
Abstract
An epidemiological paradox surrounds Salmonella enterica serovar Enteritidis. In high-income settings, it has been responsible for an epidemic of poultry-associated, self-limiting enterocolitis, whereas in sub-Saharan Africa it is a major cause of invasive nontyphoidal Salmonella disease, associated with high case fatality. By whole-genome sequence analysis of 675 isolates of S. Enteritidis from 45 countries, we show the existence of a global epidemic clade and two new clades of S. Enteritidis that are geographically restricted to distinct regions of Africa. The African isolates display genomic degradation, a novel prophage repertoire, and an expanded multidrug resistance plasmid. S. Enteritidis is a further example of a Salmonella serotype that displays niche plasticity, with distinct clades that enable it to become a prominent cause of gastroenteritis in association with the industrial production of eggs and of multidrug-resistant, bloodstream-invasive infection in Africa.
Similar content being viewed by others

Main
Salmonella enterica serovar Enteritidis (hereafter referred to as S. Enteritidis) has been a global cause of major epidemics of enterocolitis, which have been strongly associated with intensive poultry farming and egg production1. This serovar is usually considered to be a generalist in terms of host range and has a low human invasiveness index, typically causing self-limiting enterocolitis2. Following a number of interventions in the farming industry involving both improved hygiene and poultry vaccination, epidemic S. Enteritidis has been in decline in many countries, including the UK and United States3,4. S. Enteritidis has also been used extensively since the early 1900s as a rodenticide (named the 'Danysz virus'), following development at the Institut Pasteur, France. Although by the 1960s Salmonella-based rodenticides had been banned in the United States, Germany, and the UK, S. Enteritidis is still produced as a rodenticide in Cuba, under the name Biorat5.
Serovars of Salmonella that cause enterocolitis in industrialized settings are strongly associated with life-threatening invasive nontyphoidal Salmonella (iNTS) disease in sub-Saharan Africa. S. Enteritidis and Salmonella enterica serovar Typhimurium (S. Typhimurium) are the two leading causes of iNTS disease in sub-Saharan Africa6, and both are associated with multidrug resistance7. The clinical syndrome iNTS disease is associated with immunosuppression in the human host, in particular with malnutrition, severe malaria, and advanced HIV in young children and with advanced HIV in adults8. It has been estimated to cause 681,000 deaths per year9.
Salmonella is a key example of a bacterial genus in which there is a recognizable genomic signature that distinguishes between a gastrointestinal and an extraintestinal/invasive lifestyle10, whereby functions required for escalating growth in an inflamed gut are lost when the lineage becomes invasive11. To investigate whether there were distinct bacterial characteristics explaining the very different epidemiological and clinical profiles of epidemic isolates of serotype S. Typhimurium from sub-Saharan Africa and industrialized settings, whole-genome sequence investigations of this serovar were previously undertaken. These identified a new pathotype of multilocus sequence type (MLST) ST313 from sub-Saharan Africa, which differed from clades that cause enterocolitis in industrialized settings, by showing patterns of genomic degradation potentially associated with more invasive disease and differential host adaptation12,13,14,15,16,17.
In relation to S. Enteritidis, there is a growing body of literature on the evolutionary history, phylogeny, and utility of whole-genome sequence for surveillance of S. Enteritidis outbreaks18,19,20. The broadest study of the phylogeny thus far identified five major lineages but contained only two African isolates21. There have also been limited reports of isolates of S. Enteritidis from African patients living in Europe that are multidrug resistant (MDR) and display a distinct phage type (PT42)22,23. We therefore hypothesized that there are distinct lineages of S. Enteritidis circulating in both the industrialized and developing worlds—with different origins and likely distinct routes of spread—that are associated with different patterns of disease, which will display the distinct genomic signatures characteristic of differential adaptation. To investigate this hypothesis, we have collected a highly diverse global collection of S. Enteritidis isolates and compared them using whole-genome sequencing, the typing methodology giving the highest possible resolution.
Results
Isolate collection
In total, we sequenced 675 isolates of S. Enteritidis collected between 1948 and 2013. The collection originated from 45 countries and six continents (Table 1); 496 of the 675 isolates were from Africa, including 131 from South Africa, a further 353 from the rest of sub-Saharan Africa, and 12 from North Africa (Table 1). There were 343 isolates from normally sterile human sites (invasive), 124 non-invasive human isolates (predominantly from stool samples), and 40 isolates from animal, food, or environmental sources. The full metadata are described in Supplementary Table 1 and have been uploaded to the publically available database EnteroBase (see URLs).
Phylogeny
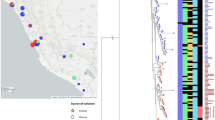
We mapped 675 S. Enteritidis genomes and 1 Salmonella enterica serovar Gallinarum (S. Gallinarum) genome to the S. Enteritidis strain P125109 reference sequence. We excluded variable regions and screened the remaining sites for SNPs. This left an alignment containing a total of 42,373 variable sites, from which a maximum-likelihood phylogeny was constructed using S. Gallinarum—a closely related serovar—as an outgroup (Fig. 1). HierBAPS was run over two rounds, which provided clear distinction among clades and clusters24. The phylogeny of S. Enteritidis showed evidence of three clades associated with epidemics, one which we have termed the 'global epidemic clade' that includes the reference PT4 isolate P125109 and two African clades, the first predominantly composed of West African isolates (labeled the 'West African clade') and the second composed of isolates predominantly originating in Central and Eastern Africa (called the 'Central/Eastern African clade'). The other clades and clusters predicted by HierBAPS, the largest of which is a paraphyletic cluster from which the global epidemic clade emerged (outlier cluster) and a further five smaller clades or clusters predicted by HierBAPS, are shown in Figure 1.

There are three epidemic clades: two African epidemic clades and a global epidemic clade. The scale bar shows nucleotide substitutions per site.
The global epidemic clade contained isolates of multiple phage types, including PT4 and PT1, which have been linked to the global epidemic of poultry-associated human enterocolitis25. It comprised 250 isolates from 28 countries, including 43 from Malawi and 82 from South Africa. These isolates were collected across a 63-year period (1948–2013). Antimicrobial susceptibility testing had been performed on 144 isolates: 104 were susceptible to all antimicrobials tested, 5 were MDR (resistant to three or more antimicrobial classes), 1 was nalidixic acid resistant, and none were extended-spectrum β-lactamase (ESBL)-producing isolates. Database comparison of the genomes from this clade showed that 221 (88%) of them contained no predicted antimicrobial resistance (AMR) genes apart from the cryptic resistance gene aac(6′)-Iy (ref. 26).
The global epidemic clade has emerged from a diverse cluster previously described by Zheng et al.27, which encompassed 131 isolates (outlier cluster in Fig. 1). In addition to being paraphyletic, this group was geographically and temporally diverse and was predominantly drug susceptible (59/71 isolates). Whereas the majority of the diversity among phage-typed isolates was present within the global epidemic clade, only this cluster contained isolates of PT14b, which was recently associated with a multiple-country outbreak of S. Enteritidis enterocolitis in Europe linked to chicken eggs from Germany28. There were also 41 isolates from South Africa in this clade, where S. Enteritidis has been a common cause of bloodstream infection, and 39 bloodstream isolates from Malawi. Database comparison of the genomes from this clade demonstrated that 122 (82%) contained no predicted AMR genes apart from the cryptic resistance gene aac(6′)-Iy.
There were two related, but phylogenetically and geographically distinct, epidemic clades that largely originated from sub-Saharan Africa. The Central/Eastern African clade included 166 isolates, all but 2 of which (from South Africa) came from this region. Of these isolates, 126 of 155 (82%) were MDR and 148 of 153 (97%) displayed phenotypic resistance to between one and four antimicrobial classes. All of these genomes contained at least five predicted resistance-conferring genes, and 128 (77%) contained nine (Table 2 and Supplementary Table 2). Of these isolates, 155 of 165 (94%) were cultured from a normally sterile compartment of a human (blood or cerebrospinal fluid) and were considered to be causing invasive disease (Table 2). The second African epidemic clade was strongly associated with West Africa, with 65 of 66 isolates coming from this region and 1 isolate from the United States. This clade was also associated with drug resistance (62 (94%) resistant to ≥1 antimicrobial class by phenotype and genotype) and human invasive disease (61 (92%)). It also included two isolates that were subtyped as PT4.
The remaining 58 isolates included in this study were extremely diverse—phylogenetically, temporally, and geographically. Only two displayed any phenotypic antimicrobial resistance, one of which was MDR. Inspection of the genomes showed that five had predicted AMR genes in addition to aac(6′)-Iy, four of which were isolated in sub-Saharan Africa. Twenty were associated with invasive human disease, and six were recovered from stool. Three isolates were from stocks of rodenticide, and these were phylogenetically remote from both the global epidemic clade and the two African epidemic clades.
To add further context to these findings, we screened the entire publically available Public Health England (PHE)-sequenced Salmonella routine surveillance collection, which includes 2,986 S. Enteritidis genomes, 265 of which were associated with travel to Africa (Supplementary Fig. 1). Within this very large collection, including 61 (2.0%) bloodstream isolates and 2,670 (89.4%) stool isolates, only 6 isolates (4 from blood culture and 1 from stool) fell within the West African clade and 1 isolate (from stool) belonged to the Central/Eastern African clade. Notably, these isolates were all either associated with travel to Africa and/or taken from patients of African origin.
It is apparent from the location of the archetypal reference isolate and archetypal phage types in the phylogeny (Supplementary Fig. 2) that the majority of S. Enteritidis studied previously belonged to the global epidemic clade associated with enterocolitis in industrialized countries. Furthermore, it is also clear that two additional, previously unrecognized S. Enteritidis lineages have emerged, largely restricted to Africa, that are strongly associated with MDR and invasive disease.
To understand how recently these Africa-associated lineages emerged, we used Bayesian Evolutionary Analysis by Sampling Trees (BEAST) to reconstruct the temporal history of the epidemic clades29. These data (Supplementary Fig. 3) estimate that the most recent common ancestor (MRCA) of the Central/Eastern African clade dates to 1945 (95% credible interval (CrI) = 1924–1951), and for the West African clade the MRCA was estimated to date from 1933 (95% CrI = 1901–1956). We estimate that the MRCA of the global epidemic clade originated around 1918 (95% CrI = 1879–1942; Supplementary Fig. 4), with a modern expansion occurring in 1976 (95% CrI = 1968–1983), while the paraphyletic cluster from which it emerged dates to approximately 1711 (95% CrI = 1420–1868).
Contribution of the accessory genome
Prophages have the potential to carry nonessential 'cargo' genes, which suggests that they confer a level of specialization to their host bacterial species, whereas plasmids may confer a diverse array of virulence factors and antimicrobial resistance30,31. Therefore, it is critical to evaluate the accessory genome in parallel with the core genome. We used 622 sequenced genomes to determine a pangenome; this analysis yielded a core genome comprising 4,076 predicted genes present in ≥90% of isolates, including all 12 recognized Salmonella pathogenicity islands as well as all 13 fimbrial operons found in the P125109 reference32. The definition for core genes was set to minimize stochastic loss of genes from the core genome due to errors in individual assemblies across such a large data set. The accessory genome consisted of 14,015 predicted genes. Of the accessory genes, 324 were highly conserved across the global and two African epidemic clades, as well as the outlier cluster. Almost all were associated with the acquisition or loss of mobile genetic elements (MGEs) such as prophages or plasmids. Prophage regions have been shown to be stable in Salmonella genomes and are potential molecular markers, the presence of which has previously been used to distinguish specific clades13,33.
The lineage-specific whole-gene differences among the major clades are summarized in Figure 2 and plotted against representatives of the four major clades in Supplementary Figure 5. The lineage-specific sequence regions included 57 predicted genes found to be unique to the global epidemic clade (Fig. 2), all of which were associated with prophage φSE20, a region shown to be essential for invasion of chicken ova and mice in a previous study34. There were a further 39 genes conserved in the global epidemic clade and the paraphyletic outlying cluster, which were absent from both African clades, 26 of which corresponded to region of difference (ROD)-21 (ref. 32). The Central/Eastern African clade contained 77 predicted genes that were absent in the other clades. Thirty-three were associated with the virulence plasmid, and a further 40 chromosomal genes were associated with a new Fels2-like prophage region (φfels-BT). The West African clade had only 15 distinct predicted genes, 11 of which were plasmid associated. The two African clades shared a further 102 genes: 48, including a leucine-rich repeat region, were associated with a new prophage region closely related to Enterobacter phage P88, 44 were associated with a Gifsy-1 prophage found in Salmonella enterica serovar Bovismorbificans (S. Bovismorbificans), and 8 were associated with a Gifsy-2 prophage that has degenerated in the P125109 reference.

The approximate positions of prophages on the chromosome are depicted, although the prophages are not drawn to scale. The number in parentheses is the number of predicted genes on each prophage.
The S. Enteritidis plasmid is the smallest of the generic Salmonella virulence plasmids at 58 kb and is unusual in that it contains an incomplete set of tra genes that are responsible for conjugative gene transfer. We reconstructed the phylogeny of the S. Enteritidis virulence plasmid backbone using reads that mapped to the S. Enteritidis reference virulence plasmid, pSENV. Of the 675 genomes, 120 (18%) lacked pSENV. The virulence plasmid phylogeny was similar to that of the chromosome, suggesting that this plasmid has been stably maintained by each lineage and diversified alongside the chromosome (Supplementary Fig. 6).
The virulence plasmids from the African clades at ∼90 kb were much larger than those present in the other clades. A representative example was extracted from Malawian isolate D7795, sequenced using long-read technology to accurately reconstruct it (PacBio sequencing; Online Methods), and denoted pSEN-BT (GenBank accession LN879484). pSEN-BT was composed of a backbone of pSENV with additional regions that were highly similar to recently sequenced fragments of a new S. Enteritidis virulence plasmid (pUO-SeVR) isolated from an African patient presenting with MDR invasive S. Enteritidis in Spain22. Plasmid pSEN-BT harbored nine AMR genes (full list in Supplementary Table 2), plus additional genes associated with virulence and a toxin/antitoxin plasmid addiction system. Of note, plasmids from the West African isolates carried the catA1 chloramphenicol resistance gene (encoding chloramphenicol acetyltransferase A1), whereas the Central/Eastern African strains carried catA2 and the tetracycline resistance gene tet(A). Like pSENV, the African virulence plasmid contained an incomplete set of tra genes and so is not self-transmissible. This was confirmed by conjugation experiments and is consistent with previous reports22,23. These observations suggest that the evolution of the S. Enteritidis plasmid mirrors that of the chromosome; it is thus not a 'novel' plasmid but in different locations in sub-Saharan Africa has acquired different AMR genes.
Multiple signatures of differential host adaptation
It has been observed in multiple serovars of Salmonella, including S. Typhi, S. Gallinarum, and S. Typhimurium ST313, that the degradation of genes necessary for the use of inflammation-derived nutrients is a marker of that lineage having moved from an intestinal to a more invasive lifestyle13,14,32,35. Accordingly, we looked for similar evidence in a representative example of an MDR, invasive Central/Eastern African clade isolate (D7795) that was isolated from the blood of a Malawian child in 2000. The draft genome sequence of D7795 closely resembled that of P125109; however, in addition to the new prophage repertoire and plasmid genes described above, it harbored a number of predicted pseudogenes or hypothetically disrupted genes (HDGs)11.
In total, there were 42 putative HDGs in D7795, many of which were found in genes involved in gut colonization and fecal shedding, as well as various metabolic processes such as biosynthesis of cobalamin, which is a cofactor for anaerobic catabolism of inflammation-derived nutrients, such as ethanolamine, following infection36. Curation of the SNPs and indels predicted to be responsible for pseudogenization across the Central/Eastern African clade and West African clade showed that 37 of 42 predicted HDGs were fixed in other representatives of the Central/East African clade, with 27 of them present in over 90% of the isolates from that clade. Relatively fewer HDGs in D7795 (19/42) were present in representatives of the West African clade, although 13 were present in ≥90% of isolates (Supplementary Table 3).
In addition to this evidence of reductive evolution in D7795, there were 363 genes containing nonsynonymous SNPs, which change the amino acid sequence and so may have functional consequences37. The two African clades were screened for the presence of nonsynonymous SNPs; 131 of the SNPs were found to be present and completely conserved across both clades, including nonsynonymous SNPs in 43 genes encoding predicted membrane proteins, 36 metabolic genes, and 23 conserved hypothetical genes (Supplementary Table 4). Furthermore, many of these nonsynonymous SNPs fell in genes within the same metabolic pathways as the HDGs (see the Supplementary Note for a detailed description). A list of some of the common traits identified among the functions of genes lost independently by D7795, S. Typhi, and S. Gallinarum is provided in Supplementary Table 5. The disproportionate clustering of mutations in membrane structures observed in the African clades is yet another sign of differential host adaptation analogous to that reported in both S. Typhi35 and S. Gallinarum32.
Biolog growth substrate platform profiling
We used the Biolog platform to generate a substrate growth utilization profile for selected S. Enteritidis isolates. Corresponding signal values for replicate pairs of a Central/Eastern African isolate (D7795) and a global epidemic isolate (A1636) were compared using principal-component analysis and found to be highly consistent. In total, 80 metabolites showed evidence of differential metabolic activity (Fig. 3). Evaluation of data from the Central/Eastern African isolate using Pathway Tools software showed that 14 of 27 (52%) pathways with evidence of decreased metabolic activity at 28 °C had a corresponding component of genomic degradation. This was also true for 12 of 30 (40%) pathways with evidence of decreased metabolic activity at 37 °C.

The figure also displays whether there are corresponding mutations in genes related to the affected metabolic pathway. NS-SNP, nonsynonymous SNP; HDG, hypothetically disrupted gene; P/S, phosphorous and sulfur sources.
Instances of reduced metabolic activity in the Central/Eastern African strain (D7795) in comparison to the global epidemic strain (A1636) included dulcitol and glycolic acid in the glycerol degradation pathway, propionic acid in the propanediol pathway, and ethylamine and ethanolamine. These are all reactions that are dependent on vitamin B12 (cobalamin), for which there was a corresponding signature of genomic degradation. Also, there was reduced activity in response to three forms of butyric acid, alloxan, and allantoic acid metabolism. Allantoin can be found in the serum of birds, but not humans, and is used as a carbon source during S. Enteritidis infection of chickens38, and HDGs relating to allantoin have been noted in S. Typhimurium ST313 (ref. 13). The full list of differences is detailed in Supplementary Tables 6 and 7. This is a further sign of decreased metabolism in the Central/Eastern African isolate in the anaerobic environment of the gut.
Chick infection model suggests divide in host range
Given the phenotypic differences observed in the genotypically distinct global and African clades, we hypothesized that these lineages could have differing infection phenotypes in an in vivo challenge model. We compared the infection profile of a member of the Central/Eastern African clade (D7795) to that of the reference global epidemic strain P125109 in an avian host. The chicken group infected with P125109 showed mild hepatosplenomegaly, consistent with infection by this Salmonella serovar, and cecal colonization (Fig. 4a–c). In contrast, the Central/Eastern African strain displayed significantly reduced invasion at 7 days post-infection (dpi) of both liver (P = 0.027) and spleen (P = 0.007); however, cecal colonization was not significantly reduced (P = 0.160). This is in marked contrast to the behavior of S. Typhimurium ST313, which is more invasive in a chick infection model12.

(a–c) Plots demonstrate the failure of Central/Eastern African clade isolate to invade chicken spleen (a) and liver (b) or to colonize chicken cecum (c) at 7 dpi (n = 24 at this time point) in comparison to the global epidemic clade. Numbers are expressed as colony forming units (CFU) per gram of tissue.
Discussion
S. Enteritidis is an example of a successful Salmonella lineage with the apparent ability to adapt to different hosts and transmission niches as and when opportunities for specialization have presented themselves. Langridge et al. recently evaluated the Enteritidis/Gallinarum/Dublin lineage of Salmonella, identifying components of the nature and order of events associated with host range and restriction39. In the present study, we have highlighted the plasticity of S. Enteritidis, providing evidence of three distinct epidemics of human disease. In addition, we show multiple additional clades and clusters that demonstrate the huge reservoir of diversity among S. Enteritidis from which future epidemics might emerge.
An important question posed by this study is why distinct clades of Salmonella emerged to become prominent causes of iNTS disease in Africa, from a serotype normally considered to be weakly invasive. The presence of a highly immunosuppressed population due to the HIV pandemic is clearly a key host factor that facilitates the clinical syndrome iNTS disease40,41. In addition to human host factors, there are two distinct African epidemic lineages that have emerged in the last 90 years. Both lineages are associated with a novel prophage repertoire, an expanded multidrug-resistance-augmented virulence plasmid, and patterns of genomic degradation with similarity to those of other host-restricted invasive Salmonella serotypes, including S. Typhi and S. Gallinarum, and to clades of S. Typhimurium associated with invasive disease in Africa13,32,35. This pattern of genomic degradation is concentrated in pathways specifically associated with an enteric lifestyle; however, it is noteworthy that, in the chick infection model, the African S. Enteritidis strain invaded the chick liver and spleen less well than the strain from the global pandemic clade. This raises the possibility that the two clades occupy different ecological niches outside the human host or that they behave differently within the human host, and screening of the very large S. Enteritidis collection from routine Salmonella surveillance by PHE supports the assertion that these lineages are geographically restricted to Africa. This study therefore indicates a need to understand what these ecological niches might be and then to define the transmission pathways of African clades of S. Enteritidis, to facilitate public health interventions to prevent iNTS disease.
The evolution of the S. Enteritidis virulence plasmid is intriguing; pSENV is the smallest of the known Salmonella virulence-associated plasmids, but in sub-Saharan Africa the plasmid has nearly doubled in size, partly through the acquisition of AMR genes. The absence of tra genes necessary for conjugal transfer indicates that MDR status has evolved either through acquisition of MGEs multiple times or through clonal expansion and vertical transmission of the plasmid to progeny. The available data suggest that the former scenario has happened twice, once in West Africa and once in Central/Eastern Africa.
Despite S. Enteritidis being reported as a common cause of bloodstream infection in Africa6,7, the Global Enteric Multicenter Study (GEMS) found that Salmonella serotypes were an uncommon cause of moderate-to-severe diarrhea in African children less than 5 years of age42. Our data associating the African lineages with invasive disease are also consistent with data presented in a recent Kenyan study comparing a limited number and diversity of S. Enteritidis isolates from blood and stool. Applying the lineages defined in this study to the genome data reported from Kenya showed that 20.4% of isolates from that study belonging to the global clade were associated with invasive disease, whereas 63.2% of the isolates in that study belonging to our Central/Eastern African clade were associated with invasive disease43. The remaining isolates were associated with cases of enterocolitis or asymptomatic carriage, confirming that the Central/Eastern African clade can also cause enterocolitis. The association of S. Enteritidis clades circulating in sub-Saharan Africa with iNTS disease may reflect the fact that their geographical distribution permits them to behave as opportunistic invasive pathogens in a setting where advanced immunosuppressive disease is highly prevalent in human populations.
In summary, two clades of S. Enteritidis have emerged in Africa, which have phenotypes and genotypes different from the strains of S. Enteritidis circulating in the industrialized world. These strains display evidence of changing host adaptation, different virulence determinants, and multidrug resistance, a parallel situation to the evolutionary history of S. Typhimurium ST313. They may have ecologies and/or host ranges different from those of global strains and have caused epidemics of bloodstream infection (BSI) in at least three countries in sub-Saharan Africa, yet they are rarely responsible for disease in South Africa. An investigation into the environmental reservoirs and transmission of these pathogens is warranted and urgently required.
Methods
Bacterial isolates.
S. Enteritidis isolates were selected on the basis of six factors: date of original isolation, antimicrobial susceptibility pattern, geographical site of original isolation, source (human (invasive versus stool), animal, or environmental), phage type (where available), and multilocus variable number tandem repeat (MLVA) type (where available). S. Enteritidis P125109 (ENA accession AM933172) isolated from a poultry farm in the UK was used as a reference32. The full metadata are detailed in Supplementary Table 1. Isolates have been attributed to regions according to United Nations statistical divisions.
Sequencing, SNP calling, construction of phylogeny, and comparative genomics.
PCR libraries were prepared from 500 ng of DNA as previously described45. Isolates were sequenced using Illumina Genome Analyzer II, HiSeq 2000, and MiSeq machines, and 150-bp paired-end reads were generated. The strains were aligned to S. Enteritidis reference genome P125109 using a pipeline developed in house at the Wellcome Trust Sanger Institute. For each isolate sequenced, the raw sequence read pairs were split to reduce the overall memory usage and allow reads to be aligned using more than one CPU. The reads were then aligned using SMALT, a hashing-based sequence aligner. The aligned and unmapped reads were combined into a single BAM file. Picard was used to identify and flag optical duplicates generated during the construction of a standard Illumina library, thus reducing possible effects from PCR bias. All of the alignments were created in a standardized manner, with the commands and parameters stored in the header of each BAM file, allowing for the results to be easily reproduced.
The combined BAM file for each isolate was used as input data in the SAMtools mpileup program to call SNPs and small indels, producing a BCF file describing all of the variant base positions46. A pseudogenome was constructed by substituting the base call at each variant or non-variant site, defined in the BCF file, in the reference genome. Only base calls with depth of coverage >4 or quality >50 were considered in this analysis. Base calls in the BCF file failing this quality control filter were replaced with an “N” character in the pseudogenome sequence.
All of the software developed is freely available for download from GitHub under an open source license, GNU GPL 3.
Phylogenetic modeling was based on the assumption of a single common ancestor; therefore, variable regions where horizontal genetic transfer occurs were excluded47,48. A maximum-likelihood phylogenetic tree was then built from the alignments of the isolates using RAxML (version 7.0.4) with a GTR + I + G model49. The maximum-likelihood phylogeny was supported by 100 bootstrap pseudoreplicate analyses of the alignment data. Clades were predicted using HierBAPS24. This process was repeated to construct the plasmid phylogeny, using reads that aligned to pSENV. To ascertain the presence of the clusters defined by HierBAPS in the PHE routine Salmonella surveillance collection, 17 isolates representing the diversity of the collection were compared against 2,986 S. Enteritidis PHE genomes. Single-linkage SNP clustering was performed as previously described50. A maximum-likelihood phylogeny showing integration of the 17 isolates with 50-SNP cluster representatives of the PHE S. Enteritidis collection was constructed as above. FASTQ reads from all PHE sequences in this study can be found under the PHE Pathogens BioProject accession (PRJNA248792).
Temporal reconstruction was performed using BEAST (version 1.8.2)51. A relaxed lognormal clock model was initially employed. The results of this model indicated that a constant clock model was not appropriate, as the posterior of the standard deviation of the clock rate did not include zero. A range of biologically plausible population models (constant, exponential, and skyline) was investigated. Skyline models can be biased by non-uniform sampling, and we observed strong similarity between the reconstructed skyline population and the histogram of sampling dates; therefore, this model was excluded. The exponential models consistently failed to converge and were excluded. Thus, for all data sets, lognormal clock and constant population size models were used. The computational expense required for this analysis precluded running estimators for model selection. However, we note that Deng et al.21 used the same models in their analysis of 125 S. Enteritidis isolates. Default priors were used except for ucld.mean, Gamma(0.001,1000), initial: 0.0001; exponential.popSize, LogNormal(10,1.5), initial: 1.
Three chains of 100 million states were run in parallel for each clade of the four major HierBAPS clades, as well as a fourth chain without genomic data to examine the influence of the prior, which in all cases was uninformative. The final results, as used here, all had effective sample sizes of over 200 and had convergence between all three runs. For the global and global outlier lineages, the data sets were not computationally feasible to analyze. We thus created three further random subsets of the data by drawing n isolates from each sampled year where n was sampled from a Poisson distribution with λ = 2. The posteriors of all subsets were extremely similar, and runs were combined to produce the final MRCA estimates.
To gain detailed insight into genomic differences, a single high-quality sequence from Malawian S. Enteritidis isolate D7795 was aligned against P125109 using ABACAS and annotated52. Differences were manually curated against the reference sequence using the Artemis Comparison Tool (ACT)53. Sections of contigs that were incorporated into the alignment but that did not align with P125109 were manually inspected and compared to public databases using BLASTN. When these regions appeared to be novel prophages, they were annotated using the phage search tool PHAST and manually curated54. To investigate whether the SNPs and/or indels that were predicted to be responsible for pseudogene formation in D7795 were unique to that isolate or conserved in both African epidemic clades, all isolates were aligned to P125109 and the relevant SNPs or indels were investigated using in silico PCR of the aligned sequences. Manual curation was performed to confirm the nature of all pseudogene-associated SNPs and indels. Nonsynonymous SNPs identified in D7795 were sorted throughout the African clades by extracting and aligning the appropriate gene sequences from P125109 and D7795. The coordinates of the nonsynonymous SNPs were then used to identify the relevant sequence and determine the nature of the base.
Accessory genome.
The pangenome for the data set was predicted using ROARY55. Genes were considered to be core to S. Enteritidis if present in ≥90% of isolates. A relaxed definition of the core genome was applied as assemblies were used to generate it and, the more assemblies one uses, the more likely it is that a core gene will be missed in one sample owing to an assembly error. The remaining genes were considered to be core to the clades or clusters predicted by HierBAPS if present in ≥75% of isolates from each clade or cluster. These genes were then curated manually using ACT to search for their presence and position in P125109 or the improved draft assembly of representative isolates of each of the other clades if not present in P125109. Any large accessory regions identified were search by BLAST against the assembled genomes of the entire collection to confirm that they were grossly intact.
Plasmid identification.
Plasmid DNA was extracted from isolate D7795 using the Kado and Liu method and separated by gel electrophoresis alongside plasmids of known size, to estimate the number and size of the plasmids present56. Plasmid conjugation was attempted by mixing 100 μl of overnight cultures of donor and recipient strains (rifampicin-resistant Escherichia coli C600) on LB agar plates and incubating overnight at 26 °C or 37 °C. The plasmid was sequenced using the PacBio platform to gain long reads and a single improved draft assembly, which was aligned against P125109 plasmid pSENV (GenBank accession HG970000). For novel regions of the plasmid from isolate D7795, genes were predicted using GLIMMER and manual annotations were applied on the basis of homology searches against public databases, using both BLASTN and FASTA. The plasmid phylogeny was reconstructed using the same methodology as for the chromosome; a maximum-likelihood phylogenetic tree was built from the alignments of the isolates using RAxML (version 7.0.4) with a GTR + I + G model.
Identification of AMR genes.
A manually curated version of the Resfinder database was used to investigate isolates for the presence of AMR genes57. To reduce redundancy, the database was clustered using CD-HIT-EST58, with the alignment length of the shorter sequence required to be 90% the length of the longer sequence. All other options were left as default values. The representative gene of each cluster was then mapped with SMALT to the assemblies of each isolate, and matches with an identity of 90% or greater were considered significant, in line with the default clustering parameters of CD-HIT-EST. Where partial matches were identified at the ends of contigs, having identity of 90% or greater with the matched region of the gene, potential AMR gene presence was recorded. To confirm the presence of these partial matches, raw sequencing reads for the pertinent isolates were mapped using SMALT to these genes to check for at least 90% identity across the entire gene.
Biolog growth substrate platform profiling.
The Biolog platform enables the simultaneous quantitative measurement of a number of cellular phenotypes and, therefore, the creation of a phenotypic profile under a variety of assay conditions59. Incubation and recording of phenotypic data were performed using an Omnilog plate reader.
In these experiments, two replicates of D7795 were compared to two replicates of a PT4-like strain at 28 °C and 37 °C to represent environmental and human temperature. Biolog plates PM1–PM4 and PM9 (carbon source (PM1 and PM2), nitrogen source (PM3), phosphor and sulfur source (PM4), and metabolism and osmotic pressure (PM9)) were used. Each well was inoculated as described below, thereby testing 475 conditions at once (each plate had one negative-control well).
The isolates were cultured overnight on LB agar at 37 °C in air to exclude contamination. Colonies were scraped off plates and dispensed into IF-0a solution (Biolog) to a cell density corresponding to 81% transmittance. For each plate used, 880 μl of this cell suspension was added to 10 ml of IF-10b GP/GP solution (Biolog) and 120 μl of dye mix G (Biolog). This was then supplemented with 1 ml of a solution of 7.5 mM D-ribose (Sigma), 2 mM magnesium chloride, 1 mM calcium chloride, 2 mM sodium pyrophosphate (Sigma), 25 μM L-arginine (Sigma), 25 μM L-methionine (Sigma), 25 μM hypoxanthine (Sigma), 10 μM lipoamide (Sigma), 5 μM nicotine adenine dinucleotide (Sigma), 0.25 μM riboflavin (Sigma), 0.005% by mass yeast extract (Fluka), and 0.005% by mass Tween-80 (Sigma). 100 μl of this mixture was dispensed into each well of the assay plate. Plates were then allowed to equilibrate in air for 5 min before being sealed in airtight bags and loaded into the Omnilog machine (Biolog). Plates were scanned every 15 min for 48 h while incubated at 28 °C or 37 °C in air. Culture under anaerobic conditions was unavailable. Two paired replicates were analyzed for each of the two isolates.
After completion of the run, the signal data were compiled and analyzed using the limma package in R described previously60. A log-transformed fold change of 0.5 controlling for a 5% false discovery rate was used as a cutoff for investigating a specific metabolite further using Pathway Tools61 and determining whether the metabolic change was related to pseudogenes and nonsynonymous SNPs in genes in the respective genomes.
In vivo infection model.
Two isolates were used in the chick models: S. Enteritidis P125109 and D7795. Unvaccinated commercial female egg-layer Lohmann Brown chicks (Domestic Fowl (Gallus gallus)) were obtained from a commercial hatchery and housed in secure floor pens at a temperature of 25 °C. Eight chicks per strain per time point were inoculated by gavage at 10 d of age and received a dose of ∼108 Salmonella CFU in a volume of 0.2 ml. Subsequently, four to five birds from each group were humanely killed at 3, 7, or 21 dpi. At postmortem, the liver, spleen, and cecal contents were removed aseptically, homogenized, serially diluted, and dispensed onto Brilliant Green agar (Oxoid) to quantify CFU as described previously62. Statistical analysis was performed using SPSS, version 20 (IBM). Kruskal–Wallis testing was used to compare bacterial loads between infected groups.
All work was conducted in accordance with the UK legislation governing experimental animals, Animals (Scientific Procedures) Act 1986, under project license 40/3652 and was approved by the University of Liverpool ethical review process before the award of the project license. The licensing procedure requires power calculations to determine minimal group sizes for each procedure to ensure results are significant. For these experiments, a group size of eight birds per time point was chosen, on the basis of a variation of 1.0 log10 in bacterial count between groups being significant along with previous experience of Salmonella infection studies. Groups were randomly selected on receipt from the hatchery; investigators conducting these experiments were not blinded to experimental group, as the current UK code of practice requires all cages or pens to be fully labeled with experimental details. No animals were excluded from the analysis. All animals were checked a minimum of twice daily to ensure their health and welfare.
Code availability.
Software is referenced and URLs are provided in the URLs section. All software is open source.
URLs.
BEAST, http://beast.bio.ed.ac.uk/; Biolog, http://www.biolog.com/; BLASTN, http://blast.ncbi.nlm.nih.gov/; EnteroBase, https://enterobase.warwick.ac.uk/; limma package, http://www.bioconductor.org/; PacBio platform, http://www.pacificbiosciences.com/; Picard, https://broadinstitute.github.io/picard/; R, http://www.R-project.org/; SMALT, http://www.sanger.ac.uk/science/tools/smalt-0; United Nations statistical divisions, http://millenniumindicators.un.org/unsd/methods/m49/m49regin.htm.
Accession codes.
Accession codes for sequencing data, including both raw sequencing reads and assembled sequences, are listed in Supplementary Table 1.
Accession codes
Accessions
BioProject
European Nucleotide Archive
NCBI Reference Sequence
Change history
24 October 2016
In the version of this article initially published, the last name of author Lars Barquist was inadvertently duplicated. The error has been corrected in the HTML and PDF versions of the article.
References
Ward, L.R., Threlfall, J., Smith, H.R. & O'Brien, S.J. Salmonella enteritidis epidemic. Science 287, 1753–1754 author reply 1755–1756 (2000).
Jones, T.F. et al. Salmonellosis outcomes differ substantially by serotype. J. Infect. Dis. 198, 109–114 (2008).
O'Brien, S.J. The “decline and fall” of nontyphoidal Salmonella in the United Kingdom. Clin. Infect. Dis. 56, 705–710 (2013).
Braden, C.R. Salmonella enterica serotype Enteritidis and eggs: a national epidemic in the United States. Clin. Infect. Dis. 43, 512–517 (2006).
Friedman, C.R., Malcolm, G., Rigau-Pérez, J.G., Arámbulo, P. III & Tauxe, R.V. Public health risk from Salmonella-based rodenticides. Lancet 347, 1705–1706 (1996).
Reddy, E.A., Shaw, A.V. & Crump, J.A. Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect. Dis. 10, 417–432 (2010).
Feasey, N.A. et al. Three epidemics of invasive multidrug-resistant Salmonella bloodstream infection in Blantyre, Malawi, 1998–2014. Clin. Infect. Dis. 61 (Suppl. 4), S363–S371 (2015).
Feasey, N.A., Dougan, G., Kingsley, R.A., Heyderman, R.S. & Gordon, M.A. Invasive non-typhoidal Salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379, 2489–2499 (2012).
Ao, T.T. et al. Global burden of invasive nontyphoidal Salmonella disease, 2010. Emerg. Infect. Dis. 21, 941–949 (2015).
Nuccio, S.P. & Bäumler, A.J. Reconstructing pathogen evolution from the ruins. Proc. Natl. Acad. Sci. USA 112, 647–648 (2015).
Nuccio, S.P. & Bäumler, A.J. Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio 5, e00929–14 (2014).
Parsons, B.N. et al. Invasive non-typhoidal Salmonella Typhimurium ST313 are not host-restricted and have an invasive phenotype in experimentally infected chickens. PLoS Negl. Trop. Dis. 7, e2487 (2013).
Kingsley, R.A. et al. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 19, 2279–2287 (2009).
Okoro, C.K. et al. Signatures of adaptation in human invasive Salmonella Typhimurium ST313 populations from sub-Saharan Africa. PLoS Negl. Trop. Dis. 9, e0003611 (2015).
Herrero-Fresno, A. et al. The role of the st313-td gene in virulence of Salmonella Typhimurium ST313. PLoS One 9, e84566 (2014).
Ramachandran, G., Perkins, D.J., Schmidlein, P.J., Tulapurkar, M.E. & Tennant, S.M. Invasive Salmonella Typhimurium ST313 with naturally attenuated flagellin elicits reduced inflammation and replicates within macrophages. PLoS Negl. Trop. Dis. 9, e3394 (2015).
Okoro, C.K. et al. Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat. Genet. 44, 1215–1221 (2012).
Allard, M.W. et al. On the evolutionary history, population genetics and diversity among isolates of Salmonella Enteritidis PFGE pattern JEGX01.0004. PLoS One 8, e55254 (2013).
den Bakker, H.C. et al. Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar Enteritidis. Emerg. Infect. Dis. 20, 1306–1314 (2014).
Taylor, A.J. et al. Characterization of foodborne outbreaks of Salmonella enterica serovar Enteritidis with whole-genome sequencing single nucleotide polymorphism–based analysis for surveillance and outbreak detection. J. Clin. Microbiol. 53, 3334–3340 (2015).
Deng, X. et al. Genomic epidemiology of Salmonella enterica serotype Enteritidis based on population structure of prevalent lineages. Emerg. Infect. Dis. 20, 1481–1489 (2014).
Rodríguez, I., Guerra, B., Mendoza, M.C. & Rodicio, M.R. pUO-SeVR1 is an emergent virulence-resistance complex plasmid of Salmonella enterica serovar Enteritidis. J. Antimicrob. Chemother. 66, 218–220 (2011).
Rodríguez, I., Rodicio, M.R., Guerra, B. & Hopkins, K.L. Potential international spread of multidrug-resistant invasive Salmonella enterica serovar Enteritidis. Emerg. Infect. Dis. 18, 1173–1176 (2012).
Cheng, L., Connor, T.R., Sirén, J., Aanensen, D.M. & Corander, J. Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol. Biol. Evol. 30, 1224–1228 (2013).
Rodrigue, D.C., Tauxe, R.V. & Rowe, B. International increase in Salmonella enteritidis: a new pandemic? Epidemiol. Infect. 105, 21–27 (1990).
Magnet, S., Courvalin, P. & Lambert, T. Activation of the cryptic aac(6)-Iy aminoglycoside resistance gene of Salmonella by a chromosomal deletion generating a transcriptional fusion. J. Bacteriol. 181, 6650–6655 (1999).
Zheng, J. et al. Genetic diversity and evolution of Salmonella enterica serovar Enteritidis strains with different phage types. J. Clin. Microbiol. 52, 1490–1500 (2014).
Inns, T. et al. A multi-country Salmonella Enteritidis phage type 14b outbreak associated with eggs from a German producer: 'near real-time' application of whole genome sequencing and food chain investigations, United Kingdom, May to September 2014. Euro Surveill. 20, 21098 (2015).
Drummond, A.J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).
Thomson, N. et al. The role of prophage-like elements in the diversity of Salmonella enterica serovars. J. Mol. Biol. 339, 279–300 (2004).
Rotger, R. & Casadesús, J. The virulence plasmids of Salmonella. Int. Microbiol. 2, 177–184 (1999).
Thomson, N.R. et al. Comparative genome analysis of Salmonella Enteritidis PT4 and Salmonella Gallinarum 287/91 provides insights into evolutionary and host adaptation pathways. Genome Res. 18, 1624–1637 (2008).
Cooke, F.J. et al. Prophage sequences defining hot spots of genome variation in Salmonella enterica serovar Typhimurium can be used to discriminate between field isolates. J. Clin. Microbiol. 45, 2590–2598 (2007).
Shah, D.H., Zhou, X., Kim, H.Y., Call, D.R. & Guard, J. Transposon mutagenesis of Salmonella enterica serovar Enteritidis identifies genes that contribute to invasiveness in human and chicken cells and survival in egg albumen. Infect. Immun. 80, 4203–4215 (2012).
Parkhill, J. et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413, 848–852 (2001).
Thiennimitr, P. et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA 108, 17480–17485 (2011).
Kingsley, R.A. et al. Genome and transcriptome adaptation accompanying emergence of the definitive type 2 host-restricted Salmonella enterica serovar Typhimurium pathovar. MBio 4, e00565–13 (2013).
Dhawi, A.A. et al. Adaptation to the chicken intestine in Salmonella Enteritidis PT4 studied by transcriptional analysis. Vet. Microbiol. 153, 198–204 (2011).
Langridge, G.C. et al. Patterns of genome evolution that have accompanied host adaptation in Salmonella. Proc. Natl. Acad. Sci. USA 112, 863–868 (2015).
Feasey, N.A. et al. Modelling the contributions of malaria, HIV, malnutrition and rainfall to the decline in paediatric invasive non-typhoidal Salmonella disease in Malawi. PLoS Negl. Trop. Dis. 9, e0003979 (2015).
Gordon, M.A. et al. Non-typhoidal Salmonella bacteraemia among HIV-infected Malawian adults: high mortality and frequent recrudescence. AIDS 16, 1633–1641 (2002).
Kotloff, K.L. et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case–control study. Lancet 382, 209–222 (2013).
Kariuki, S. & Onsare, R.S. Epidemiology and genomics of invasive nontyphoidal Salmonella infections in Kenya. Clin. Infect. Dis. 61 (Suppl. 4), S317–S324 (2015).
Betancor, L. et al. Genomic and phenotypic variation in epidemic-spanning Salmonella enterica serovar Enteritidis isolates. BMC Microbiol. 9, 237 (2009).
Harris, S.R. et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13, 130–136 (2013).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Holt, K.E. et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat. Genet. 40, 987–993 (2008).
Croucher, N.J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science 331, 430–434 (2011).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood–based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Ashton, P. et al. Revolutionising public health reference microbiology using whole genome sequencing: Salmonella as an exemplar. Preprint at. bioRxiv http://dx.doi.org/10.1101/033225 (2015).
Drummond, A.J., Suchard, M.A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Assefa, S., Keane, T.M., Otto, T.D., Newbold, C. & Berriman, M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969 (2009).
Carver, T.J. et al. ACT: the Artemis Comparison Tool. Bioinformatics 21, 3422–3423 (2005).
Zhou, Y., Liang, Y., Lynch, K.H., Dennis, J.J. & Wishart, D.S. PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352 (2011).
Page, A.J. et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693 (2015).
Kado, C.I. & Liu, S.T. Rapid procedure for detection and isolation of large and small plasmids. J. Bacteriol. 145, 1365–1373 (1981).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 (2012).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Tracy, B.S., Edwards, K.K. & Eisenstark, A. Carbon and nitrogen substrate utilization by archival Salmonella Typhimurium LT2 cells. BMC Evol. Biol. 2, 14 (2002).
Croucher, N.J. et al. Dominant role of nucleotide substitution in the diversification of serotype 3 pneumococci over decades and during a single infection. PLoS Genet. 9, e1003868 (2013).
Karp, P.D. et al. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief. Bioinform. 11, 40–79 (2010).
Salisbury, A.M., Bronowski, C. & Wigley, P. Salmonella Virchow isolates from human and avian origins in England—molecular characterization and infection of epithelial cells and poultry. J. Appl. Microbiol. 111, 1505–1514 (2011).
Acknowledgements
This work was supported by the Wellcome Trust. We would like to thank the members of the Pathogen Informatics Team and the core sequencing teams at the Wellcome Trust Sanger Institute (Cambridge, UK). We are grateful to D. Harris for work in managing the sequence data.
This work was supported by a number of organizations. The authors from the Wellcome Trust Sanger Institute were funded by Wellcome Trust award 098051. N.A.F. was supported by Wellcome Trust research fellowship WT092152MA. N.A.F., R.S.H., and this work were supported by a strategic award from the Wellcome Trust for the MLW Clinical Research Programme (101113/Z/13/Z). The authors from the Institut Pasteur were funded by the Institut Pasteur, by the Institut de Veille Sanitaire, and by the French government 'Investissement d'Avenir' program (Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence, grant ANR-10-LABX-62-IBEID). J.J. was supported by the antibiotic resistance surveillance project in the Democratic Republic of the Congo, funded by Project 2.01 of the Third Framework Agreement between the Belgian Directorate General of Development Cooperation and the Institute of Tropical Medicine (Antwerp, Belgium). S.K. was supported by NIH grant R01 AI099525-02. A.E.M. was supported by Wellcome Trust grant 098051 while at the Wellcome Trust Sanger Institute and by Biotechnology and Biological Sciences Research Council grant BB/M014088/1 at the University of Cambridge. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Study design: N.A.F., N.R.T., M.A.G., G.D., R.A.K., and J.P. Data analysis: N.A.F., N.R.T., J.H., T.J.D., T.F., L.B.B., P.W., L.L.-L., G.C.L., S.R.H., A.E.M., M.F., and M.A. Isolate acquisition and processing, and clinical data collection: N.A.F., K.H.K., J.J., X.D., C.M., S.K., C.A.M., R.S.O., F.-X.W., S.L.H., A.M.S., M.M., P.D., C.M.P., J. Cheesbrough, N.F., J. Campos, J.A.C., L.B., K.L.H., S.N., T.J.H., O.L., T.A.C., M.D.T., S.O.S., S.M.T., K.B., M.M.L., D.B.E., and R.S.H. Manuscript writing: N.A.F., J.H., N.R.T., and M.A.G. All authors contributed to manuscript editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Maximum-likelihood phylogeny placing representative isolates from the current study within the context of the diversity of S. Enteritidis genomes in the PHE collection.
Black taxa labels represent PHE 50-SNP cluster representatives, and red taxa labels represent 17 representative strains from this study. The scale bar shows mean number of substitutions per site.
Supplementary Figure 2 Maximum-likelihood phylogeny with strains of known phage type highlighted.
The scale bar shows mean number of substitutions per site.
Supplementary Figure 5 Distribution of prophage regions across the isolate collection highlighted.
Red indicates presence, and blue indicates absence. The scale bar shows mean number of substitutions per site.
Supplementary Figure 6 Maximum-likelihood phylogeny of S. Enteritidis plasmids.
The scale bar shows mean number of substitutions per site.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6 and Supplementary Note. (PDF 2555 kb)
Supplementary Tables 1–7
Supplementary Tables 1–7. (XLSX 110 kb)
Rights and permissions
About this article

Cite this article
Feasey, N., Hadfield, J., Keddy, K. et al. Distinct Salmonella Enteritidis lineages associated with enterocolitis in high-income settings and invasive disease in low-income settings. Nat Genet 48, 1211–1217 (2016). https://doi.org/10.1038/ng.3644
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3644
This article is cited by
-
Tackling non-typhoidal Salmonella with humility
Nature Microbiology (2024)
-
Geography shapes the genomics and antimicrobial resistance of Salmonella enterica Serovar Enteritidis isolated from humans
Scientific Reports (2023)
-
A Novel Strategy for Reducing Salmonella Enteritidis Cross-Contamination in Ground Chicken Meat Using Thymol Nanoemulsion Incorporated in Chitosan Coatings
Food and Bioprocess Technology (2023)
-
Highly drug resistant clone of Salmonella Kentucky ST198 in clinical infections and poultry in Zimbabwe
npj Antimicrobials and Resistance (2023)
-
Characterization of virulence factors of Salmonella isolated from human stools and street food in urban areas of Burkina Faso
BMC Microbiology (2021)
