
Abstract
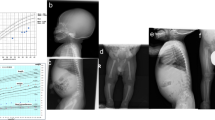
We identified four girls with a consistent constellation of facial dysmorphism and malformations previously reported in a single mother–daughter pair. Toe syndactyly, telecanthus and anogenital and renal malformations were present in all affected individuals; thus, we propose the name 'STAR syndrome' for this disorder. Using array CGH, qPCR and sequence analysis, we found causative mutations in FAM58A on Xq28 in all affected individuals, suggesting an X-linked dominant inheritance pattern for this recognizable syndrome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others


Accession codes
References
Green, A., Sandford, R. & Davison, B. J. Med. Genet. 33, 594–596 (1996).
Kohlhase, J., Wischermann, A., Reichenbach, H., Froster, U. & Engel, W. Nat. Genet. 18, 81–83 (1998).
Kohlhase, J. et al. Hum. Mol. Genet. 11, 2979–2987 (2002).
van Bokhoven, H. et al. Nat. Genet. 37, 465–467 (2005).
Spitz, R. et al. Genes Chromosom. Cancer 45, 1130–1142 (2006).
Carrel, L. & Willard, H.F. Nature 434, 400–404 (2005).
Ma, C., Papermaster, D. & Cepko, C.L. Proc. Natl. Acad. Sci. USA 95, 9938–9943 (1998).
Sicinski, P. et al. Cell 82, 621–630 (1995).
Bohm, J., Kaiser, F.J., Borozdin, W., Depping, R. & Kohlhase, J. Biochem. Biophys. Res. Commun. 356, 773–779 (2007).
Bouchard, C. et al. EMBO J. 18, 5321–5333 (1999).
Knoepfler, P.S., Cheng, P.F. & Eisenman, R.N. Genes Dev. 16, 2699–2712 (2002).
Van Esch, H. et al. Am. J. Hum. Genet. 77, 442–453 (2005).
Archer, H.L. et al. J. Med. Genet. 43, 451–456 (2006).
Acknowledgements
We thank the research subjects and their families for their participation, generosity and patience. We thank G. Scherer for critical discussion, P. Hermanns and B. Rösler for help with cell cultures, and C. Lich for technical assistance. J.K. received funding from the Deutsche Forschungsgemeinschaft (grant no. Ko1850/6-1,6-2).
Author information
Authors and Affiliations
Contributions
S.U. contributed to the clinical evalutation of cases, syndrome delineation and subject enrollment in the study. D.B. performed array CGH, mutation analysis and qPCR. W.B. performed mutation analysis on MYCN, SALL1 and SALL4 and FAM58A breakpoint cloning. F.J.K. performed co-immunoprecipitation studies. K. Buiting performed X-chromosome inactivation studies. S.K. and P.B. performed cell culture studies, siRNA knockdown experiments and proliferation assays, and contributed to the manuscript. J.B., F.B. and A.C. cloned expression constructs and performed RT-PCR. K. Borowski, K.K.-N., G.M., T.S.-M., B.S., D. Bartholdi, R.S., B.Z., and A.S.-F contributed to subject enrollment and clinical evaluation. S.U., D.B., J.B., F.J.K., K.Bu., S.K., P.B., R.S., G.M. and A.S.-F. also contributed to the manuscript. J.K. oversaw all aspects of the research and wrote major parts of the manuscript.
Corresponding author
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Methods, Supplementary Figures 1–6, Supplementary Table 1 (PDF 3653 kb)
Rights and permissions
About this article
Cite this article
Unger, S., Böhm, D., Kaiser, F. et al. Mutations in the cyclin family member FAM58A cause an X-linked dominant disorder characterized by syndactyly, telecanthus and anogenital and renal malformations. Nat Genet 40, 287–289 (2008). https://doi.org/10.1038/ng.86
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.86
This article is cited by
-
An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank
Nature Neuroscience (2021)
-
Cyclin-dependent kinases and rare developmental disorders
Orphanet Journal of Rare Diseases (2020)
-
Genetic, environmental, and epigenetic factors involved in CAKUT
Nature Reviews Nephrology (2015)
-
HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort
Pediatric Nephrology (2011)
