
Abstract
Despite extensive screening, 1–5% of cystic fibrosis (CF) patients lack a definite molecular diagnosis. Next-generation sequencing (NGS) is making affordable genetic testing based on the identification of variants in extended genomic regions. In this frame, we analyzed 23 CF patients and one carrier by whole-gene CFTR resequencing: 4 were previously characterized and served as controls; 17 were cases lacking a complete diagnosis after a full conventional CFTR screening; 3 were consecutive subjects referring to our centers, not previously submitted to any screening. We also included in the custom NGS design the coding portions of the SCNN1A, SCNN1B and SCNN1G genes, encoding the subunits of the sodium channel ENaC, which were found to be mutated in CF-like patients. Besides 2 novel SCNN1B missense mutations, we identified 22 previously-known CFTR mutations, including 2 large deletions (whose breakpoints were precisely mapped), and novel deep-intronic variants, whose role on splicing was excluded by ex-vivo analyses. Finally, for 2 patients, compound heterozygotes for a CFTR mutation and the intron-9c.1210-34TG[11–12]T5 allele—known to be associated with decreased CFTR mRNA levels—the molecular diagnosis was implemented by measuring the residual level of wild-type transcript by digital reverse transcription polymerase chain reaction performed on RNA extracted from nasal brushing.
Similar content being viewed by others

Introduction
Cystic fibrosis (CF; OMIM#219700) is one of the most common autosomal recessive disorders with a median incidence in Europe of 1 in 3500.1 The classical symptoms include chronic obstructive pulmonary disease, exocrine pancreatic insufficiency, and elevation of sodium and chloride concentrations in the sweat.2 Moreover, most CF males present with infertility as a result of congenital bilateral absence of the vas deferens.3 CF is caused by mutations in the CF transmembrane conductance regulator (CFTR; OMIM*602421) gene.4, 5 It encodes a protein that functions as a Cyclic adenosine monophosphate-activated chloride channel at the apical membrane of epithelial cells.6 To date, over 1600 mutations have been described in the CFTR gene (http://www.genet.sickkids.on.ca/cftr, accessed 22 January, 2016, excluding sequence variations that are possibly polymorphisms and variants with an unknown functional meaning). About 14% of them are classified as splicing defects, but for ~40% of these, the specific effect on pre-mRNA splicing is unknown.7 Despite extensive genetic screening, 1–5% of CF patients lack a definite molecular diagnosis, even after the sequencing of all exons and splice junctions.8
The genotype–phenotype relation in CF is very complex: some phenotypic features are mainly shaped by the CFTR genotype, whereas others are strongly influenced by modifying genetic and environmental factors. A prototypic example is the phenotypic variability associated with the (TG)m(T)n polymorphic locus at the 3′ end of CFTR intron 9 (formerly known as intron 8). This polymorphism determines the efficiency of exon-10 splicing, with alleles bearing less T repeats and more TG repeats associated with a lower rate of exon-10 inclusion.9, 10, 11 The exon-10 skipped CFTR transcript encodes a misfolded and nonfunctional protein,12 whose expression, in combination with a CF-causing mutation in trans, may be associated with monosymptomatic forms of CF, like congenital bilateral absence of the vas deferens or chronic airway diseases.13 The T5 allele is also known to modify the expressivity of the p.Arg117His mutation when in cis.14
To further complicate the CF genotype-phenotype picture, it has been suggested that mutations in genes coding for the multi-subunit amiloride-sensitive sodium channel ENaC (SCNN1A, SCNN1B and SCNN1G) could cause a CF-like phenotype.15 These genes are expressed primarily on the apical membrane of the epithelial cells of kidney, lung and colon. Importantly, in the airways, the ENaC channel activity is inhibited by CFTR. Accordingly, in CF, the ENaC activity is elevated, thus resulting in a marked increase in sodium intake into epithelial cells.16 The hypothesis is that an altered ENaC function, caused by mutations in one of these 3 genes, can ultimately mimic and/or contribute to the CF phenotype.15
In recent years, the introduction of massively parallel next-generation sequencing (NGS) has allowed the simultaneous analysis of hundred thousand DNA fragments and thus dramatically changed our approach to the genetic analysis of inherited diseases.17 The major advantage offered by NGS is the ability to produce an enormous volume of data in a time- and cost-efficient way. This, combined with the possibility to multiplex both the capture of target genomic regions and the sequencing, allows the simultaneous analysis of single genes or panel of selected genes in a large number of patients at unprecedentedly possible speed and costs. At present, commercial kits for amplicon resequencing of CFTR exons and NGS are available from different companies or have been in-house developed by research groups.18 Target capture and sequencing protocols have also been developed to screen the whole CFTR 190-kb-long genomic region. However, to our knowledge, whole-gene sequencing was applied only as a proof-of-principle approach, to search for mutations in an already characterized cohort of CF patients.19
Here, we developed a whole-gene NGS protocol from one side to assess the efficacy/sensitivity of targeted resequencing and from the other to test the method for molecular screening of CF in a panel of Italian patients with either incomplete or no genetic diagnosis. In 2 cases, absolute mRNA molecule count by digital reverse-transcription (RT) PCR assays was used to precisely quantify the residual level of the CFTR wild-type transcript, hence completing the molecular diagnosis.
Materials and methods
This study was approved by local Ethical Committees and was performed according to the Declaration of Helsinki and to the Italian legislation on sensible data recording. Signed informed consent was obtained from all participants and from parents of subjects younger than 18 years.
NGS experiments and data analysis
NGS libraries were prepared starting from 1 μg of genomic DNA from each patient, using a Nimblegen SeqCap EZ Library custom design capture kit (Roche/Nimblegen, Madison, WI, USA). Equal amounts of 6 samples were pooled prior to capture. Libraries were sequenced as 75-bp paired-end reads on an HiSeq2000 (Illumina, San Diego, CA, USA), according to the manufacturer’s protocols. Details on DNA extraction, target capture, library preparation, quality check and sequencing are reported in Supplementary Materials.
Pre-capture pooling, target-enrichment, as well as NGS were performed through an external service provider (Yale Genome Center; Yale University, USA).
Data were analyzed between November 2012 and March 2013 using an in-house developed pipeline (Supplementary Materials). Considering that indel and structural variant (SV) calling still represent a tricky step, all data were reanalyzed with the GeneSpring NGS software (Agilent Technologies, Palo Alto, CA, USA), implementing algorithms specifically developed for the identification of indels and SV (that is, 'SNP detection' and 'SV detection' options).
Ex-vivo splicing assays of selected deep-intronic variants
To generate hybrid minigene constructs, diverse CFTR regions were PCR amplified from the genomic DNA of patients and cloned into the α-globin-fibronectin hybrid plasmid (modified pBS-KS),20 as previously described.21 Splicing assays were performed by transiently transfecting 4 μg of each of the 8 recombinant vectors into HeLa cells (cultured according to standard procedures). In each experiment, an equal number of cells (2.5 × 105) were transfected with the FuGENE 6 reagent (Roche) in 6-well plates, as described by the manufacturer. Total RNA was isolated from cells 24 h after transfection, using the Eurozol kit (Euroclone, Wetherby, UK). Reverse transcription polymerase chain reactions (RT-PCRs) were carried out with primers mapping in the flanking fibronectin exonic regions of the plasmid. All primer sequences are available on request.
Absolute quantification of CFTR transcripts
RNA from nasal brushing was obtained from 2 CF patients and 9 healthy controls, and reverse transcribed as described in Supplementary Materials.
The level of wild-type CFTR transcripts was evaluated by digital RT-PCR. Reaction mixtures were prepared by combining 1 μl of cDNA with the QuantStudio 3D Digital PCR Master Mix and a custom TaqMan assay (Sigma-Aldrich, Milan, Italy), designed to amplify CFTR transcripts spanning exons 10-11. For the normalization step, a second TaqMan assay was designed to co-amplify, in the same tube, the hydroxymethylbilane synthase (HMBS) mRNA. The sequences of primers and probes used in digital RT-PCR assays are listed in Supplementary Table 1.
Digital RT-PCRs were performed on a QuantStudio 3D Digital PCR System (Thermo Fisher Scientific, Wilmington, DE, USA). Each reaction mixture was loaded onto a QuantStudio 3D Digital PCR Chip, which contains 20 000 wells, and cycled under standard conditions for 40 cycles. End-point fluorescence data were collected and analyzed using the QuantStudio 3D Digital PCR Instrument and the QuantStudio 3D AnalysisSuite according to the manufacturer’s instructions. The precision of all copy counts showed with a standard error of lower than 5%.
Results
Patients
Among individuals referred to the Cystic Fibrosis Centers of Milan and Verona in the last 5 years, 23 CF patients and one CF carrier (named CF-1 to CF-24, all of Caucasian origin) were selected for being submitted to NGS of the entire CFTR gene as well as of the coding regions and exon/intron boundaries of the SCNN1A, SCNN1B and SCNN1G genes (Supplementary Table 2).
Among selected patients, 17 had an incomplete genetic diagnosis, even after screening by conventional copy-number analysis and Sanger sequencing. In particular, for 5 of them, no mutations were identified, whereas 12 were heterozygous for only one genetic defect (2 were carrier of the F508del mutation). Finally, 3 patients (CF-22, 23 and 24) were not subjected to any previous molecular screening. In all cases, the inclusion criterion for entering the study was the availability of the DNA from both parents.
As controls, we selected 3 patients (CF-15, 17 and 18) showing a complete genetic diagnosis (all compound heterozygous for substitutions/small deletions), and one heterozygous carrier of a large deletion (CF-12) (Supplementary Table 3).
NGS sequencing statistics
For NGS analysis, we chose a custom-designed target-enrichment strategy based on the Nimblegen SeqCap EZ Library capture kit. Probes were designed on the basis of the genomic coordinates of the entire 189-kb-long CFTR gene (human genome release GRCh37/hg19, chr7:117,120,017-117,308,718), and of all exons and splicing junctions of the ENaC genes (SCNN1A, mapping on chr12p13.31, and SCNN1B and SCNN1G both located on chr16p12.2). Altogether, the target regions accounted for a total of 207 508 bp.
A pilot group of DNAs was analyzed by performing the enrichment step multiplexing six samples, and the sequencing step in a single HiSeq2000 lane. The overall coverage of the target region was >98%, with only about 3 kb of CFTR intronic repetitive sequences left uncovered, and a mean coverage of ~3500 × (in all cases the coverage was >2600 ×) (Supplementary Table 4). As the coverage was extremely high, we scaled up the number of pooled samples in the subsequent experiments: multiplexing 6 samples in the capture phase and 18 in the sequencing step, we obtained a mean depth >1150 × (in all cases the coverage was >800 ×), again with a 98% coverage of the target region (Supplementary Table 4). Unfortunately, one patient (CF-20) failed the sequencing step.
On average, each subject resulted carrier of 162 genetic variants, 24 of which having a minor allele frequency <1% in the general population.
Identification of genetic variants in CFTR
Variants were annotated by filtering them against an in-house developed database and classified in one of the following categories: (1) already known pathogenic mutations (retrieved from: the Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca/app, the Human Gene Mutation Database, HGMD, http://www.hgmd.cf.ac.uk/ac/index.php, and from the CF Centre of Milan database); (2) potentially pathogenic variants included in dbSNP135; (3) common (minor allele frequency, MAF>1%), likely not pathogenic, variants annotated in dbSNP135; (4) rare single nucleotide variations (SNVs) of unknown functional relevance; (5) novel variants. Variants annotated in group 1 were considered of immediate diagnostic relevance, whereas variations included in groups 2, 4 and 5 were selected for Sanger sequencing validation. Probably due to the obtained high coverage, all variants were confirmed (100% concordance rate).
Our pipeline allowed the identification of all pathogenic variants already detected by routine screening (18 alleles for a total of 13 different mutations), including the 21-kb deletion in the CF-12 control individual. In addition, we identified: (i) 3 point mutations that were already reported in the literature to be clinically associated with CF but were not identified by Sanger sequencing (CF-4, 10 and 11); (ii) one large deletion (CF-16, see further); (iii) 5 known mutations in the newly-screened CF patients (CF-22, 23 and 24); and (iv) 22 novel deep-intronic variants of unknown significance (Table 1; Supplementary Table 5). Finally, the poly-thymidine-guanine and poly-thymidine c.1210–34TG[11–13]T[5–9] tracts were also inspected. To this aim, the variant caller GeneSpring SNP detection was used, and it correctly assigned different alleles to all individuals (as subsequently verified by Sanger sequencing). In particular, we found 2 patients carrying the T5 pathogenic variant, thus possibly completing the diagnosis for both of them (see further; Table 1).
Overall, for 6 of the 11 patients with incomplete genetic diagnosis successfully analyzed, NGS allowed the detection of the second causative allele, corresponding to a diagnostic yield of 54.6%. Conversely, no clearly pathogenic mutation was found in the five patients negative for CFTR mutations after previous screenings.
Characterization of CFTR large deletions
Using the specific algorithm 'SV detection' implemented in the GeneSpring software, we were able to identify 2 large deletions, both present in the heterozygous state (Table 1).
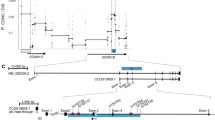
As expected, the first one was identified in individual CF-12, from one side confirming the already-available genetic diagnosis, and from the other underlining the ability of the NGS approach to detect such genetic lesions. The mutation consisted of a 21-kb-long deletion encompassing introns 1–3 of the CFTR gene (Figure 1a). The second structural variant was identified in patient CF-16: it is a 9.5-kb-long deletion involving the 3′ end of the CFTR gene (removing exons 25–27) (Figure 1b).

Identification of the 21-kb-long and the 9.5-kb-long large deletions. (a, b) The main features characterizing, respectively, the 21-kb-long and the 9.5-kb-long deletions, both identified in the heterozygous state by NGS. For each deletion, a window of the IGV software showing the alignment of the reads in the region of the mutation is shown, together with a close-up view at the 5′ deletion breakpoint (highlighting the drop in the number of reads to about half). Below the IGV window, the relevant CFTR genomic region is displayed, with exons represented by boxes and introns by lines (drawn to scale). In this scheme, the position of primers used to PCR amplify the region across deletion breakpoints, as well as the breakpoint nucleotide positions along chromosome 7, are shown. In the lower part of the panel, the sequence electropherogram of the region corresponding to the deletion junction is shown. NGS, next-generation sequencing; PCR, polymerase chain reaction. A full color version of this figure is available at the Journal of Human Genetics journal online.
Inspection of the NGS read alignment also allowed an easy definition of the deletion breakpoints for both lesions. Breakpoints were confirmed by designing couples of primers mapping immediately upstream and downstream of the breakpoints predicted by the 'SV detection' software, and PCR amplifying the mutant allele from the patient’s DNA. Concerning the 21-kb-long deletion (21 085 bp), Sanger sequence analysis of the PCR product evidenced that, while the wild-type sequence is characterized by the presence of a 4-bp-long CTTT sequence at both cleavage sites, in the deleted allele only one copy is retained. Hence, the precise location of the 5′ and 3′ breakpoints cannot be unambiguously determined, and may range between positions chr7:117,138,366-117,138,370 and chr7:117,159,443-117,159,447, respectively (Figure 1a). Concerning the 9.5-kb-long deletion (9454 bp), sequencing of the deleted allele evidenced the presence of five additional nucleotides (TAACT) between the breakpoints, as already described (Figure 1b).22
Ex-vivo characterization of deep-intronic variants
Among the 22 novel deep-intronic nucleotide substitutions, 4 variants (named V1–V4) were selected for further characterization because bioinformatically predicted to potentially interfere with normal splicing (Supplementary Table 6).
The molecular characterization of the putative V1–V4 mutations was tackled by using an ex-vivo approach, as RNA to be extracted from nasal epithelial cells of patients (or their relatives) was not available to the study. In particular, we generated recombinant minigene constructs (4 wild-type, 4 mutant; Supplementary Figure 1A) that were independently transfected in HeLa cells. Subsequent RT-PCR assays did not evidence any splicing alteration for transcripts originating from minigenes carrying the V1, V2 or V3 variants (Supplementary Figure 1B). Concerning the V4 variant, RT-PCR fragments amplified from cells transfected with the wild-type or the mutant construct were again identical, but in this case an additional band was visible for both samples. Sequencing analysis revealed that the additional band originates from the insertion of a pseudoexon of 91 nucleotides, corresponding to a fragment of CFTR intron 23 lying between positions IVS23+152 and IVS23+243. However, this unexpected pseudoexon activation is independent from the presence of the V4 variant, which was no further investigated.
Quantification of residual wild-type CFTR transcript by digital RT-PCR
Since the intron-9c.1210-34TG11-12T5 alleles are known to be associated with high rates of exon-10 skipping (and hence with low wild-type CFTR expression levels), we explored the possibility that the phenotype in patients 2 and 5 could indeed be explained by the presence of the T5 allele in combination with the corresponding CF-causing mutation in trans (that is, the splicing mutations c.1585-1G>A and c.2657+5G>A, determining the out-of-frame skipping of exons 12 and 16, respectively).23, 24, 25 To this aim, we set up digital RT-PCR assays for the absolute quantification of the wild-type CFTR mRNA, designing PCR primers to specifically amplify exon-10-containing transcripts (Figure 2a). Digital PCR26, 27 is an accurate technique allowing the absolute quantification of RNA transcripts by partitioning the PCR reaction mix over a large number of wells, so that each well contains a single copy or no copies of the target region. On the basis of the assumption of Poisson distribution of copies, the number of template copies originally present in the sample can be recalculated simply counting the number of wells in which amplification has successfully occurred.28

Evaluation of residual wild-type CFTR transcript using digital RT-PCR. Absolute expression levels were measured by digital RT-PCRs in patients CF-2 and CF-5 (that is, subjects carrying a null mutation and the T5 allele in compound heterozygosity), as well as nine healthy controls (none being carrier of the T5 allele). (a) Schematic representation of the CFTR gene between exons 10 and 12. Exons and introns are represented by boxes and lines, and are not drawn to scale. The positions of the primer couple used for the digital RT-PCR assay are shown by arrows, whereas the TaqMan probe is indicated by a horizontal line. (b) Histogram representing the results of digital RT-PCR. The number of copies of the target region is shown, normalized on the number of copies of the housekeeping HMBS mRNA (co-amplified with a second TaqMan assay). Bars represent the mean+the precision of the assay as calculated by the software.
Digital RT-PCRs were performed on RNA extracted from nasal brushing of the 2 patients and of 9 healthy controls (none carrying the T5 allele). Digital counts of the wild-type CFTR transcript resulted 58 and 76 molecules/μl for patient CF-2 and 5, respectively. These values correspond to 21 and 28% of the mean wild-type CFTR level measured in controls, in whom the number of molecules/μl ranged from 128 to 456 (Figure 2b).
Genetic variants in SCNN1B
Sequencing of SCNN1A, SCNN1B and 2SCNN1G genes led to the identification of 2 missense variants in the heterozygous state, both located in the SCNN1B gene. In particular, in patient CF-3, the c.1313A>G transition (p.Glu438Gly) was identified, whereas in patient CF-11 the c.818A>G substitution (p.Tyr273Cys) was disclosed. Both missense variants lead to an amino-acid change affecting a conserved residue (Supplementary Figure 2), and are found in European non-Finnish individuals from the Exome Aggregation Consortium (ExAC) database, both with a global frequency below 1:10 000 (http://exac.broadinstitute.org, last accessed 22 January 2016). The p.Tyr273Cys variant was classified as potentially damaging by seven of nine common programs used to predict the functional impact of identified variants (Supplementary Materials), whereas the p.Glu438Gly variant was predicted to be deleterious by 4 of 9 programs (Supplementary Table 7).
To understand the possible consequences of the newly-identified variants on the SCNN1B protein structure, the molecular model of the human protein was inspected (Supplementary Figure 2). The SCNN1B protein is characterized by the presence of 2 transmembrane domains and a wide extracellular loop (including ~70% of the whole amino-acid sequence). This loop, together with the extracellular domains of the other 2 subunits, forms a funnel that directs ions from the lumen into the pore of ENaC.29 The p.Glu438Gly mutation affects the extracellular domain of the protein, in a region well exposed to the solvent: the amino-acid substitution is predicted to change a negatively-charged residue with a small apolar one, thus possibly modifying the electrostatic signature of this region. Also the p.Tyr273Cys mutation involves the SCNN1B extracellular domain: the substitution falls within a beta strand, close to a highly-conserved Cys residue (Cys272), which is known to participate to the maintenance of the overall structure of the channel. Hence, the addition of a second Cys in this context could perturb the bond network normally characterizing the region (Supplementary Figure 2).
Discussion
In the era of the P4 medicine ('personalized', 'predictive', 'preventive' and 'participatory'), the possibility to have a complete genetic diagnosis is the fundamental prerequisite for directing therapeutic strategies. CF conforms to this paradigm, especially in the light of the many efforts made in the last few years to develop new therapies targeting specific CFTR mutations.30 At present, commercial kits for amplicon resequencing of CFTR exons using multiplex amplification, pooling of barcoded samples and NGS are available from different companies. These solutions promise to completely replace the traditional multi-step genetic testing of CF, reducing the costs of analysis and increasing the success rate of the diagnosis. Here, we explored the application of whole-gene CFTR sequencing to CF genetic testing by screening a number of 'difficult' cases, that is, patients with an incomplete diagnosis even after a preliminary extensive molecular screening. Our approach was successful both in identifying all mutations (including a large deletion) already detected by routine screening, and in ameliorating the percentage of genetically-diagnosed patients (Table 1).
Indeed, our data together with previous literature19 suggest that whole-gene resequencing can in principle be used as a 'one-step strategy' to the molecular diagnosis of CF, for a number of reasons. First, this strategy has the advantage of not suffering from allele-dropout complications, because the library preparation procedure is based on probes for the capture step rather than on PCR amplification.31, 32 This advantage is worth not only in comparison with conventional Sanger sequencing, but also with amplicons-based NGS methods. Second, our strategy should allow the identification of large intragenic deletions, duplications, and rearrangements, which may go undetected by exon-only resequencing strategies. The inspection of the NGS read alignment should also allow an immediate definition of the deletion breakpoints at the nucleotide level, a process often laborious and time consuming. Third, thanks to the possibility to multiplex samples, both at the capture and at the sequencing level, we observed an increased time efficacy of whole-gene sequencing over Sanger sequencing. In addition, the foreseeable cost reductions coupled with the possibility to further multiplex samples (we reached a mean depth of >1150) promise to make this choice even more affordable over traditional approaches. Last, even in the unfortunate event of not being able to complete a genetic diagnosis, the availability of the sequence of all CFTR intronic regions still represents a great source of information. For instance, we identified a total of 22 novel deep-intronic variants, 18 of which at the moment remain without a functional meaning (Supplementary Table 5; Supplementary Figure 1). It is conceivable that in the future the accumulation of data on intronic sequences and the introduction of high-throughput functional analyses will assign a pathogenic meaning to some of these genetic variants, further increasing the diagnostic yield.
Even after this extensive sequencing strategy, five probands did not show any mutation in CFTR, whereas six were still lacking the second mutation. Missing mutations could either reside in the few intronic regions not covered by our NGS experiment, or be one of the already identified deep-intronic variants that we did not functionally characterize. Alternatively, considering that indels and structural variants still represent a problem in the alignment procedures, it is possible that they were not properly called by the adopted software. Finally, we cannot rule out the possibility that some patients, despite their CF or CF-like appearance, are indeed phenocopies with no mutations in their CFTR gene.33 Accordingly, no mutated allele was identified in the five analyzed patients negative after conventional CFTR genetic screening. Further studies by whole-exome sequencing might help a better phenotype classification and might potentially reveal novel gene/s related to the CF phenotype.
Interestingly, it has been reported that patients with a CF-like disorder, but having mutations in only one copy of their CFTR genes, were carrier of gain-of-function mutations in the ENaC-encoding subunits.15, 34, 35 More recently, Ramos et al.,36 also suggested an oligogenic nature for CF-like phenotypes, with the description of patients having no mutations in CFTR but multiple variants in SCNN1A, SCNN1B SCNN1G, and even in the SERPINA1 gene. The inclusion of the ENaC subunit genes in our NGS design was aimed at taking into account also this variable. However, we only found 2 missense variants in the SCNN1B gene, in one case in a patient with a complete molecular diagnosis of CF (CF-11), in the other in a patient with no pathogenic mutations in CFTR (CF-3). In both cases, the functional impact of the relevant variant still remains to be clarified.
RNA analysis, even though not routinely undertaken in diagnostic laboratories, has proven to be an important step in both the genetic diagnosis of CF and in the definition of the pathogenic potential of point mutations in CFTR. For instance, RT-PCR studies on RNA extracted from nasal epithelial cells or lymphocytes have been instrumental to clarify the role of deep-intronic variants affecting splicing.7, 37, 38, 39 We used this approach to complete the genetic diagnosis of a couple of patients resulted compound heterozygotes for a 'clear' disease-causing mutation and the c.1210-34TG[11-12]T5 alleles (CF-2 and CF-5; Table 1). The use of digital RT-PCR, which is an extremely accurate and sensitive method for the absolute quantification of transcripts, allowed us to verify that both patients have an overall amount of CFTR mRNA below 30%. This value should indeed be considered as an overestimate in the case of the CF-5 patient. In fact, a proper evaluation of the level of wild-type CFTR transcripts should take into consideration all possible splicing events as well as the stability of transcripts bearing premature stop codons. In this frame, patient CF-5, who carries a splicing mutations affecting exon 16, is expected to have a lower level of the wild-type CFTR mRNA than that measured by digital RT-PCR, because the assay was designed for the specific amplification of the region encompassing exons 10–12. Hence, our results are in line with previous studies, describing a wide range of full-length CFTR transcripts (4–20%) associated with mild CF phenotypes,39, 40, 41 and hence reinforce the notion that the T5 allele can contribute to the CF phenotype in some patients.
In summary, we propose a new diagnostic protocol to be applied to the genetic diagnosis of CF: a one-shot PCR-based test followed by full-gene NGS screening (Supplementary Figure 3). The preliminary PCR-based screening is aimed at the identification of the most common CF mutation, that is, F508del, which can be easily detected by using either a simple and economic high-resolution melting analysis or other relatively low-cost genotyping methods (fluorogenic 5′ nuclease PCR assays or fluorescent fragment analysis on a capillary electrophoresis sequencer). Should this preliminary analysis be negative, the subsequent NGS step will allow the most comprehensive search for point mutations and gross alterations. In the light of the possible improvements in the programs used for mutation detection and in the hope of further cost drops, this strategy promises to be the most straightforward, convenient and rapid diagnostic protocol for CF patients.
Accession codes
References
Southern, K. W., Munck, A., Pollitt, R., Travert, G., Zanolla, L., Dankert-Roelse, J. et al. A survey of newborn screening for cystic fibrosis in Europe. J. Cyst. Fibros. 6, 57–65 (2007).
Knowles, M. R. & Durie, P. R. What is cystic fibrosis? N. Engl. J. Med. 347, 439–442 (2002).
Chillón, M., Casals, T., Mercier, B., Bassas, L., Lissens, W., Silber, S. et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N. Engl. J. Med. 332, 1475–1480 (1995).
Riordan, J. R., Rommens, J. M., Kerem, B., Alon, N., Rozmahel, R., Grzelczak, Z. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 (1989).
Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A. et al. Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080 (1989).
Collins, F. S. Cystic fibrosis: molecular biology and therapeutic implications. Science 256, 774–779 (1992).
Faà, V., Incani, F., Meloni, A., Corda, D., Masala, M., Baffico, A. M. et al. Characterization of a disease-associated mutation affecting a putative splicing regulatory element in intron 6b of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. J. Biol. Chem. 284, 30024–30031 (2009).
Sosnay, P. R., Siklosi, K. R., Van Goor, F., Kaniecki, K., Yu, H., Sharma, N. et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat. Genet. 45, 1160–1167 (2013).
Cuppens, H., Lin, W., Jaspers, M., Costes, B., Teng, H., Vankeerberghen, A. et al. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J. Clin. Invest. 101, 487–496 (1998).
Pagani, F., Buratti, E., Stuani, C., Romano, M., Zuccato, E., Niksic, M. et al. Splicing factors induce cystic fibrosis transmembrane regulator exon 9 skipping through a nonevolutionary conserved intronic element. J. Biol. Chem. 275, 21041–21047 (2000).
Buratti, E., Brindisi, A., Pagani, F. & Baralle, F. E. Nuclear factor TDP-43 binds to the polymorphic TG repeats in CFTR intron 8 and causes skipping of exon 9: a functional link with disease penetrance. Am. J. Hum. Genet. 74, 1322–1325 (2004).
Strong, T. V., Wilkinson, D. J., Mansoura, M. K., Devor, D. C., Henze, K., Yang, Y. et al. Expression of an abundant alternatively spliced form of the cystic fibrosis transmembrane conductance regulator (CFTR) gene is not associated with a cAMP-activated chloride conductance. Hum. Mol. Genet. 2, 225–230 (1993).
Schwarz, M., Gardner, A., Jenkins, L., Norbury, G., Renwick, P. & Robinson, D. Testing Guidelines for Molecular Diagnosis of Cystic Fibrosis, (Clinical Molecular Genetics Society, UK, 2009).
Ferec, C. & Cutting, G. R. Assessing the disease-liability of mutations in CFTR. Cold Spring Harb. Perspect. Med. 2, a009480 (2012).
Sheridan, M. B., Fong, P., Groman, J. D., Conrad, C., Flume, P., Diaz, R. et al. Mutations in the beta-subunit of the epithelial Na+ channel in patients with a cystic fibrosis-like syndrome. Hum. Mol. Genet 14, 3493–3498 (2005).
Wine, J. J. The genesis of cystic fibrosis lung disease. J. Clin. Invest. 103, 309–312 (1999).
Ng, S. B., Buckingham, K. J., Lee, C., Bigham, A. W., Tabor, H. K., Dent, K. M. et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 42, 30–35 (2010).
Trujillano, D., Weiss, M. E. R., Köster, J., Papachristos, E. B., Werber, M., Kandaswamy, K. K. et al. Validation of a semiconductor next-generation sequencing assay for the clinical genetic screening of CFTR. Mol. Genet. Genomic. Med. 3, 396–403 (2015).
Trujillano, D., Ramos, M. D., González, J., Tornador, C., Sotillo, F., Escaramis, G. et al. Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J. Med. Genet. 50, 455–462 (2013).
Baralle, M., Baralle, D., De Conti, L., Mattocks, C., Whittaker, J., Knezevich, A. et al. Identification of a mutation that perturbs NF1 gene splicing using genomic DNA samples and a minigene assay. J. Med. Genet. 40, 220–222 (2003).
Rimoldi, V., Straniero, L., Asselta, R., Mauri, L., Manfredini, E., Penco, S. et al. Functional characterization of two novel splicing mutations in the OCA2 gene associated with oculocutaneous albinism type II. Gene 537, 79–84 (2014).
Taulan, M., Girardet, A., Guittard, C., Altieri, J. P., Templin, C., Beroud, C. et al. Large genomic rearrangements in the CFTR gene contribute to CBAVD. BMC Med. Genet. 8, 22 (2007).
Kerem, B. S., Zielenski, J., Markiewicz, D., Bozon, D., Gazit, E., Yahav, J. et al. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc. Natl Acad. Sci. USA 87, 8447–8451 (1990).
Highsmith, W. E. Jr, Burch, L. H., Zhou, Z., Olsen, J. C., Strong, T. V., Smith, T. et al. Identification of a splice site mutation (2789 +5 G>A) associated with small amounts of normal CFTR mRNA and mild cystic fibrosis. Hum. Mutat. 9, 332–338 (1997).
Sharma, N., Sosnay, P. R., Ramalho, A. S., Douville, C., Franca, A., Gottschalk, L. B. et al. Experimental assessment of splicing variants using expression minigenes and comparison with in silico predictions. Hum. Mutat. 35, 1249–1259 (2014).
Vogelstein, B. & Kinzler, K. W. Digital PCR. Proc. Natl. Acad. Sci. USA 96, 9236–9241 (1999).
Huggett, J. F. & Whale, A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin. Chem. 59, 1691–1693 (2013).
Nuzzo, F., Paraboschi, E. M., Straniero, L., Pavlova, A., Duga, S. & Castoldi, E. Identification of a novel large deletion in a patient with severe factor V deficiency using an in-house F5 MLPA assay. Haemophilia 21, 140–147 (2015).
Edelheit, O., Hanukoglu, I., Dascal, N. & Hanukoglu, A. Identification of the roles of conserved charged residues in the extracellular domain of an epithelial sodium channel (ENaC) subunit by alanine mutagenesis. Am. J. Physiol. Renal. Physiol. 300, F887–F897 (2011).
Corvol, H., Thompson, K. E., Tabary, O., le Rouzic, P. & Guillot, L. Translating the genetics of cystic fibrosis to personalized medicine. Transl. Res. 168, 40–49 (2016).
Gnirke, A., Melnikov, A., Maguire, J., Rogov, P., LeProust, E. M., Brockman, W. et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 27, 182–189 (2009).
Blais, J., Lavoie, S. B., Giroux, S., Bussières, J., Lindsay, C., Dionne, J. et al. Risk of misdiagnosis due to allele dropout and false-positive PCR artifacts in molecular diagnostics: analysis of 30,769 genotypes. J. Mol. Diagn. 17, 505–514 (2015).
Tsui, L. C. & Dorfman, R. The cystic fibrosis gene: a molecular genetic perspective. Cold Spring Harb. Perspect. Med 3, a009472 (2013).
Azad, A. K., Rauh, R., Vermeulen, F., Jaspers, M., Korbmacher, J., Boissier, B. et al. Mutations in the amiloride-sensitive epithelial sodium channel in patients with cystic fibrosis-like disease. Hum. Mutat. 30, 1093–1103 (2009).
Fajac, I., Viel, M., Gaitch, N., Hubert, D. & Bienvenu, T. Combination of ENaC and CFTR mutations may predispose to cystic fibrosis-like disease. Eur. Respir. J. 34, 772–773 (2009).
Ramos, M. D., Trujillano, D., Olivar, R., Sotillo, F., Ossowski, S., Manzanares, J. et al. Extensive sequence analysis of CFTR, SCNN1A, SCNN1B, SCNN1G and SERPINA1 suggests an oligogenic basis for cystic fibrosis-like phenotypes. Clin. Genet. 86, 91–95 (2014).
Chillón, M., Dörk, T., Casals, T., Giménez, J., Fonknechten, N., Will, K. et al. A novel donor splice site in intron 11 of the CFTR gene, created by mutation 1811+1.6kbA—>G, produces a new exon: high frequency in Spanish cystic fibrosis chromosomes and association with severe phenotype. Am. J. Hum. Genet. 56, 623–629 (1995).
Costantino, L., Claut, L., Paracchini, V., Coviello, D. A., Colombo, C., Porcaro, L. et al. A novel donor splice site characterized by CFTR mRNA analysis induces a new pseudo-exon in CF patients. J. Cyst. Fibros. 9, 411–418 (2010).
Costantino, L., Rusconi, D., Soldà, G., Seia, M., Paracchini, V., Porcaro, L. et al. Fine characterization of the recurrent c.1584+18672 A>G deep-intronic mutation in the cystic fibrosis transmembrane conductance regulator gene. Am. J. Respir. Cell. Mol. Biol. 48, 619–625 (2013).
Chu, C. S., Trapnell, B. C., Curristin, S., Cutting, G. R. & Crystal, R. G. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat. Genet. 3, 151–156 (1993).
Rave-Harel, N., Kerem, E., Nissim-Rafinia, M., Madjar, I., Goshen, R., Augarten, A. et al. The molecular basis of partial penetrance of splicing mutations in cystic fibrosis. Am. J. Hum. Genet. 60, 87–94 (1997).
Acknowledgements
This work was supported by the Italian Cystic Fibrosis Foundation, grant numbers FFC#6/2011 and FFC#5/2015 (to s.d.), and by Italian fiscal contribution '5 × 1000' 2011 devolved to Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico. We wish to thank Carlo Castellani (Cystic Fibrosis Centre, Ospedale Civile Maggiore, Verona, Italy), Fabiola Corti and Amalia Negri (Division of Pediatrics, Livorno Hospital, Italy), and Rita Padoan (Cystic Fibrosis Centre, Spedali Civili, Brescia) for providing additional clinical data on some patients.
Author contributions
LS and GS designed the experiments and analyzed NGS data. LS performed gene expression analysis by digital PCR, functional validation of intronic variants, and mapping of deletion breakpoints. GS, RA and SD were responsible for data interpretation and drafting the manuscript. CC, MS and PM were involved in the design of the work (patient selection), data interpretation and critical revision of the manuscript. All authors read and approved the final version submitted for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Straniero, L., Soldà, G., Costantino, L. et al. Whole-gene CFTR sequencing combined with digital RT-PCR improves genetic diagnosis of cystic fibrosis. J Hum Genet 61, 977–984 (2016). https://doi.org/10.1038/jhg.2016.101
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.101
This article is cited by
-
Deep intronic mutations and human disease
Human Genetics (2017)
