
Key Points
-
Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are a newly described family of intracellular sensors of microbial infections and danger signals. NLRs detect microbial motifs such as bacterial peptidoglycan (sensed by NOD1 and NOD2) or bacterial flagellin (sensed by NLR family, CARD-domain-containing 4 (NLRC4; also known as IPAF)) and NLR family, apoptosis inhibitory protein 5 (NAIP5). NLRs also sense danger signals, including uric acid, K+ efflux, extracellular ATP, silica, asbestos and β-amyloid peptide through NLR family, pyrin domain-containing 3 (NLRP3).
-
NLRs trigger innate immune responses by inducing signalling pathways, such nuclear factor-κB, mitogen-activated protein kinases, and the caspase 1 inflammasome. This results in the activation of inflammatory cytokines and/or chemokines. NLRs also work in synergy with Toll-like receptors to potentiate signal transduction pathways.
-
Mutations in several NLR genes are associated with autoinflammatory disorders, including NOD2 (associated with Crohn's disease and Blau syndrome), NLRP3 (associated with Muckle–Wells syndrome, chronic infantile neurologic cutaneous and articular syndrome, and familial cold urticaria), NOD1 (associated with asthma, allergy and atopic eczema) and NLRP1 (associated with vitiligo).
-
NLRs have a crucial role in the detection of molecules that were initially known as adjuvants, such as muramyl peptides and complete Freund's adjuvant (sensed by NOD1 and NOD2) and aluminium hydroxide (sensed by NLRP3). On detection of these molecules, NLRs shape the immune response to antigens, highlighting the link between NLRs and adaptive immunity.
-
Because of their importance in innate immunity and adjuvanticity, NLRs and NLR-triggered pathways are promising target candidates for therapeutic strategies against autoinflammatory disorders. The recent development of interleukin 1-specific strategies against gout and Muckle–Wells syndrome illustrates the translation of NLR basic research into clinical practice.
Abstract
Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are a family of intracellular sensors that have key roles in innate immunity and inflammation. Whereas some NLRs — including NOD1, NOD2, NAIP (NLR family, apoptosis inhibitory protein) and NLRC4 — detect conserved bacterial molecular signatures within the host cytosol, other members of this family sense 'danger signals', that is, xenocompounds or molecules that when recognized alert the immune system of hazardous environments, perhaps independently of a microbial trigger. In the past few years, remarkable progress has been made towards deciphering the role and the biology of NLRs, which has shown that these innate immune sensors have pivotal roles in providing immunity to infection, adjuvanticity and inflammation. Furthermore, several inflammatory disorders have been associated with mutations in human NLRgenes. Here, we discuss the effect that research on NLRs will have on vaccination, treatment of chronic inflammatory disorders and acute bacterial infections.
Similar content being viewed by others
Main
Innate immunity to microbial pathogens relies on the specific host-receptor detection of pathogen- and danger-derived molecular signatures (collectively referred to as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), respectively). The field of innate immunity research is currently experiencing a remarkable expansion due to the identification of several families of these PAMP and DAMP sensors, which are collectively termed pattern recognition molecules1,2. The best-studied family of pattern recognition molecules is the Toll-like receptor (TLR) family, which in mammals encodes 10–12 membrane-spanning molecules with diverse specificities for PAMPs or DAMPs3. More recently, another family of pattern recognition molecules — named the nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family — has received considerable attention.
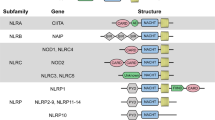
In humans, the NLR family is composed of 22 intracellular pattern recognition molecules (Fig. 1) that share a central NACHT domain (domain present in NAIP, CIITA, HET-E and TP1) and a carboxy-terminal leucine-rich repeat (LRR) region4,5,6. One subfamily of NLRs is composed of NOD1 and NOD2, which have an amino-terminal caspase recruitment domain (CARD) required to trigger nuclear factor-κB (NF-κB) signalling (Fig. 2). The NLR family, pyrin domain-containing (NLRP) proteins constitute another homogenous NLR subfamily, and current research suggests that they are essential for the induction and regulation of the caspase 1 inflammasome through their N-terminal pyrin domain7. Another important aspect of NLR biology is that a number of the genes that encode these proteins are mutated in human chronic inflammatory disorders, including crohn's disease (for NOD2), Muckle–Wells syndrome (for NLRP3), atopic disorders (for NOD1) and vitiligo (for NLRP1)5. This suggests that NLRs have a key role in linking host innate immunity to microbes and the regulation of inflammatory pathways8.

Schematic representation of the 22 members of the nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family as found in the human genome. Common to all NLRs is a central NACHT domain (domain present in NAIP, CIITA, HET-E and TP1) flanked on the carboxy-terminus by a leucine-rich repeat domain. NLRs can be divided into four subfamilies, mainly on the basis of their amino-terminal domain: (a) the sole member of the NLRA family, CIITA, is unique in that it acts as a transcription factor; (b) the NLRB subfamily consists of one member, NAIP (NLR family, apoptosis inhibitory protein), responsible for triggering interleukin 1β (IL-1β) secretion in response to intracellular flagellin; (c) members of the NLRP subfamily express an N-terminal pyrin domain and are crucial for the organization of IL-1β inflammasomes; (d) the NLRC/X subfamily members display either an N-terminal caspase recruitment domain (CARD), or an undefined domain that has no apparent homology with other proteins.

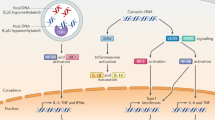
NOD1 and NOD2 detecttheintracellular peptidoglycan fragments M-TriDAP (L-Ala-D-Glu-meso-diaminopimelic acid) and MDP (muramyl dipeptide), respectively. Activation of NOD1 and NOD2 triggers the recruitment of the adaptor protein RIPK2 (receptor-interacting serine–threonine kinase 2; also known as RIP2), which activates downstream signalling, including nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs). NLR family, pyrin domain-containing 3 (NLRP3; also known as NALP3) is activated by many signals: microbes or bacterial toxins, end- or by-products of stress and danger signals (MSU (monosodium urate) and CPPD (calcium pyrophosphate dihydrate) crystals, β-amyloid fibrils and ultraviolet (UV) radiation) or xenogenous compounds (aluminium hydroxide, asbestos and silica). All these signals converge on activation of the NLRP3 inflammasome, composed of the proteins NLRP3, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)), cardinal and caspase 1. The NLRP3 inflammasome in turn triggers the cleavage of pro-interleukin 1β (pro-IL-1β) into mature IL-1β. Caspase 1 also cleaves pro-IL-18 and pro-IL-33. It remains unclear how so many signals converge to the NLRP3 inflammasome, but it has been shown that common downstream signals might be the production of reactive oxygen species (ROS) or detection of damage to the lysosomal membrane. In addition, pannexin 1 may function as a channel to mediate entry of Toll-like receptor (TLR) ligands into the cytosol, which in turn may directly trigger activation of the NLRP3 inflammasome. Of note, K+ efflux has been shown to activate the NLRP3 inflammasome, through mechanisms that remain poorly defined. This K+ efflux can be a danger signal (for example, following extracellular ATP-dependent activation of purinergic receptors (P2XRs); this can occur when ATP is released from dying cells); similarly, K+ efflux can arise from bacterial infection or the action of toxins. TLRs act independently of NLR-dependent pathways, and trigger a number of signalling cascades following the engagement of the adaptor proteins MYD88 (myeloid differentiation primary response protein 88), TRIF (TIR domain-containing adaptor protein inducing interferon (IFN)-β; also known as TICAM1), TIRAP (TIR-domain-containing adaptor protein; also known as MAL) and TRAM (TRIF-related adaptor molecule; also known as TICAM2), including NF-κB, MAPKs and type I IFN responses. TLRs have a crucial role in the induction of pro-IL-1β, as well as pro-IL-18 and pro-IL-33. LPS, lipopolysaccharide; LTA, lipoteichoic acid; PAM3CSK4, palmitoyl-3-cysteine-serine-lysine-4; TH- cell, T helper cell.
The PAMPs and DAMPs that are detected by some NLRs have recently been identified (Table 1). NOD1 and NOD2 sense peptidoglycan, a heterogeneous polymer found in the cell walls of bacteria. These two NLRs detect distinct entities within the peptidoglycan polymer: whereas NOD2 detects muramyl dipeptide (MDP), which is a motif common to Gram-positive and Gram-negative bacterial peptidoglycan9,10, NOD1 specifically detects diaminopimelic acid (DAP)-type peptidoglycan, which is found almost exclusively in Gram-negative bacteria11,12. NLRP3 (also known as NALP3 or cryopyrin) has been shown to detect a range of PAMPs (such as MDP, poly(I–C), double-stranded RNA from viruses, bacterial RNA and pore-forming toxins), as well as a number of DAMPs (such as extracellular ATP, uric acid, asbestos, silica, aluminium hydroxide and amyloid-β peptide), which suggests the existence of a common stimulus, triggered downstream of all these PAMPs and/or DAMPs (see Refs 13, 14 for recent reviews on this topic). It has been proposed that this common trigger could be stimulated by reactive oxygen species or lysosomal damage15,16,17. The other recently identified molecular patterns that trigger NLRs include flagellin (sensed by NLR family, CARD domain-containing 4 (NLRC4; also known as IPAF) and NLR family, apoptosis inhibitory protein (NAIP))18,19 and the lethal toxin of Bacillus anthracis (sensed by NLRP1) (Table 1)20. Following detection of their specific PAMP or DAMP, NLRs trigger a number of signalling pathways that, overall, contribute to the host response to microbes and xenocompounds. Several review articles have focused on NLRs and their biological function5,13,21. Indeed, this family of intracellular sensors is emerging as a crucial hub for the regulation of host responses to bacterial pathogens, inflammation and adaptive immunity.
In this Review, only the most extensively studied NLRs are discussed; however, research on other NLRs associated with disease and inflammation, such as NLRP1 (the mutation of which leads to vitiligo)22 and NLRP12 (the mutation of which results in hereditary periodic fever syndromes)23, is still emerging. In addition, mutations in NOD2 have been associated with a growing number of malignant diseases, including early-onset breast cancer24,25,26, non-Hodgkin's lymphoma27, melanoma28 and lung cancer26, and with negative outcomes following transplant surgery29,30; however, the molecular mechanisms underlying these associations are still unclear. Therefore, research in the field of NLRs has a broad clinical relevance. This article reviews literature in the field of NLR research that is pertinent to the development of improved vaccination strategies, the treatment of inflammatory disorders — for example, inflammatory bowel disease including Crohn's disease, asthma, gout and rheumatoid arthritis — and possibly acute and chronic bacterial infections. The recent exciting discoveries in NLR research mean that this emerging area of investigation may have matured to a point at which transferring our fundamental knowledge to the development of clinical interventionsis realistic and appropriate. Here, we highlight the therapeutic potential of targeting NLRs, their ligands or the pathways that they trigger.
The role of NLRs in adjuvanticity
Historically, vaccination has proved to be one of the most effective medical interventions, allowing for the control or eradication of major diseases. However, traditional vaccination strategies such as the use of live attenuated pathogens, whole inactivated organisms or inactivated toxins present certain limitations. For example, some pathogens are difficult or even impossible to grow in culture, live attenuated vaccines can cause disease in immune-compromised individuals by reverting to a more virulent strain, and whole inactivated vaccines often contain components that have unwanted side effects. Therefore, the optimization of vaccination strategies remains an area of intense clinical research.
One aspect of vaccine development concerns the immunogenic molecules and/or particles themselves: synthetic peptides, DNA and recombinant protein subunits are being tested in a number of vaccines. Although these compounds offer advantages, such as reduced toxicity, they are often poorly immunogenic when administered alone. This is particularly true for vaccines based on synthetic peptides or recombinant proteins. Consequently, another angle of investigation is to enhance the immune response to a specific antigen by developing improved vaccine adjuvants31,32 that are potent, safe and compatible with protein subunits and peptides33. Although vaccination is considered one of the most effective medical interventions, research into vaccine adjuvants has largely been an empirical exercise. In particular, the host sensors and the mechanisms of action of these compounds have not been fully characterized. Now, the discovery of the role of NLRs in the action of certain adjuvants has provided new insight for vaccine development.
It is striking that throughout the twentieth century, long before the identification of NLRs (in the past decade), there had been extensive use of vaccination strategies involving molecules that are now known as NLR ligands. Two major classes of molecules that have been used as adjuvants illustrate this observation: bacterial cell wall preparations containing peptidoglycan (Box 1), which are now known to trigger the activation of NOD1 and NOD2, and inorganic crystals such as aluminium hydroxide (Box 2), which have been recently shown to induce the activation of the NLRP3 inflammasome. These observations are particularly important with regard to the crosstalk between NLRs and TLRs that has been the subject of numerous investigations in recent years. Indeed, whereas TLR ligands are pyrogenic and crucial for the induction of inflammatory cytokines, NLR ligands have a poor capacity to trigger these pathways without a synergistic boost from TLR ligands in vitr o34,35,36,37. Despite these apparently weak stimulatory properties, NOD1 (Refs 38, 39), NOD2 (Refs 40, 41) and NLRP3 (Refs 42–44) ligands are required for the action of some immune adjuvants.Therefore, the interaction between NLRs and TLRs seems to be more crucial for adaptive immunity than previously appreciated. Because of these observations, an interesting new area of investigation for vaccine and adjuvant development will be to use combinations of immunostimulatory agents that target the pathways of different receptors45.
NOD1 ligands. Research in the 1970s identified monomeric peptidoglycan subunits as minimal structures responsible for the adjuvanticity of complete Freund's adjuvant (CFA), and peptides containing DAP were shown to have adjuvant activity38,46,47. Two synthetic derivatives of the peptides derived from DAP-containing peptidoglycan, namely FK156 and FK565, received the most attention. These peptides were shown to have activity against bacterial48,49 and viral50,51,52 challenges, as well as possessing antitumoral properties53,54,55 and acting as immune adjuvants38. Following the discovery of NOD1 as the cellular sensor of DAP-containing peptides, the adjuvant capacity of these NOD1 agonists has been studied in more detail. It has been shown that FK156 in saline solution added to an ovalbumin (OVA) antigen elicits the priming of antigen-specific T cell and B cell immunity with a predominant T helper (TH2) polarization profile39. However, in the presence of TLR agonists (lipopolysaccharide (LPS) and pam3cys4), FK156 helped to increase TH1, TH2 and TH17 responses. Moreover, when the adjuvant CFA, which contains a mixture of NOD1, NOD2 and TLR ligands, was used in NOD1-deficient mice, the induction of adaptive immunity to the antigen was suboptimal, showing that NOD1 has a central role in the adjuvant action of CFA. Finally, it was shown that, following immunization with FK156, non-haematopoietic cells provided essential signals to orchestrate the development of TH2 immunity. This provides evidence that dendritic cells are not solely responsible for integrating microbial and antigen signals to instruct adaptive immune responses. Of note, it remains unknown how NOD1 activation in non-haematopoietic cells affects dendritic-cell function and the outcome of the response to a specific antigen.
NOD2 ligands. Before MDP was identified as a PAMP recognized by NOD2 (Refs 9, 10), it had been identified as the minimum effective component of CFA in 1974 (Ref. 56) and was characterized as a general immunostimulant capable of inducing non-specific immune responses to tumours and infections. MDP was subsequently shown to have a wide range of physiological effects including pyrogenicity and somnogenicity, which probably depend on the ability of MDP to induce the secretion of pro-inflammatory cytokines, such as interleukin 1β (IL-1β) and tumour necrosis factor (TNF). Despite extensive research on MDP, the molecule was found to be too pyrogenic and arthritogenic to be used as an adjuvant in humans. Therefore, efforts were focused on developing less pyrogenic derivatives of MDP that would still have immunomodulatory properties. The several non-toxic MDP derivatives that emerged from these efforts include the adamantylamine dipeptide (AdDP)57, L18-MDP58, MDP-Lys(L18)59, murabutide (an ester derivate)60, threonyl-MDP61 and glucosaminylmuramyl dipeptide62,63. More recently, it has been shown that the adjuvanticity of MDP is NOD2 dependent, since NOD2-deficient mice cannot mount a normal humoral response after immunization with MDP plus an antigen40,41. In fact, depending on the administration context, the adjuvanticity of MDP changes64,65. In saline solution, MDP mainly enhances humoral responses40,41,66 but, when used in conjunction with lipophilic carrier systems such as liposomes, oil-in-water emulsions or some lipophilic derivatives, it induces a strong cellular immunity67.
In vitro studies have shown that MDP recognition by NOD2 results in the transcription of a large repertoire of genes, many of which are dependent on activation by NF-κB. NOD2 activation leads mainly to the production of pro-inflammatory cytokines (IL-1β, IL-6, TNF and IL-8) and chemokines (keratine-derived chemokine, regulated upon activation, normal T cells expressed and secreted (RANTES; also known as CCL5) and CXC-chemokine ligand 5 (CXCL5; also known as ENA-78))8, nitric oxide68 and antimicrobial peptides (β-defensin 2)35, and also leads to an increase in the expression of co-stimulatory and adhesion molecules69,70,71. MDP also induces the production of superoxides, prostaglandins and collagenase72. In addition, MDP as well as other muropeptides (tripeptides and disaccharide tripeptides and tetrapeptides) have been shown to synergize with TLRs73 and increase the effect of immunomodulatory molecules such as cytokines (such as interferon-γ (IFNγ), IL-1β, IL-32 and granulocyte–macrophage colony-stimulating factor (GM-CSF))74,75,76,77. All these factors are crucial for the recruitment and activation of effector cells as well as inflammatory processes that result in the establishment of an appropriate adaptive immune response, highlighting the therapeutic potential of NOD2 molecules. To date, the focus on MDP has been on its capacity to activate antigen-presenting cells, and it is seen as a molecular adjuvant that targets myeloid cells such as dendritic cells. However, unlike NOD1, it remains unknown whether the adjuvanticity of NOD2 in vivo requires non-haematopoietic cells.
Different studies have demonstrated the potential effect of MDP administration. MDP alone or in combination with other agents confers resistance to viruses (for example, HIV78, Sendai virus79, herpes simplex virus79,80,81, influenza virus82,83 and vaccinia virus80), bacteria84,85, fungi86 and tumours87,88,89. Finally, murabutide, another promising MDP derivative, has been identified as a molecule devoid of pyrogenic60 and somnogenic90 activities that could nevertheless suppress HIV type 1 (HIV-1) replication in macrophages91.
NLRP3. NLRP3 is an essential component of the inflammasome, a protein complex that promotes cleavage of pro-caspase 1 into its active form7. Active caspase 1 is an enzyme that activates various substrates including the cytokines IL-1β, IL-18 and IL-33 (Fig. 2). The current model predicts that the inflammasome regulates the release of IL-1β; this is because the production of mature IL-1β requires not only stimuli that activate NF-κB (such as LPS) for the expression of pro-IL-1b, but also a second signal from stimuli (such as NLRP3 activation) that lead to the formation of the inflammasome complex that activates caspase 1. In the laboratory context, in which pure NLR and TLR agonists are added directly to cells, these two signals are discrete; however, in the context of many microbial infections both of these signals would be simultaneously provided. Therefore, NLRP3 activation leads to direct activation and secretion of IL-1β as well as other cytokines, including IL-18 and IL-33. As IL-1β and IL-18 are key cytokines that act on numerous immune cells, this makes NLRP3 agonists useful components of vaccines and immunostimulators.
Among others, NLRP3 responds to bacterial infection with Listeria monocytogenes and Staphylococcus aureu s92, as well as to ATP92, nigericin92, maitotoxin92, MDP93, viral RNA94, imidazoquinoline95, uric acid crystals96, asbestos15, silica17 and amyloid-β16. NLRP3 also responds to aluminium salts in vitro and in vivo; however, the role of NRLP3 in aluminium hydroxide-dependent potentiation of antibody responses remains controversial (discussed below). It seems unlikely that a single molecule is capable of distinguishing such a diverse array of structurally distinct compounds. Instead, NLRP3 probably responds to a secondary danger signal that accompanies these compounds, such as an ionic flux13. However, how NLRP3 activation contributes to adjuvanticity is not fully understood. It is possible that the activation of NLRP3 as part of the inflammasome is a common event in response to several adjuvants or more generally to particulate compounds. For example, two other adjuvants, chitosan (a polysaccharide derived from chitin) and Quil A (a saponin extracted from the bark of Quillaja saponaria)97, as well as the particulate compounds silica17 and asbestos15, also stimulate NLRP3. In addition, extracellular ATP triggers NLRP3-dependent activation of caspase 1 through signalling by the purinergic P2X7 receptor on antigen-presenting cells98,99. Extracellular ATP has also been shown to augment the delayed-type hypersensitivity response to 2,4-dinitrochlorobenzene or tumour antigens100, but the dependency on NLRP3 has not been investigated. The mechanism by which MDP could activate the inflammasome is unclear. For example, in vitro, MDP has been shown to induce caspase 1 activation and consequent IL-1β secretion through its activation of NOD2 and/or NLRP3 (Refs 93, 101) or through NLRP1 (Ref. 102). In vivo, it has also been shown that MDP induces IL-1β production through either NOD2 or NLRP3 pathways103,104. Some non-toxic derivatives of MDP with intact adjuvanticity have been shown to induce lower levels of IL-1β production than MDP in viv o105. It therefore seems that inflammasome stimulation and the adjuvant activity of MDP are separable. However, it is not clear whether this activation is truly through direct sensing of MDP by NLRP3 or whether MDP generates a secondary danger signal that is sensed by NLRP3. Further investigation is required to conclusively determine whether IL-1β secretion and NLRP3 are required for mediating the adjuvant effects of MDP.
In vitro, all reports agree that aluminium hydroxide can directly activate NLRP3 to trigger the caspase 1-dependent processing of pro-IL-1β. However, whether NLRP3 is required for the aluminium hydroxide-dependent potentiation of antibody responses in vivo remains controversial. Mice deficient in NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC) or caspase 1 present defective OVA-specific IgG1 antibodies after immunization with OVA plus aluminium hydroxide in an asthma model42. Similarly, NLRP3-deficient mice develop defective OVA- or diphtheria toxin-specific IgE and IgG1 antibodies after intraperitoneal immunization with OVA plus aluminium hydroxide and diphtheria toxin plus aluminium hydroxide97. By contrast, the NLRP3 knockout mice can mount a normal OVA-specific IgG1 antibody response following intraperitoneal immunization with OVA plus aluminium hydroxide. However, this study still found a defect in OVA-specific IgE antibodies with a concomitant increase in OVA-specific IgC2c, suggesting a switch from TH2- to TH1-type immunity106. Another study suggests that NLRP3-deficient mice are able to mount a normal IgG1 response following immunization with aluminium hydroxide plus human serum albumin43; however, it does not report the human serum albumin-specific IgE level.
Another report describes an alternative model of NLRP3 activation after aluminium hydroxide injection in vivo. The authors suggest that this adjuvant causes NLRP3 activation through the production of uric acid, which activates NLRP3 in antigen-presenting cells44. This last report is in accordance with the idea that the ability of aluminium hydroxide to induce a TH2-cell polarized immune response depends on its effects on dendritic cells107, and is further supported by the observation that exposing dendritic cells to this adjuvant in vitro polarizes T cells to a TH2-type profile108. Importantly, the molecular events that are responsible for the adjuvant action of aluminium hydroxide remain largely unknown. Indeed, IL-1β seems to be dispensable for adjuvanticity mediated by aluminium hydroxide, as IL-1 receptor-deficient mice develop normal pulmonary TH2-type allergic responses after immunization with this adjuvant plus OVA109. Finally, IL-18 has been shown to facilitate aluminium hydroxide-induced IL-4 production but not to participate in the production of OVA-specific IgG1 and IgE antibodies110. Together, these partially conflicting reports of in vitro and in vivo observations suggest that, in an in vivo setting, aluminium hydroxide does not polarize T cell responses exclusively through dendritic cells, but also through other as yet undefined cell populations. The discovery of the key part played by NLRP3 in driving responses to aluminium hydroxide will provide a better understanding of the mode of action of this adjuvant.
NLRs and chronic inflammation
The association of NLRs with various chronic illnesses has received much attention, as insight into the function of these receptors is expected to lead to new therapeutic approaches. The diseases associated with NLRs are mostly chronic inflammatory disorders (Table 2), underscoring the importance of these receptors in regulating immune responses. Despite the seemingly straightforward implications of aberrant NLR activity on inflammation, the putative roles of NLRs in specific disease pathology are diverse and a complex picture is emerging. Multiple environmental and genetic factors probably work in combination with different NLRs, which results in different disorders. Although many challenges remain, there is great potential for developing new strategies for combating chronic inflammation. In fact, as discussed below, the discovery of the role of NLRP3 in several diseases has led to the development of new treatment regimens for several auto-inflammatory disorders. A better understanding of the role of NLRs in other chronic illnesses could help guide the development of drugs that complement existing therapies or that provide relief for individuals who do not respond well to existing treatments. Research in this field should also provide clues for strategies to prevent the onset of many of these illnesses.
NLRP3 mutations and autoinflammatory syndromes. NLRP3 was first described when mutations that cause muckle–Wells syndrome and familial cold urticaria were mapped to the gene encoding this protein111. Subsequent studies have also linked NLRP3 to chronic infantile neurological cutaneous and articular syndrome (CINCA; also known as neonatal-onset multisystem inflammatory disease (NOMID))112,113. The mutations associated with these diseases seem to lead to deregulation of inflammasome formation that results in increased caspase 1 activation and IL-1β release114,115. Interestingly, individuals with the same NLRP3 mutation can be affected by familial cold urticaria, CINCA or Muckle–Wells syndrome, diseases that are mainly distinguished by the severity of their symptoms; this indicates that environmental or additional genetic factors probably have a role in mediating the different pathologies associated with these mutations116,117. The range of symptoms associated with mutations of NLRP3 could also be a result of the apparent capacity of this protein to respond to many different agonists, as discussed above.
Despite the potentially complex interactions between NLRP3 and environmental factors, a rather straightforward approach that involves the use of anakinra (Kineret; Amgen) — a recombinant IL-1 receptor antagonist that is approved for the treatment of rheumatoid arthritis — has proved successful for treating diseases associated with mutations of NLRP3. Following the first reported successful treatment of two Muckle–Wells syndrome patients with anakinra118, several other trials of anakinra on Muckle–Wells syndrome patients reported improvements in clinical symptoms as well as reversal of hearing119,120,121 and vision loss122. Anakinra therapy has also proved successful for treating patients affected by familial cold urticaria123 or by CINCA124. The efficacy of anakinra for these disorders indicates that, despite differences in clinical manifestations, IL-1β overproduction is largely responsible for the pathology of these syndromes. Therefore, it seems reasonable to speculate that treatment of other diseases involving the NLRP3 inflammasome with anakinra could prove successful. In the case of gout this has proved to be true; NLRP3 has been implicated in driving the uric acid-induced inflammation associated with gout96 and it has been shown that a drug used to treat the disease, colchicine, inhibits the NLRP3 inflammasome125. Initial trials of anakinra to treat gout have also proved successful126,127, which suggests that NLRP3-driven IL-1β production has a key role in the pathogenesis of gout. Another long acting IL-1 receptor antagonist rilonacept (Arcalyst; Regeneron) is currently being tested to treat NLRP3-associated syndromes and has had promising initial results128,129. It will be interesting to see whether these drugs could be applied to other diseases that potentially involve NLRP3 such as ischaemia–reperfusion syndromes130, silicosis131 or Alzheimer's disease16.
Discovery of the role of NLRP3 in Muckle–Wells syndrome, familial cold urticaria and CINCA has therefore helped direct the development of a new strategy for treating these diseases and is also applicable to other inflammatory disorders that involve NLRP3 activation. In the future, drugs could also be developed based on small molecules that target specific domains of NLRP3. For example, the nucleotide-binding and hydrolytic activity of NLRP3 could be targeted, as ATP hydrolysis is required for NLRP3-dependent IL-1β production132. Additionally, the pyrin domain in NLRP3 could be targeted as it seems to be required for inflammasome formation and subsequent caspase 1 activation133,134,135. These drugs would have the added benefit of specifically targeting the over-activation of NLRP3. Future applications could also involve screening for NLRP3 mutations in individuals who are at risk for auto-inflammatory syndromes, and establishing therapies that prevent the onset of clinical disease altogether. In addition, understanding how environmental factors determine the clinical outcomes of these disorders will assist in the development improved therapies.
NOD2 deficiency and Crohn's disease. In 2001, two groups independently reported a frameshift mutation and two missense variants of NOD2 as the first genetic risk factors identified for Crohn's disease136,137. Subsequent studies have identified additional genes associated with Crohn's disease including the gene encoding the IL-23 receptor138 and the autophagy gene ATG16L1 (Refs 139, 140); however, NOD2 mutations have been consistently found to have the highest disease-specific risk association. Before its association with Crohn's disease, NOD2 had been described as a monocyte-restricted gene involved in NF-κB activation141. Soon after its association with Crohn's disease was discovered, NOD2 was reported to be an intracellular sensor of MDP9,10. In addition, although NOD2 tissue expression was initially thought to be restricted to monocytes and other myeloid lineage cells, subsequent studies found that, on stimulation of NF-κB, NOD2 expression could be induced in epithelial cells142,143,144. Although the implications of the expression pattern of NOD2 are not clear, these findings are in accordance with the observed increase in NOD2 expression in monocytes and epithelial cells from patients with Crohn's disease145. These findings have spurred much research aiming to delineate the precise contribution of NOD2 in the development of Crohn's disease, raising hopes for new insight into the aetiology of the disease and new therapies.
Many observations have emerged that could have implications for new treatment strategies for Crohn's disease. For example, NOD2 mutations associated with the disease result in decreased NF-κB activation in response to stimulation with MDP10,146,147, and cells from people with mutations in the gene encoding NOD2 secrete reduced amounts of cytokines in response to various stimuli10,148,149. Cells harbouring NOD2 mutations express reduced levels of pro-inflammatory cytokines such as IL-8, IL-1β and TNF in response to MDP150, TLR2 agonists151 or MDP in combination with TLR agonists149,152,153. This is an unexpected finding as Crohn's disease is generally associated with augmented expression of pro-inflammatory cytokines. The observed reduction in TNF is particularly surprising as therapy with the TNF-specific antibody infliximab (Remicade; Centocor) is effective in treating Crohn's disease patients with NOD2 mutations154,155, which indicates that TNF over-production still has a role in the pathogenesis of Crohn's disease even when NOD2 is mutated. One possible explanation is that NOD2 mutations lead to a loss of tolerance to TLR agonists. Prior stimulation of normal cells with NOD2 ligands results in decreased TNF secretion when the cells are re-stimulated with TLR agonists156,157. However, in NOD2-deficient cells, pre-stimulation with a NOD2 agonist does not inhibit TLR activation when the cells are re-stimulated with LPS and instead leads to uninhibited release of pro-inflammatory cytokines such as TNF. Other experiments involving Nod2 frameshift knock-in mice indicated that this mutation enhances inflammatory responses to MDP and increases IL-1β levels158. Although this finding is hard to reconcile with the aforementioned studies involving clinical samples, there is one report that dendritic cells from patients with the frameshift NOD2 mutation express increased levels of IL-1β in response to MDP stimulation159. Therefore, there is controversy as to whether NOD2 mutations associated with Crohn's disease are gain-of-function or loss-of-function mutations. It is possible that NOD2 activity is delicately balanced and that different mutations associated with Crohn's disease could result in either loss-of-function or gain-of-function phenotypes that ultimately result in a disruption of intestinal homeostasis.
The observation that NOD2 mutations lead to increased IL-1β production is intriguing, especially as a decreased ratio of IL-1 receptor antagonist to IL-1β has been observed in the colonic mucosa of patients with Crohn's disease160,161,162. However, the mechanism by which IL-1β could mediate the inflammation seen in the condition is not clear. A recent study found that mutations in regulatory regions of NLRP3 are linked to Crohn's disease163. These mutations appear to lower NLRP3 expression and decrease IL-1β secretion in response to LPS, further supporting a role for deregulation of IL-1β production in Crohn's disease. It is also important to note that there is a report of one patient with Crohn's disease in which anakinra worsened the symptoms of the condition164, indicating that the situation is more complicated than the overproduction of IL-1β seen in NLRP3-associated syndromes. Another interesting observation is that mutations in NOD2 result in decreased expression of the anti-inflammatory cytokine IL-10 in cells from patients with Crohn's disease stimulated with MDP and TLR agonists69,151,152. Therefore, it would seem that in Crohn's disease the reduced IL-10 production that results from NOD2 deficiency could contribute to the inflammation that is characteristic of the condition. However, a recent study found that a mutation in IL-10 is associated with ulcerative colitis but not Crohn's disease165. This may indicate that IL-10 deficiency does not directly contribute to the development of Crohn's disease and may provide some explanation as to why clinical trials with recombinant IL-10 therapy have had little success in treating individuals with this condition166.
Nonetheless, it is still possible that NOD2-dependent IL-10 deficiency has a role in exacerbating inflammation in Crohn's disease when combined with other genetic and environmental factors. As mentioned above, NOD2 stimulation has also been directly implicated in driving the polarization of T cell responses. Therefore, a loss of NOD2 signalling could result in a cytokine imbalance that drives the TH1-type profile typical of Crohn's disease. Finally, it is possible that impaired NOD2 function contributes to a reduced function of the intestinal epithelial cell barrier. The direct role of NOD2 in barrier defence has been suggested by the observation that it is highly expressed by Paneth cells — specialized innate immune cells found in intestinal crypts that are essential for promoting barrier integrity167,168,169. The ability of Paneth cells to secrete anti-microbial peptides known as α-defensins is decreased in individuals with NOD2 mutations170. In addition, the Paneth cells of NOD2-deficient mice secrete reduced amounts of cryptdin (the mouse equivalent of α-defensin), and this coincides with an increased susceptibility to oral infection with Listeria monocytogene s41. However, the possible correlation of increased susceptibility to infection with Crohn's disease is controversial. One possible mechanism would be that that impaired function of the epithelial cell barrier allows for frequent breaches of the intestinal barrier by bacteria, which ultimately leads to disturbances in the homeostasis of T cell responses to the normal flora. Another possibility is that reduced production of anti-microbial compounds changes the composition of the normal flora and promotes the survival of bacterial species that are more prone to cause inflammation. It is important to note that a recent study has questioned the link between the NOD2 frameshift mutation and α-defensin secretion171, and the contribution of NOD2 to the defence function of Paneth cells in the epithelial cell barrier remains controversial.
The examples listed above help illustrate the complex roles that NOD2 has in the development of Crohn's disease. Its functions in maintenance of the epithelial cell barrier and the correct balance of cytokines probably contribute to different aspects of chronic inflammation. Despite the multiple immune defects that result from NOD2 dysfunction, many individuals who are homozygous for NOD2 mutations are asymptomatic. In fact, in some Asian populations such as the Japanese172,173 and Chinese174,175 there is no correlation between NOD2 genotype and the risk of developing Crohn's disease. This indicates that NOD2 mutations must act together with other genetic and environmental factors to predispose individuals to develop chronic inflammation. For example, a variant of ATG16L1 has been identified that is associated with a modest increase in the risk of developing the disease. However, this variant of ATG16L1 is present in an individual who is homozygous for a NOD2 mutation, the association with Crohn's disease increases dramatically139. Therefore, it is important to consider the interaction of many environmental and genetic elements when designing new therapies.
Despite these challenges, targeting NOD2 function has potential for treating Crohn's disease. As most patients with Crohn's disease do not have NOD2 mutations, it may be feasible to stimulate NF-κB activation by administering MDP or other NOD2 agonists. Stimulation of NOD2 could prevent the onset of an inflammatory 'flare up' by improving the function of the epithelial cell barrier through enhanced secretion of α-defensin, or improve the symptoms of the disease by boosting IL-10 production and restoring the function of regulatory T cells. For example, polysaccharide A (PSA) from Bacteriodes fragilis suppresses inflammation by boosting IL-10 production176 and is important for maintaining correct T cell polarization177,178. Therefore, PSA may be a candidate for treating Crohn's disease and other inflammatory bowel diseases. Alternatively, compounds that antagonize NOD2 activation could be developed as means to attenuate episodes of inflammation in individuals in whom NOD2 over-activation has a role in mediating pathology. For example, small molecular compounds that target the caspase-activating and recruitment domain (CARD) of NOD2 are good candidates for attenuating NOD2 signalling.
Atopic disease and over-activation of NOD1 and NOD2. Mutations in NOD1 have been associated with asthma and atopic eczema179,180. These mutations also correlate with decreased levels of IgE in patients, suggesting a direct role in pathology. Similar to NOD2, NOD1 recognizes peptidoglycan and enhances TLR-mediated NF-κB activation181 (Fig. 2). However, unlike NOD2, NOD1 is expressed by most tissues182 and recognizes a meso-diaminopimelic acid (meso-DAP)-containing peptidoglycan fragment that is present in most Gram-negative bacteria and a subset of Gram-positive bacteria11,12. These observations may partially account for the different pathologies associated with NOD1 and NOD2 mutations. The mechanisms by which NOD1 mutations lead to disease are not clear. However, one NOD1 mutation that is associated with asthma is thought to lead to changes in the levels of different isoforms of NOD1. Truncated isoforms do not respond to NOD1 ligands183 and have been proposed to help silence NOD1 activation; therefore, NOD1 mutations involved in atopic diseases are thought to result in increased activation of NF-κB due to a lack of these regulatory isoforms. NOD2 mutations that are thought to result in increased NF-κB activation are associated with blau syndrome184,185 and early-onset sarcoidosis186,187,188. The discovery of the apparent common aetiology of these diseases coincided with interest in using infliximab for treatment of many atopic diseases. So far there has been some success in treating Blau syndrome with infliximab189 and a study has shown that infliximab reduces inflammation in an acute asthma mouse model190. Another approach could be to use compounds that directly inhibit the activation of NF-κB to treat diseases associated with the over-activation of NOD1 and/or NOD2.
NLR agonists for treating acute infections
One of the main functions of NLRs is to help prevent microbial infection (Table 3). However, the association between NLR dysfunction and human disease is not always clear. For example, one study found a significant association between the presence of a polymorphism in NOD1 and susceptibility to gastritis and duodenal ulcers associated with Helicobacter pylori191; however, a second report failed to show a link between NOD1 polymorphisms and H. pylori-associated gastritis and ulcers192. Another report linked NOD2 polymorphisms with susceptibility to infection with Mycobacterium tuberculosis in an African American population193, but a different study found no evidence that NOD2 polymorphisms are associated with an increased frequency of M. tuberculosis infections in a South African population194. The disparity between these studies could perhaps be attributed to the different methodologies used or to the heterogeneity of the genetic background and environmental conditions of the individuals involved. Thus, human studies have sometimes failed to show an association between NLRs and susceptibility to infection. However, in more controlled mouse or in vitro experiments, in which there is a more homogenous genetic background and greater control over environmental conditions, NLRs have been implicated in mediating inflammatory responses to a range of different bacteria. NOD1-knockout mice are more susceptible to infection with H. pylor i195 and NOD2-deficient mice are more susceptible to infection with Listeria monocytogene s41; NLRP1 is required for mediating anthrax lethal toxin-induced IL-1β production during Bacillus anthracis infection in mice102; NLRC4 is involved in sensing intracellular bacteria that possess flagella or type III secretion systems, including Salmonella tiphimuriu m18,19, Shigella flexner i196, Legionella pneumophil a197, Pseudomonas aeruginos a198,199and L. monocytogene s200; a more recently discovered NLR family member, NLRX1, has been shown to potentiate the activation of NF-κB and JUN N-terminal kinase (JNK; also known as MAPK8) in response to S. flexneri infection201; and, as mentioned above, NLRP3 stimulates IL-1β production in response to infection with L. monocytogenes and Staphylococcus aureu s92. Additionally, NLRs have been implicated in the response to viral infections: NLRP3 has been shown to induce IL-1β expression in response to Sendai virus, influenza virus94 and adenovirus202, whereas NLRX1 is a potent inhibitor of NF-κB activation and IFNβ production in response to Sendai virus and Sindbis virus203.
The examples above underscore the important role of NLRs in the response to microbial infections. This raises the question of whether NLR agonists could provide a means to boost inflammatory responses to microbial pathogens. It has also been observed that co-infection of mice with Haemophilus influenzae and Streptococcus pneumoniae enhances neutrophil killing of S. pneumoniae through a mechanism that involves NOD1 activation by H. influenzae204. NOD1 agonists have also been shown to enhance the resistance of mice to various bacterial48 and viral infections50,52,205. Therefore, it is possible to boost innate immune responses to microbes using NOD1 agonists. A new study found that individuals who are deficient in myeloid differentiation primary response protein 88 (MYD88), an adaptor protein required for TLR signalling (Fig. 2), are more susceptible to infections by pyogenic bacteria, presumably owing to constitutively weak inflammatory responses206. Could NLR activation provide an MYD88-independent means to boost NF-κB activation in these individuals? Conversely, could antagonism of NLR pathways help reduce excessive inflammation during infection? Many challenges remain before NLR agonists and/or antagonists can be used in a clinical setting to prevent and treat infection, such as determining the optimal dose, method of delivery and timing of administration of the drugs. Despite these challenges, harnessing the powerful stimulatory properties of NLRs has great potential to aid in the development of new strategies to fight infection.
Concluding remarks
Although the field of NLR research is relatively new, there have already been important advances that provide insight into vaccine and drug development (Timeline). For example, our understanding of the role of NOD1 and NOD2 in regulating the polarization of the T cell response to adjuvants presents clues on how to improve vaccines that target stimulation of cellular or humoral immunity. The discovery of the association of NLRP3 with several syndromes has led to therapy for diseases that previously had no effective treatment. The involvement of NLRP3 inflammasome activation in disorders such as ischaemia–reperfusion syndromes130, silicosis131 and Alzheimer's disease16 suggests that anakinra may be an effective therapeutic strategy and is therefore a strong candidate for clinical trials. As mentioned above, NOD1 agonists can prevent infection in animal models and consequently should be considered for certain clinical applications, such as the treatment of microbial infections that do not respond well to antibiotics. There is also interest in developing small-molecule antagonists that inhibit activation of NLRs by directly targeting specific receptor domains, such as the pyrin domain in NLRP3 or the CARD domains of NOD1 and NOD2, as such compounds would be of substantial clinical value. This interest has not yet evolved beyond the theoretical stage and many difficulties exist in developing such compounds. However, naturally occurring compounds such as PSA (from Bacteroides fragilis, which inhibits NF-κB activation) are readily available and could be tested in animal models and perhaps in clinical settings. With the advent of mass spectrometry-based proteome studies, screening large numbers of microbial products for candidates that interact with NLRs is now feasible. Therefore, like many existing drugs, the best drug candidates for targeting NLR activity may come from natural sources such as commensal or pathogenic parasites, yeast, bacteria or viruses that have evolved mechanisms to manipulate NLR pathways. The steadily growing body of knowledge on NLRs will have a crucial impact on our understanding of the mechanisms of action of immune adjuvants, as well as the pathogenesiss of inflammatory disorders and infectious diseases, and will help direct the development of new drugs in the near future.

References
Kimbrell, D. A. & Beutler, B. The evolution and genetics of innate immunity. Nature Rev. Genet. 2, 256–267 (2001).
Imler, J. L. & Hoffmann, J. A. Toll signaling: the TIReless quest for specificity. Nature Immunol. 4, 105–106 (2003).
Kawai, T. & Akira, S. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 17, 338–344 (2005).
Franchi, L. et al. Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8 (2008).
Fritz, J. H., Ferrero, R. L., Philpott, D. J. & Girardin, S. E. Nod-like proteins in immunity, inflammation and disease. Nature Immunol. 7, 1250–1257 (2006).
Werts, C., Girardin, S. E. & Philpott, D. J. TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Differ. 13, 798–815 (2006).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002).
Carneiro, L. A., Magalhaes, J. G., Tattoli, I., Philpott, D. J. & Travassos, L. H. Nod-like proteins in inflammation and disease. J. Pathol. 214, 136–148 (2008).
Girardin, S. E. et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872 (2003).
Inohara, N. et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J. Biol. Chem. 278, 5509–5512 (2003).
Girardin, S. E. et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300, 1584–1587 (2003).
Chamaillard, M. et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nature Immunol. 4, 702–707 (2003).
Petrilli, V., Dostert, C., Muruve, D. A. & Tschopp, J. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 19, 615–622 (2007).
Benko, S., Philpott, D. J. & Girardin, S. E. The microbial and danger signals that activate Nod-like receptors. Cytokine 43, 368–373 (2008).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nature Immunol. 9, 857–865 (2008).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nature Immunol. 9, 847–856 (2008).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nature Immunol. 7, 576–582 (2006).
Miao, E. A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nature Immunol. 7, 569–575 (2006).
Boyden, E. D. & Dietrich, W. F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nature Genet. 38, 240–244 (2006).
Kanneganti, T. D., Lamkanfi, M. & Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Jin, Y. et al. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 356, 1216–1225 (2007).
Jeru, I. et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl Acad. Sci. USA 105, 1614–1619 (2008).
Huzarski, T. et al. The 3020insC allele of NOD2 predisposes to early-onset breast cancer. Breast Cancer Res. Treat. 89, 91–93 (2005).
Jaworowska, E. et al. Association between early-onset breast and laryngeal cancers. Breast Cancer Res. Treat. 97, 215–219 (2006).
Lener, M. R. et al. Prevalence of the NOD2 3020insC mutation in aggregations of breast and lung cancer. Breast Cancer Res. Treat. 95, 141–145 (2006).
Forrest, M. S. et al. Polymorphisms in innate immunity genes and risk of non-Hodgkin lymphoma. Br. J. Haematol. 134, 180–183 (2006).
Debniak, T. et al. NOD2 variants and the risk of malignant melanoma. Eur. J. Cancer Prev. 14, 143–146 (2005).
Holler, E. et al. Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood 104, 889–894 (2004).
Kruger, B. et al. Impact of NOD2/CARD15 haplotypes on the outcome after kidney transplantation. Transpl. Int. 20, 600–607 (2007).
Cox, J. C. & Coulter, A. R. Adjuvants—a classification and review of their modes of action. Vaccine 15, 248–256 (1997).
Pashine, A., Valiante, N. M. & Ulmer, J. B. Targeting the innate immune response with improved vaccine adjuvants. Nature Med. 11, S63–S68 (2005).
Schijns, V. E. Immunological concepts of vaccine adjuvant activity. Curr. Opin. Immunol. 12, 456–463 (2000).
Fritz, J. H. et al. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur. J. Immunol. 35, 2459–2470 (2005).
Uehara, A., Fujimoto, Y., Fukase, K. & Takada, H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 44, 3100–3111 (2007).
Uehara, A. et al. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell Microbiol. 7, 53–61 (2005).
Tada, H., Aiba, S., Shibata, K.-I., Ohteki, T. & Takada, H. Synergistic effect of Nod1 and Nod2 agonists with Toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect. Immun. 73, 7967–7976 (2005).
Migliore-Samour, D. et al. Immunostimulating and adjuvant activities of a low molecular weight lipopeptide. C R. Seances Acad. Sci. D 289, 473–476 (1979) (in French).
Fritz, J. H. et al. Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 26, 445–459 (2007).
Magalhaes, J. G. et al. Nod2-dependent Th2 polarization of antigen-specific immunity. J. Immunol. 181, 7925–7935 (2008).
Kobayashi, K. S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Eisenbarth, S. C., Colegio, O. R., O'Connor, W., Sutterwala, F. S. & Flavell, R. A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126 (2008).
Franchi, L. & Nunez, G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1β secretion but dispensable for adjuvant activity. Eur. J. Immunol. 38, 2085–2089 (2008).
Kool, M. et al. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J. Exp. Med. 205, 869–882 (2008).
Kornbluth, R. S. & Stone, G. W. Immunostimulatory combinations: designing the next generation of vaccine adjuvants. J. Leukoc. Biol. 80, 1084–1102 (2006).
Adam, A., Ellouz, F., Ciorbaru, R., Petit, J. F. & Lederer, E. Peptidoglycan adjuvants: minimal structure required for activity. Z. Immunitatsforsch. Exp. Klin. Immunol. 149, 341–348 (1975).
Kotani, S., Watanabe, Y., Shimono, T., Narita, T. & Kato, K. Immunoadjuvant activities of cell walls, their water-soluble fractions and peptidoglycan subunits, prepared from various gram-positive bacteria, and of synthetic n-acetylmuramyl peptides. Z. Immunitatsforsch. Exp. Klin. Immunol. 149, 302–319 (1975).
Mine, Y. et al. Immunoactive peptides, FK-156 and FK-565. I. Enhancement of host resistance to microbial infection in mice. J. Antibiot. (Tokyo) 36, 1045–1050 (1983).
Mine, Y. et al. Immunoactive peptides, FK-156 and FK-565. III. Enhancement of host defense mechanisms against infection. J. Antibiot. (Tokyo) 36, 1059–1066 (1983).
Sato, Y., Maruyama, S., Kawai, C. & Matsumori, A. Effect of immunostimulant therapy on acute viral myocarditis in an animal model. Am. Heart J. 124, 428–434 (1992).
Sato, Y. & Matsumori, A. Treatment of coxsackievirus B3 myocarditis by immunoactive peptide in an animal model. Clin. Immunol. Immunopathol. 65, 65–69 (1992).
Yokota, Y., Watanabe, Y., Wakai, Y. & Mine, Y. Protective effects of immunoactive peptide, FK565 against systemic and local infections with herpes simplex virus and murine cytomegalovirus and respiratory tract infection with influenza virus in mice. J. Antibiot. (Tokyo) 45, 258–264 (1992).
Inamura, N. et al. Activation of tumoricidal properties in macrophages and inhibition of experimentally-induced murine metastases by a new synthetic acyltripeptide, FK-565. J. Biol. Response Mod. 4, 408–417 (1985).
Schultz, R. M. & Altom, M. G. Macrophage involvement in the antitumor activity of a synthetic acyltripeptide (FK-565) against experimental lung carcinoma metastases. J. Immunopharmacol. 8, 515–528 (1986).
Izumi, S. et al. Antitumor effects of novel immunoactive peptides, FK-156 and its synthetic derivatives. J. Antibiot. (Tokyo) 36, 566–574 (1983).
Adam, A., Ciorbaru, R., Ellouz, F., Petit, J. F. & Lederer, E. Adjuvant activity of monomeric bacterial cell wall peptidoglycans. Biochem. Biophys. Res. Commun. 56, 561–567 (1974).
Masek, K., Seifert, J., Flegel, M., Krojidlo, M. & Kolinsky, J. The immunomodulatory property of a novel synthetic compound adamantylamide dipeptide. Methods Find. Exp. Clin. Pharmacol. 6, 667–669 (1984).
Ishihara, C., Yamamoto, K., Hamada, N. & Azuma, I. Effect of stearoyl-N-acetylmuramyl-L-alanyl-D-isoglutamine on host resistance to Corynebacterium kutscheri infection in cortisone-treated mice. Vaccine 2, 261–264 (1984).
Matsumoto, K. et al. Stimulation of nonspecific resistance to infection induced by muramyl dipeptide analogs substituted in the gamma-carboxyl group and evaluation of Nα-muramyl dipeptide-Nɛ-stearoyllysine. Infect. Immun. 39, 1029–1040 (1983).
Chedid, L. A. et al. Biological activity of a new synthetic muramyl peptide adjuvant devoid of pyrogenicity. Infect. Immun. 35, 417–424 (1982).
Eppstein, D. A., Byars, N. E. & Allison, A. C. New adjuvants for vaccines containing purified protein antigens. Adv. Drug Del. Rev. 233–253 (1990).
Balitsky, K. P., Umansky, V. Y., Tarakhovsky, A. M., Andronova, T. M. & Ivanov, V. T. Glucosaminylmuramyl dipeptide-induced changes in murine macrophage metabolism. Int. J. Immunopharmacol. 11, 429–434 (1989).
Pimenov, A. A. et al. Activation of cellular immunity in mice under normal conditions and in tumor growth during treatment with glucosaminyl muramyl dipeptide. Vopr. Med. Khim. 36, 58–60 (1990) (in Russian).
Ellouz, F., Adam, A., Ciorbaru, R. & Lederer, E. Minimal structural requirements for adjuvant activity of bacterial peptidoglycan derivatives. Biochem. Biophys. Res. Commun. 59, 1317–1325 (1974).
Kotani, S., Watanabe, Y., Kinoshita, F., Shimono, T. & Morisaki, I. Immunoadjuvant activities of synthetic N-acetyl-muramyl-peptides or -amino acids. Biken J. 18, 105–111 (1975).
Audibert, F., Chedid, L., Lefrancier, P. & Choay, J. Distinctive adjuvanticity of synthetic analogs of mycobacterial water-soluble components. Cell. Immunol. 21, 243–249 (1976).
Parant, M. A. et al. Immunostimulant activities of a lipophilic muramyl dipeptide derivative and of desmuramyl peptidolipid analogs. Infect. Immun. 27, 826–831 (1980).
Guo, L. H., Guo, K. T., Wendel, H. P. & Schluesener, H. J. Combinations of TLR and NOD2 ligands stimulate rat microglial P2X4R expression. Biochem. Biophys. Res. Commun. 349, 1156–1162 (2006).
Kramer, M., Netea, M. G., de Jong, D. J., Kullberg, B. J. & Adema, G. J. Impaired dendritic cell function in Crohn's disease patients with NOD2 3020insC mutation. J. Leukoc. Biol. 79, 860–866 (2006).
Heinzelmann, M., Polk, H. C. Jr, Chernobelsky, A., Stites, T. P. & Gordon, L. E. Endotoxin and muramyl dipeptide modulate surface receptor expression on human mononuclear cells. Immunopharmacology 48, 117–128 (2000).
Darcissac, E. C., Bahr, G. M., Parant, M. A., Chedid, L. A. & Riveau, G. J. Selective induction of CD11a, b, c/CD18 and CD54 expression at the cell surface of human leukocytes by muramyl peptides. Cell. Immunol. 169, 294–301 (1996).
Leclerc, C. & Vogel, F. R. Synthetic immunomodulators and synthetic vaccines. Crit. Rev. Ther. Drug Carrier Syst. 2, 353–406 (1986).
Traub, S., von Aulock, S., Hartung, T. & Hermann, C. MDP and other muropeptides—direct and synergistic effects on the immune system. J. Endotoxin Res. 12, 69–85 (2006).
Saiki, I. & Fidler, I. J. Synergistic activation by recombinant mouse interferon-γ and muramyl dipeptide of tumoricidal properties in mouse macrophages. J. Immunol. 135, 684–688 (1985).
Saiki, I. et al. Synergism between human recombinant gamma-interferon and muramyl dipeptide encapsulated in liposomes for activation of antitumor properties in human blood monocytes. Cancer Res. 45, 6188–6193 (1985).
MacEwen, E. G. et al. Adjuvant therapy for melanoma in dogs: results of randomized clinical trials using surgery, liposome-encapsulated muramyl tripeptide, and granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 5, 4249–4258 (1999).
Netea, M. G. et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1β and IL-6 production through a caspase 1-dependent mechanism. Proc. Natl Acad. Sci. USA 102, 16309–16314 (2005).
Krivorutchenko, Y. L. et al. Study of the adjuvant activity of new MDP derivatives and purified saponins and their influence on HIV-1 replication in vitro. Vaccine 15, 1479–1486 (1997).
Azuma, I. et al. Stimulation of host-defense mechanism with synthetic adjuvants and recombinant cytokines against viral infection in mice. Adv. Exp. Med. Biol. 319, 253–263 (1992).
Ikeda, S., Negishi, T. & Nishimura, C. Enhancement of non-specific resistance to viral infection by muramyldipeptide and its analogs. Antiviral Res. 5, 207–215 (1985).
Ishihara, C. et al. Effect of Nα-acetylmuramyl-L-alanyl-D-isoglutaminyl-Nɛ-stearoyl-L-lysine on resistance to herpes simplex virus type-1 infection in cyclophosphamide-treated mice. Vaccine 7, 309–313 (1989).
Masihi, K. N., Brehmer, W., Lange, W., Ribi, E. & Schwartzman, S. Protective effect of muramyl dipeptide analogs in combination with trehalose dimycolate against aerogenic influenza virus and Mycobacterium tuberculosis infections in mice. J. Biol. Response Mod. 3, 663–671 (1984).
Masihi, K. N., Lange, W., Rohde-Schulz, B. & Chedid, L. Muramyl dipeptide inhibits replication of human immunodeficiency virus in vitro. AIDS Res. Hum. Retroviruses 6, 393–399 (1990).
Bertot, G. M., Becker, P. D., Guzman, C. A. & Grinstein, S. Intranasal vaccination with recombinant P6 protein and adamantylamide dipeptide as mucosal adjuvant confers efficient protection against otitis media and lung infection by nontypeable Haemophilus influenzae. J. Infect. Dis. 189, 1304–1312 (2004).
Tatara, O., Nakahama, C. & Niki, Y. Synergistic effects of romurtide and cefmenoxime against experimental Klebsiella pneumonia in mice. Antimicrob. Agents Chemother. 36, 167–171 (1992).
Khanna, R., Ahmad, S., Khan, H. M., Kumar, H. & Mahdi, A. A. Vaccination of rhesus monkeys against Plasmodium knowlesi with aqueous suspension of MDP as an adjuvant. Indian J. Malariol. 28, 99–104 (1991).
Fidler, I. J., Sone, S., Fogler, W. E. & Barnes, Z. L. Eradication of spontaneous metastases and activation of alveolar macrophages by intravenous injection of liposomes containing muramyl dipeptide. Proc. Natl Acad. Sci. USA 78, 1680–1684 (1981).
Saiki, I., Tanio, Y., Yamamoto, K. I., Yamamura, Y. & Azuma, I. Effect of quinonyl-N-acetyl muramyl dipeptide on immune responses in tumor-bearing mice. Infect. Immun. 39, 137–141 (1983).
MacEwen, E. G. et al. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J. Natl Cancer Inst. 81, 935–938 (1989).
Krueger, J. M. et al. Muramyl peptides. Variation of somnogenic activity with structure. J. Exp. Med. 159, 68–76 (1984).
Darcissac, E. C. et al. The synthetic immunomodulator murabutide controls human immunodeficiency virus type 1 replication at multiple levels in macrophages and dendritic cells. J. Virol. 74, 7794–7802 (2000).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Martinon, F., Agostini, L., Meylan, E. & Tschopp, J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr. Biol. 14, 1929–1934 (2004).
Kanneganti, T. D. et al. Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 (2006).
Kanneganti, T. D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006). References 92 and 96 report the important discoveries that NALP3 (also known as NLRP3) is a sensor of the danger signals ATP–K+ efflux and uric acid crystals, respectively.
Li, H., Willingham, S. B., Ting, J. P. & Re, F. Cutting edge: inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J. Immunol. 181, 17–21 (2008).
Surprenant, A., Rassendren, F., Kawashima, E., North, R. A. & Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738 (1996).
Sutterwala, F. S. et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 (2006).
Granstein, R. D. et al. Augmentation of cutaneous immune responses by ATPγS: purinergic agonists define a novel class of immunologic adjuvants. J. Immunol. 174, 7725–7731 (2005).
Pan, Q. et al. MDP-induced interleukin-1β processing requires Nod2 and CIAS1/NALP3. J. Leukoc. Biol. 82, 177–183 (2007).
Hsu, L. C. et al. A NOD2–NALP1 complex mediates caspase-1-dependent IL-1β secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl Acad. Sci. USA 105, 7803–7808 (2008).
Marina-Garcia, N. et al. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J. Immunol. 180, 4050–4057 (2008).
Rosenzweig, H. L. et al. Activation of NOD2 in vivo induces IL-1β production in the eye via caspase-1 but results in ocular inflammation independently of IL-1 signaling. J. Leukoc. Biol. 84, 529–536 (2008).
Parant, M. A. et al. Selective modulation of lipopolysaccharide-induced death and cytokine production by various muramyl peptides. Infect. Immun. 63, 110–115 (1995).
Kool, M. et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 181, 3755–3759 (2008). References 42, 43, 97 and 106 identify NLRP3 as a host sensor of aluminium hydroxide, a well-known adjuvant, illustrating how NLRs influence adaptive immune responses.
Brewer, J. M. (How) do aluminium adjuvants work? Immunol. Lett. 102, 10–15 (2006).
Sokolovska, A., Hem, S. L. & HogenEsch, H. Activation of dendritic cells and induction of CD4+ T cell differentiation by aluminium-containing adjuvants. Vaccine 25, 4575–4585 (2007).
Schmitz, N., Kurrer, M. & Kopf, M. The IL-1 receptor 1 is critical for Th2 cell type airway immune responses in a mild but not in a more severe asthma model. Eur. J. Immunol. 33, 991–1000 (2003).
Pollock, K. G., Conacher, M., Wei, X. Q., Alexander, J. & Brewer, J. M. Interleukin-18 plays a role in both the alum-induced T helper 2 response and the T helper 1 response induced by alum-adsorbed interleukin-12. Immunology 108, 137–143 (2003).
Hoffman, H. M., Mueller, J. L., Broide, D. H., Wanderer, A. A. & Kolodner, R. D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nature Genet. 29, 301–305 (2001).
Feldmann, J. et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 71, 198–203 (2002).
Aksentijevich, I. et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 46, 3340–3348 (2002).
Agostini, L. et al. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Dowds, T. A., Masumoto, J., Zhu, L., Inohara, N. & Nunez, G. Cryopyrin-induced interleukin 1β secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 279, 21924–21928 (2004).
Hentgen, V. et al. Intrafamilial variable phenotypic expression of a CIAS1 mutation: from Muckle–Wells to chronic infantile neurological cutaneous and articular syndrome. J. Rheumatol 32, 747–751 (2005).
Dode, C. et al. New mutations of CIAS1 that are responsible for Muckle–Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am. J. Hum. Genet. 70, 1498–1506 (2002).
Hawkins, P. N., Lachmann, H. J. & McDermott, M. F. Interleukin-1-receptor antagonist in the Muckle–Wells syndrome. N. Engl. J. Med. 348, 2583–2584 (2003).
Yamazaki, T. et al. Anakinra improves sensory deafness in a Japanese patient with Muckle–Wells syndrome, possibly by inhibiting the cryopyrin inflammasome. Arthritis Rheum. 58, 864–868 (2008).
Mirault, T. et al. Recovery from deafness in a patient with Muckle–Wells syndrome treated with anakinra. Arthritis Rheum. 54, 1697–1700 (2006).
Rynne, M., Maclean, C., Bybee, A., McDermott, M. F. & Emery, P. Hearing improvement in a patient with variant Muckle–Wells syndrome in response to interleukin 1 receptor antagonism. Ann. Rheum. Dis. 65, 533–534 (2006).
Alexander, T. et al. Successful treatment of acute visual loss in Muckle–Wells syndrome with interleukin 1 receptor antagonist. Ann. Rheum. Dis. 64, 1245–1246 (2005).
Hoffman, H. M. et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 364, 1779–1785 (2004).
Granel, B., Serratrice, J., Disdier, P. & Weiller, P. J. Dramatic improvement with anakinra in a case of chronic infantile neurological cutaneous and articular (CINCA) syndrome. Rheumatology (Oxford) 44, 689–690 (2005).
Nuki, G. Colchicine: its mechanism of action and efficacy in crystal-induced inflammation. Curr. Rheumatol Rep. 10, 218–227 (2008).
McGonagle, D. et al. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann. Rheum. Dis. 66, 1683–1684 (2007).
So, A., De Smedt, T., Revaz, S. & Tschopp, J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res. Ther. 9, R28 (2007).
Hoffman, H. M. et al. Efficacy and safety of rilonacept (interleukin-1 trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 58, 2443–2452 (2008).
Goldbach-Mansky, R. et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 58, 2432–2442 (2008).
Wanderer, A. A. Ischemic-reperfusion syndromes: biochemical and immunologic rationale for IL-1 targeted therapy. Clin. Immunol. 128, 127–132 (2008).
Cassel, S. L. et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc. Natl Acad. Sci. USA 105, 9035–9040 (2008).
Duncan, J. A. et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl Acad. Sci. USA 104, 8041–8046 (2007).
Manji, G. A. et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-κB. J. Biol. Chem. 277, 11570–11575 (2002).
Richards, N. et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 276, 39320–39329 (2001).
Srinivasula, S. M. et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 277, 21119–21122 (2002).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606 (2001).
Hugot, J. P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411, 599–603 (2001). References 136 and 137 reported the identification of NOD2 as the first susceptibility gene for Crohn's disease, helping to establish a link between NLRs and human inflammatory disorders.
Duerr, R. H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006).
Hampe, J. et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nature Genet. 39, 207–211 (2007).
Rioux, J. D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genet. 39, 596–604 (2007).
Ogura, Y. et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 276, 4812–4818 (2001).
Gutierrez, O. et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-κB activation. J. Biol. Chem. 277, 41701–41705 (2002).
Hisamatsu, T. et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 124, 993–1000 (2003).
Rosenstiel, P. et al. TNF-α and IFN-γ regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 124, 1001–1009 (2003).
Berrebi, D. et al. Card15 gene overexpression in mononuclear and epithelial cells of the inflamed Crohn's disease colon. Gut 52, 840–846 (2003).
Chamaillard, M. et al. Gene–environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc. Natl Acad. Sci. USA 100, 3455–3460 (2003).
Bonen, D. K. et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 124, 140–146 (2003).
Li, J. et al. Regulation of IL-8 and IL-1β expression in Crohn's disease associated NOD2/CARD15 mutations. Hum. Mol. Genet. 13, 1715–1725 (2004).
van Heel, D. A. et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut 54, 1553–1557 (2005).
Netea, M. G. et al. NOD2 3020insC mutation and the pathogenesis of Crohn's disease: impaired IL-1β production points to a loss-of-function phenotype. Neth. J. Med. 63, 305–308 (2005).
Netea, M. G. et al. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn's disease. Eur. J. Immunol. 34, 2052–2059 (2004).
Netea, M. G. et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J. Immunol. 174, 6518–6523 (2005).
van Heel, D. A. et al. Detection of muramyl dipeptide-sensing pathway defects in patients with Crohn's disease. Inflamm. Bowel Dis. 12, 598–605 (2006).
Mascheretti, S. et al. Response to infliximab treatment in Crohn's disease is not associated with mutations in the CARD15 (NOD2) gene: an analysis in 534 patients from two multicenter, prospective GCP-level trials. Pharmacogenetics 12, 509–515 (2002).
Vermeire, S. et al. NOD2/CARD15 does not influence response to infliximab in Crohn's disease. Gastroenterology 123, 106–111 (2002).
Kullberg, B. J. et al. Crohn's disease patients homozygous for the 3020insC NOD2 mutation have a defective NOD2/TLR4 cross-tolerance to intestinal stimuli. Immunology 123, 600–605 (2008).
Hedl, M., Li, J., Cho, J. H. & Abraham, C. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc. Natl Acad. Sci. USA 104, 19440–19445 (2007).
Maeda, S. et al. Nod2 mutation in Crohn's disease potentiates NF-κB activity and IL-1β processing. Science 307, 734–738 (2005).
Braat, H. et al. Consequence of functional Nod2 and Tlr4 mutations on gene transcription in Crohn's disease patients. J. Mol. Med. 83, 601–609 (2005).
Dionne, S., D'Agata, I. D., Hiscott, J., Vanounou, T. & Seidman, E. G. Colonic explant production of IL-1 and its receptor antagonist is imbalanced in inflammatory bowel disease (IBD). Clin. Exp. Immunol. 112, 435–442 (1998).
Andus, T. et al. Imbalance of the interleukin 1 system in colonic mucosa—association with intestinal inflammation and interleukin 1 receptor antagonist [corrected] genotype 2. Gut 41, 651–657 (1997).
Nishiyama, T., Mitsuyama, K., Toyonaga, A., Sasaki, E. & Tanikawa, K. Colonic mucosal interleukin 1 receptor antagonist in inflammatory bowel disease. Digestion 55, 368–373 (1994).
Villani, A. C. et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nature Genet. 41, 71–76 (2009).
Carter, J. D., Valeriano, J. & Vasey, F. B. Crohn disease worsened by anakinra administration. J. Clin. Rheumatol. 9, 276–277 (2003).
Franke, A. et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nature Genet. 40, 1319–1323 (2008).
Braat, H., Peppelenbosch, M. P. & Hommes, D. W. Interleukin-10-based therapy for inflammatory bowel disease. Expert Opin. Biol. Ther. 3, 725–731 (2003).
Lala, S. et al. Crohn's disease and the NOD2 gene: a role for paneth cells. Gastroenterology 125, 47–57 (2003).
Ogura, Y. et al. Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut 52, 1591–1597 (2003).
Aldhous, M. C., Nimmo, E. R. & Satsangi, J. NOD2/CARD15 and the Paneth cell: another piece in the genetic jigsaw of inflammatory bowel disease. Gut 52, 1533–1535 (2003).
Wehkamp, J. et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut 53, 1658–1664 (2004).
Simms, L. A. et al. Reduced α-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn's disease. Gut 57, 903–910 (2008).
Yamazaki, K., Takazoe, M., Tanaka, T., Kazumori, T. & Nakamura, Y. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn's disease. J. Hum. Genet. 47, 469–472 (2002).
Inoue, N. et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology 123, 86–91 (2002).
Leong, R. W. et al. NOD2/CARD15 gene polymorphisms and Crohn's disease in the Chinese population. Aliment Pharmacol. Ther. 17, 1465–1470 (2003).
Guo, Q. S., Xia, B., Jiang, Y., Qu, Y. & Li, J. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J. Gastroenterol. 10, 1069–1071 (2004).
Mazmanian, S. K., Round, J. L. & Kasper, D. L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008).
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O. & Kasper, D. L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005).
Wang, Q. et al. A bacterial carbohydrate links innate and adaptive responses through Toll-like receptor 2. J. Exp. Med. 203, 2853–2863 (2006).
Weidinger, S. et al. Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J. Allergy Clin. Immunol. 116, 177–184 (2005).
Hysi, P. et al. NOD1 variation, immunoglobulin E and asthma. Hum. Mol. Genet. 14, 935–941 (2005).
van Heel, D. A. et al. Synergistic enhancement of Toll-like receptor responses by NOD1 activation. Eur. J. Immunol. 35, 2471–2476 (2005).
Inohara, N. et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J. Biol. Chem. 274, 14560–14567 (1999).
Girardin, S. E. et al. Identification of the critical residues involved in peptidoglycan detection by Nod1. J. Biol. Chem. 280, 38648–38656 (2005).
Miceli-Richard, C. et al. CARD15 mutations in Blau syndrome. Nature Genet. 29, 19–20 (2001).
van Duist, M. M. et al. A new CARD15 mutation in Blau syndrome. Eur. J. Hum. Genet. 13, 742–747 (2005).
Kanazawa, N. et al. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J. Invest. Dermatol. 122, 851–852 (2004).
Kanazawa, N. et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-κB activation: common genetic etiology with Blau syndrome. Blood 105, 1195–1197 (2005).
Rose, C. D. et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J. Rheumatol. 32, 373–375 (2005).
Milman, N. et al. Favourable effect of TNF-α inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS 114, 912–919 (2006).
Deveci, F. et al. Evaluation of the anti-inflammatory effect of infliximab in a mouse model of acute asthma. Respirology 13, 488–497 (2008).
Hofner, P. et al. Genetic polymorphisms of NOD1 and IL-8, but not polymorphisms of TLR4 genes, are associated with Helicobacter pylori-induced duodenal ulcer and gastritis. Helicobacter 12, 124–131 (2007).
Rosenstiel, P. et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell. Microbiol. 8, 1188–1198 (2006).
Austin, C. M., Ma, X. & Graviss, E. A. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J. Infect. Dis. 197, 1713–1716 (2008).
Moller, M. et al. Host susceptibility to tuberculosis: CARD15 polymorphisms in a South African population. Mol. Cell Probes 21, 148–151 (2007).
Viala, J. et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunol. 5, 1166–1174 (2004).
Suzuki, T. et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111 (2007).
Amer, A. et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223 (2006).
Sutterwala, F. S. et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245 (2007).
Franchi, L. et al. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur. J. Immunol. 37, 3030–3039 (2007).
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A. & Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564 (2008).
Tattoli, I. et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 9, 293–300 (2008).
Muruve, D. A. et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008).
Moore, C. B. et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451, 573–577 (2008).
Lysenko, E. S. et al. Nod1 signaling overcomes resistance of S. pneumoniae to opsonophagocytic killing. PLoS Pathog. 3, e118 (2007).
Yokota, Y., Wakai, Y., Watanabe, Y. & Mine, Y. Inhibitory effect of FK-565 alone and in combination with zidovudine on retroviral infection by Friend leukemia virus in mice. J. Antibiot. (Tokyo) 41, 1479–1487 (1988).
von Bernuth, H. et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science 321, 691–696 (2008).
Freund, J., Lipton, M. M. & Morrison, L. R. Demyelination in the guinea pig in chronic allergic encephalomyelitis produced by injecting guinea pig brain in oil emulsion containing a variant of Mycobacterium butyricum. Arch. Pathol. (Chic) 50, 108–121 (1950).
Freund, J. The effect of paraffin oil and mycobacteria on antibody formation and sensitization; a review. Am. J. Clin. Pathol. 21, 645–656 (1951).
White, R. G., Bernstock, L., Johns, R. G. & Lederer, E. The influence of components of M. tuberculosis and other Mycobacteria upon antibody production to ovalbumin. Immunology 1, 54–66 (1958).
Gupta, R. K. et al. Adjuvants—a balance between toxicity and adjuvanticity. Vaccine 11, 293–306 (1993).
Tal, C. & Laufer, A. Amyloidosis in mice following injections with Freud's adjuvant, its components separately and homologous liver-adjuvant mixture. Br. J. Exp. Pathol. 41, 605–609 (1960).
Steblay, R. W. Glomerulonephritis induced in sheep by injections of heterologous glomerular basement membrane and Freund's complete adjuvant. J. Exp. Med. 116, 253–272 (1962).
Lederer, E., Adam, A., Ciorbaru, R., Petit, J. F. & Wietzerbin, J. Cell walls of Mycobacteria and related organisms; chemistry and immunostimulant properties. Mol. Cell Biochem. 7, 87–104 (1975).
Glenny, A. T., Pope, C. G., Waddington, H. & Wallace, U. The antigenic value of toxoid precipitation by potassium alum. Path. Bact. 29, 31 (1926).
Iyer, S., HogenEsch, H. & Hem, S. L. Relationship between the degree of antigen adsorption to aluminum hydroxide adjuvant in interstitial fluid and antibody production. Vaccine 21, 1219–1223 (2003).
Romero Méndez, I. Z., Shi, Y., HogenEsch, H. & Hem, S. L. Potentiation of the immune response to non-adsorbed antigens by aluminum-containing adjuvants. Vaccine 25, 825–833 (2007).
Hansen, B., Sokolovska, A., HogenEsch, H. & Hem, S. L. Relationship between the strength of antigen adsorption to an aluminum-containing adjuvant and the immune response. Vaccine 25, 6618–6624 (2007).
Nemazee, D., Gavin, A., Hoebe, K. & Beutler, B. Immunology: Toll-like receptors and antibody responses. Nature 441, E4; discussion E4 (2006).
Ren, T., Zamboni, D. S., Roy., C. R., Dietrich, W. F. & Vance, R. E. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PLoS Pathog. 2, e18 (2006).
Molofsky, A. B. et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J. Exp. Med. 203, 1093–1104 (2006).
Petrilli, V. et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 (2007).
Steimle, V., Otten, L. A., Zufferey, M. & Mach, B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75, 135–146 (1993).
Rasmussen, H. B., Kelly, M. A. & Clausen, J. Genetic susceptibility to multiple sclerosis: detection of polymorphic nucleotides and an intron in the 3' untranslated region of the major histocompatibility complex class II transactivator gene. Hum. Immunol. 62, 371–377 (2001).
Koizumi, K. et al. Single nucleotide polymorphisms in the gene encoding the major histocompatibility complex class II transactivator (CIITA) in systemic lupus erythematosus. Ann. Rheum. Dis. 64, 947–950 (2005).
Skinningsrud, B. et al. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 93, 3310–3317 (2008).
Macaluso, F. et al. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 16, 692–698 (2007).
Alimohammadi, M. et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N. Engl. J. Med. 358, 1018–1028 (2008).
McGovern, D. P. et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 14, 1245–1250 (2005).
Repique, C. J. et al. Susceptibility of mice deficient in the MHC class II transactivator to infection with Mycobacterium tuberculosis. Scand. J. Immunol. 58, 15–22 (2003).
Zhang, X., Hong, X., Deng, G. & Bai, X. Single nucleotide polymorphisms and functional analysis of class II transactivator (CIITA) promoter IV in persistent HBV infection. J. Clin. Virol. 40, 197–201 (2007).
He, Y. et al. Not polymorphism but methylation of class II transactivator gene promoter IV associated with persistent HBV infection. J. Clin. Virol. 37, 282–286 (2006).
Vinzing, M. et al. NAIP and Ipaf control Legionella pneumophila replication in human cells. J. Immunol. 180, 6808–6815 (2008).
Lamkanfi, M. et al. The Nod-like receptor family member Naip5/Birc1e restricts Legionella pneumophila growth independently of caspase-1 activation. J. Immunol. 178, 8022–8027 (2007).
Coers, J., Vance, R. E., Fontana, M. F. & Dietrich, W. F. Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol 9, 2344–2357 (2007).
Pedra, J. H. et al. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-γ axis during Anaplasma phagocytophilum infection. J. Immunol. 179, 4783–4791 (2007).
Wright, E. K. et al. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr. Biol. 13, 27–36 (2003).
Diez, E. et al. Birc1e is the gene within the Lgn1 locus associated with resistance to Legionella pneumophila. Nature Genet. 33, 55–60 (2003).
Kim, J. G., Lee, S. J. & Kagnoff, M. F. Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by Toll-like receptors. Infect. Immun. 72, 1487–1495 (2004).
Travassos, L. H. et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J. Biol. Chem. 280, 36714–36718 (2005).
Welter-Stahl, L. et al. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell. Microbiol. 8, 1047–1057 (2006).
Opitz, B. et al. Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ. Res. 96, 319–326 (2005).
Zilbauer, M. et al. A major role for intestinal epithelial nucleotide oligomerization domain 1 (NOD1) in eliciting host bactericidal immune responses to Campylobacter jejuni. Cell. Microbiol. 9, 2404–2416 (2007).
Kim, Y. G. et al. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28, 246–257 (2008).
Girardin, S. E. et al. CARD4/Nod1 mediates NF-κB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2, 736–742 (2001).
Opitz, B. et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J. Biol. Chem. 279, 36426–36432 (2004).
Ferwerda, G. et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog. 1, 279–285 (2005).
Gandotra, S., Jang, S., Murray, P. J., Salgame, P. & Ehrt, S. Nucleotide-binding oligomerization domain protein 2-deficient mice control infection with Mycobacterium tuberculosis. Infect. Immun. 75, 5127–5134 (2007).
Ferwerda, G. et al. Mycobacterium paratuberculosis is recognized by Toll-like receptors and NOD2. J. Leukoc. Biol. 82, 1011–1018 (2007).
Meinzer, U. et al. Nod2 mediates susceptibility to Yersinia pseudotuberculosis in mice. PLoS ONE 3, e2769 (2008).
Peeters, H. et al. CARD15 variants determine a disturbed early response of monocytes to adherent-invasive Escherichia coli strain LF82 in Crohn's disease. Int. J. Immunogenet. 34, 181–191 (2007).
Acknowledgements
J.G.M. received funding from Fundação para a Ciência e Tecnologia (Portugal) and the Fondation Bettencourt Schueller. The Girardin laboratory is funded by the Canadian Institutes of Health Research (MOP81360), the Crohn's and Colitis Foundation of Canada and the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Corresponding author
Related links
Related links
DATABASES
OMIM
FURTHER INFORMATION
Glossary
- Nuclear factor-κβ
-
A transcription factor activated by NLR or TLR signalling that mediates expression of cytokines and chemokines.
- Inflammasome
-
A multi-protein complex that processes pro-caspase 1 into mature caspase 1.
- Crohn's disease
-
A type of inflammatory bowel disease characterized by granulomatous inflammation in any region of the gastrointestinal tract (although most frequently occurring in the terminal ileum).
- Atopic disorders
-
Allergic hypersensitivity that can manifest as eczema, rhinitis, asthma or conjunctivitis, in which the affected organ does not come into direct contact with the allergen.
- Muramyl dipeptide
-
(MDP). A component of peptidoglycan (N-acetylmuramyl-L-alanyl-D-isoglutamine) that is specifically detected by NOD2. Synthetic MDP is frequently used in experiments as a means to directly stimulate NOD2.
- Vitiligo
-
A skin disorder that involves dysfunction of melanocytes, resulting in loss of pigmentation.
- Hereditary periodic fever syndromes
-
Rare heritable disorders that include Muckle–Wells syndrome, familial cold urticaria, and chronic infantile neurological cutaneous and articular syndrome. They are characterized by short and periodically occurring attacks of fever and severe localized inflammation that are not caused by infection.
- FK156 and FK565
-
Desmuramylpeptide (DMP) structures from peptidoglycan recognized by NOD1. FK156 (D-lactyl-L-alanyl-γ-D-glutaml-L-meso-diaminopimelyl-L-glycine) chemically mimics the structure that was originally purified from fermentation broths of Streptomyces strains. FK565 (heptanoyl-γ-D-glutamyl-L-meso-diaminopimelyl-D-alanine) is one of the synthesized derivatives of FK156. Both FK156 and FK565 are synthetically produced and used to stimulate NOD1 in mice.
- Murabutide
-
A muramyl dipeptide derivative (N-acetylmuramyl-L-alanyl-D-isoglutamine-α-n-butyl ester) that is not pyrogenic but maintains its immunostimulatory properties.
- β-defensins
-
Small antimicrobial cationic peptides that are produced primarily by epithelial cells to help prevent bacterial colonization of mucosal surfaces.
- Muropeptides
-
Molecules derived from peptidoglycan that contain an N-acetyl-muramic acid sugar residue linked to short peptides, such as muramyl dipeptide and murabutide, but not FK156 or FK565.
- Apoptosis-associated speck-like protein containing a CARD
-
A protein containing a carboxyterminal caspase-activating and recruitment domain (CARD) and an aminoterminal pyrin domain that is required for the formation of inflammasomes with various nucleotide-binding and oligomerization domain (NOD)-like receptor (NLR) family members including NLRP1, NLRP3 and NLRC4.
- Muckle–Wells syndrome
-
A type of periodic fever syndrome that causes sensorineural deafness and recurrent hives, and which can lead to amyloidosis.
- Familial cold urticaria
-
A type of hereditary periodic fever syndrome in which symptoms — such as a painful rash, fever, chills, joint pains and red eyes — develop 1–4 hours after a cold exposure.
- Chronic infantile neurological cutaneous and articular syndrome
-
A type of periodic fever syndrome (also known as neonatal-onset multisystem inflammatory disease) characterized by uncontrolled inflammation beginning during infancy that affects multiple parts of the body: rashes, arthritis and chronic meningitis that ultimately leads to neurological damage.
- Anakinra
-
A recombinant, non-glycosylated version of human interleukin 1 (IL-1) receptor antagonist that blocks the biological activity of IL-1β and was originally approved for treating rheumatoid arthritis.
- Rilonacept
-
A dimeric fusion protein consisting of the ligand-binding domains of human interleukin 1 (IL-1) receptor and the IL-1 receptor accessory protein attached to the human Fc chain. It acts as an IL-1 trap by binding to IL-1 and preventing interaction with its receptors.
- Ischaemia–reperfusion syndrome
-
Inflammation associated with different forms of ischaemia resulting from injury, stroke, surgery or other events that result in loss of blood flow to organs. Return of blood flow to affected organs is often followed by inflammation that is associated with elevated levels of interleukin 1β.
- Silicosis
-
A chronic inflammation of the lungs caused by exposure to silica dust.
- ATG16L1
-
A gene involved in the process of autophagy, whereby cellular materials are recycled, that has been implicated in modulating bacterial infections.
- Infliximab
-
A chimeric monoclonal antibody — containing a mouse derived Fab for specificity and a human Fc chain to minimize immunogenicity — that binds to tumour necrosis factor and neutralizes its biological activity.
- Ulcerative colitis
-
An inflammatory bowel disease in which chronic inflammation of the colon leads to destruction of the epithelium and formation of ulcers.
- Blau syndrome
-
A rare autosomal dominant disorder characterized by granulomatous arthritis, iritis, cranial neuropathies and skin rash.
- Early-onset sarcoidosis
-
A juvenile-onset systemic granulomatosis similar to Blau syndrome that affects mainly the skin, joints and eyes.
- JUN N-terminal kinase
-
A mitogen-activated protein kinase that phosphorylates JUN in response to various stress stimuli including cytokines, and is involved in T cell activation and apoptosis.
- Myeloid differentiation primary response protein 88
-
An adaptor protein for most Toll-like receptors that is required to activate gene transcription that is dependent on nuclear factor-κB.
Rights and permissions
About this article
Cite this article
Geddes, K., Magalhães, J. & Girardin, S. Unleashing the therapeutic potential of NOD-like receptors. Nat Rev Drug Discov 8, 465–479 (2009). https://doi.org/10.1038/nrd2783
Issue Date:
DOI: https://doi.org/10.1038/nrd2783
This article is cited by
-
A novel necroptosis related gene signature and regulatory network for overall survival prediction in lung adenocarcinoma
Scientific Reports (2023)
-
Nod1-dependent NF-kB activation initiates hematopoietic stem cell specification in response to small Rho GTPases
Nature Communications (2023)
-
Vaccine adjuvants: mechanisms and platforms
Signal Transduction and Targeted Therapy (2023)
-
Pyrimidine synthesis inhibition enhances cutaneous defenses against antibiotic resistant bacteria through activation of NOD2 signaling
Scientific Reports (2018)
-
Tumor Necrosis Factor-α in Heart Failure: an Updated Review
Current Cardiology Reports (2018)
