin. Cancer Biol. 80, 379–390 (2022).
Goggins, B. J. et al. Pharmacological HIF-1 stabilization promotes intestinal epithelial healing through regulation of α-integrin expression and function. Am. J. Physiol. Gastrointest. Liver Physiol. 320, G420–G438 (2021).
Sun, J. et al. HIF-1α overexpression in mesenchymal stem cell-derived exosomes mediates cardioprotection in myocardial infarction by enhanced angiogenesis. Stem Cell Res. 11, 1–13 (2020).
Sutthammikorn, N. et al. Topical gynura procumbens as a novel therapeutic improves wound healing in diabetic mice. Plants 10, 1122 (2021).
Catrina, S.-B. & Zheng, X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia 64, 709–716 (2021).
Schönborn, M. et al. Pro-and anti-angiogenic factors: their relevance in diabetic foot syndrome—a review. Angiology 73, 299–311 (2021).
Zhang, H. & Pollin, T. I. Epigenetics variation and pathogenesis in diabetes. Curr. Diab. Rep. 18, 121 (2018).
Xu, C. et al. Relationship between homocysteine level and diabetic retinopathy: a systematic review and meta-analysis. Diagn. Pathol. 9, 1–9 (2014).
Boykin, J. V. Jr Wound nitric oxide bioactivity: a promising diagnostic indicator for diabetic foot ulcer management. J. Wound Ostomy Cont. Nurs. 37, 25–32 (2010).
Bao, P. et al. The role of vascular endothelial growth factor in wound healing. J. Surg. Res. 153, 347–358 (2009).
Bitto, A. et al. Simvastatin enhances VEGF production and ameliorates impaired wound healing in experimental diabetes. Pharmacol. Res. 57, 159–169 (2008).
Galeano, M. et al. Recombinant human erythropoietin stimulates angiogenesis and wound healing in the genetically diabetic mouse. Diabetes 53, 2509–2517 (2004).
Ahluwalia, A. & S Tarnawski, A. Critical role of hypoxia sensor-HIF-1α in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr. Med. Chem. 19, 90–97 (2012).
Lu, Y.-Y., Wu, C.-H., Hong, C.-H., Chang, K.-L. & Lee, C.-H. Glut-1 enhances glycolysis, oxidative stress, and fibroblast proliferation in keloid. Life 11, 505 (2021).
Acknowledgements
This work is supported by the National Natural Science Foundation of China, China (22077109, 21775131 and 82073715), the HKBU SKLEBA Research Grant (SKLP_2223_P03), the Science and Technology Development Fund, Macau SAR (File no. 0007/2020/A1), SKL-QRCM(UM)-2020-2022, and the University of Macau (MYRG2019-00002-ICMS, MYRG2020-00091-ICMS and MYRG2020-00017-ICMS).
Author information
Authors and Affiliations
Contributions
G.L., D.L. and C.W. performed the experimental work and drafted the manuscript. S.L. performed the zebrafish experimental work. F.C. performed the experimental work. P.L. helped design the project. C.N.K. and W.W. performed the chemical synthesis. C.H.L, D.L.M., L.L. and S.M.Y.L. designed the projects, analyzed the experimental data, and analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, G., Li, D., Wu, C. et al. Homocysteine-targeting compounds as a new treatment strategy for diabetic wounds via inhibition of the histone methyltransferase SET7/9. Exp Mol Med 54, 988–998 (2022). https://doi.org/10.1038/s12276-022-00804-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00804-1
This article is cited by
-
Epigenetic mechanism of SET7/9-mediated histone methylation modification in high glucose-induced ferroptosis in retinal pigment epithelial cells
Journal of Bioenergetics and Biomembranes (2024)
-
Discovery and evaluation of active compounds from Xuanfei Baidu formula against COVID-19 via SARS-CoV-2 Mpro
Chinese Medicine (2023)
-
Tumor-suppressive functions of protein lysine methyltransferases
Experimental & Molecular Medicine (2023)
