
Abstract
Duchenne muscular dystrophy (DMD) is an X chromosome-linked lethal muscular disorder with progressing muscle wasting and weakness caused by mutations in the gene encoding a subsarcolemmal protein dystrophin. For a long time, there was no effective cure; however, advances in molecular biology have allowed the development of radical treatment approaches. Among them, exon-skipping therapy using antisense oligonucleotides is very promising, because it corrects the reading frame of the dystrophin-encoding gene and restores protein expression, resulting in the conversion of DMD to a clinically milder form, Becker muscular dystrophy (BMD). However, clinical trials in exon-skipping therapy did not provide satisfactory results, which may be attributed to inefficient exon skipping, low expression level of restored dystrophin and inadequate methods of muscle function evaluation. To date, exon-skipping approaches have particularly focused on the correction of the gene-reading frame. However, the problem is that the relationship between the resultant and expected phenotypes in terms of definite symptomatic improvement has not yet been elucidated. In other words, previously conducted clinical trials have not been planned based on the comprehensive assessment of genotype–phenotype relationship in BMD, which demonstrates a broad range of symptom severity depending on the functional activity of the truncated dystrophin. The analysis I present in this review strongly suggests that the development of exon-skipping therapy and its clinical trials should be based on large-cohort studies of BMD.
Similar content being viewed by others
Introduction
Muscular dystrophies are mostly hereditary disorders clinically manifested by progressive muscle wasting and weakness, and pathologically by muscle degeneration and necrosis.1 Among them, Duchenne muscular dystrophy (DMD) is the most severe and has the highest incidence. DMD is manifested as walking abnormality, usually in 2- to 5-year-old boys, which develops into whole-body skeletal muscle atrophy and weakness, and inability to walk before the 16th birthday. This condition is further aggravated by progressive deterioration of the respiratory and heart muscles, and the patients finally die of pulmonary or cardiac failure around the age of 30 years. Recently, it has been suggested that increased levels of serum creatine kinase may be a diagnostic marker of DMD in the asymptomatic stage.
DMD is caused by a mutation in the DMD gene encoding a subsarcolemmal protein dystrophin, which is localized in the Xp21 locus and is one of the longest human genes spanning 2500 kb and comprising 79 exons. Dystrophin is expressed not only in skeletal and cardiac muscles but also in smooth muscles and the central nervous system. This protein has a rod-shaped structure consisting of an actin-binding domain, a rod domain consisting of 24 spectrin-like repeat motifs, a cysteine-rich domain interacting with dystroglycan and sarcoglycan complexes, and a C-terminal domain. Dystrophin binds to dystroglycan, sarcoglycans and syntrophin, forming the dystrophin-associated glycoprotein complex, which creates a link between the cytoplasm and basement membrane through the plasma membrane of the muscle fibers. The dystrophin-associated glycoprotein complex provides membrane stabilization during muscle contraction and it has been considered to have a role in signal transduction from the extracellular space to the cell cytoplasm.2
The lack of dystrophin may cause vulnerability of the muscle plasma membrane to mechanical stress, which increases intracellular calcium concentration, activating calcium-dependent proteases such as calpain3 and inducing various proinflammatory chemokines and cytokines,4 which ultimately result in muscle degeneration and necrosis. Abnormal expression of neuronal nitric oxide synthase, aquaporin 4, sodium ion channels, L-type calcium ion channels and stretch-activated channels at the sarcolemma is suggested to be associated with the pathological mechanism underlying DMD progression.3 The resulting deregulation of degeneration–regeneration cycles in the muscle leads to increased fibrogenesis and replacement of muscle tissue with fibrous and fatty tissues.
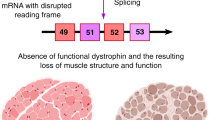
In the DMD gene, exons 3–8 and 45–55 are identified as mutational hotspot regions subjected to missense and nonsense substitutions, as well as deletions, insertions and duplications.5 The disruption of the open-reading frame by out-of-frame mutations blocks dystrophin expression, leading to DMD phenotype. However, nonsense mutations creating a premature stop codon or in-frame deletions result in the production of a truncated dystrophin, which translates into a milder disease phenotype called Becker muscular dystrophy (BMD),6 in which the severity of symptoms differs depending on the length, structure, and function of truncated dystrophin. The described genetic mechanism of muscular dystrophy, called the ‘reading frame theory,’ explains phenotypic differences in 90% of patients with dystrophinopathy.7
In recent decades, advances in the management of cardiopulmonary functions in DMD patients extended their life expectancy by more than 10 years. However, although corticosteroid therapy with rehabilitation maintains motor function,8 it does not stop the progression of the disease; in addition, the long-term use of corticosteroids can cause various adverse effects. Therefore, other treatment approaches are required.
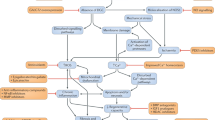
Some major therapeutic strategies based on the correction of dystrophin deficiency are shown in Figure 1. They can be directed at the recovery or complementation of dystrophin expression, which can be achieved through genetic manipulations, including exon skipping,9 gene therapy with viral vector10 and read-through therapy11 or cell transplantation.12 Other strategies are directed against the consequence of dystrophin deficiency such as membrane stabilization by copolymers,13 control of increased calcium concentration,14 reduction of inflammation and proteolysis by appropriate drugs,15, 16 inhibition of fibrosis17 and promotion of muscle regeneration.18, 19 In addition, approaches related to concomitant pathogenic mechanisms, such as utrophin induction20 or modulation of neuronal nitric oxide synthase,21 have also been proposed. Among the listed treatment strategies, genetic therapies aimed to restore dystrophin expression are considered the most promising. In this review, I summarize challenges of exon-skipping therapy and propose measures to ensure its successful implementation.

Pathogenic mechanism of muscular dystrophy and corresponding therapeutic strategies. Dystrophin deficiency makes the muscle membrane vulnerable to mechanical stress, resulting in an increase of cytosolic calcium concentration through calcium channels and activation of calcium-dependent proteases such as calpain, as well as secretion of pro-inflammatory mediators, which ultimately leads to muscle degeneration and necrosis. Muscle degeneration–regeneration cycles are impaired in the course of the disease and muscles are subsequently replaced by fibrous and adipose tissues. Radical approaches aimed at the restoration of dystrophin expression include exon skipping, gene therapy using viral vectors and read-through therapy. Palliative approaches aimed at the alleviation of dystrophin deficiency symptoms include the following: (1) reduction of membrane damage by membrane stabilizer copolymers, (2) downregulation of cytosolic calcium concentration by a calcium channel blocker TRPV2, (3) decrease of inflammation by corticosteroids and anti-inflammatory drugs and (4) reduction of fibrogenesis by angiotensin-converting enzyme. Regenerative therapies using stem cells are also extensively investigated. A full color version of this figure is available at the Journal of Human Genetics journal online.
Development of exon-skipping therapy
A number of evidences indicate significant clinical potential of exon-skipping therapy in DMD.6, 22, 23, 24 In particular, revertant fibers are rare ‘sporadic/spontaneously arising’ dystrophin-positive fibers that are observed in dystrophic muscles of DMD patients; this is explained by reading frame correction through skipping of the mutated exon or surrounding exon(s) in the hotspot region and subsequent recovery of dystrophin expression.22, 23 In addition, dystrophin-deficient phenotypes are diverse, ranging from severe DMD (total absence of dystrophin) to BMD and even asymptomatic cases characterized by the expression of truncated dystrophin proteins with different lengths.6, 24 Considering that the advances in the development of artificial nucleic acids resulted in the generation of highly efficient and relatively non-toxic antisense oligonucleotides (AOs) to be used for exon skipping,24 the exon-skipping therapy can be a promising strategy to treat DMD.
AOs are chemically synthesized DNA fragments containing ~20 bases that are designed to bind to complementary sequences of the target pre-mRNA. Various artificial chemicals have been developed to overcome DNA and RNA instability, and to achieve high levels of specific affinity, nuclease resistance and safety, as well as to simplify fabrication. Among them, 2′-O-methyl-phosphorothioate AO (2′OMeAO) and phosphorodiamidate morpholino oligomers (PMOs) exhibit the best characteristics.24, 25
Alternative splicing occurs in the spliceosome complex consisting of several proteins, small nuclear RNA and mRNA of the exon–intron consensus boundaries, and cis-elements, including exon-splicing enhancer, which recognizes and removes introns from pre-mRNA. AOs combine targeted exon–intron boundaries or exon-splicing enhancer and inhibit spliceosome coupling. In general, AOs can be designed using exon-splicing enhancer Finder (http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home); however, they may not necessarily induce efficient exon skipping25 and the skipping effect may be different in vitro and in vivo. Therefore, the AO design is not straightforward and should be optimized based on the prediction of the pre-mRNA secondary structure.26
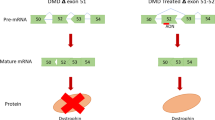
Hybridization of AOs to targeted exons induces artificial skipping of the mutated exon or surrounding exons and corrects the open-reading frame in the process of pre-mRNA splicing to mRNA, thus restoring dystrophin expression (Figure 2).24 This approach is applicable to ~90% of DMD patients, as more than one exon deletion can be targeted.13 Compared with gene therapy using viral vectors or stem cell transplantation, AOs can be regarded as drugs and ethical hurdles may be reduced.

Principle of exon 51-skipping therapy in DMD patients carrying exon 52 deletion. Deletion of exon 52 disrupts the open-reading frame of the dystrophin-encoding pre-mRNA, abrogating dystrophin expression (a). Antisense oligonucleotides (AOs) targeting exon 51 induce exon 51 skipping and correct the reading frame, restoring the expression of dystrophin in its truncated form. Accordingly, exon-skipping therapy is expected to convert a severe DMD phenotype to mild dystrophinopathy (b).
Preclinical studies of exon-skipping therapies
In preclinical studies of exon skipping, several DMD animal models have been employed,27 including dystrophin-deficient mdx mice28, 29 with a nonsense mutation in exon 23 treated with 2′OMeAO30, 31, 32, 33, 34 and PMO,35, 36 and dystrophic dogs37, 38, 39, 40, 41 with a mutation of a splice donor site in intron 6 treated with PMO targeting exons 6 and 8.42 All these studies achieved a certain degree of success in the restoration of dystrophin expression in skeletal muscles of the whole body but not in cardiac muscle. This constituted a serious flaw in AO design, considering the increase in the death rate of DMD patients from heart failure and lethal arrhythmia in recent years due to a decrease in respiratory failure-associated fatalities achieved by the mechanical ventilators.43 Therefore, a highly efficient cell-penetrating peptide-linked PMO that was able to restore dystrophin expression in both skeletal and cardiac muscles has been developed.44
Exon-skipping therapy is aimed at correcting gene mutations occurring in a small population; accordingly, the development of personalized pharmaceuticals is expensive, limiting the application of this treatment approach. Therefore, efforts are focused on the whole hotspot mutation region in the DMD gene to make exon-skipping therapy available for many DMD patients. The number of patients with mutations in the exon 45–55 hotspot region constitutes ~60% of DMD patients carrying gene deletions.45 According to the UMD–DMD database (http://www.UMD.be/DMD/), ~13% of DMD patients could be treated by exon-skipping therapy targeting exon 51.46 In addition, a preclinical study on exon 51-skipping therapy using PMO showed its efficiency in restoring dystrophin expression in skeletal muscles of exon 52-deficient mdx mice, which was accompanied by the improvement of muscle function and pathology.47, 48, 49 Furthermore, ~10% of DMD patients can be treated by exon 53-skipping therapy.50 The other exons that could be potentially targeted by exon skipping and related mutations are shown in Table 1.
Clinical trials for exon-skipping therapies
When four DMD patients with deletions in exons 48–50, 49–50, 50 or 52 were subjected to exon 51-skipping treatment, intramuscular administration of exon 51-targeting 2′OMeAO resulted in the recovery of dystrophin expression and improved magnetic resonance imaging findings in parts of the treated muscles.51 Clinical trials of drisapersen (2′OMeAO)52 and AVI-4658 (PMO)53 targeting exon 51 showed their potential as disease-modifying drugs. However, a phase III clinical trial of drisapersen conducted by BioMarin Pharmaceutical, Inc. (San Rafael, CA, USA) was ceased at the end because of the lack of the effect.54 On the other hand, phase III trials of eteplirsen (PMO) performed by Sarepta Therapeutics, Inc. (Cambridge, MA, USA) confirmed the therapeutic potential of the drug, which was subsequently approved by the U.S. Food and Drug Administration.55
Regarding exon 53 skipping, National Centre of Neurology and Psychiatry (Tokyo, Japan), and Nippon Shinyaku Co., Ltd (Kyoto, Japan), which have developed exon 53 NS-065/NCNP-01, have completed the first early exploratory clinical trial in humans (http://www.ncnp.go.jp/tmc/pressrelease_03.html). The study confirmed the restoration of dystrophin mRNA reading frame and dystrophin expression in patients receiving high drug doses without serious adverse events and the following next-phase trials are being planned. Phase I/II trials of SRP-4053 (Sarepta Therapeutics, Inc.)56 and PRO053 (BioMarin Pharmaceutical, Inc.) for exon 53 skipping are being currently conducted. Furthermore, phase I SRP-4045 (Sarepta)56 and phase I/II BMN045 (BioMarin) trials for exon 45 skipping, and a phase I/II trial of BMN044 (BioMarin) for exon 44 skipping are under development.
Problems and prospects of exon-skipping therapy for DMD
The first problem is that the Tmax (the time when the rate of absorption equals the rate of elimination) and half-life of PMOs are short: 1 h and ~1·5 h, respectively, and plasma clearance is fast. In animal models, the duration of PMO effects was 2~3 months and repeated administration was required for stable expression of dystrophin. To overcome these issues, the next-generation AO chemicals should be developed.57
The second problem is that skipping efficiency demonstrates tissue- and cell-specific differences as evidenced by significantly lower induction of dystrophin by 2′OMeAO and PMO in the heart compared with skeletal muscles. Considering that many DMD patients die of cardiac complications, the improvement of dystrophin induction efficiency in the myocardium is of paramount importance. So far, it has been considered that the intracellular uptake of AOs occurs via passive migration through the leaky membrane of dystrophin-deficient muscle cells. However, a recent study revealed that PMO intracellular delivery is dependent on the developmental stage of myogenesis rather than on diffusion through dystrophin-lacking membranes of muscle cells.58 On the other hand, cell-penetrating peptide peptide-linked PMO, which has an amphiphilic nature, can be transported into muscle by endocytosis through scavenger receptor class A after self-assembly into nanoparticles.59 This mechanism may be a key to improving the drug delivery potential of PMOs not only in skeletal but also in cardiac muscle.
The third problem is that the number of patients carrying DMD mutations within exons 45–55 is limited and a single exon-skipping approach is not practical. Some studies have reported that patients with deletions in exons 45–55 consistently present very mild or asymptomatic phenotypes.45, 60, 61 Exons 45–55 cover the mutation hotspot region and skipping an entire exon 45–55 stretch may be applied to 60% of patients with DMD45 and severe BMD.24, 62 Thus, multi-exon (45–55) skipping therapy conducted in exon 52-deficient mdx mice resulted in skipping of all targeted exons and restoration of dystrophin expression, which improved muscle function and alleviated pathology.63 Beside exons 45–55, deletion of exons 45–48 and 45–51 also conferred mild clinical phenotypes compared with that of exons 45–47 or 45–49;64 these variations in disease phenotypic severity could be explained by different structures of truncated dystrophin proteins.65 Moreover, some patients carrying a deletion of exons 3–9, which covers the second mutation hotspot region comprising exons 3–7, had mild or asymptomatic conditions even at the old age and multi-exon 3–9 skipping could be applicable to ~7% of patients with DMD.66
PRoblems of phenotypic evaluation after exon-skipping therapy for DMD
To objectively evaluate post-therapeutic muscle function recovery, revision of the current test system is strongly recommended. Thus, at present the motor activity of DMD patients is assessed by the 10 m run, 6 min walk test and the North Star Ambulatory Assessment. The reliability and validity of the 10 m run, 6 min walk test and North Star Ambulatory Assessment have been confirmed in a multi-center longitudinal study,67 and the tests were employed in clinical studies.68 However, these conventional evaluation methods have limitations, especially for children with confounding factors, because they are usually performed at the time of the patient’s visit to the hospital, which makes the examination less frequent and discontinuous, and subjective to patient’s physical and emotional conditions. In this respect, it has been proposed that the physical activity should be monitored on a daily basis using a continuously worn actigraph, which would record consecutive daily energy expenditure, muscle strength, and walking distance. Such data would more objectively reflect the changes in patient motor function in the natural environment69, 70 and, thus, would present a more reliable indicator of the treatment outcome.
It is also critically important to perform comprehensive analysis of the functional activity of the truncated dystrophin expressed owing to exon skipping. The development of exon-skipping therapy and its clinical trials have been focused on the restoration of the gene reading frame rather than on the resulting converted phenotype, which has not been adequately evaluated. For example, exon 51 skipping applicable for certain gene deletions (Table 1) is predicted to accomplish phenotypic conversion from DMD to BMD; however, the severity of the resultant phenotype has not been confirmed in previously reported cases. As BMD has a broad phenotypic spectrum, the objective results of clinical trials for exon-skipping therapies would be fully warranted by collecting and evaluating extensive clinical data on the severity of the transformed phenotype.
There are a number of nationwide databases on DMD, such as UMD–DMD (http://www.UMD.be/DMD/), Leiden Open Variation Database (http://www.dmd.nl/nmdb/home.php), TREAT-NMD DMD Global database (http://umd.be/TREAT_DMD/) or Registry of Muscular Dystrophy (http://www.remudy.jp/). Although these databases are very useful for pediatricians and neurologists, the phenotypic classification is rough: DMD, intermediate muscular dystrophy and BMD, whereas the detailed information on the clinical course of BMD is missing. To ensure the success of clinical trials for exon-skipping therapy in DMD, I strongly suggest that large-cohort studies on the genotype–phenotype relationship in BMD and its long-term clinical history should be conducted. Based on these data, further clinical trials of exon-skipping therapy would be planned and implemented.
In summary, palliative treatments such as corticosteroid therapy are limited to the improvement of muscle function and patient’s prognosis; therefore, genetic therapies such as exon-skipping therapy aimed at the restoration of functional dystrophin expression are required. However, the development of exon-skipping therapies and their conventional clinical trials have not been planned based on the comprehensive assessment of genotype–phenotype relationship in BMD. Considering that BMD demonstrates a broad range of symptom severity depending on the functional activity of the truncated dystrophin, large-cohort studies of BMD would be the first step for moving towards successful exon-skipping therapy for DMD.
References
Hoffman, E. P., Brown, R. H. & Kunkel, L. M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Sunada, Y. & Campbell, K. P. Dystrophin-glycoprotein complex: molecular organization and critical roles in skeletal muscle. Curr. Opin. Neurol. 8, 379–384 (1995).
Yeung, E. W., Whitehead, N. P., Suchyna, T. M., Gottieb, P. A., Sachs, F. & Allen, D. D. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 562, 367–380 (2005).
Porter, J. D., Khanna, S., Kaminski, H. J., Rao, J. S., Merriam, A. P., Richmonds, C. R. et al. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 11, 263–272 (2002).
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H. & Kunkel, L. M. An explanation for the phenotype differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1998).
Koenig, M., Beggs, A. H., Moyer, M., Scherpf, S., Heindrich, K. & Bettecken, T. el al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 45, 498–506 (1989).
Tuffery-Giraud, S., Beroud, C., Leturcq, F., Yaou, R. B., Hamroun, D., Michel-Calemard, L. et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 30, 934–945 (2009).
Bushby, K., Finkel, R., Birnkrant, D. J., Case, L. E., Clemens, P. R., Cripe, L. et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9, 77–93 (2010).
Yokota, T., Pistilli, E., Duddy, W. & Nagaraju, K. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert. Opin. Biol. Ther. 7, 831–842 (2007).
Okada, T. & Takeda, S. Current challenges and future directions in recombinant AAV-mediated gene therapy of Duchenne muscular dystrophy. Pharmaceuticals (Basel) 6, 813–836 (2013).
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifillis, P. et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–92 (2007).
Bajek, A., Porowinska, D., Kloskowski, T., Brzoska, E., Ciemerych, M. A. & Drewa, T. Cell therapy in Duchenne muscular dystrophy treatment: clinical trials overview. Crit. Rev. Eukaryot. Gene Expr. 25, 1–11 (2015).
Houang, E. M., Haman, K. J., Filareto, A., Perlingeiro, R. C., Bates, F. S., Lowe, D. A. et al. Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev. 2, 15042 (2015).
Iwata, Y., Katanosaka, Y., Arai, Y., Shigekawa, M. & Wakabayashi, S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum. Mol. Genet. 18, 824–834 (2009).
Ermolova, N. V., Martinez, L., Vetrone, S. A., Jordan, M. C., Roos, K. P., Sweeney, H. L. et al. Long-term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul. Disord. 24, 583–595 (2014).
Childers, M. K., Bogan, J. R., Bogan, D. J., Greiner, H., Holder, M., Grange, R. W. et al. Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of duchenne muscular dystrophy. Front. Pharmacol. 2, 89 (2012).
Morales, M. G., Cabrera, D., Céspedes, C., Vio, C. P., Vazquez, Y., Brandan, E. et al. Inhibition of the angiotensin-converting enzyme decreases skeletal muscle fibrosis in dystrophic mice by a diminution in the expression and activity of connective tissue growth factor (CTGF/CCN-2). Cell Tissue Res. 353, 173–187 (2013).
Apolinário, L. M., De Carvalho, S. C., Santo Neto, H. & Marques, M. J. Long-term therapy with omega-3 ameliorates myonecrosis and benefits skeletal muscle regeneration in Mdx mice. Anat. Rec. (Hoboken) 298, 1589–1596 (2015).
Hayashiji, N., Yuasa, S., Miyagoe-Suzuki, Y., Hara, M., Ito, N., Hashimoto, H. et al. G-CSF supports long-term muscle regeneration in mouse models of muscular dystrophy. Nat. Commun. 6, 6745 (2015).
Ricotti, V., Spinty, S., Roper, H., Hughes, I., Tejuram, B., Robinsonm, N. et al. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-Arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with Duchenne muscular dystrophy. PLoS ONE 11, e0152840 (2016).
Messina, S., Bitto, A., Vita, G. L., Aguennouz, M., Irrera, N., Licata, N. et al. Modulation of neuronal nitric oxide synthase and apoptosis by the isoflavone genistein in Mdx mice. Biofactors 41, 324–329 (2015).
Wilton, S. D., Dye, D. E., Blechynden, L. M. & Laing, N. G. Revertant fibres: a possible genetic therapy for Duchenne muscular dystrophy? Neuromuscl. Disord. 7, 329–335 (1997).
Crawford, G. E., Lu, Q. L., Partridge, T. A. & Chamberlain, J. S. Suppression of revertant fibers in mdx mice by expression on a functional dystrophin. Hum. Mol. Genet. 10, 2745–2750 (2001).
Nakamura, A. & Takeda, S. Exon-skipping therapy for Duchenne muscular dystrophy. Neuropathology 29, 494–501 (2009).
Echigoya, Y., Mouly, V., Garcia, L., Yokota, T. & Duddy, W. In silico screening based on predictive algorithms as a design tool for exon skipping of oligonucleotides in Duchenne muscular dystrophy. PLoS ONE 10, e0120058 (2015).
Nakamura, A. & Takeda, S. Mammalian models of Duchenne muscular dystrophy: pathological characteristics and therapeutic applications. J. Biomed. Biotechnol. 2011, 184393 (2011).
Bulfield, G., Siller, W. G., Wight, P. A. & Moore, K. J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA 81, 1189–1192 (1984).
Tanabe, Y., Esaki, K. & Nomura, T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 79, 91–95 (1986).
Dunckley, M. G., Manoharan, M., Villiet, P., Eperon, I. C. & Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 7, 1083–1090 (1998).
Mann, C. J., Honeyman, K., Cheng, A. J., Ly, T., Lloyd, F., Fletcher, S. et al. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl Acad. Sci. USA 98, 42–47 (2001).
Lu, Q. L., Mann, C. J., Lou, F., Bou-Gharios, G., Morris, G. E., Xue, S. A. et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 9, 1009–1014 (2003).
Lu, Q. L., Rabinowitz, A., Chen, Y. C., Yokota, T., Yin, H., Alter, J. et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl Acad. Sci. USA 102, 198–203 (2005).
Wells, K. E., Fletcher, S., Mann, C. J., Wilton, S. D. & Wells, D. J. Enhanced in vivo delivery of antisense oligonucleotides to restore dystrophin expression in adult mdx mouse muscle. FEBS Lett. 552, 145–149 (2003).
Fletcher, S., Honeyman, K., Fall, A. M., Harding, P. L., Johnsen, R. D. & Wilton, S. D. Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 8, 207–216 (2006).
Alter, J., Lou, F., Rabinowitz, A., Yin, H., Rosenfeld, J., Wilton, S. D. et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 12, 175–177 (2006).
Cooper, B. J., Winand, N. J., Stedman, H., Valentine, B. A., Hoffman, E. P., Kunkel, L. M. et al. The homologue of the Duchenne locus in defective in X-linked muscular dystrophy of dogs. Nature 334, 154–156 (1988).
Valentine, B. A., Cooper, B. J., De Lahunta, R., Valentine, B. A., Hoffman, E. P., Kunkel, L. M. et al. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J. Neurol. Sci. 88, 69–81 (1988).
Sharp, N. J. H., Kornegay, J. N., van Camp, S. D., Herbstreith, M. H., Secore, S. L., Kettle, S. et al. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics 13, 115–121 (1992).
Shimatsu, Y., Katagiri, K., Furuta, T., Nakura, M., Tanioka, Y., Yuasa, K. et al. Canine X-linked muscular dystrophy in Japan (CXMDJ . Exp. Anim. 52, 93–97 (2003).
Shimatsu, Y., Yoshimura, M., Yuasa, K., Urasawa, N., Tomohiro, M., Nakura, M. et al. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ . Acta Myol. 24, 145–154 (2005).
Yugeta, N., Urasawa, N., Fujii, Y., Yoshimura, M., Yuasa, K., Wada, M. R. et al. Cardiac involvement in Beagle-based canine X-linked muscular dystrophy in Japan (CXMDJ: electrocardiographic, echocardiographic, and morphologic studies. BMC Cardiovasc. Disord. 6, 47 (2006).
Yokota, T., Lu, Q. L., Partridge, T., Kobayashi, M., Nakamura, A., Takeda, S. et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol. 65, 667–676 (2009).
Curran, F. J. & Colbert, A. P. Ventilator management in Duchenne muscular dystrophy and postpoliomyelitis syndrome: twelve years’ experience. Arch. Phys. Med. Rehabil. 70, 180–185 (1989).
Jearawiriyapaisarn, N., Moulton, H. M., Buckley, B., Roberts, J., Sazani, P., Fucharoen, S. et al. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscle of mdx mice. Mol. Ther. 16, 1624–1629 (2008).
Beroud, C., Tuffery-Giraud, S., Matsuo, M., Hamroun, D., Humbertclaude, V., Monnier, N. et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 28, 196–202 (2007).
Bladen, C. L., Salgado, D., Monges, S., Foncuberta, M. E., Kekou, K., Kosma, K. et al. The TREAT-NMD DMD Global Database: analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36, 395–402 (2015).
Araki, E., Nakamura, K., Nakao, K., Kameya, S., Kobayashi, O., Nonaka, I. et al. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 238, 492–497 (1997).
Kameya, S., Araki, E., Katsuki, M., Mizota, A., Adachi, E., Nakahara, K. et al. Dp260 disrupted mice revealed prolonged implicit time of the b-wave in ERG and loss of accumulation of beta-dystroglycan in the outer plexiform layer of the retina. Hum. Mol. Genet. 6, 2195–2203 (1997).
Aoki, Y., Nakamura, A., Yokota, T., Saito, T., Okazawa, H., Nagata, T. et al. In-frame dystrophin following exon 51-skipping improves muscle pathology and function in the exon 52-deficient mdx mouse. Mol. Ther. 18, 1995–2005 (2010).
Servais, L., Montus, M., Guiner, C. L., Ben Yaou, R., Annoussamy, M., Moraux, A. et al. Non-ambulant Duchenne patients theoretically treatable by exon 53 skipping have severe phenotype. J. Neuromuscul. Dis. 2, 269–279 (2015).
van Deutekom, J. C., Janson, A. A., Ginjaar, I. B., Frankhuizen, W. S., Aartsma-Rus, A., Bremmer-Bout, M. et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357, 2677–2686 (2007).
Voit, T., Topaloglu, H., Straub, V., Muntoni, F., Deconinck, N., Campion, G. et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 13, 987–996 (2014).
Cirak, S., Arechavala-Gomeza, V., Guglieri, M., Feng, L., Torelli, S., Anthony, K. et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605 (2011).
Goemans, N. M., Tulinius, M., van den Hauwe, M., Kroksmark, A. K., Buyse, G., Wilson, R. J. et al. Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open-label extension study. PLoS ONE 11, e0161955 (2016).
Syed, Y. Y. Eteplirsen: first global approval. Drugs 76, 1699–1704 (2016).
Carver, M. P., Charleston, J. S., Shanks, C., Zhang, J., Mense, M., Sharma, A. K. et al. Toxicological characterization of exon skipping phosphorodiamidate morpholino oligomers (PMOs) in non-human primates. J. Neuromuscul. Dis. 3, 381–393 (2016).
Betts, C. A., Hammond, S. M., Yin, H. F. & Wood, M. J. Optimizing tissue-specific antisense oligonucleotide-peptide conjugates. Methods Mol. Biol. 867, 415–435 (2012).
Aoki, Y., Nagata, T., Yokota, T., Nakamura, A., Wood, M. J., Partridge, T. et al. High efficient in vivo delivery of PMO in to regenerating myotubes and rescue in laminin-α2 chain null congenital muscular dystrophy mice. Hum. Mol. Genet. 22, 4914–4928 (2013).
Ezzat, K., Aoki, Y., Koo, T., McClorey, G., Benner, L., Coenen-Stass, A. et al. Self-assembly into nanoparticles is essential for receptor mediated uptake of therapeutic antisense oligonucleotides. Nano Lett. 15, 4364–4373 (2015).
Nakamura, A., Yoshida, K., Fukushima, K., Ueda, H., Urasawa, N., Koyama, J. et al. Follow-up of three patients with a large in-frame deletion of exons 45-55 in the Duchenne muscular dystrophy (DMD) gene. J. Clin. Neurosci. 15, 757–763 (2008).
Taglia, A., Petillo, R., D’Ambrosio, P., Picillo, E., Torella, A., Orsini, C. et al. Clinical features pf patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol. 34, 9–13 (2015).
Nakamura, A. & Takeda, S. Exon skipping therapy for Duchenne muscular dystrophy. Lancet 378, 546–547 (2011).
Aoki, Y., Yokota, T., Nagata, T., Nakamura, A., Tanihata, J., Saito, T. et al. Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery. Proc. Natl Acad. Sci. USA 109, 13763–13768 (2012).
Nakamura, A., Shiba, N., Miyazaki, D., Nishizawa, H., Inaba, Y., Fueki, N. et al. Comparison of the phenotypes of patients harboring in-frame deletions starting at exon 45 in the Duchenne muscular dystrophy gene indicates potential for the development of exon skipping therapy. J. Hum. Genet. 62, 459–463 (2016).
Nicolas, A., Raguénès-Nicol, C., Ben Yaou, R., Ameziane-Le Hir, S., Chéron, A., Vié, V. et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 24, 1267–1279 (2015).
Nakamura, A., Fueki, N., Shiba, N., Motoki, H., Miyazaki, D., Nishizawa, H. et al. Deletion of exons 3-9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 61, 663–667 (2016).
Pane, M., Mazzone, E. S., Sivo, S., Sormani, M. P., Messina, S., D’Amico, A. et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS ONE 9, e108205 (2014).
De Sanctis, R., Pane, M., Sivo, S., Ricotti, V., Baranello, G., Frosini, S. et al. Suitability of North Star Ambulatory Assessment in young boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 25, 14–18 (2015).
Kimura, S., Ozasa, S., Nomura, K., Yoshioka, K. & Endo, F. Estimation of muscle strength from actigraph data in Duchenne muscular dystrophy. Pediatr. Int. 56, 748–752 (2014).
Nishizawa, H., Shiba, N. & Nakamura, A. Usefulness of continuous actigraph monitoring in the assessment of the effect of corticosteroid treatment for Duchenne muscular dystrophy: a case report. J. Phys. Ther. Sci. 28, 3249–3251 (2016).
Acknowledgements
This study was supported by an Intramural Research Grant (26-6) for Neurological and Psychiatric Disorders of the National Center of Neurology and Psychiatry (to AN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Nakamura, A. Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J Hum Genet 62, 871–876 (2017). https://doi.org/10.1038/jhg.2017.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.57
This article is cited by
-
Long-Term Protective Effect of Human Dystrophin Expressing Chimeric (DEC) Cell Therapy on Amelioration of Function of Cardiac, Respiratory and Skeletal Muscles in Duchenne Muscular Dystrophy
Stem Cell Reviews and Reports (2022)
-
Severe cardiac involvement with preserved truncated dystrophin expression in Becker muscular dystrophy by +1G>A DMD splice-site mutation: a case report
Journal of Human Genetics (2020)
-
Genotype–phenotype correlation in Becker muscular dystrophy in Chinese patients
Journal of Human Genetics (2018)
-
Gene Therapy for Heart Failure: New Perspectives
Current Heart Failure Reports (2018)
-
Pharmacological inhibition of REV-ERB stimulates differentiation, inhibits turnover and reduces fibrosis in dystrophic muscle
Scientific Reports (2017)
