
Abstract
Background:
Necrotizing enterocolitis (NEC) is a life-threatening gastrointestinal disease in premature infants with high mortality and morbidity with uncertain pathogenesis. Recent research focused on the role of intraluminal bacteria and lipopolysaccharide (LPS). However, an additional role of viral agents in the pathogenesis of NEC has recently been postulated. We assessed the role of polyinosinic:polycytidylic acid (pIC) mimicking viral dsRNA in contributing to the development of NEC in neonatal mice.
Methods:
Four-d-old C57BL/6J pups were stressed by asphyxia and hypothermia twice daily. Animals were either fed by formula only (FO), formula containing LPS or pIC. After 72 h, mice were euthanized, intestines harvested, and the severity of NEC was assessed.
Results:
Breastfed mice showed no evidence of NEC. Very mild NEC-like lesions were observed in mice fed by FO. Supplementation of LPS or pIC to the formula led to increased intestinal tissue damage and inflammation compared with FO in a similar manner.
Conclusion:
Our study demonstrates the ability of viral factors to induce NEC in neonatal mice even in the absence of LPS. Furthermore, we present a new mouse model of pIC-induced NEC which may be used to obtain further mechanistic insights in the pathogenesis of this disease.
Similar content being viewed by others
Main
Over the past decades, necrotizing enterocolitis (NEC) has been studied in depth due to its devastating effect on the gastrointestinal tract of preterm infants (1,2). Although several risk factors such as prematurity, enteral feeding, and hypoxia have been described, the etiology remains unclear (2). As gut bacteria may play an important role in disease pathogenesis, current animal models of NEC use intraluminal bacteria to disrupt the intestinal mucosa and activate an inflammatory cascade leading to necrosis (3). Several studies suggest that rotavirus in combination with other microorganisms can increase the intestinal vulnerability to bacterial invasion, leading to an exaggerated intestinal immune response, and therefore cause NEC (4,5,6,7). Moreover, clinical reports indicate that the colonization or infection with astro-, toro-, or norovirus have an impact on NEC severity in preterm infants (8,9,10,11). However, a possible involvement of viral vectors has not been studied in standardized mouse models so far. In this study, we used polyinosinic:polycytidylic acid (pIC) to mimic the effect of viral dsRNA in order to assess the possibility of an involvement of viral vectors in the pathogenesis of NEC.
We hypothesized that the presence of viral ligands such as dsRNA or the synthetic analog pIC in the neonatal intestine might promote an inflammatory immune response similar to that found in NEC patients.
Results
Aggravated Clinical and Macroscopic Signs for NEC in Mice Treated With pIC or Lipopolysaccharide in Contrast to Formula Only–Fed Mice
After separation from the mother, the mean weight of the pups was 2.02 g (range: 1.53–2.40 g). During the first 36 h of treatment, the weight gain of all groups was 0.07 g per mouse and decreased thereafter in all treatment groups ( Figure 1 ). At the end of the experiment (after 72 h), animals treated with pIC lost significantly more weight compared with mice fed by formula only (FO) or lipopolysaccharide (LPS) (P < 0.05).

Impaired weight gain following treatment with formula only, formula + pIC, and formula + LPS. Mice of all groups gained about 0.07 g of weight during the first 36 h of the experiment. After 36 h, the body weight decreased in all groups. Mice fed by formula only and formula supplemented with LPS lost 0.12 g (± 0.09 g and ± 0.06 g, respectively) after that point, mice fed by formula supplemented with pIC lost 0.22 g (± 0.05 g) (* P < 0.05).
Fifty-six of 57 mice (98%) survived the experimental period of 72 h. One mouse died prematurely because of a misplaced gastric feeding tube and was therefore excluded from the study. Clinically, four mice (21%) of pIC and two mice (11%) of LPS-treated animals showed signs of abdominal distension caused by luminal gases.
Macroscopically we did not see any signs of transmural inflammation (necrosis) of the intestine after treatment for 72 h. However, slight to mediate thickening of the small bowel or intraluminal blood were observed in two pIC-treated (11%) and three LPS-treated mice (17%).
pIC Exposure Induces Severe Intestinal Injury
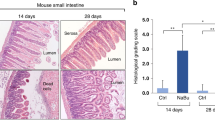
The shape of the observed intestinal lesions corresponded with those described by several authors using murine NEC models ( Figure 2 ) (12,13). The incidence of severe intestinal lesions (grade 3–4) differed significantly between breastfed mice and animals fed by formula supplemented with pIC (P < 0.01). In addition the grade of NEC differed significantly within the three treatment groups and breastfed animals (P < 0.001; Figure 3a ). Compared with breastfed animals, the grade of NEC increased significantly when formula was supplemented with pIC or LPS (P < 0.05). Animals treated with pIC showed an even higher grade of NEC compared with FO (P < 0.05).

Representative small intestinal histology slides. Induced lesions in animals treated with (b) formula only and stress, (c) pIC, and (d) LPS compared to (a) breastfed animals (hematoxylin–eosin staining, magnification: ×200 fold, scale bar = 100 µm).

Histological evaluation of NEC severity and mucosal permeability. (a) Histological evaluation (breastfed: n = 17, formula only and formula + pIC: n = 11, formula + LPS n = 10). Highest median grade of NEC in animals treated with either formula + pIC or formula + LPS compared to formula only–fed and breastfed mice (*P < 0.05). (b) Intestinal permeability (breastfed: n = 5, formula only, formula + pIC, and formula + LPS each n = 4). FITC–dextran was administrated to animals of each group 4 h prior to killing, and mucosal permeability was analyzed by measuring FITC–dextran blood plasma levels. FITC–dextran levels were significantly increased in mice treated with formula + pIC compared to breastfed animals.
In conformity with the incidence and grade of NEC, animals fed by formula supplemented with pIC showed a significant higher mucosal permeability compared with breastfed animals (P = 0.029; Figure 3b ).
No Significant Increase of Proinflammatory Chemokines in pIC- or LPS-Induced NEC
Expression levels of proinflammatory chemokines have not been shown so far in comparable studies using murine feeding NEC models (12,13,14). Whole organ ileal mRNA expression of the chemokine CXCL1 (KC) and CXCL2 (MIP-2) showed no significant increase in all treatment groups submitted to the NEC protocol compared with age-matched breastfed animals (Supplementary Figure S1 online).
Enhanced Proinflammatory Cellular Infiltration of the Small Intestine in LPS and pIC-Treated Mice Compared to FO-Fed Mice
One sample per treatment group was disregarded due to the limited number of cells; therefore seven samples per group were analyzed.
Total leukocyte numbers reached 3.7 × 105 cells ± 1.5 × 105 per small intestine in 7-d-old breastfed mice and were significantly decreased in all treatment groups (FO: 1.55 × 105 ± 7.1 × 104, P = 0.003; pIC: 1.88 × 105 ± 8.2 × 104, P = 0.013; LPS: 1.77 × 105 ± 6.1 × 104, P = 0.008; Figure 5e).
The percentage of neutrophils within the fraction of viable leukocytes in the lamina propria of breastfed animals accounted 0.7 ± 0.2% and was similar in FO-fed animals (1.1 ± 0.4%). In contrast, neutrophils were significantly increased in the lamina propria of pIC-treated (2.3 ± 1%) and LPS-treated (2.2 ± 0.8%) mice compared with FO (PpIC = 0.007 and PLPS = 0.019) and breastfed mice (PpIC < 0.001 and PLPS = 0.002; Figures 4 and 5a ). Calculated total neutrophil numbers per small intestine accounted 540 ± 340 in breastfed, 640 ± 45 in FO, 1,500 ± 990 in pIC, and 1,230 ± 530 in LPS-treated mice ( Figure 5d ).

Representative FACS plots. Following NEC induction by feeding with formula only, formula + 10 µg/g BW LPS, or formula + 10 µg/g BW pIC leukocytes were isolated from the lamina propria of the small intestine and at least 1 × 105 cells per sample analyzed by flow cytometry. Gated living CD45+, CD11b+ cells were either further gated on SSCint/hi, Ly6-G, and Ly6-C double positive cells to identify neutrophils or on SSClo, Ly6-G-, Ly6-C, and MHC II to identify intestinal monocyte/macrophage populations according to the alternative gating strategy by Bain et al. (30). Plots are representatives of seven separately analyzed intestines per group.

Phenotypic characterization of small intestinal phagocytes. (a) Significant increase of neutrophils in the lamina propria of the small intestine in animals treated with formula + pIC (10µg/g BW) and formula + LPS (10 µg/g BW) compared to formula only–fed and breastfed controls. (b) Resident macrophages, found in the “including P3, P4, P5” gate, were significantly decreased in all treatment groups. (c) In animals fed by formula supplemented with pIC (10 µg/g BW) or LPS (10 µg/g BW), the average percentage of Ly6-Chi “inflammatory monocytes” (P1 gate) was about twofold higher compared to formula only–fed and breastfed controls. (d) Calculated total cell numbers of neutrophils. (e) Living leukocytes and (f) SSClo monocytes/macrophages per small intestine (*P < 0.05; **P < 0.01; †P < 0.001).
The percentage of resident tissue monocytes/macrophage populations within the “including P3, P4, P5” gate in breastfed animals accounted 61.3 ± 10.8% and was decreased in all treatment groups in a similar manner as the total leukocyte numbers (FO: 39.8 ± 11.8%, P = 0.007; pIC: 37.1 ± 14.3%, P = 0.002; LPS 40.5 ± 10.4%, P = 0.010; Figure 5b ). However, the distribution of macrophages within the P3, P4, and P5 population did not differ in each group (data not shown). In contrast, Ly6Chi “inflammatory” P1 monocytes showed an increased percentage in pIC-treated (31 ± 15.1%) and LPS-treated (31.5 ± 6.9%) mice compared with FO (17.5 ± 4.1%, PpIC = 0.043/PLPS = 0.033) or breastfed controls (13.2 ± 1.3%; PpIC = 0.004/PLPS = 0.003; Figures 4 and 5c ). Total cell numbers of SSClo monocytes/ macrophages reached 3.74 × 104 ± 1.5 × 104 in breastfed mice, which was significantly more compared with FO-fed animals (1.46 × 104 ± 1.2 × 104; P = 0.018). Mice treated with pIC showed a total monocytes/macrophage number of 2.4 × 104 ± 1.1 × 104, which was comparable to LPS treatment (2.47 × 104 ± 1.36 × 104). Both groups did not differ significantly from either breastfed or FO-fed mice ( Figure 5f ).
Discussion
It is widely accepted that the development of NEC is promoted in the presence of different risk factors including prematurity, formula feeding, intestinal ischemia, and bacterial colonization (1,2,3).
Recent research focused on the role of intraluminal bacteria and innate immune signaling by bacterial ligands such as LPS (3,15,16,17,18). Therefore, in commonly used models of NEC induction, LPS supplementation is used to induce the inflammatory state of NEC (12,14). Neonatal TLR4 (receptor that recognizes LPS) deficient mice do not develop NEC (19).
However, TLR4 signaling does not explain the induction of NEC by gram-positive bacteria (13), yeasts, or viruses (20,21,22), which do not provide the ligand for TLR4. As the potential role of viral agents in the pathogenesis of NEC has only recently been recognized, we hypothesized that viral factors such as viral RNA could induce NEC in a neonate mouse model. Exclusively RNA viruses such as rota-, astro-, toro-, and norovirus have been associated with NEC (5,8,9,10,11,23). Among those, rotavirus, a dsRNA virus, is a well-known highly prevalent cause for severe diarrhea in infants and young children (24). We therefore used the dsRNA analog pIC mimicking viral dsRNA (25). The downstream signaling of pIC has been extensively studied. pIC typically leads to an activation of receptors recognizing ss- and dsRNA such as the endosomal receptor TLR3 and the cytosolic helicases melanoma differentiation-associated protein 5 (MDA-5) and retinoic acid inducible gene I (RIG-I) (25,26,27). McAllister et al. (27) reported that i.p. injection of pIC (30 µg/g body weight (BW)) into adult mice induces mucosal injury in a TLR3-dependent manner and proposed a TLR3 dependency in the pathogenesis of viral gastrointestinal infections. In contrast, Pott et al. (28) showed a reduced expression of TLR3 in the intestinal epithelium of neonatal mice as well as pediatric human small intestinal biopsies compared with adults. We could confirm the low transcriptional activity of TLR3 in whole organ intestinal samples of neonatal mice, which was not upregulated after pIC treatment either (Supplementary Figure S2 online). In contrast, it was previously shown that intestinal immune cells, such as macrophages, are not affected by this downregulation (28).
Our results demonstrate that luminal viral factors have the potential to induce intestinal impairment in neonatal mice with an inflammatory response similar to human NEC. We chose a pIC application of 10 µg/g BW after running a test series (ranging from 0.3 to 30 µg/g BW, data not shown) which showed that intestinal lesions are not observed below this concentration.
In addition to the increased intestinal damage and mucosal permeability, a slightly but significantly enhanced percentage of neutrophils in the intestines of mice treated by pIC and LPS vs. controls could be measured by flow cytometry. This increase corresponded to a total of about 1,000 neutrophils distributed over the entire small intestine. In combination with the low transcriptional activity of proinflammatory cytokines measured in this study, this slight increase of neutrophils is well in line with the minimal inflammation seen in experimental murine NEC by other groups (13,29).
Of note, age-matched breastfed animals showed overall higher numbers of living leukocytes compared with all treatment groups which indicate that formula feeding alone influences the maturation of the murine intestinal immune system. In line with this finding, we found that the resident monocytes/macrophages population (“including P3, P4, P5” gate according to Bain et al.) is decreased in all treatment groups compared with breastfed animals.
However, our data demonstrate an increased percentage of the Ly6Chi “inflammatory” monocyte population (P1 according to Bain et al. (30)) in the lamina propria of pIC- and LPS-treated mice, which was not found in FO-fed animals or breastfed controls. This corresponds to other studies showing an increased number of inflammatory monocytes in the small bowel of mice submitted to the NEC protocol as well as in human NEC samples (31,32). Therefore, it has been speculated that infiltrating monocytes/macrophages contribute to NEC through exaggerating the neonatal immune response by releasing proinflammatory cyto- and chemokines (33,34).
One controversy of this study is the fact that animals received breast milk prior to NEC induction, and therefore, the results may not be generalizable to classic NEC animal models which are carried out in newborn mice. These are separated during the first day of life and receive a single dose of bacteria or LPS via an orogastric route (12,13). With this protocol, breastfeeding is almost avoided, which has been shown to be protective against NEC in both humans and rodents. However, it has also been reported that this effect does not reach 100% (13,16,35).
In our model, we used 72-h-old mice and succeeded in inducing intestinal lesions by repetitive application of higher dosages of LPS compared with NEC models with newborn mice (12). Our observation that NEC induction is possible despite preceding breastfeeding is in line with results of others. The group of Hackam et al. routinely used 10-d-old mice and induced NEC over a period of 4 d (16,35). These authors reported that the time-limit for NEC induction in mice is 14–21 d, most likely due to the increasing development of the gut (36). Up to this, age the maturity of the bowel in rodents has been shown to correspond to the maturity of a human preterm neonate (37,38).
Another limitation of the current study is the lack of significant differences in the transcriptional activity of proinflammatory chemokines in the ileum (Supplementary Figure S1 online). It is most likely that the small, focal inflammatory lesions do not cause detectable changes in mRNA transcription when whole organ samples are analyzed. This might be the reason why other groups using murine feeding NEC models also failed to show increased expression levels of proinflammatory chemokines (12,13,14). Lastly, it cannot be ruled out that those “lesions” of breastfed animals scored with grade 1 and 2 were artifacts.
Taken together, our study demonstrates the ability of viral factors to induce NEC-like lesions in the absence of LPS in neonatal mice. In accordance with the literature, the inflammatory response detected in the small intestines of pIC-induced NEC in mice resembles the pathology of human NEC regarding necrotic lesions as well as infiltrating neutrophils and monocytes/macrophages (33). Furthermore, pIC-induced NEC provides a new model to study the etiology of NEC caused by viral factors to elucidate their possible role in the pathogenesis of this disease. Further mechanistic studies are needed to unravel the disastrous effect of pIC on the neonatal gut.
Methods
Neonatal NEC Mouse Model
Animal studies in this investigation were approved by the Lower Saxony State Office for Consumer Protection and Food Safety Oldenburg, Germany (registration number: 12/0769).
Breeding of C57BL/6 J mice were set by overnight mating. Seventy-two hours after birth, litters of 6 to 12 pups were separated from the mother and placed on a heating plate (37 °C). Animals were randomly divided into three groups: group 1: formula feeding, stress exposure (FO); group 2: formula feeding supplemented with pIC (Sigma-Aldrich, St Louis, MO) (10 µg/g BW) and stress exposure; group 3: formula feeding supplemented with LPS from E. coli 0127:B8 (Sigma-Aldrich) 10 µg/g BW and stress exposure (modified according to an established reference-model reported by Radulesco et al. (12)). Age-matched breastfed animals were used as controls. Beginning 3 h after separation, pups were fed every 3 h with 50 µl (~115 kcal/kg/d) of 33% Esbilac formula (PetAg, Hampshire, IL) by orogastric gavage using a 24G silicon catheter (Vygon, Aachen, Germany). To account for the increase in body weight, the feeding volume was increased daily as tolerated (50, 60, and 70 μl on days 1, 2, and 3, respectively). Mice underwent stress exposure by asphyxia (100% nitrogen for 60 s, followed by exposure to cold (4 °C) for 10 min) twice daily prior to feeding as previously described (12,13,16,35).
During the entire experiment (72 h), animals were observed every 3 h for clinical signs of NEC such as abdominal distension, apnea, rectal bleeding, and lethargy. Daily monitoring also included documentation of BW twice daily. If above-mentioned clinical signs of NEC occurred at the end of the experimental period (72 h), animals were euthanized by decapitation. The small intestines were carefully removed and visually evaluated for signs of NEC (areas of bowel necrosis, intestinal hemorrhage, perforation). In a first set of experiments (total n = 50), the entire intestine was harvested for histologic analysis. In a second set of experiments (total n = 32), small intestines were used for cell and RNA isolation. Therefore, 1 cm of the distal ileum was secured and stored in 100 µl RNAlater (Qiagen, Hilden, Germany) at −80 °C for subsequent total RNA isolation and measurement of transcriptional activity of proinflammatory cytokines. The rest of the small intestine served for the isolation of intraepithelial lymphocytes and lamina propria lymphocytes for later FACS analysis.
Mucosal Permeability
As reported in previous studies (12,39), mucosal permeability can be investigated by enteral application of fluorescein-isothiocyanate–labeled dextran (FITC–dextran) molecules (molecular weight, 73 kDa) (Sigma-Aldrich). Therefore, FITC–dextran was dissolved in formula (750 µg/g BW) and administered by orogastric gavage to mice pups of each treatment group and to age-matched breastfed control animals 4 h prior to killing. Blood samples were collected for measurement of plasma FITC–dextran levels by spectro-photo-fluorometry (Glomax Multi from Promega, Mannheim, Germany) as previously described (12).
Histologic Injury Score
The entire small bowels were placed in embedding cassettes (Roth, Karlsruhe, Germany) and immediately fixed in 10% buffered formadehyde for 3 d. Afterwards, samples were embedded in paraffin, sectioned at 4-µm thickness, and stained with hematoxylin–eosin and Masson–Goldner trichrome staining using standard protocols. Location and size of the observed lesions in the intestine were documented. Brain, liver, lung, spleen, and kidney samples were investigated for signs of tissue damage as well. Intestinal tissue injury scores were assigned by two blinded investigators based on the histologic injury scoring system described by Caplan et al. (29): NEC scores from 0 to 4 were assigned for each sample. Grade 0: intact epithelium with villus structure; 1: superficial epithelial cell sloughing; 2: mid-villous necrosis; 3: complete villous necrosis; and 4: transmural necrosis. Samples with histological scores of 3 or higher were considered positive for NEC (modified protocol of Caplan et al. (29)). The modification was made to ensure positive selection for NEC by excluding unlikely graded artificial lesions, as observed in some breastfed animals, caused by luminal air bubbles during the fixation process which might mimic “grade 2 “ lesions (Supplementary Figure S3 online).
Isolation of Lamina Propria Lymphocytes
Small intestines were processed individually, opened longitudinally, and washed three times in 20 ml 4 °C PBS-/-. They were cut into 1–2-mm-long segments followed by epithelial layer stripping in PBS-/- containing 10% fetal calf serum and 5 mmol/l EDTA for 20 min at 37 °C on an orbital shaker with 200 rpm. Intestinal pieces were washed in PBS-/- followed by enzymatic digestion in 5 ml RPMI 1640 media (Lonza, Basel, Switzerland) containing 10% fetal calf serum, 5 mmol/l N-2-hydroxyethylpiperazine-N9-2-ethanesufonic acid (PAA, Pasching, Austria) and 100 µg/ml Liberase (Roche, Basel, Switzerland) for 60 min at 37 °C and 200 rpm on an orbital shaker. The solution was centrifuged for 5 min at room temperature and 300g. Supernatants were discarded, and the pellets resuspended in 4 ml of 40% Percoll (GE Healthcare, Buckinghamshire, Great Britain). The solution was underlayed with 5 ml of 70% Percoll. The interphase, containing the lamina propria lymphocytes, was collected and subjected to flow cytometry.
Flow Cytometry
Staining was carried out according to standard protocols as previously described (40). In brief, fixable Viability Dye (eBioscience, San Diego, CA) was used to exclude dead cells, and the following antibodies (all purchased from eBioscience) were employed for staining in the presence of anti-FcRII/III mAb (clone 2.4G2; Biolegend, San Diego, CA): CD45_30F-11, CD11b_M1/70, CD11c_N418, F4/80_BM8, Ly-6C_HK1.4, Ly-6G_1A8, I-A/I-E_M5/114.15.2. Neutrophils were identified by a gating strategy according to Rose et al. (41) and intestinal monocytes/macrophages populations and nomenclature according to Bain et al. (30). Samples of at least 1 × 105 cells were acquired on a FACS CANTO II flow cytometer (BD, Franklin Lakes, NJ) and analyzed with KALUZA software (Beckman Coulter, Krefeld, Germany). Total cell numbers were calculated based on gated and total isolated cells.
Expression Analysis of Proinflammatory Chemokines
Total RNA was isolated from whole organ ileal samples using the RNeasy mini kit (Qiagen, Hilden, Germany) according to the manufacturers’ instructions. The concentration and purity was determined with a Nanodrop 2000 spectrophotometer (Thermo scientific, Waltham, MA), integrity was analyzed on a 2% agarose gel.
One microgram RNA was transcribed to cDNA using the high capacity RNA to cDNA kit (life Technologies, Darmstadt, Germany) according to manufacturers’ instructions. The mRNA expression levels of the chemokines CXCL1 (KC, Mm04207460_m1), CXCL2 (MIP-2, Mm00436450_m1), and TLR3 (Mm01207404_m1) were measured using the appropriate Taqman primer (Life Technologies). Hypoxanthine–guanine phosphoribosyltransferase (Mm01545399_m1) expression was measured for normalization, as it reaches the exponential phase at similar cycles as the target genes, a requirement of the “miqe guidelines”. qRT-PCR was performed utilizing the Applied Biosystems PCR System “Step one Plus” (Life Technologies). The following conditions were used: an initial denaturation at 95 °C for 10 minutes followed by 40 cycles at 95 °C for 15 s and at 60 °C for 60 s.
Statistical Analysis
Results are expressed as means ± SD. Significances were calculated with a one-way ANOVA and a Bonferroni post hoc test. For comparison of the NEC grades in the different groups the Kruskal–Wallis, one-way ANOVA on ranks and post hoc pairwise multiple comparison procedures (Dunn’s Method) were used. The incidence of NEC was calculated using χ2 test. P values were calculated with the software SPSS (IBM, Armonk, NY) and considered significant when < 0.05.
Statement of Financial Support
This study was funded by the German Research Foundation (DFG) (reference number LA 2828/2-1 to M.L. and reference number HO2236/8-1 to M. H.).
Disclosure
The authors declare no conflict of interest.
References
Kliegman RM, Fanaroff AA. Necrotizing enterocolitis. N Engl J Med 1984;310:1093–103.
Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med 2011;364:255–64.
Hackam DJ, Afrazi A, Good M, Sodhi CP. Innate immune signaling in the pathogenesis of necrotizing enterocolitis. Clin Dev Immunol 2013;2013:475415.
Herruzo R, Omeñaca F, García S, Diez J, Sánchez-Fauquier A. Identification of risk factors associated with nosocomial infection by rotavirus P4G2, in a neonatal unit of a tertiary-care hospital. Clin Microbiol Infect 2009;15:280–5.
Rotbart HA, Levin MJ, Yolken RH, Manchester DK, Jantzen J. An outbreak of rotavirus-associated neonatal necrotizing enterocolitis. J Pediatr 1983;103:454–9.
Sharma R, Garrison RD, Tepas JJ 3rd, et al. Rotavirus-associated necrotizing enterocolitis: an insight into a potentially preventable disease? J Pediatr Surg 2004;39:453–7.
Verdonk RC, Haagsma EB, Van Den Berg AP, et al. Inflammatory bowel disease after liver transplantation: a role for cytomegalovirus infection. Scand J Gastroenterol 2006;41:205–11.
Bagci S, Eis-Hübinger AM, Franz AR, et al. Detection of astrovirus in premature infants with necrotizing enterocolitis. Pediatr Infect Dis J 2008;27:347–50.
Clark B, McKendrick M. A review of viral gastroenteritis. Curr Opin Infect Dis 2004;17:461–9.
Lodha A, de Silva N, Petric M, Moore AM. Human torovirus: a new virus associated with neonatal necrotizing enterocolitis. Acta Paediatr 2005;94:1085–8.
Turcios-Ruiz RM, Axelrod P, St John K, et al. Outbreak of necrotizing enterocolitis caused by norovirus in a neonatal intensive care unit. J Pediatr 2008;153:339–44.
Radulescu A, Zhang HY, Yu X, et al. Heparin-binding epidermal growth factor-like growth factor overexpression in transgenic mice increases resistance to necrotizing enterocolitis. J Pediatr Surg 2010;45:1933–9.
Tian R, Liu SX, Williams C, et al. Characterization of a necrotizing enterocolitis model in newborn mice. Int J Clin Exp Med 2010;3:293–302.
Yu X, Radulescu A, Zorko N, Besner GE. Heparin-binding EGF-like growth factor increases intestinal microvascular blood flow in necrotizing enterocolitis. Gastroenterology 2009;137:221–30.
Cetin S, Ford HR, Sysko LR, et al. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem 2004;279:24592–600.
Leaphart CL, Cavallo J, Gribar SC, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol 2007;179:4808–20.
Sodhi CP, Neal MD, Siggers R, et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology 2012;143:708–718.e701–705.
Sodhi CP, Shi XH, Richardson WM, et al. Toll-like receptor-4 inhibits enterocyte proliferation via impaired beta-catenin signaling in necrotizing enterocolitis. Gastroenterology 2010;138:185–96.
Caplan MS, Simon D, Jilling T. The role of PAF, TLR, and the inflammatory response in neonatal necrotizing enterocolitis. Semin Pediatr Surg 2005;14:145–51.
Gupta S, Morris JG Jr, Panigrahi P, Nataro JP, Glass RI, Gewolb IH. Endemic necrotizing enterocolitis: lack of association with a specific infectious agent. Pediatr Infect Dis J 1994;13:728–34.
Kosloske AM, Ulrich JA. A bacteriologic basis for the clinical presentations of necrotizing enterocolitis. J Pediatr Surg 1980;15:558–64.
Mollitt DL, Tepas JJ 3rd, Talbert JL. The microbiology of neonatal peritonitis. Arch Surg 1988;123:176–9.
Bagci S, Eis-Hübinger AM, Yassin AF, et al. Clinical characteristics of viral intestinal infection in preterm and term neonates. Eur J Clin Microbiol Infect Dis 2010;29:1079–84.
Dennehy PH. Transmission of rotavirus and other enteric pathogens in the home. Pediatr Infect Dis J 2000;19:Suppl 10 ::S103–5.
Broquet AH, Hirata Y, McAllister CS, Kagnoff MF. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J Immunol 2011;186:1618–26.
Besch R, Poeck H, Hohenauer T, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 2009;119:2399–411.
McAllister CS, Lakhdari O, Pineton de Chambrun G, et al. TLR3, TRIF, and caspase 8 determine double-stranded RNA-induced epithelial cell death and survival in vivo. J Immunol 2013;190:418–27.
Pott J, Stockinger S, Torow N, et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog 2012;8:e1002670.
Caplan MS, Hedlund E, Adler L, Hsueh W. Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis. Pediatr Pathol 1994;14:1017–28.
Bain CC, Scott CL, Uronen-Hansson H, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 2013;6:498–510.
MohanKumar K, Kaza N, Jagadeeswaran R, et al. Gut mucosal injury in neonates is marked by macrophage infiltration in contrast to pleomorphic infiltrates in adult: evidence from an animal model. Am J Physiol Gastrointest Liver Physiol 2012;303:G93–102.
Pender SL, Braegger C, Gunther U, et al. Matrix metalloproteinases in necrotising enterocolitis. Pediatr Res 2003;54:160–4.
Gilchrist BF. Necrotizing Enterocolitis. 1st edn. Boca Raton, FL: CRC Press, 2000:15–76.
Maheshwari A, Kelly DR, Nicola T, et al. TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 2011;140:242–53.
Leaphart CL, Qureshi F, Cetin S, et al. Interferon-gamma inhibits intestinal restitution by preventing gap junction communication between enterocytes. Gastroenterology 2007;132:2395–411.
Sodhi C, Richardson W, Gribar S, Hackam DJ. The development of animal models for the study of necrotizing enterocolitis. Dis Model Mech 2008;1:94–8.
McElroy SJ, Weitkamp JH. Innate immunity in the small intestine of the preterm infant. Neoreviews 2011;12:e517–26.
Sangild PT. Gut responses to enteral nutrition in preterm infants and animals. Exp Biol Med (Maywood) 2006;231:1695–711.
Caplan MS, Miller-Catchpole R, Kaup S, et al. Bifidobacterial supplementation reduces the incidence of necrotizing enterocolitis in a neonatal rat model. Gastroenterology 1999;117:577–83.
Klemann C, Raveney BJ, Klemann AK, et al. Synthetic retinoid AM80 inhibits Th17 cells and ameliorates experimental autoimmune encephalomyelitis. Am J Pathol 2009;174:2234–45.
Rose S, Misharin A, Perlman H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 2012;81:343–50.
Acknowledgements
We are grateful to Dorothee Viemann (research group of experimental neonatology) for her excellent theoretical and technical assistance.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figure S1
(PDF 130 kb)
Supplementary Figure S2
(PDF 123 kb)
Supplementary Figure S3
(PDF 491 kb)
Rights and permissions
About this article

Cite this article
Ginzel, M., Yu, Y., Klemann, C. et al. The viral dsRNA analogue poly (I:C) induces necrotizing enterocolitis in neonatal mice. Pediatr Res 79, 596–602 (2016). https://doi.org/10.1038/pr.2015.261
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.261
