
Abstract
Introduction:
Intrauterine growth restriction (IUGR) is associated with high rates of neonatal morbidity. IUGR babies are often born preterm and are, therefore, exposed to antenatal glucocorticoids. Antenatal glucocorticoids significantly improve overall survival rates of preterm infants, but there is a paucity of information about their effects on IUGR Infants.
Methods:
We induced IUGR in sheep by single umbilical artery ligation (SUAL), or sham in control fetuses. To half the ewes, we administered betamethasone (BM) on d 5 (BM1) and 6 (BM2) following surgery, and collected fetal lung tissue on d 7.
Results:
SUAL alone was associated with higher circulating fetal cortisol levels (2.8 ± 0.4 vs. 1.0 ± 0.4, P = 0.001) as compared with controls but not with changes in lung morphology or surfactant protein (SP) gene expression. BM was associated with a significant reduction in lung tissue density (P = 0.048). There were no significant differences between groups in lung DNA concentration or septal crest density. SP-A, SP-B, and SP-C gene expressions were significantly increased in control and SUAL fetuses that were administered BM.
Discussion:
These results show that in SUAL fetuses, maternal BM is associated with acceleration of fetal lung structure, as occurs in normally grown fetuses, and that BM induces SP production, an effect not observed in SUAL-induced IUGR fetuses alone.
Similar content being viewed by others

Main
About 10% of North American women and 6% of European women give birth preterm, before 37-wk gestation (1). Women who are likely to give birth before 34 wk, whether spontaneously or iatrogenically, receive antenatal glucocorticoids (2). Antenatal glucocorticoids accelerate fetal lung maturation and thereby decrease the incidence of several neonatal morbidities such as respiratory distress syndrome, as well as necrotizing enterocolitis and intraventricular hemorrhage (3). Glucocorticoids promote the production of lung surfactant, accelerate the differentiation of alveolar epithelial cells, and induce the resorption of lung liquid at birth (4). However, in contrast to these beneficial effects, glucocorticoids may also inhibit alveolarization in the premature fetal lung (5) and thereby contribute to suppression of lung maturation and the subsequent development of bronchopulmonary dysplasia (6).
Intrauterine growth restriction (IUGR) is a significant complication of pregnancy associated with increased risks of perinatal mortality and morbidity, including compromised lung development such as respiratory distress syndrome (7) and long-term impaired lung function in children (8) and adults (9). In experimental studies, structural changes in the IUGR fetal lung have been reported that may underlie impaired lung function. For example, studies in sheep have found that IUGR results in fewer, larger alveoli with thicker alveolar septa, and therefore impaired blood–gas exchange, which persists into adulthood (10,11). Functionally, these structural lung effects in IUGR sheep are reflected by reduced lung compliance and increased chest wall compliance in sheep—changes that may compromise respiratory function (12).
Structural changes associated with IUGR may be similar to the alveolar arrest caused by glucocorticoids. It is, therefore, possible that higher levels of endogenous glucocorticoids in the IUGR fetus as compared with the appropriately grown fetus (13) may underlie some of these structural and functional lung changes observed in association with IUGR. This is significant because clinically the IUGR fetus has an increased risk of being born preterm (7) and is likely to be treated with antenatal glucocorticoids. However, it is not known whether antenatal glucocorticoids provide significant additive benefit to lung architecture and maturation of the IUGR fetus. In a recent review examining IUGR infants who had received glucocorticoids and those who had not, it was suggested that glucocorticoid administration does not improve neonatal outcomes following IUGR (14). In particular, there was no difference in the incidence of respiratory distress syndrome or neonatal death between IUGR infants treated with glucocorticoids and those who were not. We undertook this study to examine lung morphological changes in fetal sheep to assess the separate and combined effects of IUGR and antenatal glucocorticoids. We hypothesized that IUGR and antenatal glucocorticoids similarly regulate lung maturation in the preterm fetus and when combined, glucocorticoids would not provide additive benefit to lung maturation in growth-restricted preterm fetuses.
Results
Of the 23 ewes that underwent surgery, four single umbilical artery ligation (SUAL) fetuses and one control fetus died following surgery. Of the remaining 18 ewes, 10 received vehicle (saline) and 8 received betamethasone (BM). Thus the numbers of fetuses in each group were: control, n = 10; SUAL-alone, n = 10; control + BM, n = 8; and SUAL + BM, n = 8. All animals were used for the analysis of fetal arterial samples and body and organ weights. Histological analysis was performed on seven animals in all groups, whereas gene expression levels were measured in six animals in all groups.
Fetal Arterial Blood Gas Status
Basal blood gas and pH values were assessed on d 5, immediately before BM1. As compared with all control fetuses, basal arterial partial pressure of oxygen was significantly reduced in all SUAL fetuses (22.6 ± 1.2 mm Hg vs. 19.2 ± 0.7 mm Hg, respectively; P = 0.01). Basal glucose levels were significantly reduced in all SUAL fetuses as compared with all the controls (0.49±0.02 mmol/l vs. 0.62 ± 0.03 mmol/l, respectively; Table 1 ).
BM administration did not alter fetal oxygenation in control animals ( Table 1 ). BM administration in SUAL animals as compared with controls was associated with decreased fetal arterial partial pressure of oxygen, at the time point BM2 + 12 h (16.7±1.4 mm Hg vs. 21.2 ± 0.7 mm Hg, respectively; P = 0.02) and fetal arterial O2 saturation at BM1 + 12 h (38.6 ± 5.7% vs. 55.4 ± 4.1%, P = 0.02), BM2 + 12 h (39.1 ± 5.5% vs. 57.3 ± 2.3%, P = 0.02), and BM2 + 24 h (36.3 ± 6.6% vs. 55.8 ± 4.3%, P = 0.01). BM administration resulted in more than threefold increase in fetal arterial glucose levels in control and SUAL fetuses and more than twofold increase in lactate in control and SUAL fetuses ( Table 1 ).
Fetal Plasma Cortisol Concentration
Fetal circulating cortisol concentrations were unchanged over time in control, control + BM, and SUAL + BM fetuses (P = 0.49, P = 0.05, P = 0.65, respectively; Table 1 ). As compared with basal values, cortisol levels in SUAL-alone animals were elevated over the course of the experiment (P = 0.001; Table 1 ).
Fetal Body and Lung Weights
At the time of postmortem, 115 ± 0.4 d, fetal body weight was significantly reduced following fetal treatment (control as compared with SUAL; P = 0.01) or maternal treatment (vehicle as compared with BM; P = 0.006; Table 2 ). The fetal brain:body weight ratios were significantly increased with both fetal treatment (P = 0.005) or maternal treatment (P = 0.02; Table 2 ). Absolute fetal lung weight was significantly reduced with maternal treatment (P = 0.01) and lung:body weight ratios were significantly increased with fetal treatment (P = 0.03; Table 2 ).
Lung Morphology
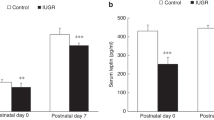
Figure 1 shows lung architectural data for all four groups of fetuses. At 0.8 gestation in fetal sheep, the percentage area of lungs occupied by lung parenchyma (tissue density) was not significantly altered by fetal treatment (P = 0.31) but was significantly reduced with maternal treatment (P = 0.048; Figure 1a ). The proportion of septal crests was not altered by fetal treatment (P = 0.09) or maternal treatment (P = 0.5; Figure 1b and Figure 2a ) and lung DNA content did not differ with fetal treatment (P = 0.24) or maternal treatment (P = 0.2; data not shown). The proportion of Ki67-positive cells within the lungs was significantly reduced following maternal treatment with BM (P < 0.001; Figure 2b and Figure 3 ).

Fetal lung architectural data. (a) Fetal lung tissue density was reduced following BM administration. (b) Septal crest density was not significantly different between treatment groups. Values are means ± SE; *P < 0.05 for maternal treatment (vehicle as compared with BM); n = 7/group. BM, betamethasone; SUAL, single umbilical artery ligation.

Histology of fetal lung tissue. (a) Photomicrographs showing Hart’s elastin staining (stained dark) and (b) Ki67 immunoreactivity (stained brown) in (A) control, (B) SUAL alone, (C) control + BM, and (D) SUAL + BM. Bar = 50 µm. BM, betamethasone; SUAL, single umbilical artery ligation.

The percentage of Ki67-positive proliferating cells within the lung was reduced following BM. Values are means ± SE; *P < 0.05 for maternal treatment (vehicle as compared with BM); n = 7/group. BM, betamethasone; SUAL, single umbilical artery ligation.
SP Gene Expression
There was no significant effect of fetal treatment on surfactant protein (SP) gene expression ( Figure 4 ). BM administration was associated with a significant increase in SP-A (P < 0.001), SP-B (P = 0.009), and SP-C (P = 0.03) mRNA expression. There was no effect of maternal treatment on SP-D mRNA expression (P = 0.41; Figure 4 ).

Normalized gene expression for SPs. (a) SP-A, (b) SP-B, (c) SP-C, and (d) SP-D are expressed as a ratio of control levels. Values are mean ± SE. *P < 0.05 for maternal treatment (vehicle as compared with BM); n = 6/group. BM, betamethasone; SP, surfactant protein; SUAL, single umbilical artery ligation.
Discussion
This study demonstrates that 1 wk of SUAL-induced IUGR alters lung weight relative to fetal body weight but does not affect lung structure. The administration of BM, in the dosing regimen used clinically, was associated with structural lung changes, in both control and SUAL fetuses. SUAL-induced IUGR alone did not alter the lung expression of SP mRNAs, whereas BM significantly increased the expression of SP-A, SP-B, and SP-C in normally grown and SUAL fetuses. Previous studies have examined the effects of either IUGR or glucocorticoid administration separately on promoting lung structure and biochemical changes, but a high proportion of human IUGR babies are born preterm (7) and exposed to antenatal glucocorticoids, and therefore this study sheds important light on their combined effects.
The duration of SUAL in this study was relatively short (1 wk) but was associated with the key features of human IUGR, namely altered fetal growth, fetal hypoxemia, and increased levels of circulating cortisol, reflecting SUAL-induced IUGR as an appropriate model of human IUGR (15,16,17). Despite increased cortisol concentrations, 1 wk of SUAL-induced IUGR alone did not alter fetal lung morphology. This is supported by a previous study in sheep in which umbilicoplacental embolization was used to induce IUGR (10). Following 20 d of umbilicoplacental embolization, tissue density and alveolar size and number were unaffected. However when these animals were studied postnatally, tissue density was increased at 8 wk of age whereas at 2 years of age the IUGR lung had fewer, larger alveoli (10,11). This suggests that while no structural changes are present in fetal tissue, experimental IUGR may induce morphological changes later in life.
SP gene expression was not altered by the SUAL procedure alone. This finding confirms a previous study in sheep that used 20 d of umbilicoplacental embolization to induce IUGR (18). However, this has not been a consistent finding. Using umbilicoplacental embolization to induce IUGR earlier in pregnancy, Gagnon and colleagues reported an increase in mRNA for SP-A and SP-B (19). In contrast, in a recent study where carunclectomy was used to induce IUGR, a reduction in fetal lung expression of SP-A, SP-B, and SP-C mRNA was reported (20). The differences between these experimental models of IUGR most likely reflect the timing of IUGR, rather than the method of inducing IUGR. Because glucocorticoids induce the production of SPs (21,22), the hypercortisolemia induced by IUGR in this and other studies (18,19,20) would be expected to increase SP mRNA expression. Another known regulator of SPs is fetal oxygenation (23). However, this effect is likely to mediate SPs when induced at a later gestational age (23), which may explain why no effect of moderate hypoxemia, induced by SUAL, was observed in our study. Thus, in the majority of animal models IUGR does not induce SP gene expression, which may underlie the impaired lung function in these infants immediately following birth.
In contrast, maternal administration of BM was associated with structural and functional changes in normally grown and growth-restricted fetuses. These effects in the normally grown fetus have been described previously (5,21,22,24), however, the relative importance of exogenous glucocorticoids in the growth-restricted fetus is poorly understood. With regard to the structural changes, these were comparable to those observed in control fetuses. This observation would suggest that antenatal steroids would be expected to reduce respiratory distress syndrome in preterm IUGR babies, as is observed in normally grown infants.
This study was undertaken in fetal sheep during the late canalicular and early saccular stage of lung development, approximating 26 wk of human lung development (25), and thus experimentally represents a common clinical scenario. At this point in development, the fetal lung consists of primitive alveoli as the airway walls thin and the surface area for gas exchange increases (26). It also represents a development stage of significant cellular proliferation. Glucocorticoids are known to inhibit cell proliferation in a number of tissues (27), including the lung (28). Thus, the reduced cell proliferation we observed in both control and SUAL fetuses treated with BM was not surprising but may result in the arrest of normal development. We also expected to observe a decrease in the proportion of septal crests in response to glucocorticoid administration (24,29). The septal crests represent the developing alveoli, and exogenous glucocorticoids decrease alveolar septa at the expense of future lung growth and development (29). The changes to alveolarization, as demonstrated by a reduction in the septal crest proportions, are thought to take longer than 48 h (30). In this study, we did not observe a reduction in septal crest number, most likely because there was a short interval between glucocorticoid exposure and tissue collection. This lack of an effect over a short (48 h) time interval has been reported previously (30). In contrast, when the interval between glucocorticoid exposure and lung assessment is longer, such as 7 d, changes to alveolarization are observed (24).
In addition to structural effects, glucocorticoids also induce maturation of the pulmonary surfactant system (22). We confirmed such effects in this study, in SPs A, B, and C in both control and SUAL fetuses. This increase confirms previous reports in normally grown animals (22). Of interest, only BM and not SUAL had an effect on SPs. This would further suggest that antenatal glucocorticoids confer lung function benefits in the preterm IUGR fetus, as they do in the normally grown preterm fetus.
The administration of glucocorticoids to the ewe resulted in fetal hyperglycemia, with circulating fetal glucose levels increased threefold. Such an effect has been documented previously in normally grown fetal sheep following glucocorticoid administration (31). It is known that glucocorticoids increase maternal plasma glucose levels (32), with maximum levels reached 8 h following administration (33). In this study, we found that fetal glucose levels were increased within 6 h after the first BM injection and maximal levels were observed following the second BM exposure. As compared with their control twins, SUAL fetuses were relatively hypoglycemic before BM administration and hyperglycemia following glucocorticoid administration was attenuated in the SUAL fetuses. This finding may be due, at least in part, to an increase in the glycolytic activity of the IUGR placenta (34) limiting transfer capability of glucose across the compromised placenta.
The time course undertaken in this study—1 wk between the onset of SUAL-induced IUGR and postmortem, incorporating BM administration on d 5 and d 6—was chosen because we had previously demonstrated that this results in asymmetric growth restriction and fetal hypoxemia (15,17) and because we wished to replicate a common clinical scenario, the preterm IUGR baby that is exposed to glucocorticoids for a short period before birth (35). We also did not want our animals to deliver in response to BM—which occurs in sheep (36), nor did we want to confound experimental outcomes with the administration of progestogen to prevent preterm labor (36). However, a limitation of this time course may be that it was not long enough to observe significant changes in some measures of lung morphometry and surfactant production.
In summary, we have shown that in SUAL fetuses, maternal BM is associated with acceleration of fetal lung structure, as occurs in normally grown fetuses, and that BM induces SP production, an effect not observed in SUAL-induced IUGR fetuses alone. Thus, although it has been reported that antenatal glucocorticoids do not improve a range of clinical outcomes in IUGR babies (14), we feel that it is premature to abandon such therapy until we better understand the risks and benefits.
Methods
Fetal Surgery
Experimental procedures were approved by the Monash Medical Centre Animal Ethics Committee. Surgery was performed on 23 twin-bearing Border-Leicester Merino crossbred ewes at ~108 d gestational age (term, 145–147 d). All ewes received intravenous ampicillin (1 g) before the induction of anesthesia with intravenous 20 mg/ kg sodium thiopentone (Pentothal; Bomac Laboratories, Auckland, New Zealand). General anesthesia was maintained with 2% isoflurane (Isoflo; Abbott, Sydney, Australia) in oxygen and nitrous oxide (70:30). Under aseptic conditions, each fetus was exteriorized to allow the insertion of sterile polyvinyl catheters containing saline into the femoral artery (inner diameter 0.8 mm, outer diameter 1.5 mm; Dural Plastics, Sydney, Australia) and the amniotic sac (inner diameter 1.5 mm, outer diameter 2.7 mm; Dural Plastics). The umbilical arteries were identified and a small incision made in the sheath surrounding the umbilical cord ~3 cm from the fetal abdomen. SUAL was performed in one twin by placing two ligatures around one of the two umbilical arteries. The cord was manipulated but not ligated in the control twin. The fetuses were then returned to the uterus and catheters were exteriorized via an incision in the flank of the ewe.
Experimental Procedures
Fetal arterial blood samples were taken each day following surgery for analysis of partial pressure of oxygen and carbon dioxide, pH, and oxygen saturation of hemoglobin using an ABL 700 blood gas analyzer (Radiometer, Copenhagen, Demark). On d 5 following surgery, ewes received either 11.4 mg BM intramuscular (termed BM1; Celestone Chronodose; Schering Plough, Sydney, Australia) or an equal volume of saline and then received a second dose of 11.4 mg BM or saline 24 h later on d 6 (termed BM2). This dosing regimen reflects the clinical use of BM (37,38). Thus, there were four treatment groups: control, SUAL-alone, control + BM, and SUAL + BM. Fetal arterial blood samples were collected 24 h before each BM dose as well as 6 h and 12 h after BM1 and BM2. On day 7, animals were killed with maternal injection of pentobarbitone (Lethabarb; Virbac, Peakhurst, Australia) and fetal lungs were collected for analysis.
Fetuses were weighed before the fetal lungs were removed, weighed, and drained of liquid. The left bronchus was ligated and random portions of the left lung were snap-frozen and stored at −70 °C. The right lung was fixed with 4% paraformaldehyde instilled at 20 cm H2O via the trachea and then postfixed in Zamboni’s fixative and processed for light microscopy.
The right lung was separated into cranial, middle, and caudal lobes and sliced at 5-mm intervals. Three slices from each lobe were chosen at random, cut into 2-cm2 sections, and embedded in paraffin wax. One block from each of the three right lung lobes was then randomly selected and sectioned as slices of 5-µm thickness before being incubated at 60 °C, dewaxed, and rehydrated. Sections were stained with hematoxylin and eosin or Hart’s resorcin–fuschin stain to identify elastin and the presence of septal crests. The percentage of area occupied by tissue was calculated by image analysis (Image-Pro Plus, Bethesda, MD) and septal crest density was measured using point counting and expressed as a proportion of tissue area (39). All counting was undertaken by a single observer (A.E.S.) blinded to the experimental group. Five fields of view for each section were used in the analyses.
Ki67 immunohistochemistry was used to identify proliferating cells (40). Antigen retrieval was undertaken in citric acid buffer (0.01 mol/l, pH 6.0) using a microwave oven (4 × 5 min) followed by incubation for 30 min. Sections were then rinsed in Tris-buffered saline (TBS; 0.1 mol/l, pH 7.6) with 0.1% Triton X-100 before endogenous peroxidase activity was blocked by incubating sections in 3% hydrogen peroxide for 10 min at room temperature. Nonspecific binding was blocked by incubating sections in 5% normal goat serum in TBS with 0.3% Triton X-100 for 30 min and then incubating overnight at 4 °C in a 1:100 dilution of rabbit monoclonal anti-Ki67 (Thermo Scientific, Barrington, IL) antibody in TBS with 2% normal goat serum and 0.1% Triton X-100. Sections were then rinsed in TBS and incubated in a 1:500 dilution of biotinylated goat anti-rabbit secondary antibody for 90 min at room temperature. Sections were again rinsed in TBS before incubation with streptavidin horseradish peroxidase (1:200) and visualization using diaminobenzidine complex. Sections were then counterstained with hematoxylin. Ki67-positive cells were counted and expressed as a percentage of the total number of cells within each field of view.
Tissue DNA concentrations were measured using a fluorometric DNA assay. Briefly, tissue samples (~500 mg) were homogenized in ice-cold sodium phosphate buffer (3 mol/l NaCl, 0.05 mol/l Na2HPO4, 0.05 mol/l NaH2PO4·2H2O, 0.002 mol/l EDTA, pH 7.4) and centrifuged for 5 min at 3,500 r.p.m. (4 °C). A 1:5 dilution of the supernatant was used to measure DNA. An EDTA solution (0.002 mol/l) and fluorochrome (Hoechst 33258; 2.5µg/ml in 0.125 mol/l NaH2PO4·2H2O, 3 mol/l NaCl, pH 7.4) were added to each standard and sample in triplicate before measuring fluorescence using an excitation wavelength of 356 nm and an emission wavelength of 480 nm.
Total RNA was extracted from fetal lung tissue using an RNeasy Midi Kit (Qiagen, Doncaster, Australia) per the manufacturer’s guidelines. cDNA was synthesized using 1 µg of DNase-treated RNA with the Superscript III First Strand Synthesis System (Invitrogen, Mulgrave, Australia). SP-A, SP-B, SP-C, SP-D, and 18S ribosomal RNA were amplified by quantitative real-time PCR using a Fast Real-Time PCR system (7900HT; Applied Biosystems, Mulgrave, Australia). Reactions containing 5 l SYBR Green (Applied Biosystems), 1 l primers (
Table 3
), 0.5 l cDNA, and 3.5 l nuclease-free water were loaded in triplicate into a 384-well plate using a liquid handling instrument (Corbett Research CAS1200; Qiagen). A control reaction containing all reagents except the cDNA template was included to test for reagent contamination. The mean threshold (CT) value for each sample was calculated and the mean CT value for 18S was subtracted (CT). The CT value was then normalized ( ) and expressed relative to the mean mRNA levels of the gene of interest in samples from the control group.
) and expressed relative to the mean mRNA levels of the gene of interest in samples from the control group.
Fetal plasma cortisol concentrations were determined using an established in-house radioimmunoassay. The assay sensitivity was 3.7 ng/ml and the inter- and intra-assay coefficients of variation were 15.1% and 8.4%, respectively. There was no cross-reactivity with BM (41).
Statistical Analysis
Data are presented as means ± SE. Statistical analyses were undertaken using SigmaPlot (Systat Software, Chicago, IL). A two-way ANOVA was undertaken on normally distributed data, with fetal treatment (control or SUAL) and maternal treatment (vehicle or BM) as fixed variables for analysis of fetal body weight, lung weight, lung:body weight ratio, tissue density, cell proliferation, DNA concentration, and septal crest densities. Data pertaining to SP mRNA levels were normalized by log-transform before parametric analysis. Post hoc least significant difference test was performed as required. Three-way repeated-measures ANOVA was used to examine fetal arterial parameters and cortisol changes over time, with a least significant difference post hoc test. Where significant interactions were detected between factors, differences between groups were isolated with one- or two-way ANOVA. Statistical significance was accepted when P < 0.05.
Statement of Financial Support
This work was funded by project grant no. 436721 from the National Health and Medical Research Council (Australia) and the Victorian Government's Operational Infrastructure Support Program.
References
Beck S, Wojdyla D, Say L, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bull World Health Organ 2010;88:31–8.
NIH Consensus Development Panel on the Effect of Corticosteroids for Fetal Maturation on Perinatal Outcomes. Effect of corticosteroids for fetal maturation on perinatal outcomes: NIH consensus development panel on the effect of corticosteroids for fetal maturation on perinatal outcomes. JAMA 1995;273:413–8.
Crowley PA . Antenatal corticosteroid therapy: a meta-analysis of the randomized trials, 1972 to 1994. Am J Obstet Gynecol 1995;173:322–35.
Liggins GC . The role of cortisol in preparing the fetus for birth. Reprod Fertil Dev 1994;6:141–50.
Willet KE, Jobe AH, Ikegami M, Newnham J, Brennan S, Sly PD . Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr Res 2000;48:782–8.
Coalson JJ . Pathology of new bronchopulmonary dysplasia. Semin Neonatol 2003;8:73–81.
Gilbert WM, Danielsen B . Pregnancy outcomes associated with intrauterine growth restriction. Am J Obstet Gynecol 2003;188:1596–9.
Greenough A, Yuksel B, Cheeseman P . Effect of in utero growth retardation on lung function at follow-up of prematurely born infants. Eur Respir J 2004;24:731–3.
Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO . Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ 1991;303:671–5.
Maritz GS, Cock ML, Louey S, Joyce BJ, Albuquerque CA, Harding R . Effects of fetal growth restriction on lung development before and after birth: a morphometric analysis. Pediatr Pulmonol 2001;32:201–10.
Maritz GS, Cock ML, Louey S, Suzuki K, Harding R . Fetal growth restriction has long-term effects on postnatal lung structure in sheep. Pediatr Res 2004;55:287–95.
Joyce BJ, Louey S, Davey MG, Cock ML, Hooper SB, Harding R . Compromised respiratory function in postnatal lambs after placental insufficiency and intrauterine growth restriction. Pediatr Res 2001;50:641–9.
Mericq V, Medina P, Kakarieka E, Márquez L, Johnson MC, Iñiguez G . Differences in expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur J Endocrinol 2009;161:419–25.
Torrance HL, Derks JB, Scherjon SA, Wijnberger LD, Visser GH . Is antenatal steroid treatment effective in preterm IUGR fetuses? Acta Obstet Gynecol Scand 2009;88:1068–73.
Miller SL, Chai M, Loose J, et al. The effects of maternal betamethasone administration on the intrauterine growth-restricted fetus. Endocrinology 2007;148:1288–95.
Miller SL, Supramaniam VG, Jenkin G, Walker DW, Wallace EM . Cardiovascular responses to maternal betamethasone administration in the intrauterine growth-restricted ovine fetus. Am J Obstet Gynecol 2009;201:613.e1–8.
Supramaniam VG, Jenkin G, Loose J, Wallace EM, Miller SL . Chronic fetal hypoxia increases activin A concentrations in the late-pregnant sheep. BJOG 2006;113:102–9.
Cock ML, Albuquerque CA, Joyce BJ, Hooper SB, Harding R . Effects of intrauterine growth restriction on lung liquid dynamics and lung development in fetal sheep. Am J Obstet Gynecol 2001;184:209–16.
Gagnon R, Langridge J, Inchley K, Murotsuki J, Possmayer F . Changes in surfactant-associated protein mRNA profile in growth-restricted fetal sheep. Am J Physiol 1999;276(3 Pt 1):L459–65.
Orgeig S, Crittenden TA, Marchant C, McMillen IC, Morrison JL . Intrauterine growth restriction delays surfactant protein maturation in the sheep fetus. Am J Physiol Lung Cell Mol Physiol 2010;298:L575–83.
Ballard PL, Ning Y, Polk D, Ikegami M, Jobe AH . Glucocorticoid regulation of surfactant components in immature lambs. Am J Physiol 1997;273(5 Pt 1):L1048–57.
Jobe AH, Nitsos I, Pillow JJ, Polglase GR, Kallapur SG, Newnham JP . Betamethasone dose and formulation for induced lung maturation in fetal sheep. Am J Obstet Gynecol 2009;201:611.e1–7.
Braems GA, Yao LJ, Inchley K, et al. Ovine surfactant protein cDNAs: use in studies on fetal lung growth and maturation after prolonged hypoxemia. Am J Physiol Lung Cell Mol Physiol 2000;278:L754–64.
Polglase GR, Nitsos I, Jobe AH, Newnham JP, Moss TJ . Maternal and intra-amniotic corticosteroid effects on lung morphometry in preterm lambs. Pediatr Res 2007;62:32–6.
Pringle KC . Human fetal lung development and related animal models. Clin Obstet Gynecol 1986;29:502–13.
Kotecha S . Lung growth for beginners. Paediatr Respir Rev 2000;1:308–13.
Quiros I, Mayo JC, Garcia-Suarez O, et al. Melatonin prevents glucocorticoid inhibition of cell proliferation and toxicity in hippocampal cells by reducing glucocorticoid receptor nuclear translocation. J Steroid Biochem Mol Biol 2008;110:116–24.
Oshika E, Liu S, Ung LP, et al. Glucocorticoid-induced effects on pattern formation and epithelial cell differentiation in early embryonic rat lungs. Pediatr Res 1998;43:305–14.
Vyas J, Kotecha S . Effects of antenatal and postnatal corticosteroids on the preterm lung. Arch Dis Child Fetal Neonatal Ed 1997;77:F147–50.
Pinkerton KE, Willet KE, Peake JL, Sly PD, Jobe AH, Ikegami M . Prenatal glucocorticoid and T4 effects on lung morphology in preterm lambs. Am J Respir Crit Care Med 1997;156(2 Pt 1):624–30.
McCallum J, Smith N, MacLachlan JN, et al. Effects of antenatal glucocorticoids on cerebral protein synthesis in the preterm ovine fetus. Am J Obstet Gynecol 2008;198:103.e1–6.
Shelton SD, Boggess KA, Smith T, Herbert WN . Effect of betamethasone on maternal glucose. J Matern Fetal Neonatal Med 2002;12:191–5.
Foglia LM, Deering SH, Lim E, Landy H . Maternal glucose levels after dexamethasone for fetal lung development in twin vs singleton pregnancies. Am J Obstet Gynecol 2008;199:380.e1–4.
Magnusson AL, Powell T, Wennergren M, Jansson T . Glucose metabolism in the human preterm and term placenta of IUGR fetuses. Placenta 2004;25:337–46.
French NP, Hagan R, Evans SF, Godfrey M, Newnham JP . Repeated antenatal corticosteroids: size at birth and subsequent development. Am J Obstet Gynecol 1999;180(1 Pt 1):114–21.
Jenkin G, Jorgensen G, Thorburn GD, Buster JE, Nathanielsz PW . Induction of premature delivery in sheep following infusion of cortisol to the fetus. I. The effect of maternal administration of progestagens. Can J Physiol Pharmacol 1985;63:500–8.
Robertson MC, Murila F, Tong S, Baker LS, Yu VY, Wallace EM . Predicting perinatal outcome through changes in umbilical artery Doppler studies after antenatal corticosteroids in the growth-restricted fetus. Obstet Gynecol 2009;113:636–40.
Wallace EM, Baker LS . Effect of antenatal betamethasone administration on placental vascular resistance. Lancet 1999;353:1404–7.
Allison BJ, Crossley KJ, Flecknoe SJ, Davis PG, Morley CJ, Hooper SB . Ventilation and oxygen: dose-related effects of oxygen on ventilation-induced lung injury. Pediatr Res 2010;67:238–43.
Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H . Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984;133:1710–5.
Bocking AD, McMillen IC, Harding R, Thorburn GD . Effect of reduced uterine blood flow on fetal and maternal cortisol. J Dev Physiol 1986;8:237–45.
Acknowledgements
We thank Alex Satragno for his surgical assistance and Tharani Sabaretnam and Jan Loose for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sutherland, A., Crossley, K., Allison, B. et al. The effects of intrauterine growth restriction and antenatal glucocorticoids on ovine fetal lung development. Pediatr Res 71, 689–696 (2012). https://doi.org/10.1038/pr.2012.19
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2012.19
This article is cited by
-
Mortality and neurological outcomes in extremely and very preterm infants born to mothers with hypertensive disorders of pregnancy
Scientific Reports (2021)
-
Antenatal corticosteroids and preterm offspring outcomes in hypertensive disorders of pregnancy: A Japanese cohort study
Scientific Reports (2020)
-
Fetal growth restriction is associated with an altered cardiopulmonary and cerebral hemodynamic response to surfactant therapy in preterm lambs
Pediatric Research (2019)
-
Altered cardiovascular function at birth in growth-restricted preterm lambs
Pediatric Research (2016)
