
Abstract
Most invasive bacterial infections are caused by species that more commonly colonize the human host with minimal symptoms. Although phenotypic or genetic correlates underlying a bacterium's shift to enhanced virulence have been studied, the in vivo selection pressures governing such shifts are poorly understood. The globally disseminated M1T1 clone of group A Streptococcus (GAS) is linked with the rare but life-threatening syndromes of necrotizing fasciitis and toxic shock syndrome1. Mutations in the GAS control of virulence regulatory sensor kinase (covRS) operon are associated with severe invasive disease, abolishing expression of a broad-spectrum cysteine protease (SpeB)2,3 and allowing the recruitment and activation of host plasminogen on the bacterial surface4. Here we describe how bacteriophage-encoded GAS DNase (Sda1), which facilitates the pathogen's escape from neutrophil extracellular traps5,6, serves as a selective force for covRS mutation. The results provide a paradigm whereby natural selection exerted by the innate immune system generates hypervirulent bacterial variants with increased risk of systemic dissemination.
Similar content being viewed by others


Main
GAS is estimated to cause ∼700 million cases of self-limited throat or skin infection each year worldwide7. Invasive GAS disease occurs in approximately 1/1,000 cases, with associated mortality of 25%7. Epidemic invasive disease is associated with the emergence of the globally disseminated GAS M1T1 clone1,8, which is distinguished from related strains by acquisition of prophages encoding virulence determinants such as superantigen SpeA and DNase Sda1 (refs. 9,10). In the M1T1 GAS clone, the transition from local to systemic infection can be linked to mutations in the two-component covRS regulator. The effect of these mutations is a distinct shift in the transcriptional profile of invasive GAS isolates compared with that of mucosal (throat) isolates3. The covRS mutation and changes in gene expression are recapitulated upon subcutaneous challenge of mice and analysis of GAS disseminating to the spleen in comparison with those in the original inoculum3. Prominent changes in the transcriptional profile of invasive GAS isolates include a strong upregulation of the DNase gene sda1 and a marked decrease in expression of the gene encoding the cysteine protease SpeB (ref. 3). Sda1 is a virulence factor that protects GAS against neutrophil killing by degrading the DNA framework of neutrophil extracellular traps (NETs)5,6. Abolishment of SpeB expression allows accumulation and activation of the broad-spectrum host protease plasmin on the GAS bacterial surface4. A clinical correlation between GAS invasive disease severity and diminished SpeB expression has been established2.
To elucidate the selection pressure for the rapid loss of SpeB expression in vivo, we compared the human M1T1 GAS isolate 5448 and its isogenic animal-passaged SpeB-negative variant 5448AP (ref. 11). DNA sequence analysis showed that 5448AP contained a single adenine base insertion at position 877 of the covS gene (Fig. 1a) and lacks SpeB production (Fig. 1b). Although it was equivalent to wild-type 5448 in expression of plasminogen receptors α-enolase12 and GAPDH13, 5448AP showed higher levels of the fibrinogen-binding M1 protein14,15 and streptokinase (Fig. 1c). Although washed 5448 and 5448AP cells bound identical amounts of human plasminogen (Fig. 1d), 5448AP accumulated significantly greater surface plasmin activity after growth in human plasma (P < 0.05; Fig. 1e). The observed phenotypes of 5448AP parallel those seen upon allelic replacement of the speB gene in the parent strain (mutant 5448ΔspeB; ref. 4), indicating that surface plasmin acquisition by 5448AP reflects the loss of SpeB. Additionally, other gene expression changes, such as the increase in streptokinase expression associated with covRS mutation3, may also contribute to surface plasmin acquisition by 5448AP (Supplementary Fig. 1 online). Compared with wild type, the 5448AP strain was hypervirulent in a subcutaneous infection model using transgenic mice expressing human plasminogen (P < 0.05; Fig. 1f and Supplementary Fig. 2 online). We undertook isogenic mutagenesis of 5448AP to construct a streptokinase-deficient strain (5448APΔska), which showed reduced virulence compared with 5448AP (P < 0.05; Fig. 1f). This observation is consistent with the reduced virulence of ska-deficient GAS previously reported16. Bacterial counts in the site of infection, the blood, the spleen and the liver of humanized plasminogen mice suggest that the enhanced virulence of strain 5448AP is a result of a widespread systemic infection after breakout from the site of local infection immediately before the death of the mice (Fig. 1g). These humanized animal model data agree with observations made in the clinical setting for GAS M1T1 strains, where mutations in covRS correlate with human invasive disease severity2,3.

(a) DNA sequence comparison of GAS strains 5448 and 5448AP confirms the presence of a 1-base adenine addition (nucleotide position 877) at the 3′ end of covS encoded by 5448AP (unfilled arrowhead). Filled arrowheads (p1 to p12), primers used for DNA sequence analysis. This insertion mutation at nucleotide 877 leads to the truncation of the CovS open reading frame at amino acid 300 from the CovS methionine start codon. Lower panel, putative conserved CovS domains: HAMP, histidine kinases/adenylyl cyclases/methyl-binding proteins/phosphatases; HisKA, histidine kinase domain (phosphoacceptor); HATPase: histidine kinase-like ATPase. Scales in base pairs (bp; upper panel bar) and amino acids (aa; lower panel bar). (b) Compared with that in GAS strain 5448, secreted SpeB protease activity is abrogated in 5448AP (n = 3; mean ± s.d.). *P < 0.05 versus 5448. (c) Western blot analysis of cell wall extracts for α-enolase, GAPDH and M1 protein and culture supernatants for streptokinase (unfilled arrowheads). Molecular mass markers (MWT) are indicated. (d) Washed 5448 and 5448AP cells bind equivalent amounts of human plasminogen (n = 3; mean ± s.d.). (e) After overnight growth at 37 °C in human plasma, 5448AP accumulates significantly higher levels of surface plasmin activity (n = 3; mean ± s.d.). *P < 0.05 versus 5448. (f) Survival curves after subcutaneous infection of humanized plasminogen transgenic mice (n = 10) with GAS strain 5448 (3.2 × 108 colony forming units (CFU) per dose; solid line), 5448AP (1.6 × 108 CFU/dose; dashed line) and 5448APΔska (1.5 × 108 CFU/dose; dotted line). (g) Bacterial counts in the site of infection, blood, spleen and liver of humanized plasminogen mice (n = 5) subcutaneously infected with GAS strain 5448 (2.6 × 107 CFU/dose; open circles) and 5448AP (4.9 × 107 CFU/dose; filled circles).
Next, we implanted infection chambers subcutaneously in mice and inoculated them with either GAS 5448 or 5448AP. After 24 h, bacteria were recovered and analyzed for SpeB and Sda1 expression. Quantitative real-time PCR analysis showed that speB expression was downregulated over 10,000-fold in 5448AP compared with that in wild-type 5448 (Fig. 2a). In contrast, we observed an over fivefold upregulation of sda1 gene expression in 5448 after 24 h in vivo growth, which matched the increased DNase expression seen in vivo for 5448AP (Fig. 2b and Supplementary Fig. 3a online). DNA degradation by 5448AP was increased compared with that of the GAS 5448 parent strain (Fig. 2c and Supplementary Fig. 3b), consistent with both the upregulation of sda1 expression and the known ability of SpeB to degrade Sda1 (ref. 11). The enhanced DNase activity of 5448AP compared with that of the 5448 parent strain was associated with clearance of NETs (Fig. 2d and Supplementary Fig. 4a online) and increased resistance to neutrophil killing (Fig. 2e). Neither streptokinase nor M1 protein contributed to NET clearance (Supplementary Fig. 4b).

Relative expression of (a) the speB gene and (b) the sda1 gene, as determined using quantitative real-time PCR (n = 3; mean ± s.d.). RNA was extracted from GAS either immediately before inoculation (0 h) or 24 h after inoculation of subcutaneous infection chambers. *P < 0.05 versus 5448 (0 h). (c) DNase expression in GAS mid–logarithmic phase culture supernatants as assessed by degradation of calf thymus DNA (control). (d) Clearance of NETs by GAS. Neutrophils were visualized using bright field microscopy, and NETs were visualized using Sytox Orange staining. Scale bar, 100 μm. (e) Killing of GAS by human neutrophils at a multiplicity of infection (GAS:neutrophils) of 1:10 (n = 3; mean ± s.d.). *P < 0.05 versus 5448. (f) The capacity of GAS strains 5448, 5448Δsda1, 5448RCsda1+, 5448Δsmez and SF370 to phase-shift to a SpeB-negative phenotype examined 3 d after subcutaneous infection of mice.
Neutrophils and NET-mediated extracellular killing have a pivotal role in antibacterial clearance at the initial site of infection5,17. We hypothesized that acquisition of the potent bacteriophage-encoded DNase Sda1 by the M1T1 clone provides the selective force for loss of SpeB expression in vivo, as the cysteine protease is capable of degrading this important neutrophil survival factor. To examine this possibility, we subcutaneously challenged C57BL/J6 mice separately with 5448 and the isogenic 5448Δsda1 mutant, predicting that absence of Sda1 would reduce the selective advantage for mutation to a SpeB-negative phenotype. Loss of SpeB expression in vivo during subcutaneous mouse infection was abrogated in the isogenic 5448Δsda1 mutant compared with wild-type 5448 (Fig. 2f; 1/500 SpeB-negative 5448Δsda1 colony versus 76/500 SpeB-negative 5448 colonies; P < 0.05). DNA sequence analysis of ten selected SpeB-negative 5448 colonies indicated that mutations in covRS may have been responsible for loss of SpeB expression (Supplementary Table 1 online), as previously reported3. We used reverse complementation to replace the mutated chromosomal locus in 5448Δsda1 with the wild-type allele to construct 5448RCsda1+. This complemented mutant regained the capacity to switch to the SpeB-negative phenotype (Fig. 2f; 45/500 SpeB-negative 5448RCsda1+ colonies; P = 0.39 versus wild-type). The isogenic mutant 5448Δsmez, derived identically to 5448Δsda1, was found to retain the capacity to phase-switch similarly to the wild-type strain 5448 (Fig. 2f; 33/500 SpeB-negative 5448Δsmez colonies; P = 0.11). These observations suggest that the phase-switching phenotype is due to allelic replacement of the sda1 gene and not due to the methodology used to construct an isogenic GAS mutant in strain 5448. The M1 serotype GAS strain SF370 is known not to encode Sda19,10. We found SF370 to have minimal capacity to switch to the SpeB-negative phenotype compared with strain 5448 (Fig. 2f; 2/500 SpeB-negative SF370 colonies; P < 0.05) consistent with the absence of Sda1 and thus lack of selective advantage for mutation to the SpeB-negative phenotype.
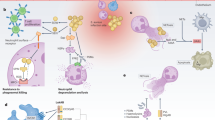
The globally disseminated Streptococcus pyogenes M1T1 clone emerged in the mid-1980s as a major cause of severe GAS invasive disease. Recent genome-scale analyses have found that in comparison with other M1 strains, the M1T1 clone has acquired two lysogenized bacteriophage genomes, encoding Sda1 and SpeA respectively9,10. Whereas the introduction of SpeA into the GAS population increases the propensity to cause streptococcal toxic shock, this study has shown that positive selection pressure in vivo is placed upon the bacteriophage-encoded virulence determinant Sda1. Loss of SpeB spares Sda1 from degradation11 and improves GAS resistance against neutrophil clearance. In vivo, the phase-shift in SpeB expression is abrogated by isogenic mutagenesis of sda1. The genetic basis for loss of SpeB expression has been previously described3. Loss of SpeB has also been shown to result in increased invasive propensity of M1T1 by the accumulation of surface-bound plasmin activity4. Therefore, we hypothesize that the bacteriophage-mediated acquisition of the sda1 gene by the ancestral M1T1 provided evolutionary selection pressure for increased neutrophil resistance through SpeB loss, which results in a hyperinvasive phenotype and can lead to severe invasive disease progression (Fig. 3). The evolution of bacterial pathogens principally occurs either through deletion events or horizontal gene transfer and acquisition18, as exemplified by the bacteriophage-mediated acquisition of the sda1 gene by M1T1. These data provide a paradigm for bacteriophage-mediated acquisition of virulence determinants and development of severe disease by otherwise benign human pathogens.

(a) After entry through the skin, GAS (blue) are able to express SpeB (required during the early stages of the infection process; black dots). Host neutrophils mount an innate immune response and entrapment of GAS in NETS (orange) begins. (b) Within the GAS population, a mutation in covRS occurs (green), resulting in loss of SpeB expression and improved resistance to killing by neutrophils. (c) Selection pressure by neutrophils results in an increase in the proportion of covRS mutant phenotype GAS within the bacterial population, improved NET clearance and neutrophil resistance. (d) Loss of SpeB expression allows the accumulation of surface plasmin activity, leading to systemic infection.
Methods
GAS strains used in this study.
Invasive GAS isolate 5448 and the isogenic animal-passaged SpeB-negative variant 5448AP have been described previously11. The isogenic mutants 5448Δsda1 (ref. 5), 5448ΔspeB (ref. 11) and GAS strain SF370 (ref. 19), have also been described previously. We used allelic exchange to precisely replace the deleted sda1 chromosomal locus in 5448Δsda1 with the wild-type sda1 gene to construct strain 5448RCsda1+ essentially as previously described5. The isogenic 5448Δsmez mutant was also constructed identically to 5448Δsda1 (ref. 5). Integrational mutagenesis of ska and emm1 was performed essentially as previously described20 using the temperature-sensitive plasmid pVE6007 (ref. 21; Supplementary Table 2 online).
Sequence analysis of covRS.
To screen GAS strains for mutations in the covRS locus, we designed 12 primers for PCR and DNA sequence analysis (Fig. 1a and Supplementary Table 3 online). Using BLASTN analysis, assembled sequences were aligned against GAS genomes and a single adenine base insertion mutation was identified at position 877 in the 5448AP covS gene, using numbering relative to the ATG start codon of 5448 covS. Other in vivo–derived, SpeB-negative GAS strain 5448 derivatives were analyzed for covRS mutations in an identical manner.
SpeB and plasminogen interaction assays.
We routinely identified SpeB-positive and SpeB-negative isolates by the Columbia skim-milk agar assay22. Quantitative SpeB assays were undertaken as previously described23. Assays of bacterial surface acquisition of plasmin from human plasma and western blot identification of α-enolase, GAPDH, streptokinase and M1 protein were conducted essentially as previously described4, with the exception that cross-specific rabbit antiserum to M protein (anti-M53) was used to identify M1 protein. GAS strain NS1133 (ref. 24) was used as an internal control for bacterial surface plasmin acquisition assays4 undertaken by incubating bacteria overnight in human plasma. Plasminogen-binding assays were conducted as previously described24.
Humanized plasminogen mice.
We backcrossed transgenic humanized plasminogen AlbPLG1 mice heterozygous for the human plasminogen transgene16 >6 times with C57BL/J6 mice. Groups of AlbPLG1 mice (n = 10) were subcutaneously infected with GAS strains 5448 and 5448AP and mortality was monitored for 10 days. Alternatively, groups of AlbPLG1 mice (n = 5) were subcutaneously infected with either 5448 or 5448AP for 48 h and the lesion (site of infection), blood, spleen and liver collected and the number of viable bacteria determined.
Real-time PCR.
In order to isolate in vivo–derived RNA, we used a subcutaneous Teflon chamber model25. We injected 100 μl of bacterial suspension into the subcutaneous chambers using sterile 25-gauge needles. At 24 h after injection, sterile 25-gauge needles were used to collect the tissue cage fluid to analyze bacterial content and SpeB status, and to extract RNA from recovered bacteria11,25. RNA was extracted from bacterial pellets using RNeasy kits, treated with DNase for 1 h to remove contaminating genomic DNA, and then recovered using RNeasy columns. Superscript II was used to reverse transcribe RNA into cDNA. We performed all Sybr-Green real-time quantitative PCR reactions using an ABI PRISM 7700 Sequence Detection System and calculated relative expression amounts using the delta-delta CT method11. Primers used for real-time PCR analysis of speB and sda1 have been previously described11,26.
DNase assays and interaction of GAS with neutrophils.
We assessed DNase activity, visualized NETs and performed neutrophil killing assays as previously described5.
SpeB phase-shift assays.
Separate cohorts of C57BL/J6 mice (n = 10) were inoculated subcutaneously with a nonlethal dose of GAS to examine the in vivo phase-shift of SpeB during infection. The inocula used in these experiments were plated out onto blood agar plates and then individual colonies tested for SpeB expression status as described above (n = 50). The 5448, 5448Δsda1, 5448RCsda1+ and 5448Δsmez inocula were found to be 100% SpeB-positive. On day 3 after infection, mice were put to death by CO2 asphyxiation and representative bacteria isolated from skin lesions4. The SpeB status of individual colonies (n = 50) was determined as described above.
Statistical analyses.
Statistical analysis of SpeB expression and status, plasminogen-binding, surface plasmin activity, quantitative real-time PCR, human neutrophil killing assays, and NET quantification were performed using a one way analysis of variance with a Dunnett's multiple comparison test. Differences were considered statistically significant at P < 0.05. Differences in survival of humanized plasminogen transgenic mice infected with GAS strains 5448, 5448AP and 5448APΔska were determined by the log-rank test. All statistical tests were performed using GraphPad Prism version 4.00.
Ethics permissions.
Permission to obtain human blood and undertake animal experiments was obtained from University of California San Diego, University of Wollongong and University of Tennessee ethics committees. Volunteers provided informed consent before blood samples were obtained.
A full description of the methods used in this manuscript is provided in the Supplementary Methods online.
Note: Supplementary information is available on the Nature Medicine website.
References
Cunningham, M.W. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13, 470–511 (2000).
Kansal, R.G., McGeer, A., Low, D.E., Norrby-Teglund, A. & Kotb, M. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect. Immun. 68, 6362–6369 (2000).
Sumby, P., Whitney, A.R., Graviss, E.A., DeLeo, F.R. & Musser, J.M. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2, 41–49 (2006).
Cole, J.N. et al. Trigger for group A streptococcal M1T1 invasive disease. FASEB J. 20, 1745–1747 (2006).
Buchanan, J.T. et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16, 396–400 (2006).
Sumby, P. et al. Extracellular deoxyribonuclease made by group A Streptococcus assists pathogenesis by enhancing evasion of the innate immune response. Proc. Natl. Acad. Sci. USA 102, 1679–1684 (2005).
Carapetis, J.R., Steer, A.C., Mulholland, E.K. & Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 5, 685–694 (2005).
Walker, M.J., McArthur, J.D., McKay, F.C. & Ranson, M. Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol 13, 308–313 (2005).
Aziz, R.K. et al. Mosaic prophages with horizontally acquired genes account for the emergence and diversification of the globally disseminated M1T1 clone of Streptococcus pyogenes. J. Bacteriol. 187, 3311–3318 (2005).
Sumby, P. et al. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192, 771–782 (2005).
Aziz, R.K. et al. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol. Microbiol. 51, 123–134 (2004).
Pancholi, V. & Fischetti, V.A. α-Enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J. Biol. Chem. 273, 14503–14515 (1998).
Pancholi, V. & Fischetti, V.A. A major surface protein on group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J. Exp. Med. 176, 415–426 (1992).
Ringdahl, U. et al. A role for the fibrinogen-binding regions of streptococcal M proteins in phagocytosis resistance. Mol. Microbiol. 37, 1318–1326 (2000).
McArthur, J.D. & Walker, M.J. Domains of group A streptococcal M protein that confer resistance to phagocytosis, opsonization and protection: implications for vaccine development. Mol. Microbiol. 59, 1–4 (2006).
Sun, H. et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 305, 1283–1286 (2004).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Ochman, H. & Moran, N.A. Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science 292, 1096–1099 (2001).
Ferretti, J.J. et al. Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 98, 4658–4663 (2001).
Nizet, V. et al. Genetic locus for streptolysin S production by group A Streptococcus. Infect. Immun. 68, 4245–4254 (2000).
Maguin, E., Duwat, P., Hege, T., Ehrlich, D. & Gruss, A. New thermosensitive plasmid for Gram-positive bacteria. J. Bacteriol. 174, 5633–5638 (1992).
Ashbaugh, C.D., Warren, H.B., Carey, V.J. & Wessels, M.R. Molecular analysis of the role of the group A streptococcal cysteine protease, hyaluronic acid capsule, and M protein in a murine model of human invasive soft-tissue infection. J. Clin. Invest. 102, 550–560 (1998).
Collin, M. & Olsen, A. Generation of a mature streptococcal cysteine proteinase is dependent on cell wall-anchored M1 protein. Mol. Microbiol. 36, 1306–1318 (2000).
McKay, F.C. et al. Plasminogen binding by group A streptococcal isolates from a region of hyperendemicity for streptococcal skin infection and a high incidence of invasive infection. Infect. Immun. 72, 364–370 (2004).
Kazmi, S.U. et al. Reciprocal, temporal expression of SpeA and SpeB by invasive M1T1 group A streptococcal isolates in vivo. Infect. Immun. 69, 4988–4995 (2001).
Aziz, R.K., Ismail, S.A., Park, H.W. & Kotb, M. Post-proteomic identification of a novel phage-encoded streptodornase, Sda1, in invasive M1T1 Streptococcus pyogenes. Mol. Microbiol. 54, 184–197 (2004).
Acknowledgements
The authors wish to thank A. Jeng and K. Chalasani for constructing the isogenic mutant 5448Δsmez, and R. Attia for assisting with real-time PCR. A. Hollands and A. Henningham are recipients of Australian Postgraduate Awards. This work was supported by the National Health and Medical Research Council of Australia 459103 (M.J.W.), US National Institutes of Health grant AI48176 (V.N.) and a Department of Employment Science and Technology (Australia) International Science Linkages grant CG001195 (M.J.W., V.N., M.K.). The authors thank G. Ellmers and R. Dinnervill for illustrating Figure 3, and M. Wilson for critically reading this manuscript.
Author information
Authors and Affiliations
Contributions
M.L.S.-S. and M.J.W. constructed strain 5448RCsda1+. A. Hollands and V.N. constructed strains 5448Δska and 5448Δemm1. M.J.W., J.N.C., J.K.K., A. Hollands and R.K.A. undertook covRS DNA sequence analysis. K.D. and G.S.C. undertook plasminogen binding assays. M.J.W., A. Hollands, J.N.C., and J.K.K. performed SpeB, surface plasmin and western blot analyses. A. Hollands, M.L.S.-S., J.N.C., J.K.K., A. Henningham, J.D.M. and M.J.W. performed survival curves and SpeB phenotype switching studies. M.J.W. and R.G.K. implanted mouse infection chambers and R.K.A., R.G.K. and M.K. undertook real-time PCR analysis. A.J.S., J.T.B., A. Hollands, M.J.W. and V.N. performed DNA NET and neutrophil killing assays. M.J.W., G.S.C., M.K. and V.N. supervised the project. M.J.W. coordinated the project. M.J.W., A. Hollands, M.L.S.-S., J.N.C., R.K.A., R.G.K., J.T.B., G.S.C., M.K. and V.N. contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4, Supplementary Tables 1–3, Supplementary Methods (PDF 504 kb)
Rights and permissions
About this article
Cite this article
Walker, M., Hollands, A., Sanderson-Smith, M. et al. DNase Sda1 provides selection pressure for a switch to invasive group A streptococcal infection. Nat Med 13, 981–985 (2007). https://doi.org/10.1038/nm1612
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1612
This article is cited by
-
Neutrophil-derived reactive agents induce a transient SpeB negative phenotype in Streptococcus pyogenes
Journal of Biomedical Science (2023)
-
Schistosome egg-derived extracellular vesicles deliver Sja-miR-71a inhibits host macrophage and neutrophil extracellular traps via targeting Sema4D
Cell Communication and Signaling (2023)
-
Moonlighting chromatin: when DNA escapes nuclear control
Cell Death & Differentiation (2023)
-
Pathogen-driven degradation of endogenous and therapeutic antibodies during streptococcal infections
Nature Communications (2023)
-
Pathogenesis, epidemiology and control of Group A Streptococcus infection
Nature Reviews Microbiology (2023)
