
Abstract
The revolution in genome sequencing technologies has enabled the comprehensive detection of genomic variations in human cells, including inherited germline polymorphisms, de novo mutations, and postzygotic mutations. When these technologies are combined with techniques for isolating and expanding single-cell DNA, the landscape of somatic mosaicism in an individual body can be systematically revealed at a single-cell resolution. Here, we summarize three strategies (whole-genome amplification, microdissection of clonal patches in the tissue, and in vitro clonal expansion of single cells) that are currently applied for single-cell mutational analyses. Among these approaches, in vitro clonal expansion, particularly via adult stem cell-derived organoid culture technologies, yields the most sensitive and precise catalog of somatic mutations in single cells. Moreover, because it produces living mutant cells, downstream validation experiments and multiomics profiling are possible. Through the synergistic combination of organoid culture and genome sequencing, researchers can track genome changes at a single-cell resolution, which will lead to new discoveries that were previously impossible.
Similar content being viewed by others
Introduction
Over the past decade, advances in DNA sequencing technology have enabled us to explore entire genome sequences in faster and cheaper ways1. As of 2020, ~30-fold human whole-genome sequences could be produced in less than a week with a cost of <$1,0001,2. With this technology, researchers are able to access the vast majority of, if not all, genetic variations in an individual for the first time3,4. Because these variations are the most important element underlying the phenotypic diversity of human individuals, their sensitive and accurate detection is critical for precision health care. More fundamentally, mutations are cumulative records of DNA damage and repair processes through the germline and somatic lineages5. Therefore, the burden and patterns of genomic variations provide deep insights into the mutational processes operative over time6.
Humans are multicellular organisms. An adult body consists of 40 trillion somatic cells developing from a single common ancestral cell, the fertilized egg7. Through a series of mitotic cell divisions, the single-cell organizes a complex multicellular system during the developmental stage. After full development and growth, many tissues are maintained via the homeostatic proliferation of adult stem cells that compensate for the loss of differentiated cells8. It is generally assumed that the genetic constitution of all the somatic cells of an individual is virtually identical and uniform. Indeed, all cells in an individual harbor 3–4 million germline polymorphisms inherited from both parents9 as well as 50–100 de novo mutations acquired during the formation of germ cells10,11 (Fig. 1a, b). However, these are not all the genetic variants present in the cellular pool of an individual. Postzygotic mutations, also known as somatic mutations, can accumulate during every mitosis from the first cell division in life onward12 (Fig. 1a, b). The concept of accumulating genomic changes was first proposed in 195913, but the incidence and underlying molecular mechanisms across cellular lineages have been poorly characterized14, primarily due to the lack of techniques for the sensitive discovery of somatic mutations restricted to single cells. In theory, the acquisition of somatic mutations occurs via independent events in each cell caused by exposure to external mutagens or endogenous mutagenic processes, such as spontaneous methylcytosine deamination, errors in DNA replication, and the misregulation of APOBEC enzymes (apolipoprotein B mRNA editing enzyme or catalytic polypeptide-like enzymes)5. These mutations are known to cause various human diseases, including not only cancers but also neurodevelopmental and immune-related disorders5,15,16,17,18,19.

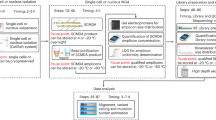
a The origin of genomic mutations together with the cellular phylogeny of an individual’s development. Four different origins (inherited, de novo, and embryonic and late stages) are represented. b The characteristics of genomic variants according to their origin. The mutation number per cell (x-axis) and the proportion of mutant cells in an individual’s body (y-axis) are illustrated. For postzygotic mutations, we assume that the endogenous mutation rate is 1 mutation per cell per cell doubling. The threshold of mutation detection (6.6% of the cell fraction) is calculated with 30-fold whole-genome sequencing. c The signal intensity of genomic variants in ordinary bulk-tissue whole-genome sequencing according to the origin of the genomic variants. WGS, whole-genome sequencing.
It is estimated that roughly 0.1–10 mutations can be acquired during every mitosis of human cells10,12,14,20. If ~1 somatic mutation arises in each of the two daughter cells of a fertilized egg, the adult body will acquire two mutations overall from the first cell division. Unlike inherited and de novo mutations, these acquired mutations are not shared by all cells in an individual body. Instead, if the two earliest cells contribute more or less equally to an adult body, each of the two mutations will be carried by one of the two cellular subsets, each constituting ~50% of the cells in an individual body (Fig. 1b). Assuming a similar mutation rate in later cell generations, four and eight mutations will arise in the four- and eight-cell stages, respectively, and the mutations will be harbored by ~25% and ~12.5% of the adult cell pool. In this scenario of the continuous accumulation of one mutation per cell per cell division, the 40 trillion cells in an individual body collectively acquire 40 trillion mutations from the most recent cell division in each somatic lineage, which far outnumbers the total bases of the human genome (~3 billion nucleotides) (Fig. 1b). Through continuous homeostatic cellular proliferation over an individual’s lifetime, the level of somatic mosaicism increases, and the genome sequences of cells become more heterogeneous.
Until recently, somatic mutations were cataloged mostly from cancer tissues21. Neoplastic cells in a cancer tissue are formed by clonal expansion from the most recent common ancestral (MRCA) cell22; thus, all the somatic mutations carried in the MRCA cell are shared by all cancer cells (Fig. 1a) if we ignore the small possibility of deletions involving previously mutated loci during tumor progression. These shared mutations can be detected by the conventional 30-fold whole-genome sequencing (WGS) of tumor bulk tissues. This technical possibility enabled an international effort to systematically catalog somatic mutations from >10,000 human cancers in the last decade23,24. By contrast, polyclonal nonneoplastic tissues are much more challenging for genome analyses, as most mutations are confined to single cells or to a very small fraction of cells in the tissue; thus, weak signals from the mutation are difficult to distinguish from ubiquitous technical errors arising from the sequencing process25 (Fig. 1c). The deep genome sequencing of polyclonal bulk tissues and duplex DNA sequencing26,27,28 are applicable for exploring somatic mutations present in a given tissue, but these mutations lack sensitivity and a single-cell resolution. Nevertheless, multiple lines of evidence now suggest that the progressive accumulation of genomic aberrations is a common feature of apparently healthy cells as they age and is the source of human diseases26,29,30. Their functional impacts and association with human diseases have not been explored due to the lack of technology facilitating systematic discoveries. With the reduction of sequencing costs, exploring somatic mosaicism by sequencing multiple genomes from the same individual has become affordable.
Recently, organoid technologies that facilitate long-term clonal expansion of stem cells were developed31,32. Organoid technologies allow adult tissue stem cells to expand and self-organize into an organotypic 3D tissue architecture. Because organoids recapitulate the cellular physiology of in vivo tissues, these models have been used to study the pathogenesis of many diseases, such as infections, genetic disorders, and cancers. Additionally, as we will further discuss below, organoids provide great opportunities for the study of somatic mutations in single cells without error-prone whole-genome amplification.
In this review article, we provide an overview of three approaches currently being used for the detection of somatic mutations accumulated in single cells. The details of each approach, along with their advantages and limitations, are discussed. The combination of organoid and genome sequencing technologies yields the most sensitive and precise mutation catalog, at the expense of intensive labor requirements. We predict that the landscape of somatic mosaicism among the various cell types of young and aged individuals or healthy and diseased individuals will be explored via the synergistic integration of these techniques.
Main text
Amount of DNA required for genome sequencing
As of 2020, Illumina platforms, such as NovaSeq, were being dominantly applied for population-scale genome sequencing. The accuracy and efficiency of DNA library preparation have been greatly improved over the last decade. Current standard library preparation kits require ~100 ng of genomic DNA as the input material. Specialized low-input DNA library kits can be used with ~1 ng of genomic DNA33, although the quality of genome sequencing is usually compromised, with lower library complexity and higher levels of GC bias and sequencing error rates34. In principle, genomic DNA from a single cell should be exclusively employed to detect somatic mutations restricted to a single cell. However, because a human diploid cell contains only ~0.006 ng (6 pg) of genomic DNA35, the direct sequencing of a single genome is physically not possible. To overcome this challenge, researchers have amplified single genomes through several strategies.
Whole-genome amplification
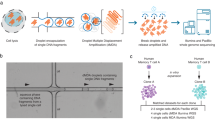
A single DNA copy can be exponentially duplicated using in vitro amplification methods, such as polymerase chain reaction (PCR). Using the whole-genome amplification (WGA) technique, picograms of single-cell genomic DNA can be amplified to the microgram scale36 (Fig. 2). In the 1980s−1990s, early PCR-based WGA strategies were designed by the ligation of specific primers to sheared DNA fragments (linker-adapter PCR)37 or by targeting specific primers to repetitive sequences in the genome38. More efficient techniques then followed, including (1) degenerate oligonucleotide-primed PCR (DOP-PCR), which is an exponential PCR method with random primers39; (2) multiple displacement amplification (MDA), which uses a highly processive and strand-displacing phi29 DNA polymerase to exponentially amplify single-stranded DNA in isothermal conditions40,41; and (3) multiple annealing and looping-based amplification cycles (MALBAC), which leads to nonspecific but quasi-linear DNA amplification by protecting the amplification products in a loop structure that is not further amplified42. More recently, the linear amplification via transposon insertion (LIANTI) method was developed, in which genomic DNA is transcribed multiple times in vitro, and the expressed RNAs are sequenced43. These techniques each have their own pros and cons, as illustrated in previous review articles36,44.

Three different strategies that can be applicable for detecting single-cell mutations are illustrated with brief experimental steps. Expected outcomes from each of the strategies are shown below with summaries of source genomes and sequence reads produced from the source. True, borderline, and false findings are highlighted with green, orange, and red boxes, respectively. LCM laser-capture microdissection.
Although WGA approaches seem straightforward for single-genome sequencing, they create a large number of amplification biases and errors, including artificial false mutations, allelic dropout, and preferential amplification of one of the two alleles (Fig. 2). For example, in single-nucleotide variant detection experiments, DOP-PCR, MDA, and MALBAC produce 1–40 artifact mutations per 100 kb43. On a genome-wide scale, these processes can generate 30,000–1,200,000 artificial point mutations, which is a much higher number than the number of true somatic mutations in single cells (usually <10,000 mutations)45. Despite the existence of many downstream bioinformatics strategies, it is quite challenging to filter out the excessive number of experimental artifacts46. In addition, due to frequent allelic dropout, a high fraction of true variants (21–76%) can be lost during the WGA process47. Moreover, amplification noise interferes with the accurate detection of DNA copy number changes36, particularly when the length of amplifications/deletions is short (<100 kb), so true somatic structural variations are difficult to detect.
Despite these limitations, WGA-based single-genome sequencing has been applied to explore single-genome mutations, particularly in nonproliferating cells such as neurons48,49,50. Although sequences are sometimes heavily contaminated by errors during WGA steps, some features of these genomes, such as the mutational burden and functional mutations shared by multiple cells, have been explored by using these techniques46.
Microdissection of clonal patches
Similar to cancer tissues, some adult stem cells can proliferate more quickly than neighboring stem cells in normal tissues, generating a ‘clonal patch’. For instance, in the normal intestinal crypt, a few equivalent stem cells continuously compete for residence51 (Fig. 2). Among these cells, one clone may produce more progeny in a given time resulting from either a stochastic mechanism or acquired competence induced by somatic genetic/epigenetic alterations52, or both. The generation of phenotypic diversity through the continuous acquisition of new alterations and the natural selection of more effectively proliferating clones is the main tumorigenic process in normal tissues5.
Under these circumstances, the microscopic structure is continuously converted into single-clone compartments over time. The territory of a stem cell can spread even further through rounds of dynamic fission and fusion of its crypts53. Although the frequency in various tissue types is uncertain, clonal patches are known to form in many tissue types, such as the liver54, uterus52, placenta55, and other pan-body tissues56. As in cancer tissues, the cells in a clonal patch share all somatic mutations acquired in the genome of the MRCA cell. Using specialized technologies such as laser capture microdissection (LCM), these patches can be physically separated, followed by WGS without error-prone WGA33. In this way, somatic mutations in normal tissues can be efficiently detected.
In practice, microdissected tissues are not always single clones but usually include multiple clones56, either because the size of the clonal structure is smaller than the dissected structure or because the excised tissue overlaps the boundaries between multiple clonal patches. Clonality in excised tissue can be inferred from genome sequences using the number of discovered somatic mutations and their variant allele fractions (VAFs)56. Monoclonal patches show a substantial number of somatic mutations, with a VAF peak of ~50%. Oligoclonal patches provide many somatic mutations with a VAF distribution substantially lower than 50% according to the proportion of the leading clone. Polyclonal patches, composed of many clones, none of which achieve clonal dominance, yield very few mutations.
Despite these limitations, LCM has shown the ability to scalably detect somatic mutations in nonneoplastic cells. For instance, Lee-Six et al. dissected 571 colonic crypts using LCM followed by WGS57. The VAF distribution clearly suggested that ~90% of the crypts were clonalized at a certain time in the life cycle, estimated to be 5.5 years earlier than the age at dissection. The mutation rate of adult stem cells in crypts was 43.6 somatic base substitutions per year, and ~1% of the adult stem cells in the apparently normal colorectal crypts harbored known cancer driver mutations, such as AXIN2 and PIK3CA. Morphologically normal bladder58,59, liver54, endometrium52, and pan-body tissues of human individuals56 have been explored by similar techniques.
Clonalization of single cells
Mammalian cells accurately duplicate their genomes during every cell division using high-fidelity DNA polymerase in combination with DNA repair mechanisms. Relative to the error-prone ex vivo WGA approach mentioned above, the accuracy of intracellular genome duplication is considerably higher (at least 1,000 times)20,43,60. Therefore, the in vitro clonal expansion of single cells to hundreds of cells may be an option for amplifying single-cell DNA for WGS (Fig. 2).
To implement these strategies, researchers have applied various cell culture techniques, such as classical 2D culture, induced pluripotent stem cell (iPSC) culture, and organoid culture technologies. Some cell types, such as hematopoietic stem cells61, bronchial epithelial cells62, and fibroblasts63, can be successfully clonalized using primary 2D culture techniques. However, unlike cancer cell lines, most primary human cells are difficult to expand in vitro using the classical technique, particularly when a single cell is isolated. Alternatively, the iPSC technique has the advantages of fast growth and ease of cloning. Using this approach, somatic mutations in single cells due to exposure to carcinogens were systematically explored64. However, the conditions required to induce dedifferentiation are not close to physiological conditions, and this step thus constitutes a strict selective bottleneck that may be permissive to a specific group of cells. For example, the efficiency of iPSC establishment from differentiated cells is ~1% under the best conditions65,66.
For the clonal expansion of single cells in various human tissues, organoid technologies have many advantages over cell culture methods. Most importantly, various human cell types can be physiologically expanded using this technology. From the first report involving the small intestine67, primary culture conditions for at least 15 tissue stem cells31, including colon, pancreas, liver, prostate, stomach, fallopian tube, uterus, placenta, skin, breast, and lung cells, have been thoroughly established68,69,70,71,72,73,74,75,76,77,78,79,80,81,82. By applying these techniques, adult single stem cells can be rapidly expanded in chemically defined media with high efficiency. Feeder cell-free culture conditions prevent potential DNA contamination, which could cause inaccurate genome analysis. Because organoid culture conditions mimic physiological tissue environments, the long-term culture of a clone (>1 year) is possible while maintaining genomic stability83.
Pros and cons of the approaches
Each approach has its own advantages and drawbacks when applied to single-genome analyses (Table 1). WGA is technically simple, straightforward and applicable to nondividing cells but suffers from a large number of false-positive and false-negative calls. The quality of WGA genome sequences should be thoroughly investigated. The compromised genome coverage (allelic dropout) and levels of sequencing artifacts under this approach can be partially explored with downstream bioinformatic techniques, such as analyses of the genome-wide variation of sequencing read depth, the proportion of discordant paired-end reads, and the presence of error signatures84. The microdissection technique is accurate and easy to scale up but requires preclonalized anatomical structures, which are not always present in human tissues. As briefly mentioned above, the clonality of patches can be investigated according to the distribution of VAFs in somatic mutations56. Genomes in polyclonal patches cannot be further investigated due to the loss of sensitivity in the mutation detection, as mentioned above. Compared with these two approaches, in vitro clonalization requires more complicated steps, such as single-cell dissociation and proliferation, and is thus more expensive, labor-intensive, and time-consuming. In particular, the single-cell dissociation step in the organoid culture method is technically sophisticated, requiring extensive trypsinization and FACS sorting85. Single-cell proliferation is not rapid in certain cell types even in organoid cultures, as observed for alveolar type 2 cells of the lung (3–4 doublings every 2 weeks)79. Moreover, clonalization is limited to dividing cells, and if the process is not carefully controlled, culture-associated mutations can contaminate the genome86. As in WGA and microdissection, these features of the clonalization technique can be confirmed by downstream bioinformatics QC steps85. Despite these limitations, clonal expansion is a unique way to sensitively capture entire genomic loci from physically isolated single cells. Using this approach, the most accurate and sensitive catalog of somatic mutations, including base substitutions, indels, copy number changes, and structural variations, can be obtained. Moreover, this approach produces viable cells that can be used for further experimental analyses to validate the insights obtained from genomic analyses87. For example, the transcriptomes and epigenomes of a specific clone, as well as phenotypes of the cells, such as proliferation rate, tumor-forming ability, and resistance to specific chemicals, can be further explored at a single-clone resolution with the established cells.
Applications of the clonal organoid technique
Cellular phylogenies in early embryonic development
The WGS of clonalized organoids was first applied to reconstruct the early developmental phylogenies of many somatic cells collected from various murine tissues (Fig. 3a)88. As the mutations arising in an embryonic cell are inherited by its descendant cells, these mutations can be used as cellular barcodes to reveal their early founder cells. Behjati and colleagues established 25 clonal organoids from stomach, small intestine, colon, and prostate tissues of two individual mice and identified 200–1,200 base substitutions in each clone via WGS88. Using a few mutations shared by multiple clones, the researchers reconstructed the early phylogenies of the somatic cells. These efforts demonstrated that (1) the two cells of the two-cell stage mouse embryo do not contribute equally to adult cells and that (2) anatomically adjacent cells in adult tissue are not always developmentally closer than distant cells88. Most importantly, the work suggested that similar approaches may be applicable to human cells63 for understanding human embryogenesis, where the direct investigation is not possible.

a By reconstructing cellular phylogenies using somatic mutations as barcodes, clonal dynamics in early development can be traced. b The genome-wide mutational burden and signatures can be investigated in normal and diseased cells when clonal organoids are derived from healthy and diseased tissues. c The functional impact of a specific gene mutation can be investigated via the genetic engineering of organoids followed by sequencing. d By exposing organoids to carcinogens, their mutagenic impacts can be accurately assessed. e Organoid technologies enable multiomics profiling because a large number of progeny cells, which share similar genomic and phenotypic backgrounds, can be produced.
Mutational burden of healthy and pathologic cells
Clonal organoids can be used to count somatic mutations accumulated in normal aged human cells (Fig. 3b). For example, Blokzijl and colleagues established clonal organoids from the human small intestine, colon, and liver tissues, followed by WGS45. The authors found that normal cells also accumulate a considerable number of somatic mutations as an individual ages. Given the observed mutational burden, these cells were estimated to endogenously accumulate ~40 base substitutions per year. The patterns of the mutations, or mutational signatures, suggested two intrinsic mechanisms (termed COSMIC SBS1 and SBS589) underlying the age-related somatic mutations90.
Furthermore, the technique revealed the genomic differences of intestinal cells chronically exposed to pathologic environments. For example, gut epithelial cells in the colon of ulcerative colitis patients (chronic inflammatory bowel disease) were established as clonal organoids and sequenced91. Compared to control cells in healthy individuals, inflamed epithelial cells showed enriched somatic mutations in the genes of the IL-17 signaling pathway; this conclusion is supported by an independent study conducted through microdissection92.
The combination of clonal organoids and WGS has been employed to compare the mutational burden in normal and cancer cells in the same patient at an absolute single-cell resolution93. As expected, tumor cells carry several times more somatic mutations than normal colorectal cells; this is caused by additional mutational processes specifically present in neoplastic cells. More interestingly, tumor tissues harbor a very high level of intratumoral genome heterogeneity, with thousands of unique mutations in the single tumoral clones that are responsible for the clone-specific differences in responses to anticancer drugs. Although organoid-based techniques are being extensively applied to cancer tissues94, tumor organoids of some cancer types, such as lung adenocarcinomas, cannot yet be easily established95.
Genomic effects of specific genetic alterations
The possibility of long-term culture in organoids enables the investigation of the functional and mutational impacts of specific gene mutations in controlled experimental conditions (Fig. 3c). For example, to functionally validate the enrichment of clones with an impaired IL-17 pathway in the colorectal epithelium of ulcerative colitis patients91,92,96, the NFKBIZ gene was truncated by CRISPR–Cas9 editing in clonal colon organoids91. Indeed, the NFKBIZ mutant clones showed a higher proliferation rate than normal clones under culture conditions that mimicked the inflammatory tissue environment. This implies that clones with an impaired IL-17 pathway have a selective advantage in specific microenvironments.
Lynch syndrome is a representative hereditary cancer syndrome caused by inherited mutations in genes that affect DNA mismatch repair, including MLH1, MSH2, MSH6, and PMS297. Patients with Lynch syndrome frequently develop carcinomas of the colon, endometrium, stomach, and many other organs. To investigate the impact of the dysregulation of genes in nonneoplastic cells, Drost and colleagues performed MLH1 knockout in colon organoids and cultured them to obtain multiple clonal organoids87. Two months of MLH1−/− organoid culture resulted in a significantly higher number of base substitutions and short indels, with characteristic mutational signatures in the specific sequence contexts.
Genomic impact of exogenous mutagens
The microbiota is associated with colon cancers98,99. Among the many bacterial species in the microbiota, the direct mutagenic effects of colibactin-producing E. coli were revealed by the exposure of organoids to the bacteria followed by clonalization and WGS100 (Fig. 3d). Colibactin increased the total number of somatic base substitutions and indels, which showed characteristic mutational patterns in the cocultured organoids. These signatures were validated in several cancer samples, including colorectal, head and neck, and urinary tract cancers, suggesting a mutagenic impact of the bacteria in these cancer types as well.
Cytotoxic chemotherapy is commonly used to treat metastatic cancers by inducing mutagenesis in cancer cells. The cytotoxic agents also affect normal cells. To investigate the genomic effects of one chemotherapeutic agent, 5-fluorouracil (5-FU), Christensen et al. treated normal intestinal organoids with 5-FU and then sequenced the organoids after clonalization101. Characteristic T > G substitutions in the CTT context (termed COSMIC SBS1789) were revealed in 5-FU-treated intestinal organoids, and they were also observed in 5-FU-treated cancers.
Similar approaches have been applied to reveal the mutagenic effects of ionizing radiation (IR) (Fig. 3d). IR is a strong carcinogen, but its characteristic mutational patterns have not been comprehensively identified89. Irradiated clonal organoids have a substantially higher mutational burden according to specific mutational signatures of short deletions and genomic rearrangements102.
Simultaneous multiomics profiling
In tumors, RNA expression and DNA methylation have been frequently investigated together with mutation identification103,104. More recently, the 3D genome organization has been correlated with genomic rearrangements105. By using the clonal organoid technique, such a multidimensional approach is possible in nonneoplastic normal cells (Fig. 3e). Notably, the integration of multiomics datasets can provide deep insights into the causative changes and treatment targets in human diseases106. However, primary tissues are not ideal for multiomics studies because (1) only a limited amount of material can be prepared, (2) intermixed signals are usually obtained from heterogeneous cell types, and (3) the long-term storage of the tissues is challenging. Organoids are more advantageous for this purpose because they provide pure signals from target cell types and can be readily cryopreserved. As organoid technology produces large numbers of progeny cells that are more than sufficient for WGS, a fraction of the cells can be reserved for further experiments after somatic mutation discovery. Once genome analyses reveal an interesting pattern, such as APOBEC-mediated hypermutations and/or complex genomic rearrangements, as observed under chromothripsis107, we can trace the origin and regulatory mechanisms of the mutational processes as well as their phenotypic consequences via clone-specific downstream analyses, such as transcriptome, epigenome and proteome profiling as well as experimental phenotyping. If necessary, more progeny cells can be easily produced via the further repopulation of the clonal line.
Future directions
Genomics is a big data science and is becoming much larger in scale108. The first personal genome was reported in 2007109, and genome sequences from more than 500,000 human genomes had been produced as of 2016. It would not be very surprising, in fact, if the entire human population were to be sequenced in the next several decades. In parallel, an individual may be sequenced multiple times, at least for medical purposes, for instance, to longitudinally trace the genomic evolution of cancer tissues110 or to decompose the genomic heterogeneity of a specific individual63,111.
For the last decade, whole-exome and genome sequencing have been widely used for disease gene discovery studies, and many novel causal pathogenic genes of sporadic genetic diseases have been found112. However, success rates in ‘pinpointing’ causal mutations or mechanisms are estimated to be roughly 30−50%, even for clinically indisputable genetic disorders113. Indeed, there is a substantial level of ‘missing causal mutation’, which is not in accord with the current concept of the homogeneous genomes of individuals15,114.
The sequencing of single genomes provides the ultimate resolution in personal genomics. As an individual age, stochastically acquired genetic and epigenetic alterations make their somatic cells more heterogeneous, which is likely to lead to the generation and progression of human diseases. In principle, the formation, tissue distribution, and relative contribution of somatic mosaicism are stochastic personal events and can thus be variable across individuals. We first need to record the mutational and clonal complexity of human tissues on a massive scale to extract common features and principles. Technically, as mentioned throughout this manuscript, the landscape of somatic mosaicism is becoming fundamentally explorable, as seen in recent studies involving clones from diverse anatomical regions of human bodies63,111.
Importantly, cultural and ethical issues should be considered in this context. Methods of classical biology, such as the use of animal models, are not applicable here because it is not plausible to engineer hundreds of somatic mutations in even single cells, and the species-specific context between the endogenous mutation rate and lifespan cannot be fundamentally simulated in animal models. To reveal the direct association between single-cell mutations and human diseases and the trajectory of its longitudinal and spatial changes over time, a large number of human tissue specimens, not only from diseased but also from apparently healthy specimens, should be thoroughly investigated. To this end, social agreements and legal doctrines should be developed for donating precious diseased tissue (from both surgery and biopsy), organ transplant, and postmortem specimens for scientific research. Because genome sequencing involves individual privacy and confidentiality concerns, modalities for adequately protecting such information should be developed.
Although the mission is still ambitious, the technical advances mentioned above pave the way for the systematic decomposition of genomic heterogeneities in an individual at a single-cell resolution. The sensitive and precise detection of somatic mosaicism, not only in humans but also in many other model organisms, will be a major direction for future genetic studies and will be associated with genetic diseases.
References
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Levy, S. E. & Myers, R. M. Advancements in next-generation sequencing. Annu. Rev. Genom. Hum. Genet. 17, 95–115 (2016).
Durbin, R. M. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Stratton, M. R., Campbell, P. J. & Futreal, P. A. The cancer genome. Nature 458, 719–724 (2009).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Bianconi, E. et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 40, 463–471 (2013).
Schultz, M. B. & Sinclair, D. A. When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development 143, 3–14 (2016).
Shastry, B. S. SNP alleles in human disease and evolution. J. Hum. Genet. 47, 561–566 (2002).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016).
Veltman, J. A. & Brunner, H. G. De novo mutations in human genetic disease. Nat. Rev. Genet. 13, 565–575 (2012).
Ju, Y. S. et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714–718 (2017).
Szilard, L. On the nature of the aging process. Proc. Natl Acad. Sci. USA 45, 30–45 (1959).
Martincorena, I. & Campbell, P. J. Somatic mutation in cancer and normal cells. Science 349, 1483–1489 (2015).
D’Gama, A. M. & Walsh, C. A. Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 21, 1504–1514 (2018).
Lee, J. S. et al. Germline gain-of-function mutation of STAT1 rescued by somatic mosaicism in immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like disorder. J. Allergy Clin. Immunol. 145, 1017–1021 (2020).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Jaiswal, S. Clonal hematopoiesis and nonhematologic disorders. Blood 136, 1606–1614 (2020).
Lee, J. H. Somatic mutations in disorders with disrupted brain connectivity. Exp. Mol. Med. 48, e239 (2016).
Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl Acad. Sci. USA 107, 961–968 (2010).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Nowell, P. C. The clonal evolution of tumor cell populations. Science 194, 23–28 (1976).
The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Ding, L. et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell 173, 305–320.e10 (2018).
Fox, E. J., Reid-Bayliss, K. S., Emond, M. J. & Loeb, L. A. Accuracy of next generation sequencing platforms. Gener. Seq. Appl. 1, 1000106 (2014).
Martincorena, I. et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Schmitt, M. W. et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl Acad. Sci. USA 109, 14508–14513 (2012).
Kennedy, S. R. et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 9, 2586–2606 (2014).
Martincorena, I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018).
Yizhak, K. et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364, eaaw0726 (2019).
Schutgens, F. & Clevers, H. Human organoids: tools for understanding biology and treating diseases. Annu. Rev. Pathol. 15, 211–234 (2020).
Kim, J., Koo, B. K. & Knoblich, J. A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell. Biol. 21, 571–584 (2020).
Ellis, P. et al. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat. Protoc. 16, 841–871 (2020).
Aigrain, L., Gu, Y. & Quail, M. A. Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays—a systematic comparison of DNA library preparation kits for Illumina sequencing. BMC Genomics 17, 458 (2016).
Baumer, C., Fisch, E., Wedler, H., Reinecke, F. & Korfhage, C. Exploring DNA quality of single cells for genome analysis with simultaneous whole-genome amplification. Sci. Rep. 8, 7476 (2018).
Gawad, C., Koh, W. & Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188 (2016).
Troutt, A. B., McHeyzer-Williams, M. G., Pulendran, B. & Nossal, G. J. Ligation-anchored PCR: a simple amplification technique with single-sided specificity. Proc. Natl Acad. Sci. USA 89, 9823–9825 (1992).
Blainey, P. C. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 37, 407–427 (2013).
Telenius, H. et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13, 718–725 (1992).
Dean, F. B., Nelson, J. R., Giesler, T. L. & Lasken, R. S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 11, 1095–1099 (2001).
Spits, C. et al. Whole-genome multiple displacement amplification from single cells. Nat. Protoc. 1, 1965–1970 (2006).
Zong, C., Lu, S., Chapman, A. R. & Xie, X. S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626 (2012).
Chen, C. et al. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189–194 (2017).
de Bourcy, C. F. A. et al. A quantitative comparison of single-cell whole genome amplification methods. PLoS One 9, e105585 (2014).
Blokzijl, F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016).
Dou, Y., Gold, H. D., Luquette, L. J. & Park, P. J. Detecting somatic mutations in normal cells. Trends Genet. 34, 545–557 (2018).
Huang, L., Ma, F., Chapman, A., Lu, S. & Xie, X. S. Single-Cell whole-genome amplification and sequencing: methodology and applications. Annu. Rev. Genom. Hum. Genet. 16, 79–102 (2015).
Lodato, M. A. et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018).
Huang, A. Y. et al. Parallel RNA and DNA analysis after deep sequencing (PRDD-seq) reveals cell type-specific lineage patterns in human brain. Proc. Natl Acad. Sci. USA 117, 13886 (2020).
Breuss, M. W. et al. Somatic mosaicism in the mature brain reveals clonal cellular distributions during cortical development. Preprint at https://www.biorxiv.org/content/10.1101/2020.08.10.244814v1 (2020).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007).
Moore, L. et al. The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646 (2020).
Nicholson, A. M. et al. Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell 22, 909–918 (2018).
Brunner, S. F. et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 574, 538–542 (2019).
Coorens, T. H. H. et al. Inherent mosaicism and extensive mutation of human placentas. Nature 592, 80–85 (2021).
Moore, L. et al. The mutational landscape of human somatic and germline cells. Nature 597, 381–386 (2021).
Lee-Six, H. et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019).
Lawson, A. R. J. et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82 (2020).
Li, R. et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 370, 82–89 (2020).
Brody, Y. et al. Quantification of somatic mutation flow across individual cell division events by lineage sequencing. Genome Res. 28, 1901–1918 (2018).
Lee-Six, H. et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478 (2018).
Yoshida, K. et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020).
Park, S. et al. Clonal dynamics in early human embryogenesis inferred from somatic mutation. Nature 597, 393–397 (2021).
Kucab, J. E. et al. A Compendium of mutational signatures of environmental agents. Cell 177, 821–836 (2019).
Huangfu, D. et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat. Biotechnol. 26, 1269–1275 (2008).
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Huch, M. et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 32, 2708–2721 (2013).
Huch, M. et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 494, 247–250 (2013).
Karthaus, W. R. et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175 (2014).
Bartfeld, S. et al. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148, 126–136 (2015).
Kessler, M. et al. The Notch and Wnt pathways regulate stemness and differentiation in human fallopian tube organoids. Nat. Commun. 6, 8989 (2015).
Turco, M. Y. et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 19, 568–577 (2017).
Turco, M. Y. et al. Trophoblast organoids as a model for maternal–fetal interactions during human placentation. Nature 564, 263–267 (2018).
Boonekamp, K. E. et al. Long-term expansion and differentiation of adult murine epidermal stem cells in 3D organoid cultures. Proc. Natl Acad. Sci. USA 116, 14630–14638 (2019).
Sachs, N. et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 38, e100300 (2019).
Youk, J. et al. Three-dimensional human alveolar stem cell culture models reveal infection response to SARS-CoV-2. Cell Stem Cell 27, 905–919 (2020).
Salahudeen, A. A. et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 588, 670–675 (2020).
Katsura, H. et al. Human lung stem cell-based alveolospheres provide insights into SARS-CoV-2-mediated interferon responses and pneumocyte dysfunction. Cell Stem Cell 27, 890–904 (2020).
Dekkers, J. F. et al. Modeling breast cancer using CRISPR-Cas9-mediated engineering of human breast organoids. J. Natl Cancer Inst. 112, 540–544 (2020).
Hu, H. et al. Long-term expansion of functional mouse and human hepatocytes as 3D organoids. Cell 175, 1591–1606 (2018).
Huch, M. et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015).
Kim, H. et al. FIREVAT: finding reliable variants without artifacts in human cancer samples using etiologically relevant mutational signatures. Genome Med. 11, 81 (2019).
Jager, M. et al. Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat. Protoc. 13, 59 (2017).
Kuijk, E. et al. The mutational impact of culturing human pluripotent and adult stem cells. Nat. Commun. 11, 2493 (2020).
Drost, J. et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238 (2017).
Behjati, S. et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513, 422–425 (2014).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Alexandrov, L. B. et al. Clock-like mutational processes in human somatic cells. Nat. Genet. 47, 1402–1407 (2015).
Nanki, K. et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 577, 254–259 (2020).
Olafsson, S. et al. Somatic evolution in non-neoplastic IBD-affected colon. Cell 182, 672–684 (2020).
Roerink, S. F. et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature 556, 457–462 (2018).
van de Wetering, M. et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945 (2015).
Dijkstra, K. K. et al. Challenges in establishing pure lung cancer organoids limit their utility for personalized medicine. Cell Rep. 31, 107588 (2020).
Yokoyama, A. et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature 565, 312–317 (2019).
Lynch, H. T. & de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 348, 919–932 (2003).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017).
Gagnaire, A., Nadel, B., Raoult, D., Neefjes, J. & Gorvel, J.-P. Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat. Rev. Microbiol. 15, 109–128 (2017).
Pleguezuelos-Manzano, C. et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 580, 269–273 (2020).
Christensen, S. et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat. Commun. 10, 4571 (2019).
Youk, J. et al. Mutational impact and signature of ionizing radiation. Preprint at https://www.biorxiv.org/content/10.1101/2021.01.12.426324v1 (2021).
The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Akdemir, K. C. et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 52, 294–305 (2020).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83 (2017).
Yi, K. & Ju, Y. S. Patterns and mechanisms of structural variations in human cancer. Exp. Mol. Med. 50, 1–11 (2018).
Stephens, Z. D. et al. Big data: astronomical or genomical? PLoS Biol. 13, e1002195 (2015).
Levy, S. et al. The diploid genome sequence of an individual human. PLoS Biol. 5, e254 (2007).
Lee, J.-K. et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol. 35, 3065–3074 (2017).
Coorens, T. H. H. et al. Extensive phylogenies of human development inferred from somatic mutations. Nature 597, 387–392 (2021).
Koboldt, D. C., Steinberg, K. M., Larson, D. E., Wilson, R. K. & Mardis, E. R. The next-generation sequencing revolution and its impact on genomics. Cell 155, 27–38 (2013).
Frésard, L. & Montgomery, S. B. Diagnosing rare diseases after the exome. Cold Spring Harb. Mol. Case Stud. 4, a003392 (2018).
Biesecker, L. G. & Spinner, N. B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 14, 307–320 (2013).
Acknowledgements
We thank Tae-Woo Kim and Yohan An (KAIST) for their comments. This work was supported by the Suh Kyungbae Foundation (SUHF-18010082 to Y.S.J.), the National Research Foundation (NRF) of Korea funded by the Korean Government (NRF-2020R1A3B2078973 to Y.S.J.) and the Human Frontier Science Program (RGY0071/2018).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Young Seok Ju is the founder and CEO of GENOME INSIGHT Inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Youk, J., Kwon, H.W., Kim, R. et al. Dissecting single-cell genomes through the clonal organoid technique. Exp Mol Med 53, 1503–1511 (2021). https://doi.org/10.1038/s12276-021-00680-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00680-1
This article is cited by
-
Somatic genetic variation in healthy tissue and non-cancer diseases
European Journal of Human Genetics (2023)
-
Widespread somatic L1 retrotransposition in normal colorectal epithelium
Nature (2023)
-
Asymmetric Contribution of Blastomere Lineages of First Division of the Zygote to Entire Human Body Using Post-Zygotic Variants
Tissue Engineering and Regenerative Medicine (2022)
-
Methods in organoids: a model that goes beyond our imagination
Experimental & Molecular Medicine (2021)
