
Abstract
Epigenetic deregulation is involved in acute myeloid leukemia (AML) pathogenesis and epigenetic targeting drugs are in clinical trial. Since the first results with histone-deacetylase inhibitors in AML are controversial, novel single and combined treatments need to be explored. It is tempting to combine chromatin-targeting drugs. SUV39H1, the main methyl-transferase for lysine 9 tri-methylation on histone H3, interacts with oncogenes involved in AML and acts as a transcriptional repressor for hematopoietic differentiation and immortalization. We report here that pharmacological inhibition of SUV39H1 by chaetocin induces apoptosis in leukemia cell lines in vitro and primary AML cells ex vivo, and that it interferes with leukemia growth in vivo. Chaetocin treatment upregulates reactive oxygen species (ROS) production as well as the transcription of death-receptor-related genes, in a ROS-dependent manner, leading to death receptor-dependent apoptosis. In addition to its direct inhibition by chaetocin, SUV39H1 is indirectly modulated by chaetocin-induced ROS. Accordingly, chaetocin potentiates other anti-AML drugs, in a ROS-dependent manner. The decryption of a dual mechanism of action against AML involving both direct and indirect SUV39H1 modulation represents an innovative read-out for the anticancer activity of chaetocin and for its synergy with other anti-AML drugs, suggesting new therapeutic combination strategies in AML.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) groups a number of diseases defined both by the presence of blasts in the bone marrow and deficient hematopoiesis. The prognosis is unfavorable with a long-term overall survival of <30% for patients younger than 60 years, and 10–20% for older patients, treated with conventional intensive chemotherapy.1, 2 Over the past few years, the interest of targeted therapies such as tyrosine-kinase pathway inhibition was pointed out, even though the clinical benefits are presently limited.3 Significant advance has been made with evidence that epigenetic regulation is involved in tumorigenesis.4, 5, 6 Notably, epigenetic alterations were identified as a driving phenomenon for AML and myelodysplastic syndromes.7, 8, 9 Strong evidence demonstrates that aberrant epigenetic regulation, such as silencing of tumor suppressor genes, is used by cancer cells to escape growth control and death mechanisms.10 Several drugs able to alter the epigenetic status of a cell have shown promising results for cancer treatment and DNA methyl-transferase inhibitors have been approved by the Food and Drug Administration and European Medicines Agency for the treatment of myelodysplastic syndromes.11, 12 Moreover, several histone-deacetylase inhibitors (HDACi) exhibit significant anti-tumor activity and acceptable toxicity profiles in in vitro, ex vivo and in vivo models, and are now approved or in clinical trials in monotherapy or in combination with others drugs.13 HDACi induces TNF-related apoptosis-induced ligand-dependent cell death,14, 15 thereby exerting tumor-selective death in myeloid leukemias. In addition to histone and non-histone acetylation, histone methylation has also been shown to be significant in the epigenetic control of gene expression through the establishment of stable gene-expression patterns. Histone tails can be mono- and di-methylated on arginine, and mono-, di- and tri-methylated on lysine residues. Lysine methylation can be linked to activation or repression of transcription, depending on the modified site. So far, more than 20 lysine- and 9 arginine-histone methyl-transferase (HMT) have been identified in humans, many of which are involved in leukemia such as the MLL1 fusion proteins and the human homolog of yeast Dot1.16
Despite the fact that HMTs are emerging as compelling targets for drug discovery efforts, particularly for the treatment of AML, only recently the investigation for selective inhibitors of these enzymes has begun. Direct inhibitors of HMTs have been reviewed recently.17
A specific inhibitor of lysine HMT has been described recently. Chaetocin is a fungal mycotoxin specifically inhibiting the variegation 3-9 homolog 1 (SUV39H1) enzyme.18 Chaetocin displayed anticancer properties against multiple myeloma via oxidative stress induction.19 SUV39H1 is the main HMT responsible for the accumulation of histone H3 containing a tri-methyl group at its lysine 9 position (H3K9me3) in heterochromatin. SUV39H1 interacts with proto-oncogenes such as AML1, EVI-1 and PML-RARα, which play critical roles in the development of AML, influencing the transcriptional repression of target genes involved in hematopoietic differentiation and bone marrow immortalization.20
We evaluated the pharmacological inhibition of SUV39H1 by chaetocin as a new potential anti-AML approach and explored the related mechanism(s) of action. Our results identify chaetocin as an anti-leukemia agent acting through a dual inhibitory mechanism that involves reactive oxygen species (ROS) production, and modulation of SUV39H1 activity in a ROS-dependent manner, and leads to death receptor-mediated cell death. We further highlight the interest of combining epidrugs for the treatment of AML, including SUV39H1 targeting agents.
Materials and methods
Reagents
Chaetocin, staurosporine and L-N-acetyl cysteine (L-NAC) were purchased from Sigma (St Louis, MO, USA); SAHA and MS-275 from Alexis Biochemical (Farmingdale, NY, USA); anti-CD34+ and anti-cytochrome c antibodies, as well as Z-VAD-fmk, Annexin V and 7-aminoactinomycin D (7-AAD) were from BD Pharmingen (San Jose, CA, USA); and carboxy-2′,7′-dichlorofluorescein was from Invitrogen (Carlsbad, CA, USA).
HMT assay
HMT analyses were performed according to the supplier's suggestions (BPS). Briefly, recombinant SUV39H1 or SUV39H1 cell extracts were incubated for 1 h at room temperature in 96-well plates pre-coated with histone H3 peptide substrate in 50 μl of HMT buffer containing S-adenosyl-methionine as the methyl donor in the presence or absence of chaetocin. Next, primary antibody, which recognizes methylated K9 residue of histone H3, was added. Finally, the plate was treated with a horseradish peroxidase-labeled secondary antibody, followed by addition of the horseradish peroxidase substrate to produce chemiluminescence measured using a chemiluminescence reader (Tecan, Lyon, France).
Cell culture, viability and apoptosis assay
Human leukemia cell line U937 was obtained from ATCC (Rockville, MD, USA) and was cultured in RPMI 1640 medium supplemented with 10% fetal calf serum (Gibco BRL, Villebon sur Yvette, France).
Since the toxicity of chaetocin on SL-2 insect cells was reported to be highly dependent on the initial cell density when chaetocin is added to the culture,18 exponentially growing cells were seeded at 2 × 106 cells per ml, followed by drug treatment for 24–72 h. Cell outgrowth was measured using the Cell Titer Glo Kit (Promega, Madison, WI, USA) using a FLUOstar optima luminometer (BMG Labtech, Ortenberg, Germany).
Annexin V/7AAD staining was carried out according to the manufacturer's protocol (BD Pharmingen). Briefly, after drug treatment, cells were washed with cold phosphate-buffered saline (PBS), resuspended with binding buffer (10 mM HEPES, pH 7.4, 140 mM, 2.5 mM CaCl2) before transferring 2 × 105 cells to 5 ml tube and adding 5 μl of Annexin V-fluorescein isothiocyanate and 5 μl of 7AAD. After 15 min incubation time in the dark, binding buffer (400 μl) was added to each tube and cells were analyzed using a Beckman Dickinson LSR II flow cytometer (Beckman Dickinson, Le Pont de Claix, France).
Western blotting
The ApoAlert Cell Fractionation Kit supplied by BD Bioscience Clontech (Palo Alto, CA, USA) was used according to the manufacturer's instructions. Briefly, cells were centrifuged at 600 g for 5 min at 4 °C and resuspended in 800 μl of fractionation buffer (BD Bioscience Clontech). Cells were passed 20 times through a 22-gauge needle and the homogenates were centrifuged at 700 g for 10 min and at 10 000 g for 25 min at 4 °C. Equal amounts of protein (15 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After immunoblotting with anti-cytochrome c antibodies, polyvinylidene fluoride membranes were stripped and reprobed with anti-β-tubulin, ensuring equal protein loading.
For histone detection, U937 cells were harvested and washed twice with ice-cold PBS 1 × . Then, cells were lysed in Triton Extraction Buffer (PBS containing 0.5% Triton X-100 (v/v), 2 mM phenylmethylsulfonyl fluoride, 0.02% (w/v) NaN3) at a cellular density of 107 cells per ml for 10 min on ice, with gentle stirring. After a brief centrifugation at 2000 r.p.m. at 4 °C, the supernatant was removed and the pellet was washed in half the volume of Triton Extraction Buffer and centrifuged before. The pellet was resuspended in 0.2 M HCl at a cell density of 4 × 107 cells per ml and acid extraction was proceeded overnight at 4 °C on rolling table. The day after the samples were centrifuged at 2000 r.p.m. for 10 min at 4 °C, the supernatant was removed and protein concentration was determined using the Bradford assay. About 10 μg of acid-extracted protein were loaded on 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose. The nitrocellulose was incubated with anti-mono-methyl- and tri-methyl-K9H3 (Abcam, Paris, France) and the analyses were performed according to the supplier's suggestions. Histone H4 and H3 (Abcam) were used as equal loading control.
Transfection, miRNA interference
The BLOCK-iT Pol II miR RNAi Expression Vector Kits (Invitrogen) was used to clone microRNA (miRNA) sequences contained within the engineered pre-miRNA into pcDNA 6.2-GW/EmGFP-miR expression vector to generate expression clone pcDNA.rSUV39h1-miRs (miR SUV). Briefly, Invitrogen's RNAi design algorithm on line as the miRNA target finder and design tool (http://rnaidesigner.invitrogen.com/rnaiexpress/) was used for designing the SUV39h1 pre-miRNA sequences. We selected three human candidate inserts, which derived from SUV39H1 mRNA sequences, to minimize possible nonspecific reactivity with target sequences. These inserts targeting the SUV39H1 mRNA were cloned into an expression vector plasmid to construct recombinant plasmid pcDNA.rSUV39h1-miR-1, -2 and -3 (miR SUV1, SUV2, SUV3). As a control, miR C including no insert targeting SUV39H1 mRNA was constructed, too. Recombinant plasmid constructs were verified by DNA sequencing. Plasmid purification kit was purchased from Qiagen (Valencia, CA, USA), and the miRNA forward sequencing primer (5′-GGCATGGACGAGCTGTACAA-3′) and reverse sequencing primer (5′-CTCTAGATCAACCACTTTGT-3′) were provided by Invitrogen.
miR C/SUV or dominant-negative FADD (FADD-DN)/mut constructions were transfected into U937 cells via Nucleofector Technology, as recommended by the manufacturer (Amaxa GmbH, Köln, Germany). Briefly, 5 × 106 cells were resuspended in 100 μl Cell Line Nucleofector Solution V and nucleofected with 2 μg of each vector using program V-001, which are preprogrammed into the Nucleofector device (Amaxa GmbH). Following nucleofection, the cells were immediately mixed with 500 μl of pre-warmed RPMI 1640 cell culture medium and transferred into six-well plates containing 1.5 ml RPMI 1640 medium per well. Cells were incubated at 37 °C for 24 h before analysis.
Cell cycle analysis
The cells were harvested and resuspended in staining solution containing RNAse A, propidium iodide (50 μg/ml), sodium citrate (0.1%) and NP40 (0.1%) in PBS 1 × for 30 min in the darkness. Cell cycle distribution was assessed with a FACScalibur flow cytometer using the Cell Quest software (Becton Dickinson, Milan, Italy) and analyzed by standard procedures using the same software and the ModFit LT version 3 software (Verity, Topsham, ME, USA).
Analysis of caspases activity
The activities of individual caspase were determined using commercially available kits. Cells were homogenized in Lysis Buffer (BioVision, Mountain View, CA, USA) on ice. After centrifugation, the supernatants were collected for a further analysis of caspase activity. Caspase-8 and -9 activities were determined using a Fluorometric Assay Kit (BioVision), whereas the activity of caspase-3/7 was determined using luminescence Caspase-Glo Assays (Promega) according to the manufacturer's instruction.
Low-density array-based real-time reverse transcriptase-PCR
Total RNA was isolated from cultured cells using Trizol reagent, as recommended by the manufacturer (Invitrogen). Reverse transcriptase reaction was performed using 2 μg of total RNA and Moloney murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA, USA). The resulting cDNA was then quantified using the TaqMan method.
For TaqMan low-density array (Applied Biosystems), primer pairs for 17 TNFSF ligands, 24 receptors and several control genes were incorporated into a low-density array (Assay on Demand; Applied Biosystems), and developed as described previously.21 Briefly, 5 μl cDNA (equivalent to 100 ng of total RNA) was mixed with TaqMan Universal Mix (Applied Biosystems) and loaded into one sample port. Thermal cycler conditions were as follows: 2 min at 50 °C, 15 s at 95 °C and 60 s at 60 °C for 40 cycles. Capture of fluorescence was recorded on the ABI Prism 7900HT scanner, and the CT was calculated for each assay using the Sequence Detection System software 2.1 (Applied Biosystems). Normalization of quantitative-polymerase chain reaction (PCR) assays was conducted using the CT value of the GAPDH endogenous control. Samples were then converted to a fold change ratio described using standard ΔCT formula, where ΔCT=CT target−CT average endogenous controls. Thereafter, ΔΔCT values were calculated by subtracting the ΔCT value of each target from the ΔCT of the calibrator (untreated samples). Clustering of quantitative-PCR data was conducted by Pearson's correlation and visualized using the program TIGR Multiexperiment Viewer (http://www.tigr.org/software/tm4/mev.html).
PCR was carried out using 25 μl cDNA and 20 μl of reaction mix containing 12.5 μl of TaqMan universal PCR Master Mix (Roche, Rosny sous Bois, France) (dATP, dCTP, dGTP and dUTP, MgCl2, AmpliTaqGold, Amperase uracil-N-glycosylase), 200 nM of TaqMan probes corresponding to the studied gene, 400 nM of each primer and 50 nM of GAPDH primers and TaqMan probe. TaqMan probes and primers were purchased from Applied Biosystems. Data were collected using an ABI Prism 7900 HT Sequence Detection System (Applied Biosystems) for 40 cycles (95 °C for 15 s, 60 °C for 1 min) after an initial step of 50 °C for 2 min and 95 °C for 10 min. The relative amount of mRNA was calculated as described above. All data were generated in triplicate and expressed as mean±s.d.
ROS analysis
To measure intracellular ROS levels, cells grown at 70% confluence were collected and suspended in PBS. The oxidation-sensitive fluorescent probe dihydroethidium (Invitrogen) was added at 5 μM. After 30 min of incubation under protection from light, the cell suspension was subjected to flow cytometry analysis.
Patient samples
Fresh AML cells were obtained from bone marrow of 27 patients upon informed consent, using Ficoll–Hypaque density-gradient centrifugation. Blasts were immediately cryopreserved in fetal calf serum supplemented with 10% methyl sulfoxide. All patients were diagnosed according to WHO classification at the Haematology Department of Institut Paoli Calmettes (Marseille, France). Leukemias were characterized in terms of morphology (French–American–British classification), karyotype, immunophenotyping and FLT3 gene mutation (internal tandem duplication or tyrosine kinase domain mutation). Bone marrow samples contained more than 80% leukemic blasts after processing. This project has been reviewed and approved by the Internal Review Board of Institut Paoli Calmettes, in accord with the Declaration of Helsinki.
In vivo mice experiments
Xenograft study using U937 cells were performed in NOD/Shi-scid IL2rgamma(null)-severe combined immunodeficiency disease mice maintained under specific pathogen-free conditions in individual ventilated cages with acidified water. Cells (0.2 × 106 in 100 μl) were injected intravenously (retro orbital vein) in anesthetized animals. Injected mice were randomly assigned to receive chaetocin (0.5 mg/kg) or vehicle (5% dimethyl sulfoxide in H2O) intraperitoneally for 2 weeks (eight doses). All animal procedures were performed in accordance with protocols approved by the local committee for Animal Experimentation (Comité d’Etique de Marseille pour l’expérimentation animale). At 16 days after injection, mice were killed and the femurs and spleens were collected. Bone marrow and spleen cells were collected and subjected to flow cytometry. Samples were mixed with mouse immunoglobulin G to block nonspecific staining and were incubated with an appropriate volume of indicated antibodies for 30 min on ice. The mixture was depleted of erythrocytes and was fixed in Lyzis Solution (BD Pharmingen). U937 human cells were examined by double staining with fluorescein isothiocyanate-conjugated anti-human CD45 antibody (Beckman Coulter, Marseille, France) and allophycocyanin-conjugated anti-mouse CD45 antibody (Cliniscience, Richboro, PA, USA).
Synergy determination
Synergy between chaetocin and arabinoside C (AraC) or Vorinostat was assessed using an experimentally determined constant molar ratio of chaetocin and AraC (1:40) and of chaetocin and Vorinostat (1:20). To establish the most effective concentration range for each agent, dose–response curves were generated and effective dose 50 values were calculated using nonlinear regression analysis. In subsequent experiments, cells were treated with serial dilutions of each drug individually and with both drugs simultaneously in a fixed molar ratio. The Calcusyn software (Biosoft, Cambridge, UK) was used to determine whether any synergy existed between the agents using the Chou and Talalay method,22 which takes into account both potency (effective dose 50) and the shape of the dose–effect curve. Accordingly, the combination index (CI) was calculated and analyzed as follows: CI<1, CI=1 and CI>1 indicate synergism, additive effect and antagonism, respectively.
Supplementary Materials and methods for the Supplementary figures accompany the paper on the leukemia website (http://www.nature.com/leu).
Results
SUV39H1 inhibition induces caspase-dependent apoptosis in human leukemia cells
As reported previously, the fungal mycotoxin chaetocin induces sub-micromolar inhibition of recombinant SUV39H1 activity in vitro (Figure 1a and Greiner et al.18), at which doses it can selectively decrease global histone H3-K9 tri- but not mono-methylation in U937 leukemia cell cultures (Figure 1b and Greiner et al.18) or K562 cell culture (Supplementary Figure 1A) after 24 h of treatment. Localized inhibition of H3-K9 tri-methylation was also observed by chromatin immunoprecipitation, followed by PCR amplification of p15INK4B and E-cadherin promoter region (Supplementary Figure 1B), as described previously.23 Chaetocin treatment resulted in cell cycle arrest in the S phase (Figure 1c), and dose-dependent impairment of cell viability (Figure 1d), which was also highly dependent on the initial cell density when chaetocin is added to the culture (data not shown), in agreement with previous reports.18 The consequences of the inhibition of SUV39H1 activity or expression on cell viability was further analyzed. Chaetocin treatment of U937 cells induced dose-dependent Annexin V staining, with 100 nM chaetocin inducing Annexin V staining in more than 50% of treated cells (Figure 1e), consistent with Annexin V staining being an early marker of cell death. Similarly, the knockdown of SUV39H1 expression by three distinct miRNA sequences (miR SUV 1–3) was always associated with increased Annexin V staining, as compared with either control or scrambled miRNA constructs (Figure 1f). Of note, Annexin V staining levels inversely correlated with the efficiency of SUV39H1 knockdown (Figure 1f, compare lower panel to upper panel), supporting the existence of a causal link between SUV39H1 expression and/or activity, and cell survival. The impact of the SUV39H1 miRNA construct on global H3K9 tri-methylation was also verified (Supplementary Figure 1C). Consistent with Annexin V staining results, chaetocin treatment induced executioner caspase-3/7 activity in a dose-dependent manner (Figure 2a) as well as the activation of both the caspase-8 and -9 pathways (Figure 2b), and accumulation of cytosolic cytochrome c as evidence of mitochondrial damage (Figure 2c). Inhibition of caspase activity by the general caspase inhibitor Z-VAD-fmk demonstrated the essential requirement for caspase activation in chaetocin-induced apoptosis (Figure 2d). In addition to U937 cells, chaetocin also activated caspase-3/7 activity in other leukemia cell lines (for example, HL60 and NB4; see Supplementary Figure 2A), which was associated with caspase activity-dependent apoptosis (Supplementary Figure 2B).

Inhibition of SUV39H1 activity and expression induces leukemia cell death by apoptosis. (a) Chaetocin induces sub-micromolar inhibition of SUV39H1 HMT activity. Recombinant SUV39H1 (2 ng in lanes 2–5) was incubated in HMT buffer containing histone H3 peptide substrate and S-adenosyl-methionine as the methyl donor in the presence of cheatocin (500 nM and 1000 nM in lanes 4 and 5) or diluent (lane 3), followed by HMT assay. Methylated K9 residue of the histone H3 peptide was quantified by enzyme-linked immunosorbent assay. Baseline level is indicated as the level of methylated K9 residue of the histone H3 peptide in the absence of recombinant SUV39H1 (lane 1). (b) Chaetocin reduces cellular H3K9me3 levels. U937 cells were exposed for 6 or 24 h with vehicle or chaetocin (500 and 1000 nM). Histones were extracted and H3K9me1, H3K9me3 and H3 levels were analyzed by western blotting, as indicated. Band intensity was quantified using ImageJ, and normalized to H3 levels in untreated cells. The resulting values are indicated. (c) Chaetocin induces cell cycle arrest in the S phase. U937 cell were treated for 24 h with vehicle or chaetocin (500 nM); cell-cycle profiles were analyzed by flow cytometry. Bars represent the mean±s.d. (d) Chaetocin induces dose-dependent reduction of cell viability. U937 cells (1 × 106/ml) were incubated with chaetocin (0–1000 nM) for 24 h and apoptosis was assessed using CellTiter Glo (Promega). Bars represent the mean±s.d. (e) Chaetocin induces dose-dependent apoptosis. U937 cells were incubated with chaetocin (0–1000 nM) for 24 h and apoptosis was assessed by flow cytometry using Annexin V/7AAD staining. Bars represent the mean±s.d. (f) SUV39H1 cellular depletion induces apoptosis. U937 cells were co-transfected with SUV39H1-myc and specific SUV39H1 (miR SUV1, miR SUV2, miR SUV3) or control (miR C) green fluorescence protein (GFP) encoding miRNA constructs. Reduced SUV39H1 expression was verified by western blotting and compared with cells transfected with irrelevant miR constructs (LACZ, Afad2015, upper panel). Apoptosis was determined in GFP-positive cells by Annexin V/7AAD staining (mid-panel). Representative results from four independent experiments are shown. Bars represent the mean±s.d. with standard deviation (lower panel). *P<0.05.

Chaetocin induces caspase-dependent apoptosis. (a) Chaetocin induces caspase-3/7 activity. U937 cells were treated for 24 h with chaetocin, in the presence or absence of Z-DEVD-FMK caspase-3 inhibitor (20 μM), as indicated. Caspase-3/7 activity was assessed using a proluminescent caspase-3/7 DEVD-aminoluciferin substrate; results are presented as relative light unit (RLU). Bars represent the mean±s.d. (b) Chaetocin induces both caspase-8 and -9 activities. U937 cells were treated for 24 h with chaetocin (100 nM), caspase-8 and 9 activities were assessed using pro-fluorescent LETD and LEHD tetrapeptide sequences, respectively. Results are presented as percent fluorescent cells. Bars represent the mean±s.d. (c) Western blot analysis of cytochrome c release. U937 cells were treated for 8 h with chaetocin (500 nM) or staurosporine (5 μM), as indicated, and cytoplasmic proteins were analyzed. Ratio indicates relative levels as determined after normalization to β-tubulin. (d) Inhibition of caspase activity prevents chaetocin-induced cell death. U937 cells were treated for 24 h with chaetocin (500 nM) in the presence or absence of Z-VAD-fmk (20 μM); cell viability was determined by excluding Annexin V- and 7AAD-positive cells. Results shown are representative of three independent experiments. Bars represent the mean±s.d.
Chaetocin-mediated apoptosis requires the death receptor pathway
To gain mechanistic insights into chaetocin-induced apoptosis, the transcriptional modulation of 48 pro-apoptotic and housekeeping genes by chaetocin treatment was next explored by time-course real-time PCR using TaqMan low-density array. As shown in Figure 3a, 100 nM chaetocin treatment induced and/or upregulated death receptor signaling molecule mRNAs, such as that of Fas, FasL, TNF-related apoptosis-induced ligand, DR4 and TL1A, as well as of the cell cycle inhibitor p21/WAF1, which correlated in a time-dependent manner with cell cycle arrest and caspase-dependent apoptosis induced by similar doses of chaetocin (Figures 1 and 2).15, 24, 25 Despite the induction of p21/WAF1 mRNA and protein levels in chaetocin-treated cells (Figure 3a and Supplementary Figure 3A) and the abrogation of Entinostat-induced G1 arrest,15 stable knockdown of p21/WAF1 expression did not prevent chaetocin-induced apoptosis (Supplementary Figure 3B and C). In contrast, the sensitivity to 50–200 nM chaetocin-induced apoptosis was strongly reduced in cells expressing a FADD-DN, but not in cells expressing an inactive, mutated form (DN-FADD Mut). Entinostat-induced apoptosis was similarly antagonized by the FADD-DN construct, as reported previously (Figure 3b).15

Chaetocin-induced apoptosis requires the TNF death receptor pathway. (a) TaqMan low-density Aaray analysis of U937 cells treated with chaetocin (100 nM) for 6–24 h. Results are presented as expression ratios relative to the internal control (GAPDH) transcript levels. Normalized expression data were log 2 transformed and scaled to solvent (dimethyl sulfoxide)-treated cells given an arbitrary value of 1. Cluster analyses show these values and are scaled from −3 (green) to 5 (red). Black indicates no change. Hierarchical clustering was processed with the TIGR Multiexperiment Viewer software using Pearson’s correlation. (b) U937 cells expressing the FADD-DN construct protein are resistant to chaetocin-induced apoptosis. U937 cells were transfected with pB-FADD-DN-WT or pB-FADD-DN-MUT, followed by treatment for 24 h with chaetocin (50–200 nM) or Entinostat (2.5 μM). Percent cell viability was determined by excluding Annexin V- and 7AAD-positive cells. Results shown are representative of three independent experiments. Bars represent the mean±s.d. Transfection efficiencies were >50% of GFP-positive cells, as determined using the pMax construct.
Modulation of chaetocin-induced apoptosis and SUV39H1 activity by ROS
Activation of the death receptor pathway and preliminary evidence of mitochondrial damage, such as caspase-9 activation and the cytosolic accumulation of cytochrome c, led us to investigate the induction of oxidative stress in chaetocin-treated U937 cells. Flow cytometry assays showed a dose-dependent induction of ROS by treatment with 20–500 nM chaetocin (Figure 4a), which was fully prevented by co-treatment with L-NAC, a ROS scavenger (Figure 4b). Thus, chaetocin-induced ROS correlated in a time-dependent manner with both chaetocin-induced cell-death, L-NAC blockade of chaetocin-induced apoptosis (Figure 4c) and cell cycle arrest in the S phase (Figure 4d).

Chaetocin-induced apoptosis and cell cycle arrest require oxidative stress induction. (a) Intracellular accumulation of ROS induced by chaetocin (50–1000 nM) for 24 h is prevented by L-NAC (5 mM) co-treatment. Intracellular ROS accumulation was determined by carboxy-2′,7′-dichlorofluorescein staining and flow cytometry. Results are presented as fold induction of mean fluorescence intensity, relative to untreated cells, and are representative of three independent experiments. (b) Inhibition of ROS production prevents chaetocin-induced cell death. U937 cells were treated for 24 h with chaetocin (20–1000 nM) in the presence or absence of L-NAC (5 mM); cell viability was determined by excluding Annexin V- and 7AAD-positive cells. Results shown are representative of three independent experiments. Bars represent the mean±s.d. (c) Modulation of cell cycle progression by chaetocin (500 nM) and L-NAC (5 mM) co-treatment for 24 h; cell cycle profiles were analyzed by flow cytometry. Histograms represent the percentage of cells in different phases of the cell cycle, as indicated. Bars represent the mean±s.d.
Co-treatment with L-NAC inhibited chaetocin-induced upregulation of mRNA encoding for p21/WAF1, death receptors and ligands (Supplementary Figures 4A and B), strongly suggesting that ROS production preceded and/or was required for the modulation of these targets by chaetocin. Consistently, L-NAC prevented chaetocin modulation of H3-K9 tri-methylation (Figure 5a), and partially restored SUV39H1 inhibition by chaetocin (Figure 5b). The fact that L-NAC restored chaetocin-mediated H3-K9 tri-methylation downregulation in intact cells, yet only partially prevented cell-derived SUV39H1 inhibition by chaetocin in the in vitro methylation assay, suggests that modulation of H3-K9 tri-methylation by chaetocin is not exclusively dependent on SUV39H1 activity. Of particular interest, basal cell-derived SUV39H1 activity was upregulated by L-NAC treatment, thus suggesting that ROS levels modulate SUV39H1 methyl-transferase activity.

Inhibition of ROS production modulates SUV39H1 activity. (a) U937 cells were treated for 24 h with chaetocin (500–1000 nM) in the presence or absence of L-NAC (5–20 mM), histones were extracted and anti-H3K9me3 levels were analyzed by western blotting. Loading of the histones was assessed by anti-H3 western blotting. Values represent relative H3K9me3 levels after quantification and normalization to anti-H3 levels. (b) U937 cells were incubated for 24 h with chaetocin (1000 nM) and NAC (20 mM); nuclear extracts were used to examine HMT activity. Results are presented as percent activity. Bars represent the mean±s.d. *P<0.005; **P<0.002; ***P<0.001.
Chaetocin-induced apoptosis in primary acute myeloid leukemia samples and in in vivo xenograft models
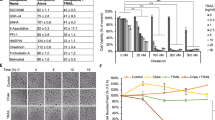
The impact of chaetocin on apoptosis was also studied ex vivo in primary blasts from individuals with AML (Table 1). Of the 27 AML cultures that were analyzed, more than 96% responded to chaetocin treatment by undergoing apoptosis, in a dose-dependent manner, for chaetocin concentration ranging from 100 to 1000 nM (Figure 6a). However, there was no correlation with the pre-treatment variables (that is, French–American–British classification, white blood cell at diagnosis, cytogenetics or molecular findings) or the initial response to induction chemotherapy (Figure 6b). Notably, although extensive apoptosis was induced in AML blasts, CD34+ normal myeloid progenitor samples from four normal donors showed moderate sensitivity to 1000 nM chaetocin treatment (Figure 6c). These data suggested that chaetocin-based treatment of AML affected blasts predominantly as compared with normal cells. Early CD34+/CD38− progenitor cells, sorted and compared with the CD33+ blasts of the same patient, showed comparable sensitivity to chaetocin treatment (Figure 6d).

Chaetocin induces anti-leukemia activity ex vivo and in vivo. (a) Unsorted leukocytes obtained from 27 patients with AML (Table 1) were cultured ex vivo and treated with chaetocin (100–1000 nM) for 24 h; cell viability was determined by excluding Annexin V- and 7AAD-positive cells (***P<0.0001). (b) Hierarchical clustering of data presented in (a), using the TIGR Multiexperiment Viewer software and Euclidean distance. The color code is indicated on the right. (c) CD34+ hematopoietic progenitor cells of four healthy donors were cultured ex vivo and treated for 24 h with chaetocin (1000 nM); percent viable cells was determined by excluding Annexin V- and 7AAD-positive cells and compared with the 27 AML patients blasts analyzed in (a) (**P<0.001). (d) The CD38−/CD34+ early progenitors and CD33+ blasts from the same patient were sorted and cultured ex vivo. Leukocytes were treated for 24 h with chaetocin (20–200 nM); percent apoptosis was determined by Annexin V and 7AAD staining. (e) In vivo experimental design. U937 cells were injected intravenously to NOD/Shi-scid IL2rgamma(null) mice, followed by intraperitoneal injection of chaetocin (0.5 mg/kg) for 16 days. At day 16, bone marrow (BM), blood and spleen were collected and processed for cytofluorometry. (f) Unsorted BM leukocytes from mock- and chaetocin-treated animals were analyzed by anti-human (h) and anti-mouse (m) CD45 staining and cytofluorimetry. One representative FACScan is presented for each group of animals. (h) Percent anti-human CD45+-positive cells in BM leukocytes from mock- and chaetocin-treated animals, with control isotypic staining (*P<0.01).
To assess the anti-leukemic activity of chaetocin in vivo, NOD/Shi-scid IL2rgamma(null)-severe combined immunodeficiency disease mice were retro-orbitally injected with U937 cells, and were treated three times weekly with intra-peritoneal administration of chaetocin (Figure 6e). The mice which were killed 16 days post-xenograft displayed a normalized weight of 96%, and 91% of their initial weight, for control mice and chaetocin-treated mice, respectively. The bone marrow and blood of each mouse were examined by flow cytometry. Human U937 xenografted cells were discriminated from the mouse leukocytes by human CD45+ versus mouse CD45+ staining (Figure 6f). Preliminary in vivo experiments demonstrated the anti-proliferative activity of chaetocin on U937 cells with human CD45+-labeled cells reduced from 47 to 30% in the bone marrow of NOD/Shi-scid IL2rgamma(null)-severe combined immunodeficiency disease mouse xenografts (Figure 6g). In all, <0.1% human CD45+-labeled U937 cells could be detected in the blood of control and chaetocin-treated animals (data not shown).
Evidence of the efficiency of combination therapy
The relevance of chaetocin treatment in combination therapies was next explored together in association with Vorinostat, a potent HDACi (for a review see Marks and Breslow26), and AraC, an important drug used against AML (for a review see Robak and Wierzbowska27). The anti-leukemic action of chaetocin was potentiated by both Vorinostat (Figure 7a; for a review see Marks and Breslow26), a potent HDACi, and AraC (Figure 7b), an important drug used against AML (for a review see Robak and Wierzbowska27). The optimal molar ratio for these drug combinations was determined experimentally using the CellTiter Glo assay (Figures 7a and b). To establish the most effective concentration range for each agent, dose–response curves were generated and effective dose 50 values (the concentration required to kill 50% of the cells) were calculated using nonlinear (sigmoidal) regression analysis. In subsequent experiments, cells were treated with serial dilutions of each drug individually and with both drugs simultaneously in a fixed ratio. Analysis of the combined drug effects was made using the median effect method.22 Briefly, this analysis involved plotting the dose–response curves for each drug and multiple diluted fixed-ratio combinations, and then calculation of the CI. As shown in Figure 7, synergy (CI<1) was observed for the combination of chaetocin with AraC in the full range of tested drug concentrations (Figure 7c), and additivity (CI≈1) to synergism was observed for the combination of chaetocin with Vorinostat, depending on the drug concentrations (Figure 7d).

Combination therapy of chaetocin with chemotherapeutic drugs in vitro. (a, b) U937 cells (1 × 106/ml) were incubated for 48 h in the presence of chaetocin (0.4 μM–6.25 nM) and (a) AraC (16–0.25 μM), or (b) Vorinostat (8–0.125 μM) by serial twofold dilutions of each drug individually or drugs combinations at a constant equipotent ratio, as indicated. Cell viability was determined using CellTitreGlo; results are presented as relative light units. Bars represent the mean±s.d. (c, d) The existence of synergy between chaetocin and AraC or Vorinostat was evaluated using the median-effect principle proposed by Chou and Talalay.22 Calculations of the CI were performed with the CalcuSyn software. The CI plot showing values <1 occurred for all tested drug combinations of chaetocin with AraC, indicating synergism for this drug combination (c), whereas CI were <1 or close to 1 for a wide range of drug concentrations for the combination of chaetocin with Vorinostat, indicating synergism or additive effect (d). (e) U937 cells (1 × 105/ml) were treated for 48 h in the presence of chaetocin (1–5 nM), Vorinostat (1 μM), AraC (50 nM) and NAC (5 mM), as indicated. Cell viability was determined using CellTitreGlo; results are presented as percent viable cells. Bars represent the mean±s.d. (f) Schematic representation of chaetocin-induced apoptosis in AML cells. Inhibition of Suv39H1 by chaetocin upregulates pro-apoptotic TNF superfamily members (TNF-related apoptosis-induced ligand, FasL and TL1A) as well as the cell cycle inhibitor p21/WAF1, and induces a cell cycle blockade and apoptosis. Chaetocin-induced apoptosis can be blocked by dominant-negative FADD TNF receptor and caspase inhibition, but not by p21/WAF1 knock down. Downregulation of Suv39H1 expression similarly induces apoptosis. Chaetocin also induces ROS production, which in turn can modulate Suv39H1 activity, upregulate pro-apoptotic TNF family members and p21/WAF1, and induce cell blockade and apoptosis. Inhibition of ROS production by L-NAC prevents all these chaetocin-mediated effects, except for the modulation of Suv39H1 activity, which is only partial.
Following seeding U937 cells at low cell density (0.1 × 106 cells per ml) in 96-well plates to increase the sensitivity of cells to chaetocin as described previously,18 even very low concentration of chaetocin (1–5 nM) potentiated Vorinostat and AraC anti-leukemic action, which was prevented by L-NAC co-treatment (Figure 7e). The fact that L-NAC specifically prevented the potentiating effect of chaetocin on both Vorinostat or AraC anti-leukemic activity strongly supports an essential role of oxidative stress in chaetocin-mediated action.
Discussion
The involvement of epigenetic alterations in the pathogenesis of AML has been suggested 14 years ago,28, 29 and epigenetic targeting drugs have significant clinical activity against AML and high-risk myelodysplasia (myelodysplastic syndromes). DNA methyl-transferase inhibitors show an overall response rate ranging from 40 to 60%, but with low incidence of complete remission and a high rate of progression even during treatment. Interestingly, these results seem independent of cytogenetic risk factors and are associated with low toxicity, allowing a broader use of these therapies even in elderly patients.12 Of note, results with HDACi, used in monotherapy against AML, have been disappointing.30, 31 Two main clinical issues remain to be addressed for the use of epidrugs: response rate optimization and incorporation into curative strategies. Combination therapies using DNA methyl-transferase inhibitors and other epidrugs, such as HDACi, are currently investigated with promising results,32 such as the Vorinostat-based combination treatment in high-risk myelodysplastic syndromes.33 However, in AMLs, the data are limited and the results controversial.31, 34
Clearly, new approaches are needed. So far, several lysine and arginine methyl-transferases35, 36 have been identified in humans, many of which have been associated with cancerogenesis.37, 38, 39 Targeting HMT in cancer may represent a promising approach. Indeed, the involvement of HMTs recruited to regulate the transcription of hematopoietic genes such as EVI-1, AML1 and PML-RARα during leukemogenesis would be in favor of the use of an HMT-targeted therapy.40, 41, 42 In support of the HMT-based therapy, aberrant gene silencing results from increased SUV39H1 occupancy of target promoter regions.23 SUV39H1 downregulation inhibits the colony formation activity of EVI-1-expressing cells, underlining a role for SUV39H1 in EVI-1-mediated bone marrow immortalization and leukemogenesis.20 Apoptosis induction following SUV39H1 knockdown further supports an important role for this enzyme in leukemic cell survival (Figure 1f).
The fact that chaetocin can induce apoptosis of leukemia cells, similarly to SUV39H1 knockdown, suggests both that SUV39H1 is a potential molecular target for cancer treatment and that pharmacological inhibition of SUV39H1 could be used against cancer. Indeed, chaetocin displayed anti-leukemic activity in vitro, in AML blasts ex vivo and in mice xenograft assays in vivo. The clear significance of ROS generation for chaetocin activity (Figure 5) suggests a dual action of chaetocin both via direct modulation of SUV39H1 activity18 and ROS production. The fact that (i) chaetocin has been reported to be a competitive inhibitor of thioredoxine (Trx) for Trx reductase activity (TrxR1);43 (ii) TrxR/Trx pathway is central in limiting ROS production; and (iii) chaetocin may exert anticancer effects via ROS generation (Isham et al.19 and this study) indicates that TrxR1 inhibition by chaetocin may at least partially explain ROS-mediated anticancer activity. However, this hypothesis does not exclude the role of the direct inhibition of SUV39H1 in chaetocin-induced apoptosis. Indeed, as is the case of chaetocin treatment, depletion of SUV39H1 induces apoptosis (Figure 1f), indicating that SUV39H1 represents an important target in AML. Given that L-NAC only partially restored the inhibition of SUV39H1 by chaetocin (Figure 5b), whereas apoptosis induction by chaetocin is fully abolished by L-NAC (Figure 4b), indicates that ROS production might in part be indirectly responsible of the enzymatic inhibition of chaetocin. An attractive hypothesis to explain this ‘indirect’ modulation of SUV39H1 by chaetocin-produced ROS relies on its recently described potential to target the hypoxia-inducible factor-1α/p300 interaction by a zinc ejection mechanism.44 The SUV39-type methyl-transferases such as SUV39H1 all share a C-terminal region bearing three conserved cysteine residues essential for the HMT activity.45, 46 The cysteine-rich domain contributes to S-adenosyl-L-methionine (AdoMet/SAM) cofactor and peptide substrate binding by forming a zinc binding site in conjunction with a conserved cysteine near the active site.47 The selective SUV39H1 inhibition by chaetocin supports this hypothesis. Indeed, chaetocin has been identified as a specific inhibitor of SUV39-type methyl-transferases, with no effect on HMT such as E(z), PRSET7 or SET7/9 that do not belong to the SU(VAR)39 class of enzymes, and lack the C-terminal cysteine-rich domain.18 The drop in SUV39H1 activity observed with thiol-reactive substances such as DTT at high doses18 also supports this type of mechanism. Of course, the ‘direct’ mechanism of chaetocin action on SUV39H1 remains to be defined by structural analysis. Given that it has been reported that chaetocin competes with the S-adenosyl-methionine methyl donor cofactor, whose binding site is formed in part by the cysteine-containing C-terminal part of SUV39H1,18 this competition might impact both directly on SUV39H1 activity and indirectly on the cysteine residues exposition to oxidation by ROS. Only a detailed structural analysis of the mechanism of action of chaetocin on SUV39H1 will better define its dual activity.
Accumulating evidence suggests that, compared with their normal counterparts, many types of cancer cells have increased levels of ROS.48 A moderate increase in ROS levels can promote cell proliferation and differentiation, whereas excessive amounts of ROS can cause oxidative damage to lipids, proteins and DNA. Therefore, manipulating ROS levels may selectively kill cancer cells, yet under persistent intrinsic oxidative stress, many cancer cells become resistant to such treatments. Several studies have indicated that modulation of ROS may influence the pathogenesis and treatment of AML.49, 50, 51 Chaetocin both induces and requires ROS production for its anti-leukemia activity, which might represent relevant insight for anticancer approaches. This anticancer action also relies on the activation of the death receptor pathway in cancer cells, as strongly suggested by the interfering action of caspase inhibitors (Figure 2), as well as molecularly by the FADD-DN blockade (Figure 3b). Although ROS production seems to be the causal element of regulation, the exact connection between ROS and death receptors remains to be defined. The fact that chaetocin combined with an HDACi (Vorinostat) or the chemotherapy agent AraC improves the anticancer activity of the single treatment highlights the interest of developing combination therapies targeting these mechanisms. Given that this complementation is counteracted by L-NAC-, chaetocin-related compounds might provide a distinct, ROS-dependent, alternative strategy for the treatment of AML, including for patients resistant to HDACi or demethylating agents.
Altogether, our results propose chaetocin-related targeting of SUV39H1 as a new therapeutic strategy in AML and underline the role of SUV39H1 activity in AML pathogenesis. Yet, variable responses to chaetocin treatment were noted among 27 analyzed cultures without correlation with pre-treatment variables. Future studies will be important to delineate the molecular basis of the observed variable responses and to establish the specific contribution of chaetocin molecular targets to the described anti-leukemia activity.
References
Goldstone AH, Burnett AK, Wheatley K, Smith AG, Hutchinson RM, Clark RE . Attempts to improve treatment outcomes in acute myeloid leukemia (AML) in older patients: the results of the United Kingdom Medical Research Council AML11 trial. Blood 2001; 98: 1302–1311.
Rowe JM, Neuberg D, Friedenberg W, Bennett JM, Paietta E, Makary AZ et al. A phase 3 study of three induction regimens and of priming with GM-CSF in older adults with acute myeloid leukemia: a trial by the Eastern Cooperative Oncology Group. Blood 2004; 103: 479–485.
Sanz M, Burnett A, Lo-Coco F, Lowenberg B . FLT3 inhibition as a targeted therapy for acute myeloid leukemia. Curr Opin Oncol 2009; 21: 594–600.
Eden A, Gaudet F, Waghmare A, Jaenisch R . Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003; 300: 455.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW et al. Induction of tumors in mice by genomic hypomethylation. Science 2003; 300: 489–492.
Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005; 37: 391–400.
Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O’Keefe C et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009; 113: 1315–1325.
Figueroa ME, Skrabanek L, Li Y, Jiemjit A, Fandy TE, Paietta E et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood 2009; 114: 3448–3458.
Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X et al. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol 2010; 28: 605–613.
Boultwood J, Wainscoat JS . Gene silencing by DNA methylation in haematological malignancies. Br J Haematol 2007; 138: 3–11.
Gurion R, Vidal L, Gafter-Gvili A, Belnik Y, Yeshurun M, Raanani P et al. 5-Azacitidine prolongs overall survival in patients with myelodysplastic syndrome—systematic review and meta-analysis. Haematologica 2010; 95: 303–310.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 2010; 28: 562–569.
Altucci L, Clarke N, Nebbioso A, Scognamiglio A, Gronemeyer H . Acute myeloid leukemia: therapeutic impact of epigenetic drugs. Int J Biochem Cell Biol 2005; 37: 1752–1762.
Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med 2005; 11: 71–76.
Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P et al. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 2005; 11: 77–84.
Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM et al. hDOT1L links histone methylation to leukemogenesis. Cell 2005; 121: 167–178.
Cole PA . Chemical probes for histone-modifying enzymes. Nat Chem Biol 2008; 4: 590–597.
Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A . Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat Chem Biol 2005; 1: 143–145.
Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC . Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 2007; 109: 2579–2588.
Goyama S, Nitta E, Yoshino T, Kako S, Watanabe-Okochi N, Shimabe M et al. EVI-1 interacts with histone methyltransferases SUV39H1 and G9a for transcriptional repression and bone marrow immortalization. Leukemia 2010; 24: 81–88.
Vire B, de Walque S, Restouin A, Olive D, Van Lint C, Collette Y . Anti-leukemia activity of MS-275 histone deacetylase inhibitor implicates 4-1BBL/4-1BB immunomodulatory functions. PLoS One 2009; 4: e7085.
Chou TC, Talalay P . Quantitative analysis of doseeffect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55.
Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR . Reexpression of epigenetically silenced AML tumor suppressor genes by SUV39H1 inhibition. Oncogene 2010; 29: 576–588.
Castellano R, Vire B, Pion M, Quivy V, Olive D, Hirsch I et al. Active transcription of the human FASL/CD95L/TNFSF6 promoter region in T lymphocytes involves chromatin remodeling: role of DNA methylation and protein acetylation suggest distinct mechanisms of transcriptional repression. J Biol Chem 2006; 281: 14719–14728.
Cherrier T, Suzanne S, Redel L, Calao M, Marban C, Samah B et al. p21(WAF1) gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 2009; 28: 3380–3389.
Marks PA, Breslow R . Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 2007; 25: 84–90.
Robak T, Wierzbowska A . Current and emerging therapies for acute myeloid leukemia. Clin Ther 2009; 31: 2349–2370.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB . Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996; 93: 9821–9826.
Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood 1998; 91: 2985–2990.
Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 2008; 112: 981–989.
Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008; 111: 1060–1066.
Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res 2006; 66: 6361–6369.
Silverman LR, Verma A, Odchimar-Reissig R, Cozza A, Najfeld V, Licht JD et al. A phase I/II study of vorinostat, an oral histone deacetylase inhibitor, in combination with azacitidine in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). Initial results of the phase I trial: A New York Cancer Consortium. J Clin Oncol 2008; 26 (Suppl): abstract 7000.
Raffoux E, Labarthe A, Cras A, Recher C, Turlure P, Marolleau JP et al. Epigenetic therapy with 5-azacitidine, valproic acid, and ATRA in patients with high-risk AML or MDS: results of the French VIVEDEP Phase II study. 50th ASH Annual Meeting 2008.
Qian C, Zhou MM . SET domain protein lysine methyltransferases: structure, specificity and catalysis. Cell Mol Life Sci 2006; 63: 2755–2763.
Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S . Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther 2007; 113: 50–87.
Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 2003; 100: 11606–11611.
Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008; 14: 355–368.
Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008; 10: 1431–1439.
Cattaneo F, Nucifora G . EVI1 recruits the histone methyltransferase SUV39H1 for transcription repression. J Cell Biochem 2008; 105: 344–352.
Carbone R, Botrugno OA, Ronzoni S, Insinga A, Di Croce L, Pelicci PG et al. Recruitment of the histone methyltransferase SUV39H1 and its role in the oncogenic properties of the leukemia-associated PML-retinoic acid receptor fusion protein. Mol Cell Biol 2006; 26: 1288–1296.
Chakraborty S, Sinha KK, Senyuk V, Nucifora G . SUV39H1 interacts with AML1 and abrogates AML1 transactivity. AML1 is methylated in vivo. Oncogene 2003; 22: 5229–5237.
Tibodeau J, Benson L, Isham C, Owen W, Bible K . The anticancer agent chaetocin is a competitive substrate and inhibitor of thioredoxin reductase. Antioxid Redox Signal 2009; 11: 1097–1106.
Cook KM, Hilton ST, Mecinovic J, Motherwell WB, Figg WD, Schofield CJ . Epidithiodiketopiperazines block the interaction between hypoxia-inducible factor-1alpha (HIF-1alpha) and p300 by a zinc ejection mechanism. J Biol Chem 2009; 284: 26831–26838.
Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000; 406: 593–599.
Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU et al. Structure of the neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell 2002; 111: 117–127.
Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU et al. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell 2003; 12: 177–185.
Trachootham D, Alexandre J, Huang P . Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 2009; 8: 579–591.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010; 17: 225–234.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010; 115: 2749–2754.
Callens C, Coulon S, Naudin J, Radford-Weiss I, Boissel N, Raffoux E et al. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. J Exp Med 2010; 207: 731–750.
Acknowledgements
We thank Anne-Odile Hueber for providing the FADD-DN construct, Valérie Depraetere-Ferrier for critical reading and Yves Toiron and Nadège Delacourt for invaluable help for studies on synergism. CH was supported by a MRT fellowship, and CR and GS by fellowships from INCa and PT by the Foundation Monahan; AN was supported by EU (Contract No. 518417). This work was supported by funds from the INSERM, ANRS, INCA (Institut National de Recherche conre le Cancer), ARC (Association de Recherche contre le Cancer) and AIRC (Associazione Italiana per la ricerca contro il cancro, EU (APOSYS 200767 and LSHC-CT2005-518417).
Author contributions
HC performed and designed the research, analyzed data and wrote the manuscript. AN performed and designed the research, and analyzed data. RC, SG and AR performed the research and analyzed data. TP/NV/LA and YC designed the research, analyzed data and wrote the manuscript. LA and YC share senior authorship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Leukemia website
Rights and permissions
About this article
Cite this article
Chaib, H., Nebbioso, A., Prebet, T. et al. Anti-leukemia activity of chaetocin via death receptor-dependent apoptosis and dual modulation of the histone methyl-transferase SUV39H1. Leukemia 26, 662–674 (2012). https://doi.org/10.1038/leu.2011.271
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2011.271
Keywords
This article is cited by
-
SUV39H1 regulates the progression of MLL-AF9-induced acute myeloid leukemia
Oncogene (2020)
-
ROS-mediated inactivation of the PI3K/AKT pathway is involved in the antigastric cancer effects of thioredoxin reductase-1 inhibitor chaetocin
Cell Death & Disease (2019)
-
The fungal metabolite chaetocin is a sensitizer for pro-apoptotic therapies in glioblastoma
Cell Death & Disease (2019)
-
The histone H3K9 methyltransferase SUV39H links SIRT1 repression to myocardial infarction
Nature Communications (2017)
-
Chaetocin reactivates the lytic replication of Epstein-Barr virus from latency via reactive oxygen species
Science China Life Sciences (2017)

{kind=link}
{kind=link}
{kind=link}
{kind=link}